

Abstract
B cell differentiation driven by microbial antigens leads to production of anti-microbial antibodies, such as those neutralizing viruses, bacteria or bacterial toxin, that are class-switched (IgG and IgA) and somatically hypermutated (maturation of the antibody response) as well as secreted in large volume by plasma cells. Similar features characterize pathogenic antibodies to self-antigens in autoimmunity, reflecting the critical role of class switch DNA recombination (CSR), somatic hypermutation (SHM) and plasma cell differentiation in the generation of antibodies to not only foreign antigens but also self-antigens (autoantibodies). Central to CSR/SHM and plasma cell differentiation are AID, a potent DNA cytidine deaminase encoded by Aicda, and Blimp-1, a transcription factor encoded by Prdm1. B cell-intrinsic expression of Aicda and Prdm1 is regulated by epigenetic elements and processes, including DNA methylation, histone posttranslational modifications and non-coding RNAs, particularly miRNAs. Here, we will discuss: B cell-intrinsic epigenetic processes that regulate antibody and autoantibody responses; how epigenetic dysregulation alter CSR/SHM and plasma cell differentiation, thereby leading to autoantibody responses, as in systemic lupus; and, how these can be modulated by nutrients, metabolites and hormones through changes in B cell-intrinsic epigenetic mechanisms, which can provide to be therapeutic targets in autoimmunity.
Introduction: Maturation of the antibody and autoantibody response
The generation of pathogenic autoantibodies in organ-specific and systemic autoimmunity is governed by mechanisms similar to those informing the generation of neutralizing antibodies against viruses, bacteria or microbial toxins [1]. Like these antibodies to foreign antigens, those to self-antigens (autoantibodies) are class-switched and somatically mutated, the products of a “maturation” of the antibody response. This starts once naive B cells encounter their cognate antigen and are activated via BCR, CD40 or TLR and cytokine receptors. Activated B cells proliferate and differentiate, generally in germinal centers (GCs) or outside GCs (extrafollicular sites), to undergo CSR and SHM, thereby giving rise to class-switched and hypermutated B cells, plasma cells and memory B cells [2,3]. Extrafollicular GC-like B cell reactions occur in both T-dependent and T-independent responses, and exhibit high levels of CSR and SHM [4,5]. By changing the IgH constant region (Cμ to Cγ, Cα or Cε), CSR endows an antibody with different biological effector functions, e.g., IgMs are limited to the intravascular torrent, IgGs can extravasate and reach virtually all body districts and IgAs dominate the response at barrier tissues. By inserting mainly point-mutations, SHM provides antibodies with the structural correlates for selection of higher-affinity mutants by antigen [2]. Class-switched and somatically hypermutated antibodies are secreted at high rates by B cell-derived plasmablasts and plasma cells. As the primary mechanisms for the maturation of autoantibody responses, CSR and SHM underpin both organ-specific, e.g., Hashimoto’s thyroiditis or myasthenia gravis, as well as systemic autoimmunity, e.g., systemic lupus erythematosus (SLE) [6], in which autoantibodies are primarily IgG, somatically hypermutated, high affinity and secreted in large amounts by an expanded plasma cell pool [6,7].
By introducing double-stranded breaks and single-stranded nicks in Ig locus DNA, AID, encoded by AICDA/Aicda, critically initiates the first step in the cascade of events that lead to CSR/SHM [8–11]. As a potent mutator, it is largely restricted to targeting the Ig locus – AID off-targeting (outside the Ig locus) can cause genome instability, often promoting neoplastic transformation [12]. AID expression is B cell-restricted and B cell differentiation stage-specific [11]. It is undetectable in resting B cells, is induced at high levels in activated B cells in antibody responses and subsides to undetectable levels in resting memory B cells and plasma cells. AID is also greatly upregulated in activated autoreactive B cells that make class-switched and somatically hypermutated autoantibodies, such as those in systemic lupus [13]. As terminally differentiated elements, plasma cells do not proliferate but secrete large amounts of antibodies or autoantibodies. For their differentiation and maintenance, they are dependent on the master transcription factor (TF) Blimp-1, encoded by PRDM1/Prdm1, as well as Irf4 and Xbp1 TFs [14]. Blimp-1 expression level is low or nil in B cells and increases in plasmablasts to plasma cells. These home to unique niches in bone marrow, where they can survive for months to years. Marrow long-lived plasma cells can maintain a durable layer of antibodies against previously experienced microbial pathogens or self-antigens in the case of autoantibodies [14,15]. Additional long-term persistence of recallable high levels of antibodies or autoantibodies is provided by memory B cells, which are reactivated and expanded upon re-exposure to cognate foreign or self-antigens [5,16].
Epigenetic mechanisms, such as DNA methylation, histone post-translational modifications and non-coding RNAs (ncRNA), regulate gene expression, thereby modulating cell functions without changes in DNA sequence [17]. In differentiating B cells, they enable and modulate genetic programs to regulate expression and activity of AID and Blimp-1, thereby modulating CSR/SHM and plasma cell differentiation to inform the antibody and autoantibody response [18–26]. In autoimmune responses, epigenetic mechanisms modulate T cell differentiation and functions, particularly of regulatory T cells, as well as macrophages and dendritic cells [27–32]. The epigenetics of these cells are beyond the scope of this review, which we will focus on the epigenetics of B cells. Here, we will attempt to provide a framework of how B cell-intrinsic epigenetic factors and processes modulate CSR/SHM and plasma cell differentiation, with a focus on regulation of AID and Blimp-1 by DNA methyltransferases (Dnmts) and Tet (ten-eleven translocation) methylcytosine dioxygenases, histone deacetylases (HDACs), HDAC inhibitors (HDIs) and non-coding (nc)RNAs, particularly miRNAs. We will conclude by discussing recent findings on how these epigenetic elements and processes can be potential targets of new therapeutic approaches to alleviate antibody-dependent autoimmunity.
Epigenetics processes and peripheral B cell differentiation
AID expression and targeting, and, therefore, CSR/SHM are regulated by B cell-intrinsic epigenetic processes, such as DNA methylation and histone post-translational modifications, and mediators, such as miRNAs. Similar B cell-intrinsic epigenetic processes and mediators regulate Blimp-1 expression as well as other TFs that drive plasma cell differentiation, such as Irf4 and Xbp1.
DNA methylation and demethylation critically regulate cell-type specific and context-dependent gene expression [33,34]. Throughout bone marrow B cell development and peripheral differentiation, the B cell genome undergoes progressive demethylation [35–37]. Methylation of cytidine to 5-methylcytosine (5mC) in (mainly) CpG islands of gene promoters and enhancer regions is catalyzed by DNA methyltransferases (Dnmts) and acts to silence gene transcription [23,33]. 5mC is converted to unmethylated state through passive or active demethylation. While passive demethylation occurs through replication dilution, active DNA demethylation, i.e., direct removal of the methyl group, is mediated by Tet (Tet1, Tet2, Tet3) proteins, which oxidize 5mC to 5-hydroxymethylcytosine (5hmC) [33]. 5hmC is enriched at active enhancers and promoters of highly transcribed genes, correlating with chromatin accessibility, yet depleted from the transcription start site (TSS) [33,38]. Tet2 and Tet3 are expressed in B lineage cells [26,37,39], with Tet2 being predominant and correlating with AID and Blimp-1 expression in B cells undergoing CSR/SHM and plasma cell differentiation (our unpublished data) (Figure 1). This together with Dnmt1 requirement for GC formation indicates the importance of maintaining an underlying methylome in such B cells [40,41]. The methylation status of the five regulatory elements located between −29-Kb to +5-Kb of Aicda TSS (collectively referred to as the Aicda super-enhancer) mediates Aicda transcriptional regulation upon B cell activation [26]. The Aicda super-enhancer includes Tet-responsive TetE1 and TetE2 cis-elements, which recruit Tet2 as facilitated by Batf, an AP-1 TF, to support active (5hmC) DNA demethylation, sustain super-enhancer accessibility and promote Aicda induction [26]. Impairment of Tet2 function in GC B cells correlates with decreased AID levels and reduced CSR [42].
Figure 1. B cell differentiation stage-specific expression of Sirt1, Tet2, AID and Blimp-1.
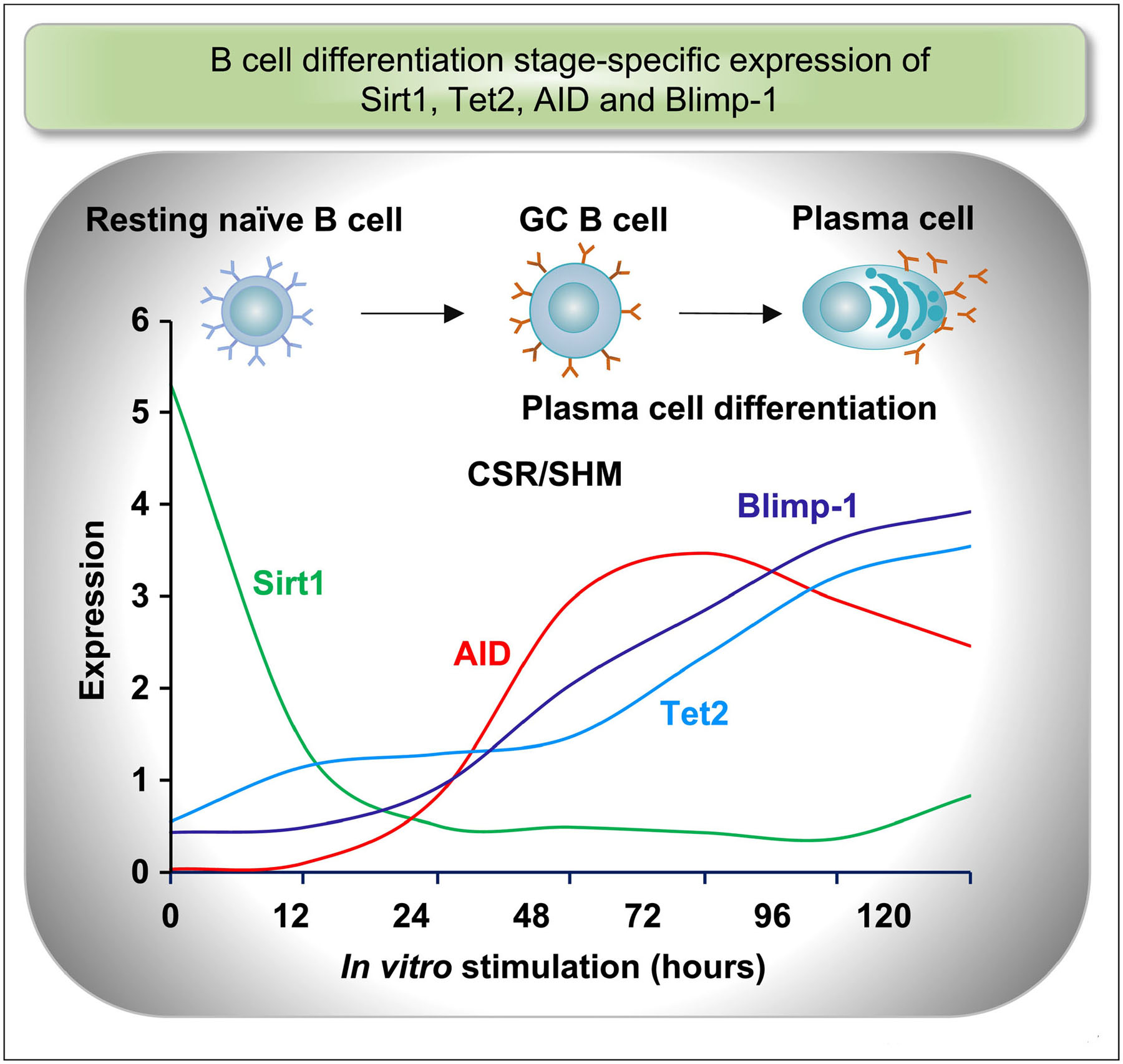
Bi-phasic B cell differentiation and associated dynamic changes in gene expression. B cell CSR/SHM precedes plasma cell differentiation, concomitant with induction of AID and Blimp-1, respectively. AID upregulation coincides with downregulation of Sirt1 transcript and protein. AID and Blimp-1 induction tracks expression of Tet2 protein. In B cells induced to express AID and Blimp-1, Tet2 protein, but not Tet2 transcript, is increased, likely through post-translational modifications that enhance the stability of this epigenetic mediator.
Large-scale DNA demethylation, up to 10% of their DNA methylome, occurs during B cell differentiation to plasma cells, with DNA hypomethylation being prevalent in Prdm1, which is enriched in binding motifs for B cell-expressed TFs, including Oct2, NF-κB, IRF4 and AP-1 [22,23]. In bone marrow long-lived plasma cells, epigenetic programming involving extensive Dnmt3A-mediated de novo DNA methylation at enhancer elements to suppress expression of over 1,000 gene loci, including Pax5, Bcl6 and Aicda [23]. Thus, epigenetic DNA hypomethylation and hypermethylation promotes and restricts plasma cell differentiation.
Histone post-translational modifications can exert diverse effects on gene expression by altering chromatin structure for selective recruitment of TFs and targeting of other elements. They include phosphorylation of serine or threonine, methylation of lysine or arginine, as well as acetylation, ubiquitylation and sumoylation of lysine. Resting (naïve) B cells display inactive chromatin structure and transcriptional repression, with genome-wide histone H3 methylation, particularly H3K9(me3) and H3K27(me3). In such B cells, the Aicda locus is fully quiescent and histone H3 acetylation, a well-defined marker of permissive chromatin, particularly at H3K14ac and H3K27ac, occurs at low levels [25,43,44]. Engagement of BCR and CD40 and/or TLR activates naïve B cells by inducing them to acquire permissive histone marks H3K4me3 and H3K9ac/K14ac, with significant increased H3 acetylation of the Aicda enhancer and promoter regions [25,43,44]. This is concomitant with appearance of combinatorial histone mark H3K4acS10ph, which is specifically read by 14-3-3 adaptors to target AID to Ig locus switch region AGCT motifs [19,45]. Histone acetylation is a dynamic process mediated by HATs and HDACs. HATs play an important role in early B cell development and peripheral differentiation, with monocytic leukemia zinc finger (MOZ) protein implicated in GC dark zone B cell expansion and GCN5 promoting expression of membrane IgM and Irf4 [46,47]. HDACs comprise “classical” Class I, II, and IV HDACs and “non-classical” Class III HDACs or Sirtuins (Sirt). Classical HDACs depend on Zn2+ as a cofactor, Sirtuins on NAD+ [48]. HDAC Class III Sirt1 actively deacetylates acetyl-lysine in multiple histones, including H3K4ac, H3K9Ac, H3K14ac and H3K36ac, to facilitate chromatin compaction and silence gene transcription [49]. Sirt1, the most abundantly expressed Sirtuin in B cells, regulates AID in a B cell differentiation stage-specific fashion. In resting B cells, high levels of Sirt1 mediate extensive histone H3 hypoacetylation across the Aicda locus, thereby keeping AID expression in check [25]. B cell activation induces profound downregulation of Sirt1 with concomitant AID upregulation resulting from decreased Sirt1 recruitment to Aicda promoter and increased H3K9Ac and H3K14Ac acetylation (Figure 1) [25].
Acquisition of permissive histone modifications, including H3K4me1 in active promoters and H3K4me3 in distal enhancers, is associated with changes of methylome and gene expression in differentiating plasma cells [23]. In these cells, loss of repressive histone modifications, such as H3K27me3, occurs at select loci, particularly genes encoding regulators of cell division and metabolism. Such repressive modifications are catalyzed mainly by EZH2 [24], which is differentially recruited by Bcl-6 and Blimp-1 to those gene loci [50,51] as TF dictating B cell fate decisions through recruitment of epigenetic factors. In B cells, inhibition of EZH2 increases expression of genes regulators of cell division and metabolism and premature Prdm1 transcription and Blimp-1 expression, thereby promoting plasma cell differentiation [52]. Prdm1 expression is regulated by transcription activators and repressors. It is dependent on acetylation and methylation of H3K9 at a Maf recognition element (MARE) in intron 5 of this gene. GC B cell stage-specific Prdm1 expression is repressed by Bach2 in cooperation with HDAC3-containing co-repressor complexes through this gene locus reduced histone acetylation [53]. Indeed, TBL1XR1, a core component of these HDAC3-containing SMRT/NCOR1 complexes, plays a pivotal role in GC B cells, preferentially interacting with BCL6, and to a lesser extent Bach2, to repress Prdm1 gene expression and restrict plasma cell differentiation [54].
Non-coding RNAs are involved in regulation of B cell differentiation and function. miRNAs, a category of small ncRNAs, regulate gene expression post-transcriptionally by hampering translation through targeting mRNA 3′-untranslated region (3’UTR) for mRNA degradation. Naïve, GC, plasma cells and memory B cells show distinct miRNA expression patterns [55]. In activated B cells, the deletion of Dicer, an RNase III endonuclease crucial for miRNA biogenesis, impairs biogenesis of multiple miRNAs, which directly regulate CSR/SHM and plasma cell differentiation [56]. miR-155, miR-181b, miR-361 and miR-26a bind to evolutionarily conserved Aicda mRNA 3′UTR target sites to silence Aicda expression [21,57–61]. These miRNAs, particularly miR-155, the most abundant in resting B cells, are downregulated upon B cell activation by CSR-inducing stimuli to allow for AID expression [57,58]. And, ablation of Aicda 3′UTR miR-155-target site increases B cell Aicda mRNA and AID protein level, thereby upregulating CSR [57,58]. Aicda 3′UTR also contains multiple binding sites for miR-181b, which is expressed at high levels in resting B cells and decreases in B cells upregulating AID to undergo CSR [59]. Thus, miR-181b would inhibit premature AID expression and its downregulation would allow for proper AID transcriptional activation at early time points, while miR-155 would restrict AID expression at a later stage of B activation [60].
Prdm1 mRNA contains a long (>2,000 nt) 3’UTR, which is targeted by multiple miRNAs, including miR-9, miR-23b, miR-30, miR-125b, miR-127 and let-7 [20,62–64]. Overexpression of miR-125b inhibits B cell differentiation to plasma cell by impairing Blimp-1 expression through targeting Prdm1 and Irf4 3’UTRs [65]. miR-30c can also downregulate Xbp1 [66], which governs late events in plasma cell differentiation. Thus, dynamic changes in miRNAs control Prdm1, Irf4 and Xbp1 genes, which are critical for B cell differentiation to plasma cell. Long non-coding RNAs (lncRNAs) are potentially important regulators of B cell development and peripheral differentiation, but their roles in these cells are still poorly understood [67,68]. Thousands of lncRNAs have been identified across multiple stages of B cell development and activation [67]. Twenty percent of these lncRNAs are promoter-associated RNAs or enhancer-associated RNAs (eRNAs). In activated B cells, expression of many lncRNA closely correlates with the nearest coding gene, thereby pointing to tandem lncRNA/gene locus functional units [67], as epitomized by lncRNAs role in regulating expression of the adjacent IgH locus in human memory B cells [69].
Epigenetic and metabolic regulation of the autoantibody response
In addition to providing energy source and biosynthetic building blocks for growth, metabolites instruct cell gene expression, and distinct metabolic pathways regulate immune cell activation, differentiation and survival [70]. Indeed, cross-talks among external environment, B cell signal transduction pathways and TFs rely on metabolites as important mediators [71], thereby pointing to a metabolite-driven epigenetic regulation of B cell differentiation to modulate antibody and autoantibody responses.
Tet2 promotes AID and Blimp-1 expression through active DNA demethylation of Aicda and Prdm1 loci as well as of other elements in GC B cells [26,72,73]. In activated B cells inducted to undergo CSR, Tet2 transcription remains flat, while Tet2 protein levels increase significantly due their stabilization by post-translational phosphorylation, acetylation and glycosylation (our unpublished data). Genome-wide DNA hypomethylation occurs in systemic autoimmune diseases [7,17,74,75], as emphasized by lupus discordance in monozygotic twins, in whom the affected twin displays decreased B cell DNA methylation and increased AID expression [76]. Accordingly, prolonged treatment of hypertensive patients with DNA methylation inhibitors, such as hydralazine, can result in autoantibody production and a lupus-like syndrome. Conversely, B cell-intrinsic deletion of Tet2/Tet3 or Tet2 alone undercuts CSR/SHM and plasma cell differentiation [26,42,72] (our unpublished data). Tet2 would couple B cell metabolic status with effective epigenetic functions, as its transcription level and activity are determined by an array of metabolic factors and changes, such a Vitamin A (retinol), Vitamin C (Ascorbate), Fe2+ (co-factor) and α-ketoglutarate (α-KG), a tricarboxylic acid (TCA) cycle metabolite. Mechanistically, retinoic acid enhances 5hmC through activation of TET2 transcription, whereas vitamin C potentiates TET2 activity and 5hmC production through enhanced Fe2+ recycling [91]. Indeed, Fe2+ withdrawal dampens Tet2-mediated conversion of 5mC to 5hmC [77]. Increased α-KG concentration from excess glucose activates Tet2, thereby enhancing genome-wide 5hmC levels [78]. Conceivably, α-KG, and, likely, vitamin C enhance Tet2-mediated active DNA demethylation in B cells, as α-KG does in stem cells [79]. On the other hand, (pathological) accumulation of 2-hydroxyglutarate (2-HG), succinate and fumarate, metabolites structurally similar to α-KG, leads to a competitive inhibition of Tet2 activity [80], which would result in hypermethylation of Aicda as well as other gene loci. Thus, inhibition of Tet2 activity by inhibition or subtraction of its co-factor(s) can conceivably represent an appealing therapeutic approach to autoantibody-mediated systemic autoimmunity involving genomic hypomethylation, such as in lupus.
Sirt1 controls, through deacetylation, the intracellular localization, stability and activity of histone and non-histone proteins, making this Class III HDAC an important mediator of multiple epigenetic functions [81]. Under physiological conditions, Sirt1 is highly expressed in resting naïve B cells, profoundly downregulated in activated B cells induced to undergo CSR/SHM, and reset at high levels in memory B cells and plasma cells. Sirt1 critically contributes to quiescent B cell homeostasis by keeping Aicda transcription in check through a three-pronged mechanism [25] (Figure 2): (i) deacetylation of Aicda promoter histones, which prevents chromatin decondensation, thereby making promoters inaccessible to TFs; (ii) deacetylation of Dnmt1, which activates this DNA demethylase to methylate the Aicda promoter; and (iii) deacetylation of the p65 component of NF-κB, inactivating this B cell transcription factor, which together with HoxC4 is critical for Aicda transcription [82]. B cells induced to undergo CSR/SHM downregulate Sirt1, leading to upregulation of AID expression [25]. Similarly, B cells in lupus mice and patients show highly reduced intrinsic Sirt1/SIRT1 expression concomitant with enhanced Aicda/AICDA transcription and heightened AID levels [25]. Accordingly, intrinsic B cell deletion leads to production of class-switched ANA autoantibodies and lupus-like symptoms in C57BL/6 mice with a non-autoimmune background [25]. Upon activation, Sirt1 can integrate multiple epigenetic mechanisms to dampen AID expression [25]. Boosting Sirt1 activity by SRT1720 dampens AID expression and CSR/SHM in T-dependent and T-independent antibody responses. Further, in lupus-prone MRL/Faslpr/lpr mice, Sirt1 activation inhibits anti-dsDNA and anti-histone IgG1 and IgG2a autoantibody responses, thereby reducing IgG1/IgG2a kidney deposition and glomerular damage [25]. Finally, as shown in myelodysplastic syndrome stem and progenitor cells, Sirt1 can deacetylate Tet2 protein and restore Tet2 activity [83], thereby further contributing to Sirt1-mediated modulation of the antibody and autoantibody response.
Figure 2. Sirt1 modulates modulation AID expression through deacetylation of B cell histone and non-histone proteins.
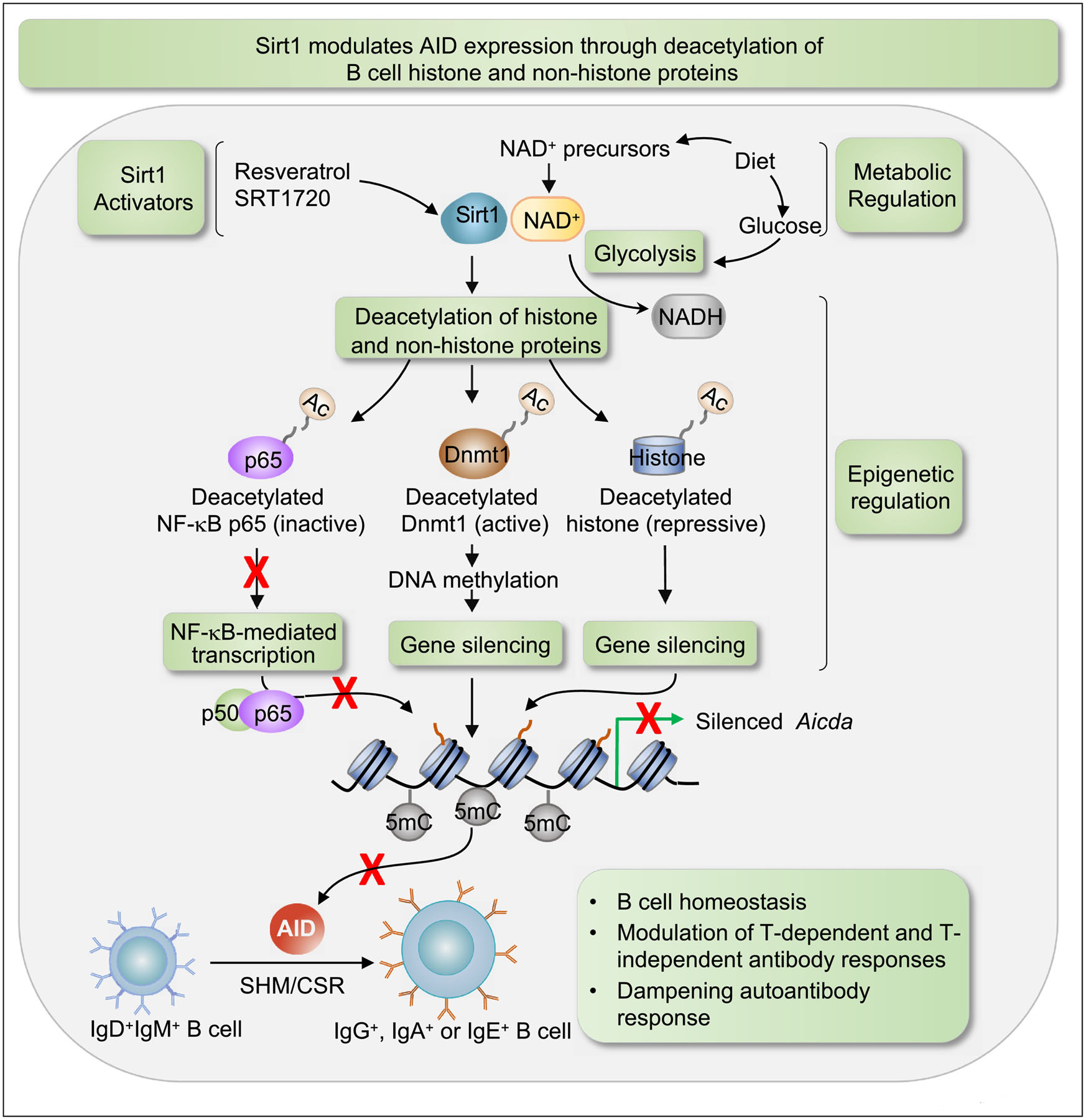
By deacetylating histone and non-histone proteins, the NAD+-dependent class III HDAC Sirt1 exerts a three-pronged repression of Aicda: (1) by deacetylating histones in the Aicda promoter, Sirt1 directly represses Aicda expression; (2) by deacetylating p65, Sirt1 reduces the activity of NF-kB, which together with HoxC4 is a key activator of Aicda promoter; (3) by deacetylating Dnmt1, Sirt1 enhances the DNA methylation activity of this methyltransferase, leading to an increased DNA methylation of the Aicda promoter, thereby further repressing Aicda expression. As intracellular levels of free NAD+ are readily influenced by the balance of nutritional availabilities and the metabolic state of the cell, the NAD+ dependence places Sirt1 at the interface of metabolism and epigenetic regulation. NAD+ precursors, available in food, increase cellular NAD+ level and enhance Sirt1 activity, while glucose, which metabolized through glycolysis converts NAD+ to NADH, lowers cytosolic NAD+/NADH ratio and Sirt1 activity. In addition, some naturally occurring and synthetic compounds, such as resveratrol and SRT1720 which facilitate NADH oxidation to NAD+ and increase Sirt1 affinity for NAD+ and acetylated substrates, function as Sirt1 activators.
As intracellular levels of free NAD+ are readily influenced by the balance of nutritional availabilities and metabolic cell state, NAD+ dependence places Sirt1 at a central interface of metabolism and epigenetic regulation. Consistently, lupus patients with severe clinical outcomes exhibit expanded populations of CD38high T cells and decreased NAD+ levels. This is in agreement with the nucleosidase activity of CD38, which is also highly expressed on activated human B cells as well as plasma cells [92]. Reduced cellular NAD+ concentration, resulting from glycolytic conversion of NAD+ to NADH, leads to decreased Sirt1 activity, increased acetylation of Aicda promoter histones, Dnmt1 and NF-κB p65, thereby enhancing Aicda expression and CSR [25]. By contrast, increased NAD+ concentration, as provided by a nicotinamide rich diet [84], would lead to Sirt1 activation and abortive maturation of antibody and autoantibody responses. Thus, Sirt1 induction by a metabolic cofactor or boost by an activator, e.g., SRT1720, would be a promising therapeutic approach to autoantibody-mediated systemic autoimmunity and other states of B cell hyperreactivity. In this vein, it would be important to determine whether Sirt1 activation is effective in dampening activation and/or differentiation of “double negative” CD27− IgD− T-bet+ (CD11chi) ABC B cells. These B cells are expanded in lupus, multiple sclerosis and rheumatoid arthritis patients as well as in autoimmune mice, correlate with disease severity, and would give rise to self-antigen reactive plasma cells upon co-stimulation with TLR ligands and IL-21 [85,86].
HDIs (valproic acid, VPA; Panobinostat, Farydak; vorinostat, SAHA, Zolinza; Trichostatin-A, TSA or romidepsin, Istodax) treatment of lupus-prone MRL/Faslpr/lpr mice reduces autoantibody levels, autoreactive plasma cell numbers, nephritis and dampen autoimmunity [20,87]. HDIs exert direct and B cell-intrinsic epigenetic effects even at moderate concentrations. In normal C57BL/6 mice, VPA, a well-characterized HDI, effectively downregulates AID and Blimp-1 by upregulating the level of miR-155, miR-181b and miR-361 which target AICDA/Aicda as well as miR-23b, miR-30a and miR-125b which target PRDM1/Prdm1, by boosting histone acetylation of respective host genes [20,63]. This miRNA downregulation of AID and Blimp-1 is dose-dependent and leads to profound inhibition of class-switched and somatically mutated T-dependent (NP-CGG) and T-independent (NP-LPS) antibody responses [20,63]. Through a similar mechanism and in a B cell-intrinsic fashion, VPA impairs the autoantibody response in lupus-prone MRL/Faslpr/lpr mice, by reducing ANA IgG levels, autoreactive plasma cell numbers and nephritis [20,87]. As inhibitor of B cell intrinsic AID and Blimp-1 (and Xbp1) expression and restrictor of generation of class-switched high-affinity autoantibodies, HDIs would provide to be potentially effective therapeutics in lupus patients.
Epigenetic HDAC inhibition by dietary catabolites, namely SCFAs, plays an important role in homeostasis and physiological regulation of antibody and autoantibody responses (Figure 3). Butyrate, propionate and acetate are the main SCFAs produced by microbiota processing of dietary “resistant” fibers (which escaped digestion by host enzymes in the upper gut) in the cecum and proximal colon [88]. While acetate is mostly consumed by colonocytes, butyrate and propionate are reabsorbed in the colon and routed into the circulatory torrent, making it possible for these SCFAs to mediate their epigenetic effects not only on intestinal B cells (Peyer’s patches, lamina propria), but also in other districts of the body, thereby conditioning antibody production systemically.
Figure 3. Epigenetic modulation of antibody and autoantibody responses by microbiota-derived SCFAs.
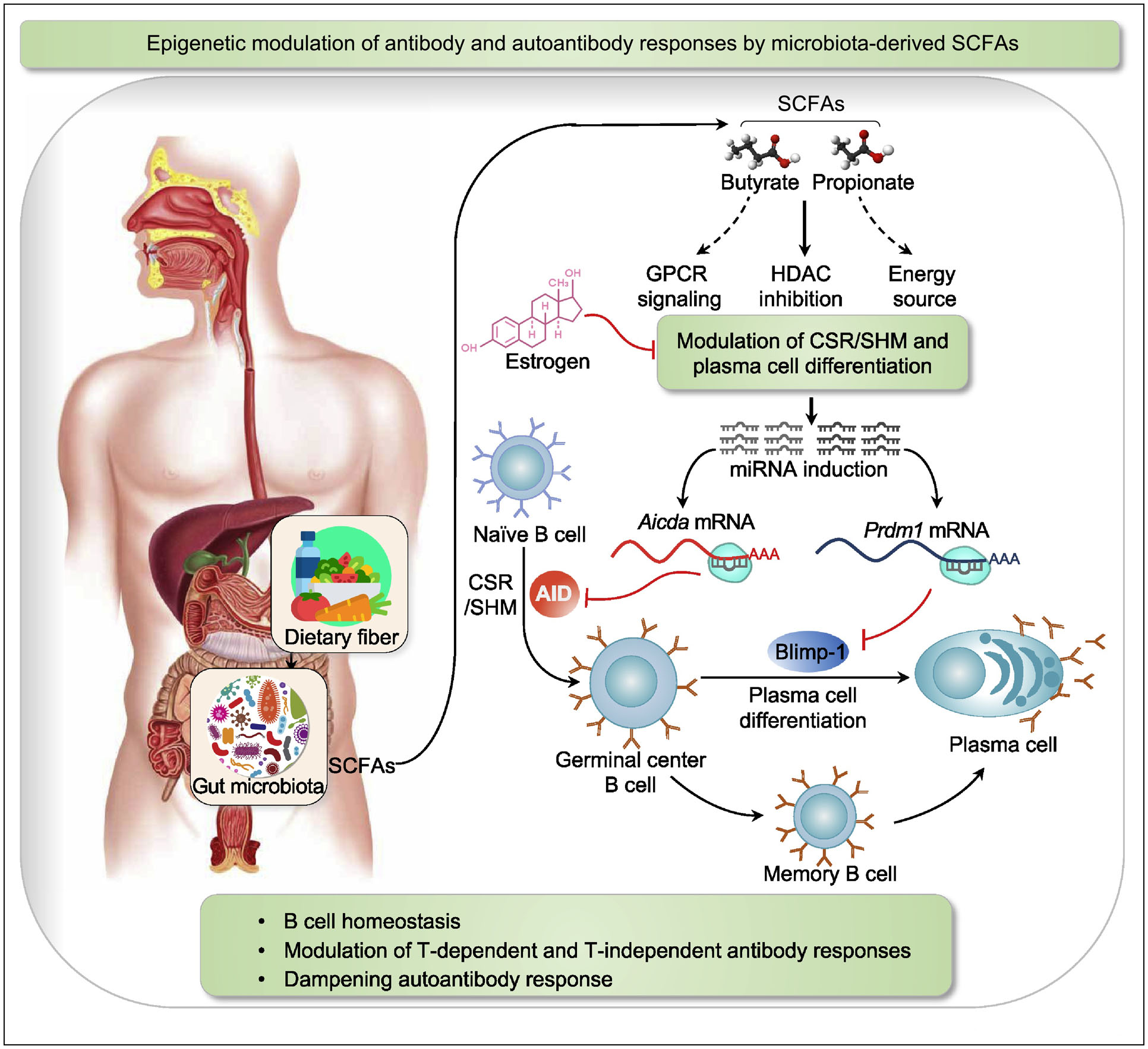
Resistant dietary fibers are fermented by gut commensal bacteria to yield SCFAs, such as butyrate and propionate. Beyond their GPCR-signaling-capacity and use as an energy source, butyrate and propionate display potent HDAC inhibitory functions. In B cells, butyrate and propionate act as HDIs to effect chromatin decondensation and induce the expression of select miRNAs that target the 3’ UTR region of Aicda and Prdm1 transcripts. Such miRNAs silence the expression of AID and Blimp-1, and reduce CSR/SHM and plasma cell differentiation, thereby inhibiting antibody and autoantibody responses. The HDI-mediated downregulation of AID expression and its impact on maturation of antibody and autoantibody responses are reversed by estrogen. Estrogen’s reversion of HDI-mediated inhibition of AID occurs at least partially through downregulation of B cell miR-26a.
Butyrate and propionate directly inhibit Class I, II and IV HDACs, effectively hyperacetylating histones, enhancing chromatin accessibility and activating gene expression [64]. In human and mouse B cells, these SCFAs act over a broad physiological range and in a dose-dependent fashion to modulate AID and Blimp levels, thereby restricting CSR/SHM and plasma cell differentiation. Like VPA, butyrate and propionate enhance histone acetylation host genes of select miRNAs that target AICDA/Aicda and PRDM1/Prdm1 [64]. In activated B cells, butyrate virtually doubles miR-155 expression, making this Aicda-targeting miRNA account for 50% of total miRNA pool [64]. Administration of SCFAs to lupus-prone MRL/Faslpr/lpr and NZB/WF1 mice effectively decreases IgG autoantibody titers, thereby ameliorating skin lesions and dampening kidney immunopathology [64], thereby suggesting a potential therapeutic use of SCFAs in lupus (Figure 4). Intestinal dysbiosis is a frequent hallmark of lupus patients, exhibiting an altered microbiota composition as compared to healthy subjects, particularly with decreased Firmicutes and increased Bacteroidetes [94]. Indeed, in healthy subjects, the gut Firmicutes/Bacteroidetes ratio has been positively correlated with SCFAs concentrations, consistent with Firmicutes ability to process dietary resistant fibers into butyrate, propionate and acetate [93]. Thus, intestinal dysbiosis in lupus alters production of SCFAs, thereby contributing to pathological autoantibody responses through dysregulation of AID and Blimp-1 in B cells. The direct activity of SCFAs on B cells would critically contribute to maintain a steady state balance between tolerance to commensal bacteria and immunity to pathogens for gut and systemic immune homeostasis.
Figure 4. Sirt1 and SCFAs dampen autoantibody response.
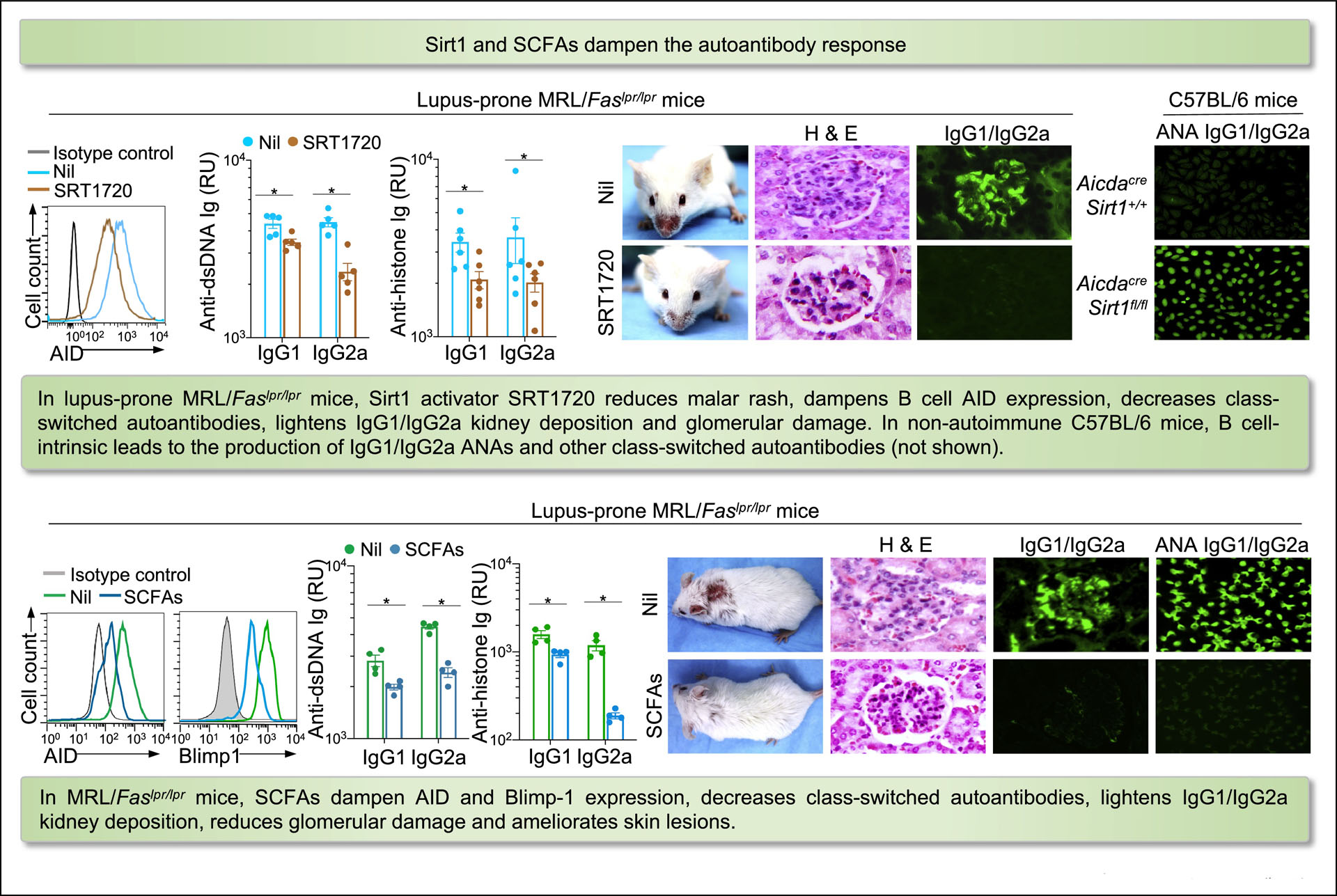
In lupus-prone MRL/Faslpr/lpr mice, Sirt1 activator SRT1720 reduces malar rash, dampens B cell AID expression, decreases class-switched autoantibodies, lightens IgG1/IgG2a kidney deposition and glomerular damage. In non-autoimmune C57BL/6 mice, B cell-intrinsic Sirt1 deletion leads to production of IgG1/IgG2a ANAs and other class-switched autoantibodies. In MRL/Faslpr/lpr mice, SCFAs dampen AID and Blimp-1 expression, reduce class-switched autoantibodies, lighten IgG1/IgG2a kidney deposition, and inhibit glomerular damage and skin lesions.
Estrogen’s contribution to the immunopathogenesis of lupus is consistent with the 9:1 female-to-male sex ratio of lupus incidence, the typical disease onset during the child-bearing years and the increased risk of disease flares in women administered exogenous estrogens [61]. As a central B cell intrinsic factor, estrogen contributes to the dysregulation of multiple epigenetic mechanisms in lupus B cells, including upregulation of Tet2. Indeed, estrogen boosts Tet2 transcription by binding of estrogen-ER complexes to evolutionarily conserved estrogen response elements (EREs) in the Tet2 promoter (our unpublished data). Estrogen can counteract or even reverse SCFA-mediated modulation of antibody and autoantibody responses [61]. This effect is mediated in great part by estrogen’s downregulation of miR-26a, whose level is boosted by SCFAs to target and downregulate Aicda transcripts. Further, miR-26a modulates the expression of Tet2 [89], which regulates AID level by active DNA demethylation of the Aicda 5’ super-enhancer [26]. The intertwined activities of estrogen and miR-26a may provide an explanation for the stronger response of women to viral and bacterial vaccines, and the female bias in autoantibody-mediated lupus autoimmunity.
Conclusions
Epigenetic changes and factors are critical in guiding B cell development and modulating B cell differentiation processes which are central to the maturation of the antibody and autoantibody response. In spite of recent advances, important questions on the nature and role of such epigenetic changes and factors remain to be thoroughly answered. For instance, class-switched high-affinity B cells to foreign antigens or self-antigens are generated through CSR/SHM in GCs and extra-GC sites. After selection for survival and expansion, such B cells undergo differentiation to plasma cells or memory B cells. Much needs to be understood how epigenetic processes determine whether a post-GC or post-extrafollicular B cell differentiates to plasma cell or memory B cell, and what underpins the re-differentiation of a memory B cell to plasma cell [5,16]. Similarly, much light needs to be shed on the nature of resting naïve B cells that seed extrafollicular expansion of pathogenic effector B cells, such as those recently identified in lupus patients. Such B cells are epigenetically poised by a distinct methylation status, which includes enrichment in motifs accessible to AP-1 and EGR TFs synergizing with T-bet [86]. This and other knowledge of epigenetic changes in autoantibody and autoantibody responses has been derived mainly from analysis of disease-associated epigenetic patterns in ex vivo human samples, studies of genetically modified mice or autoimmune mouse strains. However, an experimental and fully functioning in vivo model of the human immune system is needed to fully understand epigenetic mechanisms impact those underpinning the human antibody and autoantibody response. Although humanized NSG mice have shown some promise as model of systemic autoimmunity [90], they have had fallen short of fully supporting the maturation of human antibody responses to foreign and self-antigens. A novel humanized mouse has become available that upon hormonal conditioning provides a robust in vivo platform that allows for generation of mature human T-dependent and T-independent antibody responses and lends itself to in vivo modeling of autoimmune disease (our unpublished data). Such new generation of humanized mice can provide accurate and effective models for investigating immunization strategies and candidate vaccines against bacterial and viral pathogens. Also, they are currently used to identify potential therapeutic targets among epigenetic factor coactivators, including metabolites, and inhibitors, such as naturally occurring SCFAs, in systemic autoantibody-mediated autoimmunity.
Keywords.
B cell-intrinsic epigenetic mechanisms and factors regulate AID and Blimp-1 expression.
Tet2 actively demethylates Aicda super-enhancer elements to derepress Aicda transcription.
Sirt1 uses a three-pronged deacetylation mechanism to downregulate Aicda expression.
Microbiota-derived SCFAs suppress antibody and autoantibody responses through induction of select miRNAs that downregulate Aicda and Prdm1 expression.
Metabolic factors regulate Sirt1 and Tet2 activity, thereby modulating CSR/SHM and plasma cell differentiation in antibody response to foreign and self-antigens (autoantibodies).
Acknowledgements
We apologize that owing to space limitations only a fraction of the relevant literature was cited in this review article. This work was supported by NIH grants R01 AI 105813, R01 AI 079705, T32 AI138944 and the Alliance for Lupus Research Target Identification in Lupus Grant ALR 641363 (P.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no conflict of interest.
Reference and recommended reading
Papers of interest are highlighted as:
• of special interest.
•• of outstanding interest.
- 1.Elkon K, Casali P: Nature and functions of autoantibodies. Nat Rev Rheumatol 2008, 4:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Silva NS, Klein U: Dynamics of B cells in germinal centres. Nat Rev Immunol 2015, 15:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dufaud CR, McHeyzer-Williams LJ, McHeyzer-Williams MG: Deconstructing the germinal center, one cell at a time. Curr Opin Immunol 2017, 45:112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenks SA, Cashman KS, Woodruff MC, Lee FE, Sanz I: Extrafollicular responses in humans and SLE. Immunol Rev 2019, 288:136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cyster JG, Allen CDC: B cell responses: cell interaction dynamics and decisions. Cell 2019, 177:524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suurmond J, Diamond B: Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 2015, 125:2194–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsokos GC: Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol 2020, 21:605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T: Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102:553–563. [DOI] [PubMed] [Google Scholar]; •• This is the first demonstration that AID, which was identified and isolated by the same investigators, is essential for both CSR and SHM.
- 9.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, et al. : Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 2000, 102:565–575. [DOI] [PubMed] [Google Scholar]
- 10.Casali P, Pal Z, Xu Z, Zan H: DNA repair in antibody somatic hypermutation. Trends Immunol 2006, 27:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Zan H, Pone EJ, Mai T, Casali P: Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol 2012, 12:517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM: Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol 2016, 16:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zan H, Zhang J, Ardeshna S, Xu Z, Park SR, Casali P: Lupus-prone MRL/Faslpr/lpr mice display increased AID expression and extensive DNA lesions, comprising deletions and insertions, in the immunoglobulin locus: concurrent upregulation of somatic hypermutation and class switch DNA recombination. Autoimmunity 2009, 42:89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This demonstrates that DNA double stranded breaks, deletions, insertions and chromosomal translocations, often involving oncogenes and oncogene repressors, are a genome-wide occurrence in lupus B cells, as a result of persistently elevated expression of AID in such lymphocytes.
- 14.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM: The generation of antibody-secreting plasma cells. Nat Rev Immunol 2015, 15:160–171. [DOI] [PubMed] [Google Scholar]
- 15.Kometani K, Kurosaki T: Differentiation and maintenance of long-lived plasma cells. Curr Opin Immunol 2015, 33:64–69. [DOI] [PubMed] [Google Scholar]
- 16.Tomayko MM, Allman D: What B cell memories are made of. Curr Opin Immunol 2019, 57:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hedrich CM, Tsokos GC: Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol Med 2011, 17:714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li G, Zan H, Xu Z, Casali P: Epigenetics of the antibody response. Trends Immunol 2013, 34:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li G, White CA, Lam T, Pone EJ, Tran DC, Hayama KL, Zan H, Xu Z, Casali P: Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14-3-3 adaptors and AID to specify antibody class-switch DNA recombination. Cell Rep 2013, 5:702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study provides a mechanism for epigenetic targeting of AID and CSR. The combinatorial H3K9acS10ph histone modification specifically marks the IgH locus switch regions set to recombine and directly recruits 14-3-3 adaptors for AID stabilization to mediate CSR.
- 20.White CA, Pone EJ, Lam T, Tat C, Hayama KL, Li G, Zan H, Casali P: Histone deacetylase inhibitors upregulate B cell microRNAs that silence AID and Blimp-1 expression for epigenetic modulation of antibody and autoantibody responses. J Immunol 2014, 193:5933–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrates that through B cell-intrinsic epigenetic miRNA upregulation, HDIs dampen AID and Blimp-1 expression to dampen class-switched and hypermutated antibody and autoantibody responses.
- 21.Zan H, Casali P: Epigenetics of peripheral B-cell differentiation and the antibody response. Front Immunol 2015, 6:631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barwick BG, Scharer CD, Bally APR, Boss JM: Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol 2016, 17:1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study provides mechanistic insight into the cell-division coupled transcriptional and epigenetic reprogramming and suggest DNA hypomethylation reflects the cis-regulatory history of plasma cell differentiation.
- 23.Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, Haines RR, Bally APR, Kohlmeier JE, Boss JM: B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun 2018, 9:1900. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows that de novo DNA methylation limits B cell activation, represses plasma cell chromatin state to regulates plasma cell differentiation.
- 24.Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, Bradley JE, Randall TD, Boss JM, Scharer CD: EZH2 represses the B cell transcriptional program and regulates antibody-secreting cell metabolism and antibody production. J Immunol 2018, 200:1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study shows that EZH2 is essential in facilitating epigenetic changes that regulate antibody secreting cell fate, function and metabolism.
- 25.Gan H, Shen T, Chupp DP, Taylor JR, Sanchez HN, Li X, Xu Z, Zan H, Casali P: B cell Sirt1 deacetylates histone and non-histone proteins for epigenetic modulation of AID expression and the antibody response. Sci Adv 2020, 6:eaay2793. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study demonstrates that by deacetylating histone and nonhistone proteins (Dnmt1 and NF-κB p65), Sirt1 critically contributes to quiescence of B cell homeostasis by keeping Aicda transcription in check through a three-pronged deacetylation mechanism. It also shows that Sirt1 transduces metabolic cues into epigenetic changes to play an important B cell-intrinsic role in modulating antibody and autoantibody responses.
- 26.Lio CJ, Shukla V, Samaniego-Castruita D, Gonzalez-Avalos E, Chakraborty A, Yue X, Schatz DG, Ay F, Rao A: TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci Immunol 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows that Tet2 and Tet3 enhance AID expression by mediating 5hmC modifications at the Aicda superenhancer.
- 27.Phan AT, Goldrath AW, Glass CK: Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity 2017, 46:714–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellmeier W, Seiser C: Histone deacetylase function in CD4(+) T cells. Nat Rev Immunol 2018, 18:617–634. [DOI] [PubMed] [Google Scholar]
- 29.Kurotaki D, Kawase W, Sasaki H, Nakabayashi J, Nishiyama A, Morse HC 3rd, Ozato K, Suzuki Y, Tamura T: Epigenetic control of early dendritic cell lineage specification by the transcription factor IRF8 in mice. Blood 2019, 133:1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tay RE, Olawoyin O, Cejas P, Xie Y, Meyer CA, Weng QY, Fisher DE, Long HW, Brown M, Kim HJ, et al. : Hdac3 is an epigenetic inhibitor of the cytotoxicity program in CD8 T cells. J Exp Med 2020, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. : Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 2020, 52:825–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohkura N, Yasumizu Y, Kitagawa Y, Tanaka A, Nakamura Y, Motooka D, Nakamura S, Okada Y, Sakaguchi S: Regulatory T cell-specific epigenomic region variants are a key determinant of susceptibility to common autoimmune diseases. Immunity 2020, 52:1119–1132 e1114. [DOI] [PubMed] [Google Scholar]
- 33.Wu X, Zhang Y: TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 2017, 18:517–534. [DOI] [PubMed] [Google Scholar]
- 34.Izzo F, Lee SC, Poran A, Chaligne R, Gaiti F, Gross B, Murali RR, Deochand SD, Ang C, Jones PW, et al. : DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat Genet 2020, 52:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows that DNA methylation landscape shapes the topography of hematopoietic differentiation and supports a model in which genome-wide methylation changes are transduced to differentiation skews through biases in CpG enrichment of the transcription factor-binding motif.
- 35.Kulis M, Merkel A, Heath S, Queiros AC, Schuyler RP, Castellano G, Beekman R, Raineri E, Esteve A, Clot G, et al. : Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet 2015, 47:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study shows that CpG methylation changed extensively during B cell differerntiation.
- 36.Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS, Wang Q, Imbusch CD, Serva A, Koser SD, et al. : DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet 2016, 48:253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lio CW, Zhang J, Gonzalez-Avalos E, Hogan PG, Chang X, Rao A: Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife 2016, 5:e18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Ozark PA, Smith ER, Zhao Z, Marshall SA, Rendleman EJ, Piunti A, Ryan C, Whelan AL, Helmin KA, et al. : TET2 coactivates gene expression through demethylation of enhancers. Sci Adv 2018, 4:eaau6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orlanski S, Labi V, Reizel Y, Spiro A, Lichtenstein M, Levin-Klein R, Koralov SB, Skversky Y, Rajewsky K, Cedar H, et al. : Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc Natl Acad Sci USA 2016, 113:5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study shows that a Tet2/Tet3 conditional knockout at early stages of B-cell development prevents B cell lineage-specific programmed demethylation events. This lack of demethylation affects the expression of nearby B cell lineage genes by impairing enhancer activity, thus causing defects in B-cell differentiation and function.
- 40.Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, Ballon G, Yang SN, Weinhold N, Reimers M, et al. : DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 2011, 118:3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teater M, Dominguez PM, Redmond D, Chen Z, Ennishi D, Scott DW, Cimmino L, Ghione P, Chaudhuri J, Gascoyne RD, et al. : AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis. Nat Commun 2018, 9:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoeler K, Aufschnaiter A, Messner S, Derudder E, Herzog S, Villunger A, Rajewsky K, Labi V: TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. FEBS J 2019, 286:3566–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kieffer-Kwon KR, Nimura K, Rao SSP, Xu J, Jung S, Pekowska A, Dose M, Stevens E, Mathe E, Dong P, et al. : Myc regulates chromatin decompaction and nuclear architecture during B cell activation. Mol Cell 2017, 67:566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows that upon B cell stimulation chromatin decondensation facilitates B cell transcriptome amplification by three differentially regulated steps. B cell activation amplifies chromatin acetylation and methylation. Histone acetylation leads to spreading of chromatin nanodomain clusters, which decompact in a Myc and ATP synthesis-dependent manner.
- 44.Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, et al. : Regulation of AID expression in the immune response. J Exp Med 2007, 204:1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, et al. : 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nat Struct Mol Biol 2010, 17:1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Good-Jacobson KL, Chen Y, Voss AK, Smyth GK, Thomas T, Tarlinton D: Regulation of germinal center responses and B-cell memory by the chromatin modifier MOZ. Proc Natl Acad Sci U S A 2014, 111:9585–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kikuchi H, Nakayama M, Kuribayashi F, Imajoh-Ohmi S, Nishitoh H, Takami Y, Nakayama T: GCN5 is essential for IRF-4 gene expression followed by transcriptional activation of Blimp-1 in immature B cells. J Leukoc Biol 2014, 95:399–404. [DOI] [PubMed] [Google Scholar]
- 48.Seto E, Yoshida M: Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 2014, 6:a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu WW, Wu B, Liu WR: Sirtuins 1 and 2 are universal histone deacetylases. ACS Chem Biol 2016, 11:792–799. [DOI] [PubMed] [Google Scholar]
- 50.Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, Busslinger M: Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 2016, 17:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM, et al. : EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell 2016, 30:197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scharer CD, Barwick BG, Guo M, Bally APR, Boss JM: Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat Commun 2018, 9:1698. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Chromatin accessibility does not always correlate with transcriptional status. This study shows that a subset of genes, including Prdm1, in naive B cells display accessible promoters in the absence of transcription and are marked by H3K27me3, an EZH2 catalyzed repressive modification. Such genes encode regulators of cell division and metabolism.
- 53.Tanaka H, Muto A, Shima H, Katoh Y, Sax N, Tajima S, Brydun A, Ikura T, Yoshizawa N, Masai H, et al. : Epigenetic regulation of the Blimp-1 gene (Prdm1) in B cells involves Bach2 and histone deacetylase 3. J Biol Chem 2016, 291:6316–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venturutti L, Teater M, Zhai A, Chadburn A, Babiker L, Kim D, Beguelin W, Lee TC, Kim Y, Chin CR, et al. : TBL1XR1 mutations drive extranodal lymphoma by inducing a pro-tumorigenic memory fate. Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Jima DD, Jacobs C, Fischer R, Gottwein E, Huang G, Lugar PL, Lagoo AS, Rizzieri DA, Friedman DR, et al. : Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood 2009, 113:4586–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu S, Guo K, Zeng Q, Huo J, Lam KP: The RNase III enzyme Dicer is essential for germinal center B-cell formation. Blood 2012, 119:767–776. [DOI] [PubMed] [Google Scholar]
- 57.Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN: MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 2008, 28:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]; • One of the first two papers showing that microRNAs regulate Aicda expression.
- 58.Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, Di Virgilio M, Reina San-Martin B, Heidkamp G, Schwickert TA, et al. : MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 2008, 28:630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]; • One of the first two papers showing that microRNAs regulate Aicda expression.
- 59.de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, He L, Ramiro AR: miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med 2008, 205:2199–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zan H, Tat C, Casali P: MicroRNAs in lupus. Autoimmunity 2014, 47:272–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Casali P, Shen T, Xu Y, Qiu Z, Chupp DP, Im J, Xu Z, Zan H: Estrogen reverses HDAC inhibitor-mediated repression of Aicda and class-switching in antibody and autoantibody responses by downregulation of miR-26a. Front Immunol 2020, 11:491. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study shows that estrogen reverses HDI-mediated downregulation of AID expression and CSR through selective modulation of miR-26a.
- 62.Nie K, Gomez M, Landgraf P, Garcia JF, Liu Y, Tan LH, Chadburn A, Tuschl T, Knowles DM, Tam W: MicroRNA-mediated down-regulation of PRDM1/Blimp-1 in Hodgkin/Reed-Sternberg cells: a potential pathogenetic lesion in Hodgkin lymphomas. Am J Pathol 2008, 173:242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen T, Sanchez HN, Zan H, Casali P: Genome-wide analysis reveals selective modulation of microRNAs and mRNAs by histone deacetylase inhibitor in B cells induced to undergo class-switch DNA recombination and plasma cell differentiation. Front Immunol 2015, 6:627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanchez HN, Moroney JB, Gan H, Shen T, Im JL, Li T, Taylor JR, Zan H, Casali P: B cell-intrinsic epigenetic modulation of antibody responses by dietary fiber-derived short-chain fatty acids. Nat. Commun 2020, 11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows that gut microbiota derived SCFAs butyrate and propionate act directly, in a dose-dependent fashion and over a broad physiological range, on human and mouse B cells to downregulate AID and Blimp expression, thereby restricting CSR/SHM and plasma cell differentiation through epigenetic modulation of AICDA/Aicda and PRDM1/Prdm1 transcription.
- 65.Gururajan M, Haga CL, Das S, Leu CM, Hodson D, Josson S, Turner M, Cooper MD: MicroRNA-125b inhibition of B cell differentiation in germinal centers. Int Immunol 2010, 22:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Byrd AE, Aragon IV, Brewer JW: MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J Cell Biol 2012, 196:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brazao TF, Johnson JS, Muller J, Heger A, Ponting CP, Tybulewicz VL: Long noncoding RNAs in B-cell development and activation. Blood 2016, 128:e10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]; • By using RNA-Seq and de novo transcript assembly, this study identified in 11 stages of B-cell development and activation 4,516 lncRNAs, including some enhancer-associated eRNAs whose expression closely correlated with the nearest coding gene.
- 68.de Lima DS, Cardozo LE, Maracaja-Coutinho V, Suhrbier A, Mane K, Jeffries D, Silveira ELV, Amaral PP, Rappuoli R, de Silva TI, et al. : Long noncoding RNAs are involved in multiple immunological pathways in response to vaccination. Proc Natl Acad Sci USA 2019, 116:17121–17126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moroney JB, Vasudev A, Pertsemlidis A, Zan H, Casali P: Integrative analysis of coding, non-coding transcriptome and chromatin accessibility reveals a distinct gene expression and epigenetic landscape in human memory B cells. Nat Commun 2020, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jellusova J: Metabolic control of B cell immune responses. Curr Opin Immunol 2020, 63:21–28. [DOI] [PubMed] [Google Scholar]
- 71.Boothby M, Rickert RC: Metabolic regulation of the immune humoral response. Immunity 2017, 46:743755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, Rivas MA, Pawlikowska P, Armand M, Mouly E, et al. : TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Discov. 2018, 8:1632–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows Tet2-linked enhancer hydroxymethylation and transcriptional regulate genes that mediate GC exit, such as PRDM1.
- 73.Rosikiewicz W, Chen X, Dominguez PM, Ghamlouch H, Aoufouchi S, Bernard OA, Melnick A, Li S: TET2 deficiency reprograms the germinal center B cell epigenome and silences genes linked to lymphomagenesis. Sci Adv 2020, 6:eaay5872. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This shows that TET2 deficiency leads to DNA hypermethylation of regulatory elements in GC B cells that are associated with silencing of the respective genes.
- 74.Ballestar E: Epigenetic alterations in autoimmune rheumatic diseases. Nat Rev Rheumatol 2011, 7:263–271. [DOI] [PubMed] [Google Scholar]
- 75.Portela A, Esteller M: Epigenetic modifications and human disease. Nat Biotechnol 2010, 28:1057–1068. [DOI] [PubMed] [Google Scholar]
- 76.Ulff-Moller CJ, Asmar F, Liu Y, Svendsen AJ, Busato F, Gronbaek K, Tost J, Jacobsen S: Twin DNA methylation profiling reveals flare-dependent interferon signature and B cell promoter hypermethylation in systemic lupus erythematosus. Arthritis Rheumatol 2018, 70:878–890. [DOI] [PubMed] [Google Scholar]
- 77.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y: Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466:1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang H, Lin H, Xu H, Zhang L, Cheng L, Wen B, Shou J, Guan K, Xiong Y, Ye D: TET-catalyzed 5-methylcytosine hydroxylation is dynamically regulated by metabolites. Cell Res 2014, 24:1017–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB: Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518:413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. : Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jing H, Lin H: Sirtuins in epigenetic regulation. Chem Rev 2015, 115:2350–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, Xu Z, Mai T, Casali P: HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol 2009, 10:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun J, He X, Zhu Y, Ding Z, Dong H, Feng Y, Du J, Wang H, Wu X, Zhang L, et al. : SIRT1 activation disrupts maintenance of myelodysplastic syndrome stem and progenitor cells by restoring TET2 function. Cell Stem Cell 2018, 23:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. : The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 2012, 15:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, Rahman S, Zerrouki K, Hanna R, Morehouse C, et al. : IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat Commun 2018, 9:1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scharer CD, Blalock EL, Mi T, Barwick BG, Jenks SA, Deguchi T, Cashman KS, Neary BE, Patterson DG, Hicks SL, et al. : Epigenetic programming underpins B cell dysfunction in human SLE. Nat Immunol 2019, 20:1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• By determining the DNA methylome, chromatin accessibility profile and transcriptome in five human B cell subsets, including a newly defined effector B cell subset, from subjects with SLE and healthy controls, this study defines a differentiation hierarchy for the subsets and elucidates the epigenetic and transcriptional differences between effector and memory B cells.
- 87.Waibel M, Christiansen AJ, Hibbs ML, Shortt J, Jones SA, Simpson I, Light A, O’Donnell K, Morand EF, Tarlinton DM, et al. : Manipulation of B-cell responses with histone deacetylase inhibitors. Nat Commun 2015, 6:6838. [DOI] [PubMed] [Google Scholar]
- 88.Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F: From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 2016, 165:1332–1345. [DOI] [PubMed] [Google Scholar]
- 89.Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D’Alessio AC, Hysolli E, Guo Y, Yao H, Megyola CM, et al. : An extensive network of TET2-targeting microRNAs regulates malignant hematopoiesis. Cell Rep 2013, 5:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gunawan M, Her Z, Liu M, Tan SY90, Chan XY, Tan WWS, Dharmaraaja S, Fan Y, Ong CB, Loh E, et al. : A novel human systemic lupus erythematosus model in humanised mice. Sci Rep 2017, 7:16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hore TA, von Meyenn F, Ravichandran M, Bachman M, Ficz G, Oxley D, Santos F, Balasubramanian S, Jurkowski TP, Reik W: Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naive pluripotency by complementary mechanisms. Proc Natl Acad Sci U S A 2016, 113:12202–12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katsuyama E, Suarez-Fueyo A, Bradley SJ, Mizui M, Marin AV, Mulki L, Krishfield S, Malavasi F, Yoon J, Sui SJH, et al. : The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep 2020, 30:112–123 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TM, Comelli EM: Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes 2014, 4:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, Turroni F, Gonzalez S, Suarez A, Gueimonde M, et al. : Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 2014, 5:e01548–01514. [DOI] [PMC free article] [PubMed] [Google Scholar]