

We enrolled prospectively and randomized 296 patients, and compared NGS panel versus clinical‐based Sanger for the diagnosis of AIDs. NGS panel allowed better diagnostic yield (10.1%) compared to Sanger (4.1%). We concluded that targeted NGS improved the diagnosis and global care of patients with AIDs.
Keywords: genetic testing, hereditary autoinflammatory diseases, Sanger sequencing, targeted next‐generation sequencing
Summary
The aim of this study was to compare the effectiveness of the gene‐panel next‐generation sequencing (NGS) strategy versus the clinical‐based gene Sanger sequencing for the genetic diagnosis of autoinflammatory diseases (AIDs). Secondary goals were to describe the gene and mutation distribution in AID patients and to evaluate the impact of the genetic report on the patient’s medical care and treatment. Patients with AID symptoms were enrolled prospectively and randomized to two arms, NGS (n = 99) (32–55 genes) and Sanger sequencing (n = 197) (one to four genes). Genotypes were classified as ‘consistent/confirmatory’, ‘uncertain significance’ or ‘non‐contributory’. The proportion of patients with pathogenic genotypes concordant with the AID phenotype (consistent/confirmatory) was significantly higher with NGS than Sanger sequencing [10 of 99 (10·1%) versus eight of 197 (4·1%)]. MEFV, ADA2 and MVK were the most represented genes with a consistent/confirmed genotype, whereas MEFV, NLRP3, NOD2 and TNFRSF1A were found in the ‘uncertain significance’ genotypes. Six months after the genetic report was sent, 54 of 128 (42·2%) patients had received effective treatment for their symptoms; 13 of 128 (10·2%) had started treatment after the genetic study. For 59 of 128 (46%) patients, the results had an impact on their overall care, independent of sequencing group and diagnostic conclusion. Targeted NGS improved the diagnosis and global care of patients with AIDs.
Introduction
Systemic autoinflammatory diseases (AIDs) are a group of disorders characterized by deregulation of the innate immune system resulting in excessive inflammation. The inflammatory attacks are mediated by neutrophils and monocytes and by cellular signalling pathways involving mainly interleukin 1 (IL‐1) and IL‐6, but also tumour necrosis factor alpha (TNF‐α), interferon type 1 (IFN‐1) or the nuclear factor kappa B (NF‐κb) transcription factor.
AIDs are commonly distinguished from autoimmune diseases because they lack pathogenic autoantibodies, autoreactive T lymphocytes. The disease course depends upon the gene and the diagnosis: patients affected by AIDs present continuous or recurrent elevated acute‐phase reactants. Manifestations usually begin in early infancy or adolescence. Typical AID patients manifest fever, arthritis, skin rash and serositis (pleura, peritoneum, pericardium). Clinical signs and severity are extremely variable. These variations probably reflect the large range of possible causative AID genes and the signalling pathway. The large clinical spectrum contributes to misdiagnoses. Monogenic hereditary AIDs are considered rare diseases (< 1/2000 individuals) [1].
The year 1997 was a milestone in our knowledge of the AID pathophysiology. The first gene involved in AIDs was discovered by two consortia 21 years ago [2]. Mediterranean fever (MEFV) was identified in families who shared the same familial Mediterranean fever (FMF) phenotype. FMF is the most common AID and affects almost exclusively people with Mediterranean ancestries (Armenian, Turkish, Sephardic Jew, Maghrebian, etc.). The disease usually starts during childhood and is characterized by recurrent fever, arthritis, cutaneous rash and biological inflammation during attacks. Patients with FMF respond favourably to colchicine. However, lack of anti‐inflammatory treatment for several years may be life‐threatening because of the risk of amyloid A amyloidosis, resulting in chronic end‐stage renal failure.
Three other hereditary recurrent fevers are well described. Cryopyrin‐associated periodic syndromes (CAPSs) are caused by dominant mutations in NLR family pyrin domain containing 3 (NLRP3) and characterized by deregulation of IL‐1 activity. CAPSs include familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS) and chronic infantile neurological, cutaneous, articular (CINCA) syndrome, also known as neonatal‐onset multi‐system inflammatory disease (NOMID). The three diseases have in common recurrent fever, joint involvement, urticaria and neurosensory impairment, with a wide range of symptoms, from mild (FCAS) to most severe (CINCA).
Dominant mutations in TNF receptor superfamily member 1A (TNFRSF1A) are responsible for TNF receptor‐associated syndrome (TRAPS). Patients manifest febrile episodes lasting from a few days to a few weeks, joint involvement, myalgia, skin lesions and serositis.
Finally, mevalonate kinase deficiency (MKD) is caused by recessive mutations in mevalonate kinase (MVK) and results in fever, vomiting, diarrhoea and skin rash, most often with a neonatal onset. In its most severe form, mevalonic aciduria (MA), neurological complications are frequently observed.
Since 1997, the list of genes involved in AIDs has been growing significantly [3]. More than 40 genes are now identified as responsible for AIDs [4, 5].
Autoinflammatory medical advances are due in part to progress in genetic research and diagnosis. The discovery of new genes has highlighted new cellular signalling pathways involved in the inflammatory reaction (e.g. inflammasome, NF‐κB, IFN pathways). Understanding the physiology of AIDs leads to the development of new therapies. Genetic testing is essential for AID diagnosis, and then initiating appropriate treatment. Today, a large number of undiagnosed patients receive non‐specific anti‐inflammatory therapies, such as corticosteroids or immunomodulation therapy. In addition, mutational screening may not be exhaustive if it focuses on a limited number of known genes and/or mutational hot‐spots. Indeed, regardless of the sequencing method used, genetic tests are inconclusive for 50–75% of patients with clinical criteria of AIDs [6, 7].
A positive diagnosis depends upon the patient’s phenotype, the gene‐sequencing strategy and the sensitivity of the test (allowing or not the detection of low‐level mosaicism, for example) and the classification used for variant pathogenicity. The development of new sequencing approaches, such as next‐generation sequencing (NGS), has allowed for investigating many different genes simultaneously.
To improve the performance of molecular diagnoses of AIDs, our laboratory has elaborated a strategy to simultaneously screen a panel of AID genes. The main objective of this work was to (1) formally evaluate, in a large cohort of patients with clinical suspicion of an AID, the potential added value of systematic NGS of a panel of genes over Sanger sequencing for genes requested on the basis of clinical criteria by the prescribing physician. Our two secondary analyses were (2) to evaluate the distribution of genes and mutations in these patients and (3) to compare, for both sequencing approaches, the impact of the genetic report on the patient’s medical care and treatment.
Materials and methods
Consent
This prospective study received full ethical approval from the regional committee for the protection of individuals (no. ID‐RCB: 2015‐A00362‐47) and was registered at ClinicalTrials.gov (ID: NCT02976948). Each patient (or legal guardian if relevant) was fully informed and gave written consent for DNA analysis and participation in the study. With clinicians of reference centres for AIDs, we had developed a clinical form common for all AIDs to be provided with genetic diagnosis requests. This form collects epidemiological data and includes a list of clinical symptoms and biological markers frequently encountered in AIDs (Supporting information, Fig. S1). The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Study design
We prospectively recruited consecutive patients referred to our laboratories for genetic diagnosis of AID. We did not select them for further specific clinical criteria and only checked whether they fulfilled the following inclusion criteria: symptoms of AIDs [more than three attacks, elevated C‐reactive protein level, age of onset before age 30 years (French reference centre’s criteria), request for routine Sanger genetic diagnosis (one to four genes), a clinical form correctly completed and a signed informed consent form to participate in the study]. A reference centre physician approved each inclusion. We excluded patients with an inflammatory disease other than AIDs (neoplasia, autoimmune disease, infection, etc.), an initial request for targeted AID gene‐panel testing (NGS method) and ethnicity Armenian, Turkish, Sephardic and Arabic when mentioned (to limit bias towards FMF patients).
The patients fulfilling the inclusion criteria were randomized to two groups using Clinsight (Ennov, Paris, France). Randomization was performed using a 1 : 2 ratio, blocks of variable size, and stratified on the age of onset and sex of the patients. The random allocation sequences were computer‐generated and were implemented by a data manager who was not otherwise involved in the trial.
Patients in the first group had a genetic test as requested by the clinician (clinic‐based prescription of one to four genes). Patients in the second group benefited from NGS sequencing with a high‐throughput‐targeted AID gene panel including the genes requested.
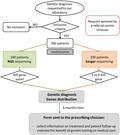
Clinicians were contacted by e‐mail 6 months after we sent the genetic report for their patient(s). They had to complete an online form to evaluate the patient’s treatment and its efficiency before and after the genetic test, the effect of the genetic report on the medical care of their patient and the clinician’s perception of the genetic diagnosis (Supporting information, Fig. S2). We sent reminder e‐mails the next month if necessary. The study design is summarized in Fig. 1.
Fig. 1.
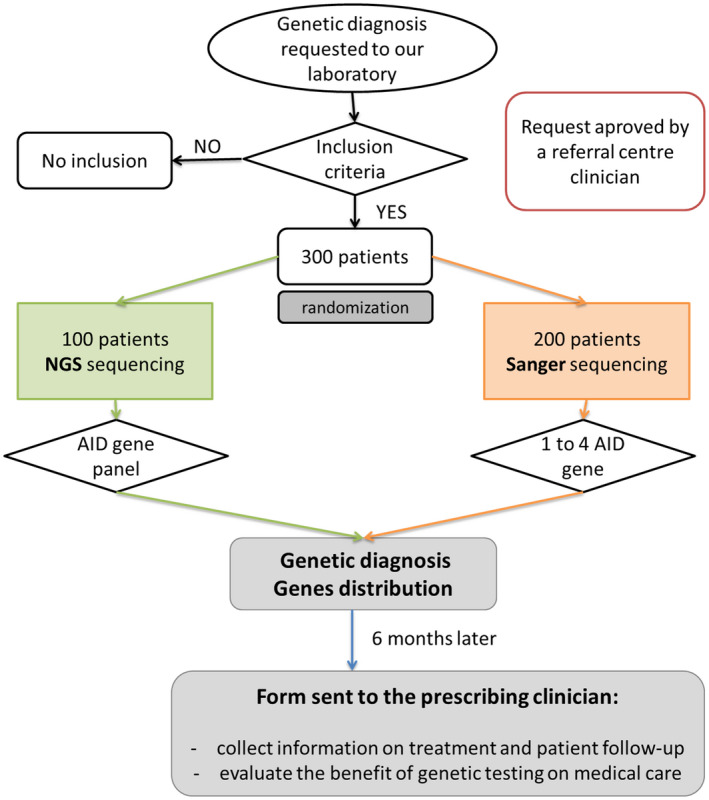
Study design.
Genetic analysis
Sanger sequencing
Two different amplicons were generated for each exon to circumvent the risk of allele dropout. Coding exons and exon–intron junctions were sequenced in both directions by using ABI PRISM Big Dye Terminator version 3.1, the Ready‐Reaction Cycle‐Sequencing kit and ABI 3130 XL (Applied Biosystems, Foster City, CA, USA). Primer sequences used for Sanger sequencing are available on request.
Targeted AID gene‐panel NGS
Our targeted panel contains genes involved in autoinflammatory syndromes. Illumina Design Studio was used to design a panel targeting 32 genes [8]. An updated version was available on 28 June 2016 because novel genes were described, which resulted in a final list of 55 AID genes (Supporting information, Table S1). For this second design, we used the Agilent Online Sure Design tool [9]. Coding regions, plus 25 base pairs (bp) flanking each exon were included in each design. Libraries were prepared by using SureSelect Target Enrichment Capture custom kits (Agilent, Santa Clara, CA, USA). Sequencing reactions were run on MiSeq or NextSeq500 equipment (Illumina, San Diego, CA, USA). Point mutations were systematically confirmed by Sanger sequencing and copy number variations were investigated by quantitative polymerase chain reaction (qPCR).
qPCR
When relevant (e.g. when a single clearly pathogenic variant was detected in recessive disease or when we suspected a large deletion/duplication by NGS), each coding exon was analysed by real‐time qPCR with a LightCycler 480 thermocycler (Roche, Basel, Switzerland).
Segregation analysis
Phasing was performed when DNA from family members was available, after obtaining informed consent.
We paid close attention to the coverage data: for each sample, we checked the Phred quality score, total number of reads, percentage of reads mapped to the target region, percentage of bases in a specific region, thresholds for coverage and distribution of forward and reverse strand reads using SeqNext (JSI Medical Systems, New York, NY, USA) [10]. Coverage was assessed for each sample. The cut‐off for data reanalysis was coverage < 90% at ×40.
Bioinformatics analysis and pathogenicity assessment of identified variants
Variants were analysed by using SeqScape (Applied Biosystems), SeqNext (JSI Medical Systems) and standard in‐silico tools. These latter tools included the Alamut software pipeline (Interactive Biosoftware, Rouen, France) for missense mutations [Grantham variation/Grantham deviation (GVGD), sorting intolerant from tolerant (SIFT), Polyphen2, MutationTaster, Grantham score) and MaxEntScan (MES), human splice finder (HSF) and neural network splice (NNSplice) for splice variant. Minor allele frequency was obtained by using the allele frequency database browsers 1000 Genomes Project and GnomAD/ExAC.
The identified variants were individually assessed and classified into pathogenicity groups by American College of Molecular Genetics guidelines [11]: class 1, clearly not pathogenic; class 2, unlikely to be pathogenic; class 3, uncertain significance; class 4, likely to be pathogenic; class 5, clearly pathogenic.
When possible, pathogenicity was retrieved from a reputable source (e.g. guidelines for hereditary recurrent fevers [12]) or from the autoinflammatory‐specific locus database Infevers [13]. The description of variants conforms to Human Genome Variant Society recommendations [14].
The genetic diagnosis was considered ‘confirmatory’ or ‘consistent’ if we detected one class 4 or 5 variant for dominant monogenic hereditary diseases; two paired‐class 4 or 5 variants for recessive monogenic hereditary diseases; or genotype/phenotype concordance. It was considered ‘uncertain’ with detection of single or multiple sequence variants of uncertain significance (VUS) only or heterozygous mutations for autosomal recessive disorders. Otherwise, it was considered ‘non‐contributory’ in all other cases.
Statistical tests
Sample size calculation
We expected to identify a mutation for 25% of patients with our new genetic diagnostic strategy and for 10% with the current strategy. The minimal number of participants required to demonstrate such a difference with power of 90%, a bilateral alpha risk of 5% and randomization performed according to a ratio of 1 : 2 (1 NGS/2 conventional Sanger method) was 300.
Comparison of the two sequencing strategies
We used the χ2 test or Fisher’s exact test when expected cell frequencies were less than 5 to compare the two sequencing strategies in terms of age classes, sex ratio, percentage of genetic diagnosis and patient care after genetic results were received. P < 0·05 was considered statistically significant. We used SAS version 9.1 (SAS Institute, Cary, NC, USA) for statistical analysis.
Results
A total of 300 patients were enrolled into the study from 29 September 2015 to 28 December 2017. Four patients were excluded after randomization (genetic testing cancelled by the ordering physician or exclusion criteria met after randomization). Among the remaining 296 participants, 197 were randomized to the Sanger sequencing strategy and 99 to the NGS panel strategy (Fig. 2). The details of the gene panel are provided in Supporting information, Table S1.
Fig. 2.
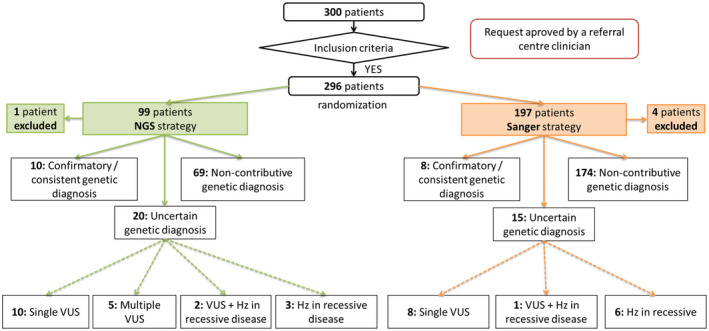
Flow‐chart of participants in the study. NGS = next‐generation sequencing; VUS = variant of uncertain significance; Hz = heterozygous variant.
Diagnostic yield: NGS versus Sanger sequencing for a given clinical suspicion
The genotypes were confirmatory or consistent in 18 of 296 (6%) patients overall (Table 1). With the Sanger strategy, eight of 197 (4·1%) patients had a confirmatory or consistent genotype (Fig. 3a). With the NGS strategy, 10 of 99 (10·1%) had a confirmatory or consistent genotype (Fig. 3b). The initial Sanger prescription was consistent with the final diagnosis in six of 10 NGS strategy patients with a confirmatory or consistent genotype. These diagnostic yields significantly differed between the two groups (P = 0.040). Overall, 35 of 296 (11·8%) patients had an ‘uncertain significance’ genotype, 15 of 197 (7·6%) with the Sanger strategy and 20 of 99 (20·2%) with the NGS strategy (Fig. 2, Table 2). Also, 53 of 296 (17.9%) patients had at least one variant identified in the AID genes, more patients with the NGS than Sanger strategy: 30 of 99 (30·3%) and 23 of 197 (11·6%) (P ≤ 0·001). Clinical signs of patients with a confirmatory or consistent genotype and an uncertain significance genotype are available in supplementary tables (Supporting information, Tables S2 and S3).
Table 1.
Variants and genotypes of patients with consistent or confirmatory genotype of autoinflammatory diseases (AIDs) by sequencing strategy: Sanger or next‐generation sequencing (NGS) (n = 18)
| Patients | Gene | Mutation c. | Mutation p. | rs ID | MAF (GnomAD) | Zygosity | Genotype interpretation |
|---|---|---|---|---|---|---|---|
| Sanger strategy | |||||||
| 18 | TNFRSF1A | c.361C>T | p.(Arg121Trp) | rs104895276 | n.a. | Heterozygous | Consistent |
| 20 | MVK | c.709A>T | p.(Thr237Ser) | rs104895366 | 1.804e‐5 | Compound heterozygous | Confirmatory |
| MVK | c.1129G>A | p.(Val377Ile) | rs28934897 | 0.001591 | |||
| 23 | MEFV | c.442G>C | p.(Glu148Gln) | rs3743930 | 0.06564 | Compound heterozygous | Consistent |
| MEFV | c.2082G>A | p.(Met694Ile) | rs28940578 | 0.0001263 | |||
| 27 | ADA2 | c.144del | p.(Arg49Glyfs*4) | rs199614299 | 3.25e‐5 | Compound heterozygous | Confirmatory |
| ADA2 | c.1348G>T | p.(Gly450Cys) | n.a. | 1.218e‐5 | |||
| 50 | MVK | c.709A>T | p.(Thr237Ser) | rs104895366 | 1.804e‐5 | Compound heterozygous | Confirmatory |
| MVK | c.1129G>A | p.(Val377Ile) | rs28934897 | 0.001591 | |||
| 59 | MEFV | c.1772T>C | p.(Ile591Thr) | rs11466045 | 0.01082 | Compound heterozygous | Consistent |
| MEFV | c.2080A>G | p.(Met694Val) | rs61752717 | 0.0002669 | |||
| 242 | MEFV | c.2040G>C | p.(Met680Ile) | rs28940580 | 0.0001056 | Compound heterozygous | Consistent |
| MEFV | c.442G>C | p.(Glu148Gln) | rs3743930 | 0.06564 | |||
| 288 | NOD2 | c.1759C>T | p.(Arg587Cys) | rs104895479 | 3.231e‐5 | Heterozygous | Confirmatory |
| NGS strategy | |||||||
| 54 | NLRC4 | c.188_189insT | p.(Glu64Argfs*4) | n.a. | n.a. | Heterozygous | Confirmatory |
| 63 | MVK | c.37A>C | p.(Lys13Gln) | n.a. | n.a. | Compound heterozygous | Confirmatory |
| MVK | c.1129G>A | p.(Val377Ile) | rs28934897 | 0.001591 | |||
| 66 | MEFV | c.1105C>T | p.(Pro369Ser) | rs11466023 | 0.01471 | Compound heterozygous | Consistent |
| MEFV | c.1105C>T | p.(Pro369Ser) | rs11466023 | 0.01471 | |||
| 83 | MEFV | c.442G>C | p.(Glu148Gln) | rs3743930 | 0.06564 | Compound heterozygous | Consistent |
| MEFV | c.2082G>A | p.(Met694Ile) | rs28940578 | 0.0001263 | |||
| 108 | NLRC4 | c.530C>G | p.(Thr177Ser) | n.a. | n.a. | Heterozygous | Confirmatory |
| 112 | DDX58 | c.1723C>T | p.(Arg575*) | rs371404578 | 1.627e‐5 | Heterozygous | Consistent |
| 141 | ADA2 | c.427delA | p.(Ile143Serfs*41) | n.a. | n.a. | Compound heterozygous | Confirmatory |
| ADA2 | c.973‐2A>G | SAV | rs139750129 | 0.0001266 | |||
| 164 | PSTPIP1 | c.748G>C | p.(Glu250Gln) | rs28939089 | n.a. | Heterozygous | Confirmatory |
| 166 | TNFAIP3 | c.(?‐1)_(*1_?)del | HTZ deletion | n.a. | n.a. | Heterozygous | Confirmatory |
| 203 | ADA2 | c.476G>A | p.(Cys159Tyr) | rs774636844 | 4.061e‐6 | Compound heterozygous | Confirmatory |
| ADA2 | c.1358A>G | p.(Tyr453Cys) | rs376785840 | 8.658e‐5 | |||
HTZ‐MAR = heterozygous mutation associated with autosomal recessive disorders; SAV = splice acceptor variant; n.a. = not available; MAF = minor allele frequency.
Fig. 3.
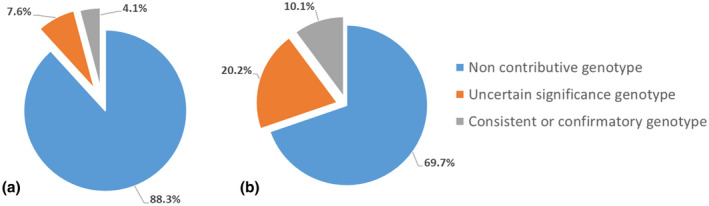
Diagnostic yields for each group. (a) Sanger sequencing strategy (197 patients); (b) next‐generation sequencing (NGS) strategy (99 patients).
Table 2.
Variants and genotypes of patients with uncertain significance genotype (n = 35)
| ID patients | Gene | mutation c. | mutation p. | rs ID | MAF (GnomAD) | Zygosity | Interpretation |
|---|---|---|---|---|---|---|---|
| Sanger strategy | |||||||
| 9 | NLRP3 | c.592G>A | p.(Val198Met) | rs121908147 | 0.008460 | Heterozygous | VUS |
| 70 | NLRP3 | c.592G>A | p.(Val198Met) | rs121908147 | 0.008460 | Heterozygous | VUS |
| 81 | TNFRSF1A | c.362G>A | p.(Arg121Gln) | rs4149584 | 0.01290 | Heterozygous | VUS |
| 109 | ADA2 | c.139G>A | p.(Gly47Arg) | rs202134424 | 2.437e‐5 | Heterozygous | HTZ‐MAR |
| 113 | MEFV | c.2230G>T | p.(Ala744Ser) | rs61732874 | 0.001753 | Heterozygous | HTZ‐MAR |
| 121 | NOD2 | c.1177C>T | p.(Arg393Cys) | rs140716236 | 0.0001479 | Heterozygous | VUS |
| 122 | TNFRSF1A | c.362G>A | p.(Arg121Gln) | rs4149584 | 0.01290 | Heterozygous | VUS |
| 127 | MEFV | c.1772T>C | p.(Ile591Thr) | rs11466045 | 0.01082 | Heterozygous | VUS |
| 136 | NOD2 | c.2883‐2A>G | SAV | rs564226539 | 2.442e‐5 | Heterozygous | VUS |
| 164 | MEFV | c.2080A>G | p.(Met694Val) | rs61752717 | 0.0002669 | Heterozygous | HTZ‐MAR |
| 206 | MEFV | c.1772T>C | p.(Ile591Thr) | rs11466045 | 0.01082 | Heterozygous | VUS |
| 232 | MEFV | c.2084A>G | p.(Lys695Arg) | rs104895094 | 0.005894 | Heterozygous | HTZ‐MAR |
| NLRP3 | c.2107C>A | p.(Gln703Lys) | rs35829419 | 0.03861 | Heterozygous | VUS | |
| 233 | MEFV | c.2084A>G | p.(Lys695Arg) | rs104895094 | 0.005894 | Heterozygous | HTZ‐MAR |
| 237 | MEFV | c.2080A>G | p.(Met694Val) | rs61752717 | 0.0002669 | Heterozygous | HTZ‐MAR |
| 275 | MEFV | c.2080A>G | p.(Met694Val) | rs61752717 | 0.0002669 | Heterozygous | HTZ‐MAR |
| NGS strategy | |||||||
| 5 | MEFV | c.1105C>T | p.(Pro369Ser) | rs11466023 | 0.01471 | Heterozygous | VUS |
| 19 | NLRP3 | c.592G>A | p.(Val198Met) | rs121908147 | 0.008460 | Heterozygous | VUS |
| 31 | IL10RA | c.883C>T | p.(Pro295Ser) | rs201777547 | 0.0001912 | Heterozygous | HTZ‐MAR |
| 99 | NLRP3 | c.592G>A | p.(Val198Met) | rs121908147 | 0.008460 | Heterozygous | VUS |
| 105 | AP1S3 | c.318T>A | p.(Asn106Lys) | n.a. | n.a. | Heterozygous | VUS |
| NOD2 | c.2507T>C | p.(Ile836Thr) | rs763192145 | 4.062e‐5 | Heterozygous | VUS | |
| 113 | TNFRSF1A | c.362G>A | p.(Arg121Gln) | rs4149584 | 0.01290 | Heterozygous | VUS |
| 129 | NLRP3 | c.404G>A | p.(Arg135His) | n.a. | n.a. | Heterozygous | VUS |
| 133 | MEFV | c.442G>C | p.(Glu148Gln) | rs3743930 | 0.06564 | Heterozygous | VUS |
| NLRP3 | c.2107C>A | p.(Gln703Lys) | rs35829419 | 0.03861 | Heterozygous | VUS | |
| 135 | MEFV | c.442G>C | p.(Glu148Gln) | rs3743930 | 0.06564 | Heterozygous | VUS |
| MEFV | c.1105C>T | p.(Pro369Ser) | rs11466023 | 0.01471 | Heterozygous | VUS | |
| 138 | NOD2 | c.2883‐2A>G | SAV | rs564226539 | 2.442e‐5 | Heterozygous | VUS |
| 139 | NLRP3 | c.1556T>C | p.(Ile519Thr) | n.a. | n.a. | Heterozygous | VUS |
| 151 | NOD2 | c.2050C>T | p.(Arg684Trp) | rs5743276 | 0.0003952 | Heterozygous | VUS |
| NOD2 | c.2722G>C | p.(Gly908Arg) | rs2066845 | 0.01085 | Heterozygous | VUS | |
| NOD2 | c.3056G>T | p.(Arg1019Leu) | rs5743295) | 3.972e‐5 | Heterozygous | VUS | |
| IL10RA | c.698T>G | p.(Val233Gly) | rs138929400 | 0.001378 | Heterozygous | HTZ‐MAR | |
| 183 | CARD14 | c.2870_2872delAGG | p.(Glu957del) | n.a. | 3.23e‐5 | Heterozygous | VUS |
| 187 | PSTPIP1 | c.203C>T | p.(Thr68Met) | rs201872851 | 0.001518 | Heterozygous | VUS |
| 225 | PSTPIP1 | c.1213C>T | p.(Arg405Cys) | rs201253322 | 0.0005487 | Heterozygous | VUS |
| 231 | ADAR | c.3401A>G | p.(Tyr1134Cys) | rs150284449 | 9.74e‐5 | Heterozygous | HTZ‐MAR |
| MEFV | c.442G>C | p.(Glu148Gln) | rs3743930 | 0.06564 | Heterozygous | VUS | |
| PSTPIP1 | c.1213C>T | p.(Arg405Cys) | rs201253322 | 0.0005487 | Heterozygous | VUS | |
| 232 | NLRP3 | c.2107C>A | p.(Gln703Lys) | rs35829419 | 0.03861 | Heterozygous | VUS |
| TNFRSF1A | c.362G>A | p.(Arg121Gln) | rs4149584 | 0.01290 | Heterozygous | VUS | |
| 235 | MEFV | c.442G>C | p.(Glu148Gln) | rs3743930 | 0.06564 | Heterozygous | VUS |
| NLRP12 | c.1054C>T | p.(Arg352Cys) | rs199881207 | 0.0003863 | Heterozygous | VUS | |
| 236 | MEFV | c.2080A>G | p.(Met694Val) | rs61752717 | 0.0002669 | Heterozygous | HTZ‐MAR |
| 241 | RNASEH2B | c.529G>A | p.(Ala177Thr) | rs75184679 | 0.001361 | Heterozygous | HTZ‐MAR |
HTZ‐MAR = heterozygous mutation associated with autosomal recessive disorders; SAV = splice acceptor variant; n.a. = not available; VUS = variants of uncertain significance.
Distribution of genes and mutations identified in AIDs
Nine different genes were identified in confirmatory or consistent genotypes. Their distribution is shown in Fig. 4a. The most commonly mutated genes were MEFV, ADA2 and MVK. For patients with an uncertain significance genotype, variants in 12 genes were identified (Fig. 4b). Genes most represented in these patients were MEFV, NLRP3, NOD2 and TNFRSF1A. All the identified mutations were submitted to Infevers, the international registry of hereditary autoinflammatory disorder mutations (https://infevers.umai‐montpellier.fr/web/) [13].
Fig. 4.
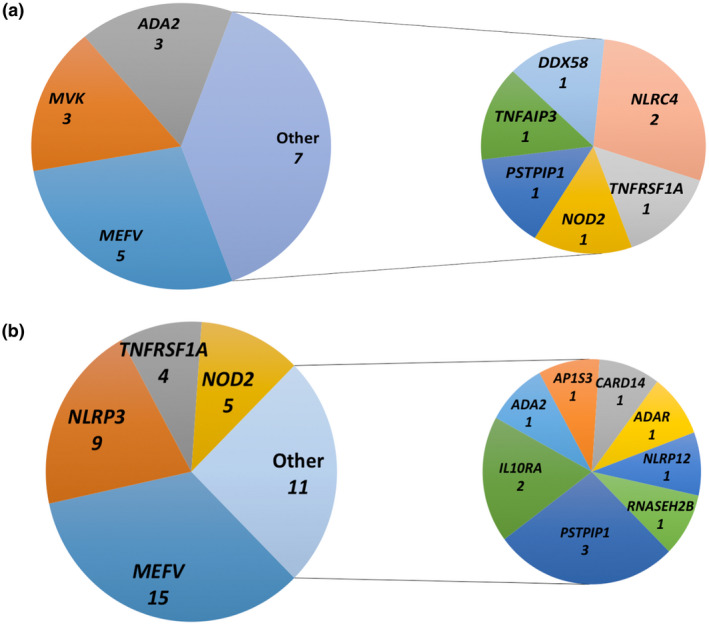
Distribution of the autoinflammatory disease (AID) genes identified (a) in the 18 patients with confirmatory or consistent genotype; (b) in the 35 patients with uncertain significance genotypes.
Impact of the genetic report on patients’ medical care and treatment
We collected responses from 134 of 300 (44·7%) prescribing doctors. Six clinicians responded that their patients were lost to follow‐up or had died (Sanger strategy, n = 4; NGS strategy, n = 2).
Treatment was fully effective for 54 of 128 (42·2%) patients and did not differ by sequencing strategy – Sanger, 35 of 87 (40·2%) patients; NGS, 19 of 41 (46·3%) patients (P = 0·513) – or when patients were analysed by their genetic status.
Receipt of the genetic report resulted in the initiation of effective treatment for only 13 of 128 (10·2%) patients and did not differ by sequencing strategy – Sanger, eight of 87 (9·2%) patients; NGS, five of 41 (12·2%) patients (P = 0.755) – or by genetic status. Physicians acknowledged that the genetic report resulted in improved medical care for 59 of 128 (46·1%) patients, with no significant difference by sequencing strategy (P = 0·732).
Discussion
The aim of this study was to compare the effectiveness of the gene‐panel NGS strategy versus the gold‐standard Sanger sequencing for the genetic diagnosis of AIDs. Secondary goals were to describe the gene and mutation distribution in AID patients and to evaluate the impact of the genetic report on the patient’s medical care and treatment. The proportion of patients with pathogenic genotypes concordant with the AID phenotype was significantly higher after NGS than Sanger sequencing, and targeted NGS improved the diagnosis and global care of patients with AIDs.
NGS sequencing is known to perform better than conventional Sanger sequencing (i.e. one or a few genes), but this has never been demonstrated formally in patients with clinical suspicion of an AID. One clear exception is MEFV exon 10 sequencing in AID patients with recurrent fever and parents from an at‐risk ethnicity, which results in more than 70% genetic confirmation and is time‐ and cost‐efficient [15]. Therefore, we excluded Mediterranean patients with a clinical FMF and patients with undefined AID and with an initial NGS prescription from the study.
As expected, we demonstrated a clear enhancement of the diagnostic yield when switching from Sanger sequencing to NGS (4 versus 10%, P = 0·04). This result is consistent with our previous diagnostic yield estimation (10%) for patients directly referred to NGS [16]. At present, the Sanger technique remains cheaper and faster in expert laboratories, which are generally small‐ and medium‐sized. A way to improve the rate of confirmatory genetic diagnosis in AIDs could be a wider use of existing diagnostic criteria or discussions within multi‐disciplinary staff meetings. Preliminary evidence‐based classifications have been proposed for periodic fevers (FMF, MKD, CAPS and TRAPS) [17] and recently deficiency of adenosine deaminase 2 (DADA2) [18].
However, the performance of NGS seems to be lower than reported in the literature. The diagnosis rate approached 30% in two available studies [7, 19]. The design and inclusion criteria of these studies probably explain this apparent discrepancy. These studies considered VUS such as R92Q for TNFRSF1A or V198M for NLRP3 as confirmatory results [19]. If we consider uncertain significance genotypes, our results are very similar to the literature. However, in a more recent report, the yield was similar to ours [20].
There is a high prevalence in our cohort of MEFV diagnoses in the ‘consistent/confirmatory genotypes’ group (Sanger n = 3, NGS n = 2), although we aimed to exclude Mediterranean patients. However, these genotypes cannot account for the demonstrated superiority of NGs strategy in this study, as more of these patients were in the Sanger group.
A second objective of this study was to evaluate the distribution of genes and mutations in this population. We identified 16 different AID genes among the 53 patients harbouring at least one mutation (class ≥ 3). The most frequent were, in decreasing order of frequency, MEFV, NLRP3, NOD2, TNFRSF1A, ADA2 and MVK, PSTPIP1 and NLRC4. FMF, MKD, CAPS and TRAPS are often coined ‘the most common recurrent hereditary fevers’ because these are the ones whose implicated gene has been known for the longest time. In this study (Fig. 4a,b), we provide probably more accurate figures of the relative involvement of the genes present in our panel in unselected AID patients. Our results corroborate the rarity of monogenic AIDs in adult and paediatric populations. Among the four best‐known recurrent‐fever genes, MEFV was the most frequently mutated, even despite efforts to limit recruitment bias. ADA2 was the second most frequent (7·5%). Studies over time will tell whether this is a true score or a possible cohort effect, because DADA2 was discovered as a new AID during the time of this study. The three other recurrent fevers were extremely rare, as in previous reports. We did not identify any patient with CAPS, although 62% of the patients we included were under age 15 years at disease onset. This finding probably represents a cohort effect rather than defective technique sensitivity, because we had identified patients with as low as 6% NLRP3 mosaicism using the same NGS panel in previous analyses (unpublished personal data).
The third aim of this study was to collect information on treatment and patient follow‐up at least 6 months after receipt of the genetic results by the clinician to evaluate the benefit of genetic testing on medical care. Clinicians declared a benefit of this genetic test on global medical care for 59 of 128 (46·1%) patients, and more specifically the initiation of treatment [13 of 128 (10·2%)], but with no difference by strategy or final diagnosis. This medical care included initiation of targeted therapies, improved disease prognosis and better genetic counselling.
The limitation of this study is the low contribution of the clinicians to the survey, and the results must be considered preliminary. Aside from the expected lack of time of the clinicians, one explanation is possibly that therapeutic management is not clearly established for AIDs caused by the most recently identified genes.
In conclusion, we formally compared the diagnosis rate with conventional Sanger sequencing and NGS with a targeted gene panel in rare monogenic AIDs. We improved our confirmatory diagnoses from 4 to 10% (150%) with NGS high‐throughput sequencing. The gene distribution in our cohort confirms the genetic heterogeneity and rarity of AIDs but provides an initial refinement of their respective frequencies. Clinicians acknowledged a general beneficial effect of genetic diagnosis on patient care.
Disclosures
On behalf of all authors, the corresponding author states that there are no conflicts of interest.
Author contributions
G. S., I. K. P., T. M., S. A. and I. T. designed the study. G. S., G. B. and M. R. analysed the data. M. R. wrote the draft. G. S., I. K. P., G. B. and I. T. edited the manuscript.
Supporting information
Figure S1. Clinical form for requesting genetic diagnosis.
Figure S2. Questionnaire sent to the clinicians 6 months after the genetic report.
Table S1. Details of autoinflammatory disease genes included in the next‐generation sequencing panel version 1 and version 2.
Table S2. Clinical characteristics of patients with confirmatory or consistent genotype of autoinflammatory diseases by sequencing strategy (n = 18).
Table S3. Clinical characteristics of patients with uncertain genotype by sequencing strategy (n = 35).
Acknowledgements
This work was supported by an internal call ‘young researchers’ of the hospital university centre of Montpellier [NCT02976948]. We thank Laura Smales for English editing.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Orphanet [internet]. Available at: https://www.orpha.net/consor/cgi‐bin/Education_AboutRareDiseases.php?lng%A0=%A0FR&lng=EN. Accessed September 24, 2020.
- 2. French FMF Consortium . A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31. [DOI] [PubMed] [Google Scholar]
- 3. Sarrabay G, Barat‐Houari M, Annakib S, Touitou I. The autoinflammatory diseases: a fashion with blurred boundaries! Semin Immunopathol 2015; 37:359–62. [DOI] [PubMed] [Google Scholar]
- 4. de Jesus AA, Goldbach‐Mansky R. Genetically defined autoinflammatory diseases. Oral Dis 2016; 22:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. The International Society of Systems Auto Inflammatory Diseases (ISSAID) [internet]. Available at: https://infevers.umai‐montpellier.fr/Classification_AID/page1.html. Accessed September 24, 2020.
- 6. Russo RAG, Brogan PA. Monogenic autoinflammatory diseases. Rheumatology 2014; 53:1927–39. [DOI] [PubMed] [Google Scholar]
- 7. Omoyinmi E, Standing A, Keylock A et al Clinical impact of a targeted next‐generation sequencing gene panel for autoinflammation and vasculitis. PLOS ONE 2017; 12:e0181874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Illumina DesignStudio [internet]. Available at: https://www.illumina.com/informatics/sample‐experiment‐management/custom‐assay‐design.html. Accessed September 24, 2020.
- 9. Sure Design‐ Agilent Technologies‐ Genomics [internet]. Available at: https://earray.chem.agilent.com/suredesign/. Accessed September 24, 2020.
- 10. SeqNext Software – JSI medical system [internet]. Available at: http://www.jsi‐medisys.de/products/seqnext. Accessed September 24, 2020.
- 11. Richards S, Aziz N, Bale S et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet 2015; 17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Gijn ME, Ceccherini I, Shinar Y et al New workflow for classification of genetic variants’ pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). J Med Genet 2018; 55:530–7. [DOI] [PubMed] [Google Scholar]
- 13. Touitou I. Infevers, the registry of Hereditary Auto‐inflammatory Disorders Mutations [internet]. Available at: https://infevers.umai‐montpellier.fr/web/. Accessed September 24, 2020.
- 14. den Dunnen JT, Dalgleish R, Maglott DR et al HGVERSUS recommendations for the description of sequence variants: 2016 update. Hum Mutat 2016; 37:564–9. [DOI] [PubMed] [Google Scholar]
- 15. Touitou I. The spectrum of familial Mediterranean fever (FMF) mutations. Eur J Hum Genet EJHG 2001; 9:473–83. [DOI] [PubMed] [Google Scholar]
- 16. Boursier G, Rittore C, Georgin‐Lavialle S et al Positive Impact of expert reference center validation on performance of next‐generation sequencing for genetic diagnosis of autoinflammatory diseases. J Clin Med 2019; 8:1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Federici S, Sormani MP, Ozen S et al Evidence‐based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis 2015; 74:799–805. [DOI] [PubMed] [Google Scholar]
- 18. Rama M, Duflos C, Melki I et al A decision tree for the genetic diagnosis of deficiency of adenosine deaminase 2 (DADA2): a French reference centres experience. Eur J Hum Genet EJHG 2018; 26:960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao Q, Lacbawan F, Li J. Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic. Semin Arthritis Rheum 2016; 45:633–7. [DOI] [PubMed] [Google Scholar]
- 20. Karacan İ, Balamir A, Uğurlu S et al Diagnostic utility of a targeted next‐generation sequencing gene panel in the clinical suspicion of systemic autoinflammatory diseases: a multi‐center study. Rheumatol Int 2019; 39:911–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clinical form for requesting genetic diagnosis.
Figure S2. Questionnaire sent to the clinicians 6 months after the genetic report.
Table S1. Details of autoinflammatory disease genes included in the next‐generation sequencing panel version 1 and version 2.
Table S2. Clinical characteristics of patients with confirmatory or consistent genotype of autoinflammatory diseases by sequencing strategy (n = 18).
Table S3. Clinical characteristics of patients with uncertain genotype by sequencing strategy (n = 35).
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.