

TLR4 is usually not assumed to be involved in Mycoplasma recognition. In this study we showed that the Mycoplasma pneumoniae‐derived lipids did interact with TLR4 which subsequently initiates the activation of NLRP3 inflammasome and formation of a positive feedback loop between autophagy and NF‐κB signalling cascade, ultimately promoting proinflammatory cytokines production in macrophages.
Keywords: autophagy, lipid, Mycoplasma pneumonia, NF‐κB, NLRP3, Toll‐like receptor 4
Summary
Mycoplasma pneumoniae is an obligate pathogen that causes pneumonia, tracheobronchitis, pharyngitis and asthma in humans. It is well recognized that membrane lipoproteins are immunostimulants exerting as lipopolysaccharides (LPS) and play a crucial role in the pathogenesis of inflammatory responses upon M. pneumoniae infection. Here, we report that the M. pneumoniae‐derived lipids are another proinflammatory agents. Using an antibody‐neutralizing assay, RNA interference or specific inhibitors, we found that Toll‐like receptor 4 (TLR‐4) is essential for M. pneumoniae lipid‐induced tumour necrosis factor (TNF)‐α and interleukin (IL)‐1β production. We also demonstrate that NLR family pyrin domain containing 3 inflammasome (NLRP3) inflammasome, autophagy and nuclear factor kappa B (NF‐κB)‐dependent pathways are critical for the secretion of proinflammatory cytokines, while inhibition of TLR‐4 significantly abrogates these events. Further characterization revealed that autophagy‐mediated inflammatory responses involved the activation of NF‐κB. In addition, the activation of NF‐κB promoted lipid‐induced autophagosome formation, as revealed by assays using pharmacological inhibitors, 3‐methyladenine (3‐MA) and Bay 11‐7082, or silencing of atg5 and beclin‐1. These findings suggest that, unlike the response to lipoprotein stimulation, the inflammation in response to M. pneumoniae lipids is mediated by the TLR‐4 pathway, which subsequently initiates the activation of NLRP3 inflammasome and formation of a positive feedback loop between autophagy and NF‐κB signalling cascade, ultimately promoting TNF‐α and Il‐1β production in macrophages.
Introduction
Mycoplasma pneumoniae is an important pathogen of the human respiratory tract and can cause primary atypical pneumonia, tracheobronchitis, pharyngitis and asthma [1]. This pathogen causes upper and lower respiratory illnesses in all age groups and accounts for 4–8% of community‐acquired bacterial pneumonia cases in adults, presenting incidence rates ranging from 20 to 70% during epidemics of sporadic M. pneumoniae in China [2]. M. pneumoniae produces various virulent factors, such as membrane lipoproteins, polysaccharides and invasive nucleases, which cause a series of pathophysiological changes [3, 4, 5]. Because M. pneumoniae has no cell wall, its cell membrane is the major structure that interacts with the host [3]. Previous studies have shown that membrane lipoproteins (or lipid‐associated membrane proteins) of M. pneumoniae are important proinflammatory molecules [6, 7]. Classical membrane lipoproteins contain an N‐terminal diacylglycerol cysteine structure, and this particular lipid moiety is the basis for the proinflammatory activity following an M. pneumoniae infection [8]. The host immune system recognizes mycoplasmal lipoproteins mainly through Toll‐like receptor (TLR)‐2; specifically, through the formation of TLR‐2/‐6 and TLR‐1/‐2 heterodimers [9]. The activated TLRs can recruit adaptor molecules, such as myeloid differentiation primary response 88 (MyD88) and MyD88 adapter‐like protein (Mal) to its intracellular region, leading to the activation of mitogen‐activated protein kinase (MAPK), nuclear factor kappa B (NF‐κB) and activator protein 1 (AP‐1), which induce the production of several proinflammatory cytokines, chemokine, prostaglandins and other inflammatory mediators from monocytes, macrophages and natural killer (NK) cells in order to eliminate M. pneumoniae [10, 11, 12].
Although it is well recognized that the membrane lipoproteins are the main inflammation‐inducing components in M. pneumoniae infection, many aspects of this inflammatory response remain unexplained. The first intriguing issue comes from the fact that although some non‐pathogenic mycoplasma species have a classical diacylglycerol cysteine structure on the N‐terminal of their membrane lipoproteins, they cannot induce any host inflammatory response in vitro and in vivo [13].The second proof originates from a recent article by Takashi et al., who used live M. pneumoniae to infect TLR‐2‐deficient (TLR‐2−/−) macrophages, and unexpectedly observed that the M. pneumoniae could induce the TLR‐2−/− macrophages to secrete cytokines [14]; however, this cytokine‐inducing activity was diminished when both TLR‐2−/− and TLR‐4−/− macrophages were used, indicating that TLR‐4 might also participate in cytokine secretion during M. pneumoniae infection. This finding also signifies the existence of some active components on the M. pneumoniae cell membrane that can bind to TLR‐4 to trigger inflammatory pathways. However, the exact ligands of TLR‐4 that activate these pathways are still unknown.
It is well established that TLR‐4 recognizes lipopolysaccharides (LPS) of Gram‐negative bacteria as a ligand [15]. In addition to LPS, other lipids on the cell membrane of various pathogens have also been shown to recognize and bind TLR‐4 to induce the production of a variety of inflammatory mediators [16]. Due to the lack of a cell wall, mycoplasma cells do not have either LPS or lipid A, but their cell membrane is equipped with abundant lipids in the form of glycolipids or phosphoglycolipids, although the majority of their biological functions are poorly understood [17]. Studies showed that some mycoplasma‐derived lipids are not only involved in the adhesion of mycoplasma and invasion of host cells, but are also important inflammatory agents for activating monocytes and macrophages [17]. For example, glycolipid MfGL‐I and MfGL‐II of M. fermentans can up‐regulate the secretion of TNF‐α, nitric oxide (NO) and prostaglandin E2 by monocytes or glial cells [18, 19, 20]. In addition, mpn483 in the M. pneumoniae genome can encode a glycosyltransferase to catalyse the synthesis of various glycolipids and is closely related to the occurrence of Guillain–Barré syndrome (a peripheral multiple neuropathy syndrome), which occurs in children and young people [21]. Taken together, these findings suggest that mycoplasmal lipids may be important pathogenic components that influence host immune functions through unknown mechanisms. However, the functions of M. pneumoniae‐derived lipids have not been fully characterized.
In the present study, we demonstrate that exposure of mouse macrophages to M. pneumoniae lipids activates the TLR‐4 signal and increases the production of tumour necrosis factor (TNF)‐α and interleuklin (IL)‐1β mediated by nuclear factor kappa B (NF‐κB) and autophagy. We further demonstrated that autophagy promotes inflammatory responses through the regulation of NF‐κB signalling and that the activation of NF‐κB, in turn, promotes autophagosome formation. This positive feedback loop between autophagy and NF‐κB may play an important role in lipid‐induced inflammatory responses.
Materials and Methods
Reagents and antibodies
Macrophage‐activating lipopeptide 2 (MALP‐2) was purchased from Enzo Lifesciences (ALX‐162‐027‐C050; Plymouth Meeting, PA, USA). Phosphate‐buffered saline (PBS), Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from gibco (Grand Island, NY, USA). Rabbit monoclonal antibodies against TLR‐2, TLR‐4, LC3B (D11, no. 3868), Beclin‐1, Atg5, p65, caspase‐1, ASC (apoptosis associated speck‐like protein containing a CARD) and β‐actin, and horseradish peroxidase (HRP)‐conjugated secondary antibodies were products of Cell Signaling Technology (Danvers, MA, USA). Anti‐TLR‐4 and anti‐TLR‐2 neutralizing antibodies were obtained from Novus (Littleton, CO, USA). Fluorescein isothiocyanate (FITC) and Alexa 488‐conjugated secondary antibody was purchased from Abcam (Cambridge, UK). H2DCFH‐DA, 3‐methyladenine (3‐MA), chloroquine (CQ), Bay11‐7821, LPS and N‐acetyl‐l‐cysteine (NAC) were procured from Sigma‐Aldrich (St Louis, MO, USA).
M. pneumoniae culture and lipids extraction
The M. pneumoniae strain, M129 (ATCC 29342), was purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured at 37°C in pleuropneumonia‐like organism (PPLO) broth (BD Biosciences, San Jose, CA, USA) containing 15% FBS, 0·25% glucose, 0·25% yeast extract (Oxoid), 1000 U/ml penicillin G and 0·005% phenol red at pH 7.6 for approximately 5–7 days until the medium colour changes from red to orange. Lipids were extracted as described previously [22, 23]. Briefly, the organisms were harvested by centrifugation at 10 000 g for 20 min at 4°C and washed three times with PBS. Next, the pellets were resuspended in chloroform : methanol (2 : 1) for 18 h at 25°C; the supernatants thus obtained were harvested by centrifugation (6000 g for 20 min at 4°C) and the extracts were dried using a rotary evaporator. Subsequently, each dried residue was redissolved in chloroform : methanol (2 : 1) and the non‐lipid contaminants in the preparation were removed employing a column of Sephadex G‐25 with chloroform : methanol (19 : 1, water‐saturated fraction) [24]. Later, the eluate was dried using a rotary evaporator, and the remaining residue was designated as ‘M. pneumoniae lipid’. The extracted lipids were subjected to thin‐layer chromatography and then verified by iodine vapour as described previously [21]. Finally, endotoxins present in the lipids were assessed using a PyroSep™ Endotoxin Specific Measurement Kit (Fujifilm, Japan), according to the manufacturer’s instructions.
Cell culture and stimulation with lipids
The mouse macrophage cell line RAW264.7 was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM supplemented with 10% heat‐inactivated FBS, 2 mmol/l L‐glutamine and 100 μg/ml penicillin/streptomycin. Bone marrow‐derived macrophages (BMDMs) were obtained from the femurs and tibias of C57BL/6 mice and cultured for 6–8 days, as described earlier [25]. Cells were maintained in a humidified atmosphere at 37°C and 5% CO2. For stimulation experiments, macrophages were seeded in serum‐free medium in 24‐ or six‐well plates (1 × 106/ml) and allowed to grow overnight. The cells were stimulated with lipids at various concentrations for the indicated time intervals.
Lactate dehydrogenase (LDH) analysis for evaluating cytotoxicity
LDH released from lipid‐treated cells were measured for the estimation of cytotoxicity. The cells were seeded into a 96‐well plate and incubated with different concentration of lipids for 24 h. Next, the supernatants were collected and assayed via an LDH Cytotoxicity Assay Kit (Promega, Madison, WI, USA), as recommended by the manufacturer. LDH leakage was calculated as the ratio of treatment‐induced LDH to maximum LDH release according to the following formula: cytotoxicity (%) = experimental LDH release/maximum LDH release × 100%.
Determination of cytokines by enzyme‐linked immunosorbent assay (ELISA)
After stimulation, cell culture supernatants were collected by centrifugation. Concentrations of TNF‐α and IL‐1β were measured using a commercial ELISA kit (eBioscience, CA, USA), according to the manufacturer’s instructions.
Immunofluorescence staining for LC3 puncta, ASC (apoptosis associated speck‐like protein containing a CARD)‐speck and nuclear translocation of NF‐κB
The macrophages were seeded onto coverslips in a 24‐well plate, and fixed and permeabilized as described previously [26]. After the samples were incubated in a blocking buffer for 1 h at room temperature, the cells were incubated with primary antibodies for LC3B, ASC or p65 overnight at 4°C, followed by incubation with Alexa 488‐conjugated antibody for another 1 h. Nuclei were stained with 1 μg/mL 4′‐6‐diamidino‐2‐phenylindole (DAPI) for 15 min at 37°C, and images were obtained using an immunofluorescence microscope (Ts2R; Nikon, Tokyo, Japan).
Western blot analysis for TLRs, LC3B, Beclin‐1 and Atg5 expression
Cell lysates were prepared in RIPA buffer and the total protein concentration was measured using a BCA Protein Assay Reagent kit (Pierce/ThermoFisher Scientific, Waltham, MA, USA). Equal amounts of the total proteins were separated using sodium dodecyl sulphate–polymerase chain reaction (SDS‐PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Burlington, MA, USA). After blocking in bovine serum albumin (BSA) for 1 h at room temperature, the membrane was incubated with specific primary antibodies for TLR‐2, TLR‐4, LC3B, Beclin‐1, Atg5 and β‐actin (1 : 1000) overnight at 4°C, after which the membrane was incubated with HRP‐conjugated secondary antibodies (1 : 2000) for 2 h at 37°C. Finally, the immunoreactive bands were detected by the ECL system (G: BOX XX8; Syngene, Frederick, MD, USA), and the band intensity was measured by densitometric analysis using Image J software.
Quantitative real‐time PCR
Total RNA was isolated using TRIzol reagent (Life Technologies, Paisley, UK), according to the manufacturer’s instructions. The cDNA obtained from 0·5 μg total RNA was used as a template for PCR amplification, as previously described. Real‐time PCR was performed using GoTaq® quantitative PCR (qPCR) Master Mix (Promega) in the LightCycler 96® instrument (Roche, Basel, Switzerland). All experiments were carried out in triplicate. Data were analyzed with the 2−ΔΔCq method [27]. The specific primer sequences were designed as follows: NLRP3 forward: 5′‐CCTACCCAAACCCACCAGT‐3′ and reverse: 5′‐TTCTTTCGGATGAGGCTGCTTA‐3′; and β‐actin forward: 5′‐CCCATCTATGAGGGTTACGC‐3′ and reverse: 5′‐TTTAATGT CACGC ACG ATTTC‐3′.
Measurement of intracellular reactive oxygen species (ROS) accumulation
The intracellular ROS level was determined using H2DCFH‐DA molecular probes, following the manufacturer’s instructions (Sigma‐Aldrich). Briefly, treated macrophages were incubated with 10 mM H2DCFH‐DA for 35 min at 37°C, then washed thrice with PBS. Fluorescence intensity was calculated by immunofluorescence microscopy (Ts2R; Nikon).
Transient transfection with small interfering siRNAs
Specific RNAs (siRNAs) of TLR‐2, TLR‐4, beclin‐1 and atg5 were synthesized by RiboBio Co. Ltd). Macrophages were cultured in six‐well plates until the cells reached a confluence of 30–50%, after which the cells were transfected with a specific siRNA using a transfection reagent according to the manufacturer’s instructions (RiboBio Co. Ltd). The medium was replaced by complete DMEM medium after 6 h of incubation. Subsequently, the cells were incubated for 48 h at 37°C and the silencing efficiency was determined by Western blot analyses.
Statistical analysis
All experiments were repeated three times independently, and the data are represented as mean ± standard error of the mean (s.e.m). spss version 18.0 software was used for statistical analysis, and one‐way analysis of variance followed by Tukey’s post‐hoc test was utilized to compare different groups. P < 0·05 was considered statistically significant.
The data that support the findings of this study are available in the Supporting information of this article.
Results
M. pneumoniae lipids induce macrophages to secrete proinflammatory cytokines
It has been demonstrated that the active immune‐stimulating components of mycoplasma are the membrane‐bound lipoproteins [28]. To determine the effect of M. pneumoniae‐derived lipids on proinflammatory cytokine expression, macrophages were stimulated with different concentrations of the lipids for 24 h, and subsequently the secretion of proinflammatory cytokines TNF‐α and IL‐1β were measured by ELISA. The results demonstrated that the lipids could not significantly induce the production of TNF‐α and IL‐1β at concentrations of less than 30 μg/ml. However, the concentrations of TNF‐α and IL‐1β were significantly increased when the lipids concentrations exceeded 30 μg/ml (Fig. 1a,b). To rule out the possibility that the cytokine‐inducing activity is evoked by the non‐lipid contaminants, the lipids were digested by proteinase K, nuclease or lipase, respectively. As expected, treatment with proteinase K or nuclease failed to decrease the lipid‐induced TNF‐α and IL‐1β production. By contrast, treatment with lipase apparently abolished the lipid‐induced cytokine response (Supporting information, Figs. S1–S3). Subsequently, to exclude the subsequent cytotoxic effect of lipids on the macrophages, LDH leakage in response to 0~150 μg/ml of lipids was assayed. Figure 1c shows that 0~120 μg/ml of lipids did not exert a cytotoxicity effect, but a high concentration of lipids (≥ 150 μg/ml) could significantly increase the LDH leakage rate during 24 h compared to the vehicle control. The results demonstrate that lipids within the range of 0~120 μg/ml can induce macrophages to produce TNF‐α and IL‐1β in a concentration‐dependent manner.
Fig. 1.

M. pneumoniae‐derived lipids induce generation of tumour necrosis factor (TNF)‐α and interleukin (IL)‐1β from macrophages. (a,b) Seeded bone marrow‐derived macrophages (BMDMs) were either stimulated with lipids (1, 10, 30, 60 and 120 μg/ml) for 24 h or left unstimulated [100 ng/ml lipopolysaccharide (LPS) and 5 ng/ml macrophage‐activating lipopeptide 2 (MALP‐2) were used as positive controls]. The cell culture supernatants were collected and the concentrations of TNF‐α (a) and IL‐1β (b) were determined by enzyme‐linked immunosorbent assay (ELISA). (c) Cells were stimulated by 1, 10, 30, 60, 120 and 150 μg/ml of lipids for 24 h; lactate dehydrogenase (LDH) leakage was assayed in the supernatants and cytotoxicity was expressed as a percentage of LDH release, determined by colorimetry. The data represent three independent experiments [mean ± standard error of the mean (s.e.m.)]; *P < 0·05 compared with the negative group.
Requirement of TLR‐4 for M. pneumoniae lipid‐induced proinflammatory cytokine secretion in macrophages
TLRs play an important role in the recognition of mycoplasma by the host and then initiating a rapid defensive mechanism, and it has been demonstrated that both TLR‐2 and TLR‐4 are expressed on bone marrow‐derived macrophages [29, 30]. To investigate whether the lipid‐induced proinflammatory cytokine production was mediated through these TLRs, macrophages were pretreated with 10 μg/ml of anti‐TLR‐2 or TLR‐4 neutralizing antibody for 1 h, and then stimulated with 120 μg/ml lipids for another 24 h. The results showed that the TLR‐2 agonist MALP‐2– induced TNF‐α and IL‐1β secretion was inhibited by pretreatment with anti‐TLR‐2 neutralizing antibody; however, no significant effects were observed on LPS and lipids (Fig. 2a). In contrast, macrophages pretreated with anti‐TLR‐4 neutralizing antibodies markedly inhibited lipid‐ and LPS‐induced proinflammatory cytokine production, but there was no significant impact on MALP‐2 stimulated cells (Fig. 2b), thereby indicating that TLR‐4 rather than TLR‐2 is required for lipid‐induced proinflammatory responses. This hypothesis was further supported by the finding that transfection of TLR‐4 small interfering RNA (siTLR‐4) significantly attenuated TNF‐α and IL‐1β production induced by lipids, but transfection of siTLR‐2 did not. The results suggest that production of the TNF‐α and IL‐1β induced by the M. pneumoniae lipids in macrophages depends upon the action of TLR‐4.
Fig. 2.

M. pneumoniae lipid‐induced proinflammatory cytokine production in macrophages is mediated via Toll‐like receptor (TLR)‐4. (a,b) Bone marrow‐derived macrophages (BMDMs) were treated with 10 μg/ml of anti‐TLR‐2 (a) or anti‐TLR‐4 (b) neutralizing antibody at 37°C for 1 h, and then treated with lipids (120 μg/ml), lipopolysaccharide (LPS) (100 ng/ml) or macrophage‐activating lipopeptide 2 (MALP‐2) (5 ng/ml) for 24 h. The secreted levels of tumour necrosis factor (TNF)‐α and interleukin (IL‐1β) were determined by enzyme‐linked immunosorbent assay (ELISA). (c) BMDMs cells were transfected with TLR‐2, TLR‐4 or scrambled small interfering RNA (siRNA) and the interference efficiency was detected by Western blot (upper panel). Following stimulation with lipids (120 μg/ml) for 24 h, the concentrations of TNF‐α and IL‐1β were determined by ELISA. The data represent three independent experiments [mean ± standard error of the mean (s.e.m.)]; *P < 0·05 denotes significant difference between the compared groups.
M. pneumoniae lipids induce proinflammatory cytokine production via NF‐κB activation
NF‐κB has been shown to regulate the expression of proinflammatory cytokines in human monocytes and macrophages [31, 32]. To investigate whether the NF‐κB plays a role in lipid‐induced cytokines responses, macrophages were treated with lipids for 6 h. Immunofluorescence results showed that lipids could induce the translocation of NF‐κB p65 subunit from the cytosol to the nucleus (Fig. 3a,b), accompanied by down‐regulation of inhibitor of NF‐κB (IκB) (Fig. 3c). Pretreatment with an anti‐TLR‐2 neutralizing antibody did not have a significant effect on the nuclear translocation of NF‐κB p65 subunit and expression of IκB; by contrast, the anti‐TLR‐4 neutralizing antibody could significantly inhibit the nuclear translocation of p65 and degradation of IκB. Moreover, pretreatment of cells with the NF‐κB inhibitor Bay 11‐7082 drastically attenuated the lipid‐induced generation of TNF‐α and IL‐1β (Fig. 3d). Taken together, these findings demonstrate that the induction of proinflammatory cytokine secretion depends upon NF‐κB activation.
Fig. 3.

Nuclear factor kappa B (NF‐κB) is required for Mycoplasma pneumoniae lipid‐induced tumour necrosis factor (TNF)‐α and interleukin (IL)‐1β secretion. RAW264.7 cells (2 × 106 cells/ml) were cultured and separately preincubated with anti‐Toll‐like receptor (TLR‐2) or anti‐TLR‐4 neutralizing antibody (10 μg/ml) for 1 h on coverslips in a 24‐well plate, following treatment with lipids for 6 h. Immunofluorescence was used to detect the translocation of NF‐κB (a) and data were quantified by counting more than 50 cells in triplicate (b) or the cell lysates were subjected to Western blot analysis using anti‐inhibitor NF‐κB (IκB) antibody (c). (d) Bone marrow‐derived macrophages (BMDMs) were pretreated with 5~10 μmol/l of the NF‐κB inhibitor Bay 11‐7082 for 40 min. Later, enzyme‐linked immunosorbent assay (ELISA) was used to determine the concentration of TNF‐α and IL‐1β before and after treatment with lipids. The data represent three independent experiments [mean ± standard error of the mean (s.e.m.)]; *P < 0·05 denotes significant difference between the compared groups.
M. pneumoniae lipids enhance intracellular ROS accumulation to trigger NLRP3 inflammasome activation
Several lines of evidence have indicated that the NLRP3 inflammasome is a critical component of the innate immune system, which contributes to induce IL‐1β production upon M. pneumoniae infection, and that ROS is an endogenous activator for NLRP3 inflammasome activation [33, 34]. Therefore, we investigated whether NLRP3 inflammasome and ROS were involved in IL‐1β secretion induced by M. pneumoniae lipids. As shown in Fig. 4a, M. pneumoniae lipids induced the expression of NLRP3 mRNA in a dose‐dependent manner; however, the induction of mRNA was abolished by Bay 11‐7082, an inhibitor of NF‐κB, indicating that NF‐κB is involved in M. pneumoniae lipid‐induced NLRP3 up‐regulation. Considering that NLRP3 inflammasome is activated by ROS under certain conditions, we examined whether activation of NLRP3 occurs via ROS formation. To accomplish this, we measured ROS generation using H2DCFH‐DA, a reagent that is oxidised into the fluorescent molecule dichlorofluorescein (DCF) by ROS and can be readily measured by fluorescence microscopy. Figure 4b clearly demonstrates that treatment of macrophages with M. pneumoniae lipids induced a significantly increase in ROS levels, while pretreatment with anti‐TLR‐4 neutralizing antibody suppressed the ROS production, but anti‐TLR‐2 neutralizing antibodies exhibited no such effect. These results indicated that the up‐regulation of intracellular ROS levels by M. pneumoniae lipids is mediated by TLR‐4. Conversely, we observed markedly increased caspase‐1 activation by these lipids, as revealed by Western blot analysis that quantified the active p10 subunit of caspase‐1, a domain indicator of the conversion of pro‐caspase‐1 to enzymatically active mature caspase‐1. Conversely, an ROS scavenger, NAC, significantly abrogated the lipid‐induced caspase‐1 activation (Fig. 4c). Moreover, we also observed that treatment with M. pneumoniae lipids could reorganize the cytoplasmic NLRP3 complex formation of the punctate structures (ASC specks); however, pretreatment of the cells with NAC markedly decreased the number of ASC specks by converting them into single specks (Fig. 4d,e). These data are in agreement with the decrease in IL‐1β production in NAC‐pretreated macrophages (Fig. 4f). Collectively, these results demonstrate that M. pneumoniae lipid‐induced IL‐1β production is mediated through the ROS‐triggered NLRP3 inflammasome activation.
Fig. 4.

M. pneumoniae lipids activate NLR family pyrin domain containing 3 inflammasome (NLRP3) inflammasome to induce interleukin (IL‐1β) secretion. (a) Bone marrow‐derived macrophages (BMDMs) were cultured in 24‐well plates and treated with a nuclear factor kappa B (NF‐κB) inhibitor Bay 11‐7082 for 40 min, and then stimulated with lipids for 18 h; transcriptional levels of NLRP3 mRNA were detected by quantitative real‐time polymerase chain reaction (PCR). (b) BMDMs were pretreated with anti‐Toll‐like receptor (TLR)‐2 or anti‐TLR‐4 neutralization antibody for 1 h. Later, the cells were stimulated with lipids for 12 h and labelled with H2DCFDA at 37°C for 30 min. The intracellular fluorescence intensity was measured using an immunofluorescence microscope (×40). (c–e) BMDMs were pretreated with 10 mmol/l N‐acetyl‐l‐cysteine (NAC) for 4 h, and then stimulated with 120 μg/ml lipids for 6 h. (c) The expression of the p10 domain of the active caspase‐1 was measured by Western blot; the formation of ASC (apoptosis associated speck‐like protein containing a CARD) specks (×100) was captured by fluorescence microscopy (d), the number of ASC specks was counted over 100 cells in each group (e); the levels of IL‐1β in the supernatants were detected by enzyme‐linked immunosorbent assay (ELISA) (f). The results are representative of similar findings from at least three separate experiments. *P < 0·05 denotes significant differences between the compared groups.
Autophagy is involved in M. pneumoniae lipid‐induced proinflammatory cytokine production
Autophagy has been shown to participate in the induction of inflammatory responses upon live M. pneumoniae infection in TLR‐2−/− macrophages [14]. To determine whether autophagy is necessary for lipid‐induced proinflammatory cytokine generation, we investigated the effect of the lipids on macrophage autophagy. Immunofluorescence results showed that LC3 puncta were significantly increased in macrophages following stimulation with the lipids for 6 h (Fig. 5a,b). In addition, Western blot demonstrated that incubation with the M. pneumoniae lipids also increased the expression of autophagic marker proteins, such as LC3B, Beclin‐1 and Atg5, thereby confirming the induction of autophagy by lipids (Fig. 5c). To further determine whether TLR‐4 was required for lipid‐induced autophagy, RAW264.7 cells were pretreated with the corresponding neutralizing antibodies. As speculated, the formation of LC3 puncta was blocked after anti‐TLR‐4 neutralizing antibody pretreatment, while no significant change was obtained for TLR‐2 antibody (Fig. 5a). To rule out the possibility that increased LC3B was attributable to lysosomal dysfunction caused by LC3B accumulation, we next blocked autophagosome–lysosome fusion using chloroquine (CQ), an inhibitor of lysosome acidification [35]. As shown in Fig. 5d, pretreatment with CQ further promoted the accumulation of LC3B in the lipid‐stimulated RAW264.7 cells, which excluded the possibility of decreased lysosomal fusion and degradation. The lipids could also down‐regulate the multifunctional autophagic degradation molecule, p62, while blocking the autophagosome–lysosome fusion with CQ rescued p62 levels (Fig. 5e). These results confirm that the lipids promote autophagic degradation. To further ensure the role of autophagy in lipid‐induced inflammatory responses, macrophages were pretreated with 3‐MA, an inhibitor of autophagy, by blocking autophagosome formation via inhibition of the classic III PI3K [36]. As shown in Fig. 5f, preincubation with 3‐MA effectively attenuated the induction of TNF‐α and IL‐1β. This down‐regulation of the proinflammatory cytokines was further confirmed by knockdown of atg5 or beclin‐1 by siRNA (Fig. 5g). These results suggest that TLR‐4/autophagy pathway played an essential role in M. pneumoniae lipid‐induced proinflammatory response.
Fig. 5.

Autophagy mediated the production of cytokines induced by M. pneumoniae lipids in macrophages. (a) RAW264.7 cells were seeded on 24‐well plates and were pretreated with anti‐Toll‐like receptor (TLR)‐2 or anti‐TLR‐4 neutralizing antibodies for 1 h, followed by incubation with the lipids (120 μg/ml) for 6 h. Immunofluorescence was used to detect the formation of LC3 puncta using anti‐LC3 antibody and fluorescein isothiocyanate (FITC)‐labelled secondary antibody (green, ×100), and nuclei of the macrophages were stained with 4′‐6‐diamidino‐2‐phenylindole (DAPI) (blue). (b) The number of LC3 puncta in each cell was counted over 100 cells in each group. (c) Cells were harvested at 6h after stimulation with different concentrations of lipids (30, 60 and 120 μg/ml), and the expression of LC3B, Beclin‐1 or Atg5 was detected by Western blot. *P < 0·05 denotes significant differences compared with the negative group. (d,e) RAW264.7 cells were pretreated with chloroquine (20 μmol/l) for 1 h, followed by 6 h of incubation with the lipids, and the expression of LC3 (d) or p62 (e) was measured using Western blot. (f) Bone marrow‐derived macrophages (BMDMs) were pretreated with 5 mmol/l 3‐methyladenine (3‐MA) for 1 h, and then stimulated by the lipids for 24 h. The tumour necrosis factor (TNF)‐α and interleukin (IL‐1β) concentrations in the culture medium were measured by enzyme‐linked immunosorbent assay (ELISA). (g) BMDMs were transfected with atg5 and beclin‐1 small interfering RNA (siRNA) for 48 h after 24 h incubation with the lipids (120 μg/ml). The TNF‐α and IL‐1β concentrations in the cell culture medium were measured using ELISA. The data represent three independent experiments [mean ± standard error of the mean (s.e.m.)]; *P < 0·05 denotes significant difference between the compared groups.
M. pneumoniae lipids induce a positive feedback loop between autophagy and NF‐κB in macrophages
As autophagy and NF‐κB play crucial roles in M. pneumoniae lipid‐induced proinflammatory response, we were interested in investigating the signalling cascade between autophagy and NF‐κB. To accomplish this, RAW264.7 cells were pretreated with 3‐MA, and subsequently the nuclear translocation of NF‐κB was evaluated by immunofluorescence. Results showed that 3‐MA significantly attenuated the cytoplasmic NF‐κB translocation to the nucleus after treatment with M. pneumoniae lipids (Fig. 6a,b). To further block the autophagic pathway, autophagy‐related genes, atg5 or beclin‐1, were eliminated with siRNA. The results showed that the knockdown of atg5 or beclin‐1 dramatically abrogated the lipid‐triggered NF‐κB activation (Fig. 6c,d). These data indicate that the lipid‐activated NF‐κB signalling, at least in part, is autophagy‐dependent. Notably, the administration of the NF‐κB inhibitor Bay 11‐7082 also significantly abolished lipid‐induced LC3B expression (Fig. 6e) and puncta formation (Fig. 6f,g). These data imply a positive feedback loop between autophagy and NF‐κB signalling pathway in M. pneumoniae lipid‐induced macrophages.
Fig. 6.

Nuclear factor kappa B (NF‐κB) participated in M. pneumoniae lipid‐induced autophagy. (a) RAW264.7 cells were pretreated with 5 mmol/l autophagy inhibitor, 3‐methyladenine (3‐MA), for 1 h and subsequently treated with the lipids (120 μg/ml) for 6 h. Immunofluorescence was performed to detect p65 subunit nuclear translocation of NF‐κB (×100). (b) Data were quantified by counting more than 50 cells in triplicate. (c) RAW264.7 cells were transfected with atg5 and beclin‐1 small interfering RNA (siRNA) for 48 h and then treated with the lipids (120 μg/ml) for 6 h. Immunofluorescence assay was performed to detect NF‐κB nuclear translocation (×100), and data were quantified by counting more than 50 cells in triplicate (d). (e) RAW264.7 cells were pretreated with NF‐κB inhibitor, Bay 11‐7082 (30 μmol/l) for 40 min followed by stimulation with the lipids (120 μg/ml) for 6 h; expression of LC3B and the LC3 puncta was measured by Western blot and immunofluorescence (f,g), respectively. The data shown represent three independent experiments.
Discussion
It is generally recognized that membrane‐bound lipoproteins of M. pneumoniae play a key role in mycoplasma‐induced inflammatory responses mediated by TLR‐2 both in vitro and in vivo [37]. However, this is not always the case. In the current study, we extracted lipids from M. pneumoniae membranes and investigated their inflammation‐inducing activity using macrophages. The present work provides three major findings. First, M. pneumoniae lipids can induce macrophages to secrete TNF‐α and IL‐1β, and this cytokine‐inducing activity is dependent of TLR‐4 but not of TLR‐2. Secondly, TLR‐4‐mediated autophagy is involved in inflammatory responses through NF‐κB signalling. Moreover, activation of NF‐κB also promotes lipid‐induced autophagy. Finally, activated TLR‐4 enhances intracellular ROS accumulation in co‐operation with NF‐κB to promote expression and assembly of NLRP3 inflammasome, ultimately promoting maturation and secretion of IL‐1β. Therefore, these findings are an important supplement to the traditional perspective that the innate immune system recognizes mycoplasma only via TLR‐2/‐6 or TLR‐1/‐2, and offers a new pathogenic mechanism for M. pneumoniae.
TLR‐2 is a predominant pattern recognition receptor, which recognizes invading M. pneumoniae by forming a heterodimer with TLR‐1 or TLR‐6 [37, 38]. It is generally believed that TLR‐4 recognizes LPS of Gram‐negative bacteria and does not play a role in M. pneumoniae recognition. However, some unexpected findings by Takashi’s group confirm that TLR‐4 is also involved in the induction of proinflammatory cytokines in response to live M. pneumoniae in TLR‐2 knock‐out macrophages [14]. These data suggest that potent proinflammatory moieties exist on the surface of M. pneumoniae, which serve as ligands for TLR‐4. Considering that LPS recognized by TLR‐4 are composed of lipids, we could reasonably speculate that the M. pneumoniae‐derived lipids may be potential ligands of TLR‐4. Based on this fact, we used a chloroform/methanol method to extract lipids from M. pneumoniae cell membrane and performed an in‐depth analysis of their effects on inflammatory response. Our results clearly showed that low doses of such lipids could not significantly induce the secretion of TNF‐α and IL‐1β in macrophages; however, the cytokines increased significantly when the lipids concentration increased to nearly 60 μg/ml. This could be explained by the possibility that the affinity between the lipids and TLR‐4 is relatively low. This hypothesis is in accordance with Takashi’s findings [14]. In their report they found that, although M. pneumoniae deficient in cytoadherence could not induce the generation of the TNF‐α in the TLR‐2 knock‐out macrophages, macrophages could still be induced to produce cytokines with a prolonged time of infection or when a high concentration of M. pneumoniae mutant strain was administered. It appears that cytoadherence per se did not induce a cytokine response in the TLR‐2 knock‐out macrophages; however, it could enhance the contact between M. pneumoniae and TLR‐4, resulting in the activation of TLR‐4‐mediated inflammatory pathway [14]. As expected, the anti‐TLR‐2 and siRNA administration did not affect M. pneumoniae lipid‐induced cytokines, but markedly abrogated MALP‐2‐induced cytokine production, because TLR‐2 acts as the unique receptor for MALP‐2. In addition, blocking of TLR‐4 significantly reduced the M. pneumoniae lipid‐induced inflammatory responses. Based on the above results, we can conclude that M. pneumoniae lipid‐induced secretion of proinflammatory cytokines is mediated by TLR‐4. Nevertheless, our study still lacks direct evidence to propose that TLR‐4 is a receptor for M. pneumoniae‐derived lipids, as the crude extract of these lipids was a mixture of neutral lipids (triglycerides, diglycerides and cholesterol), intermediate (glycolipids) polarity and polar lipids (phosphatides) [39]. Hence, the exact molecule serving as a ligand for TLR‐4 needs to be further confirmed.
When TLR‐4 is activated it can recruit the adaptor proteins, Mal and MyD88, to the intracellular Toll/interleukin‐1 receptor (TIR) domain and ultimately activate the MAPKs and NF‐κB to regulate the expression of inflammation‐related genes [40, 41]. The most common form of NF‐κB consists of a homodimer or heterodimer of RelA (p65) and p50 [42]. Under physiological conditions, NF‐ κB binds to its inhibitor and is located in the cytoplasm. After activation of extracellular signal cascades, such as TLR‐4, it can cause serine phosphorylation of the regulatory site of IκB subunit, which leads to degradation of the IκB subunit by ubiquitination modification, thus releasing NF‐κB dimer to translocate to the nucleus [43, 44]. In this study, we also demonstrated that the lipids induce nuclear translocation of NF‐κB accompanied by degradation of IκB. Inhibition of TLR‐4 significantly weakened the lipid‐induced NF‐κB activation, but inhibition of TLR‐2 did not. These results suggest that the activation of NF‐κB by M. pneumoniae‐lipids is through TLR‐4‐ rather than the TLR‐2‐dependent pathway. In addition, ELISA results revealed that the lipid‐induced expressions of TNF‐α and IL‐1β were significantly reduced when the cells were pretreated with the NF‐κB inhibitor. Taken together, these results suggest that M. pneumoniae lipid‐induced secretion of TNF‐α and IL‐1β is dependent upon TLR‐4/NF‐κB.
An inflammasome represents a multi‐protein complex that protects the host against the invading pathogens through induction of proinflammatory factors, among which the NLRP3 inflammasome plays the most important role [25, 45, 46]. The NLRP3 inflammasome complex is comprised of NLRP3, ASC and caspase‐1 [45]. Under a resting state, the expression of NLRP3 is very low in mammalian cells. Activation of NLRP3 is decided by two signals. The first signal requires the expression of pro‐IL‐1 protein and NLRP3 mRNA through the stimulation of pattern recognition receptors, such as TLR‐4, and this process usually requires the involvement of NF‐κB. Once the pro‐IL‐1 protein undergoes translational and post‐translational modification, a second signal is required for the inflammasome complex assembly, such as potassium efflux, ROS and intracellular ATP [45, 47].These factors lead to NLRP3‐ASC oligomerization and recruit caspase‐1 to the complex. The recruited caspase‐1 subsequently undergoes autocatalysis and produces p10 and p20 heterodimers, which generate enzymatically active caspase‐1 to cleave the precursor pro‐IL‐1 into its mature secreted form [45, 47]. Our results clearly confirm that M. pneumoniae lipids induce the NLRP3 mRNA transcription by NF‐κB, and formation of characteristic ASC specks, accompanied with an increase in the active p10 subunits of caspase‐1. These data suggest that the lipids trigger the NLRP3 inflammasome complex assembly after NF‐κB activation, because TLR‐4 can activate intracellular tyrosine protein kinases such as Btk and NADPH oxidase. The latter is an important enzymatic source for the production of ROS under pathological conditions. Therefore, we further investigated the role of ROS associated with NLRP3 activation in response to M. pneumoniae lipids. We found that the levels of intracellular ROS significantly increased when treated with the lipids. The formation of ROS was not affected by pretreatment with TLR‐2 neutralizing antibody, but was significantly reduced after TLR‐4 antibody was administered. In addition, pretreatment with the ROS scavenging agent, NAC, significantly retarded the lipid‐induced caspase‐1 activation and ASC speck formation as well as reduced IL‐1β secretion, suggesting that NLRP3 was involved in lipid‐induced IL‐1β maturation and secretion. Nevertheless, we still cannot rule out the possibility that the M. pneumoniae lipids enter the cytoplasm through the mechanism of endocytosis after being recognized by TLR‐4 and then activate NLRP3 through other mechanisms, or both, because it has been found that the M. salivarium‐derived lipopeptide FSL‐1 could enter the macrophages through an internalization mechanism, and the endosomes thus formed could interact with lysosomes to activate NLRP3 [48]. In future, we will further investigate the activation mechanism of NLRP3 following administration of M. pneumoniae lipids.
Another novel aspect of our studies is that an autophagic pathway is involved in the lipid‐induced cytokines production [49]. Autophagy is an important way to combat infections by pathogenic micro‐organisms [50]. Previous studies have demonstrated that a wild‐type M. pneumoniae infection could induce autophagy in macrophages through TLR‐4 [14]. Our study confirmed that the lipids also induce the formation of LC3 puncta and expression of LC3B, Beclin‐1 and Atg5. The autophagy response was not affected when the cell was pretreated with anti‐TLR‐2 neutralizing antibody; however, it was affected by the anti‐TLR‐4 neutralizing antibody, indicating that the M. pneumoniae lipid‐induced autophagy is TLR‐4‐dependent. Activation of TLR‐4‐mediated autophagy in the lipid‐stimulated macrophages compelled us to test whether autophagy is linked to inflammatory response. Using 3‐MA, we detected a decreased secretion of cytokines and activation of NF‐κB; similar trends were also obtained by knock‐down of autophagy‐related genes atg5 and beclin‐1 using siRNA. Surprisingly, inhibition of NF‐κB also dramatically attenuated the lipid‐induced autophagosome formation. These data indicate that the autophagic pathway plays a positive role in the regulation of inflammatory response, and also forms a positive feedback loop with NF‐κB to mediate M. pneumoniae lipid‐induced inflammatory effects. Nonetheless, the cross‐talk between autophagy and NF‐κB signalling cascades remains to be elucidated. It has been demonstrated that Beclin‐1 expression is required by NF‐κB, and IκB kinase (IKK) is implicated in autophagy activation [51]. In this study, We found that chloroquine treatment by itself can increase the autophagic adaptor protein p62 and LC3B expression in RAW264.7 cells. In addition, p62 is involved in the regulation of NF‐κB signalling [49], and NF‐κB activation in turn induced p62 expression, thus forming a positive feedback loop [52]. Although we detected a down‐regulated expression of p62 after stimulation with the M. pneumoniae lipids, we failed to detect any association between p62 and NF‐κB (data not shown), which needs further investigation.
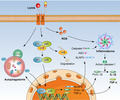
In conclusion, our results support a model in which TLR‐4 is important to induce autophagy in M. pneumoniae lipid‐mediated inflammatory response. Activated TLR‐4 not only promotes the NLRP3 inflammasome assembly by induction of ROS production, but also establishes a positive feedback loop between autophagy and NF‐κB signalling cascade, leading to the up‐regulation of proinflammatory cytokines (Fig. 7). However, there is a special form of phagocytosis in the cell, known as LC3‐associated phagocytosis (LAP), which differs from classical autophagy both mechanistically and functionally [53]. Either autophagy or LAP can up‐regulate the expression of LC3B; therefore, we cannot conclude whether autophagy or LAP or both are activated upon stimulation with the M. pneumoniae lipids. In future studies, we will purify active components of M. pneumoniae lipids and characterize the ligands of TLR‐4 in these lipids.
Fig. 7.

Schematic diagram illustrating the proposed signalling pathway involved in M. pneumoniae lipid‐induced cytokine secretion in mouse macrophages. M. pneumoniae lipids activate the Toll‐like receptor (TLR)‐4 signal to induce nuclear factor kappa B (NF‐κB) activation, NLR family pyrin domain containing 3 inflammasome (NLRP3) inflammasome assembly and autophagosome formation. The autophagy‐mediated inflammatory responses relied on the activation of NF‐κB and, in turn, the activation of NF‐κB enhances the formation of autophagosome induced by lipids. The positive feedback loop between NF‐κB signalling and autophagy induction ultimately induces tumour necrosis factor (TNF)‐α and interleukin (IL‐1β) production in mouse macrophages.
Disclosures
The authors declare no conflicts of interest.
Author contributions
Original idea: X. X. Y.; experimental procedures: H. D. L., J. H., L. S. C., R. H. L., Y. H. Z. and C. M. Z.; writing, review and editing: L. M. Q., Y. W. C. and Y. M. W.
Supporting information
Fig. S1. Quantification of TNF‐α and IL‐1β in BMDMs stimulated with proteinase K‐digested lipids. The Seeded BMDMs were either stimulated with 120μg/mL of purified lipids or proteinase K‐digested lipids (0·1 mg/ml proteinase K for 1 h at 37°C followed by 100°C for 15 min to inactivate the enzyme) for 24 h. The cell culture supernatants were collected and the concentrations of TNF‐α and IL‐1β were determined by ELISA. *P < 0·05 denotes significant difference between the compared groups.
Fig. S2. Nuclease digestion failed to decrease M. pneumoniae lipids‐induced TNF‐α and IL‐1β production in mouse macrophages. BMDMs were seeded on 24‐well plates and were stimulated with 120 μg/mL of lipids or nuclease‐pretreated lipids (80 pμg/mL of RNase and 100 μg/mL DNase for 4h at 37°C) for 24 h. The cell culture supernatants were collected and the concentrations of TNF‐α and IL‐1β were determined by ELISA. *P < 0·05 denotes significant difference between the compared groups.
Fig. S3. Lipase abrogates M. pneumoniae lipids‐induced TNF‐α and IL‐1β production. BMDMs were cultured in 24‐well plates and incubated with 120μg/mL of M. pneumoniae lipids or lipase digested‐lipids (0·1 mg/mL lipoprotein lipase for 6h at 37 °C) for 24 h. The amounts of TNF‐α and IL‐1β in the culture medium were determined by ELISA. *P < 0·05 denotes significant difference between the compared groups.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31670177, 31970177), Natural Science Foundation of Hunan Province (no. 2019JJ40253), Construct Program of the ‘Double First‐Class discipline’ of University of South China (2019SYL) and the Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study (2015‐351).
Contributor Information
X. You, Email: youxiaoxing@usc.edu.cn.
Y. Wu, Email: yimouwu@sina.com.
Data availability statement
The data that support the findings of this study are available in the Supporting information of this article.
References
- 1. Ramasamy K, Balasubramanian S, Manickam K et al Mycoplasma pneumoniae community‐acquired respiratory distress syndrome toxin uses a novel keled sequence for retrograde transport and subsequent cytotoxicity. MBio 2018; 9:e01663‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bajantri B, Venkatram S, Diaz‐Fuentes G. Mycoplasma pneumoniae: a potentially severe infection. J Clin Med Res 2018; 10:535–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Daubenspeck JM, Jordan DS, Simmons W, Renfrow MB, Dybvig K. General N‐and O‐linked glycosylation of lipoproteins in mycoplasmas and role of exogenous oligosaccharide. PLOS ONE 2015; 10:e0143362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Somarajan SR, Kannan TR, Baseman JB. Mycoplasma pneumoniae Mpn133 is a cytotoxic nuclease with a glutamic acid‐, lysine‐ and serine‐rich region essential for binding and internalization but not enzymatic activity. Cell Microbiol 2010; 12:1821–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimizu T. Inflammation‐inducing factors of Mycoplasma pneumoniae . Front Microbiol 2016; 7:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haodang L, Lianmei Q, Ranhui L et al HO‐1 mediates the anti‐inflammatory actions of Sulforaphane in monocytes stimulated with a mycoplasmal lipopeptide. Chem Biol Interact 2019; 306:10–8. [DOI] [PubMed] [Google Scholar]
- 7. Qin L, Chen Y, You X. Subversion of the immune response by human pathogenic mycoplasmas. Front Microbiol 2019; 10:1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shimizu T. Pathogenic factors of mycoplasma. Nihon Saikingaku Zasshi 2015; 70:369–74. [DOI] [PubMed] [Google Scholar]
- 9. He J, You X, Zeng Y, Yu M, Zuo L, Wu Y. Mycoplasma genitalium‐derived lipid‐associated membrane proteins activate NF‐kappaB through toll‐like receptors 1, 2, and 6 and CD14 in a MyD88‐dependent pathway. Clin Vaccine Immunol 2009; 16:1750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee KE, Kim KW, Hong JY, Kim KE, Sohn MH. Modulation of IL‐8 boosted by Mycoplasma pneumoniae lysate in human airway epithelial cells. J Clin Immunol 2013; 33:1117–25. [DOI] [PubMed] [Google Scholar]
- 11. Takeuchi O, Kaufmann A, Grote K et al Cutting edge: preferentially the R‐stereoisomer of the mycoplasmal lipopeptide macrophage‐activating lipopeptide‐2 activates immune cells through a toll‐like receptor 2‐ and MyD88‐dependent signaling pathway. J Immunol 2000; 164:554–7. [DOI] [PubMed] [Google Scholar]
- 12. You X, Wu Y, Zeng Y, Deng Z, Qiu H, Yu M. Mycoplasma genitalium‐derived lipid‐associated membrane proteins induce activation of MAPKs, NF‐kappaB and AP‐1 in THP‐1 cells. FEMS Immunol Med Microbiol 2008; 52:228–36. [DOI] [PubMed] [Google Scholar]
- 13. Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev 1998; 62:1094–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shimizu T, Kimura Y, Kida Y et al Cytadherence of Mycoplasma pneumoniae induces inflammatory responses through autophagy and toll‐like receptor 4. Infect Immun 2014; 82:3076–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schappe MS, Szteyn K, Stremska ME et al Chanzyme TRPM7 mediates the Ca2+ influx essential for lipopolysaccharide‐induced toll‐like receptor 4 endocytosis and macrophage activation. Immunity 2018; 48:59–74.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oliveira AC, Peixoto JR, de Arruda LB et al Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositol phospholipids and higher resistance to infection with T. cruzi . J Immunol 2004; 173:5688–96. [DOI] [PubMed] [Google Scholar]
- 17. Kornspan JD, Rottem S. The phospholipid profile of mycoplasmas. J Lipids 2012; 2012:640762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rottem S. Unique choline‐containing phosphoglycolipids in Mycoplasma fermentans . Chem Phys Lipids 2016; 194:94–100. [DOI] [PubMed] [Google Scholar]
- 19. Ben‐Menachem G, Rottem S, Tarshis M, Barash V, Brenner T. Mycoplasma fermentans glycolipid triggers inflammatory response in rat astrocytes. Brain Res 1998; 803:34–8. [DOI] [PubMed] [Google Scholar]
- 20. Salman M, Deutsch I, Tarshis M, Naot Y, Rottem S. Membrane lipids of Mycoplasma fermentans . FEMS Microbiol Lett 1994; 123:255–60. [DOI] [PubMed] [Google Scholar]
- 21. Klement ML, Ojemyr L, Tagscherer KE, Widmalm G, Wieslander A. A processive lipid glycosyltransferase in the small human pathogen Mycoplasma pneumoniae: involvement in host immune response. Mol Microbiol 2007; 65:1444–57. [DOI] [PubMed] [Google Scholar]
- 22. Novik G, Gamian A, Francisco Jda C, Dey ES. A novel procedure for the isolation of glycolipids from Bifidobacterium adolescentis 94 BIM using supercritical carbon dioxide. J Biotechnol 2006; 121:555–62. [DOI] [PubMed] [Google Scholar]
- 23. Razin S, Prescott B, Caldes G, James WD, Chanock RM. Role of glycolipids and phosphatidylglycerol in the serological activity of Mycoplasma pneumoniae . Infect Immun 1970; 1:408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wells MA, Dittmer JC. The use of sephadex for the removal of nonlipid contaminants from lipid extracts. Biochemistry 1963; 2:1259–63. [DOI] [PubMed] [Google Scholar]
- 25. Segovia JA, Chang TH, Winter VT et al NLRP3 is a critical regulator of inflammation and innate immune cell response during Mycoplasma pneumoniae infection. Infect Immun 2018; 86:e00548‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu Z, Xie D, Chen Y et al TLR2 mediates autophagy through ERK signaling pathway in Mycoplasma gallisepticum‐infected RAW264.7 cells. Mol Immunol 2017; 87:161–70. [DOI] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 2001; 25:402–8. [DOI] [PubMed] [Google Scholar]
- 28. Christodoulides A, Gupta N, Yacoubian V, Maithel N, Parker J, Kelesidis T. The role of lipoproteins in mycoplasma‐mediated immunomodulation. Front Microbiol 2018; 9:1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X, Zhang Y, Yin B, Liang J, Jiang F, Wu W. Toll‐like receptor 2 (TLR2) and TLR4 mediate the IGA immune response induced by Mycoplasma hyopneumoniae . Infect Immun 2019; 88 10.1128/iai.00697-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muxel SM, Acuña SM, Aoki JI, Zampieri RA, Floeter‐Winter LM. Toll‐like receptor and MIRNA‐LET‐7e expression alter the inflammatory response in Leishmania amazonensis‐infected macrophages. Front Immunol 2018; 9:2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF‐κB signaling in PMA‐stimulated THP‐1 monocytes. Biomed Pharmacother 2016; 82:167–72. [DOI] [PubMed] [Google Scholar]
- 32. Haque MA, Jantan I, Harikrishnan H. Zerumbone suppresses the activation of inflammatory mediators in LPS‐stimulated U937 macrophages through MyD88‐dependent NF‐κB/MAPK/PI3K‐Akt signaling pathways. Int Immunopharmacol 2018; 55:312–22. [DOI] [PubMed] [Google Scholar]
- 33. Xu Y, Li H, Chen W et al Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PLOS ONE 2013; 8:e77955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Choi SY, Lim JW, Shimizu T, Kuwano K, Kim JM, Kim H. Reactive oxygen species mediate Jak2/Stat3 activation and IL‐8 expression in pulmonary epithelial cells stimulated with lipid‐associated membrane proteins from Mycoplasma pneumoniae . Inflamm Res 2012; 61:493–501. [DOI] [PubMed] [Google Scholar]
- 35. Mauthe M, Orhon I, Rocchi C et al Chloroquine inhibits autophagic flux by decreasing autophagosome–lysosome fusion. Autophagy 2018; 14:1435–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi CS, Shenderov K, Huang NN et al Activation of autophagy by inflammatory signals limits IL‐1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13:255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shimizu T, Kida Y, Kuwano K. A dipalmitoylated lipoprotein from Mycoplasma pneumoniae activates NF‐kappa B through TLR1, TLR2, and TLR6. J Immunol 2005; 175:4641–6. [DOI] [PubMed] [Google Scholar]
- 38. Shimizu T, Kida Y, Kuwano K. Triacylated lipoproteins derived from Mycoplasma pneumoniae activate nuclear factor‐kappaB through toll‐like receptors 1 and 2. Immunology 2007; 121:473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pollack JD, Somerson NL, Senterfit LB. Isolation, characterization, and immunogenicity of Mycoplasma pneumoniae membranes. Infect Immun 1970; 2:326–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu J, Lu C, Liu Z, Zhang P, Guo H, Wang T. Schizandrin B protects LPS‐induced sepsis via TLR4/NF‐κB/MyD88 signaling pathway. Am J Transl Res 2018; 10:1155–63. [PMC free article] [PubMed] [Google Scholar]
- 41. Wang DD, Pan WJ, Mehmood S, Cheng XD, Chen Y. Polysaccharide isolated from Sarcodon aspratus induces RAW264.7 activity via TLR4‐mediated NF‐κB and MAPK signaling pathways. Int J Biol Macromol 2018; 120:1039–47. [DOI] [PubMed] [Google Scholar]
- 42. Lecoq L, Raiola L, Chabot PR et al Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF‐κB and transcription regulatory factors. Nucleic Acids Res 2017; 45:5564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giridharan S, Srinivasan M. Mechanisms of NF‐κB p65 and strategies for therapeutic manipulation. J Inflamm Res 2018; 11:407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mudipalli A, Li Z, Hromchak R, Bloch A. NF‐kappaB (p65/RelA) as a regulator of TNFalpha‐mediated ML‐1 cell differentiation. Leukemia 2001; 15:808–13. [DOI] [PubMed] [Google Scholar]
- 45. Bose S, Segovia JA, Somarajan SR, Chang TH, Kannan TR, Baseman JB. ADP‐ribosylation of NLRP3 by Mycoplasma pneumoniae CARDS toxin regulates inflammasome activity. mBio 2014;5 10.1128/mbio.02186-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang J, Li Y, Wang J, Wu Q, Yan H. Polydatin suppresses the development of lung inflammation and fibrosis by inhibiting activation of the NACHT domain‐, leucine‐rich repeat‐, and PYD‐containing protein 3 inflammasome and the nuclear factor‐κB pathway after Mycoplasma pneumoniae infection. J Cell Biochem 2019; 120:10137–44. [DOI] [PubMed] [Google Scholar]
- 47. Saeki A, Sugiyama M, Hasebe A, Suzuki T, Shibata K. Activation of NLRP3 inflammasome in macrophages by mycoplasmal lipoproteins and lipopeptides. Mol Oral Microbiol 2018; 33:300–11. [DOI] [PubMed] [Google Scholar]
- 48. Sugiyama M, Saeki A, Hasebe A et al Activation of inflammasomes in dendritic cells and macrophages by Mycoplasma salivarium . Mol Oral Microbiol 2016; 31:259–69. [DOI] [PubMed] [Google Scholar]
- 49. Pan H, Zhang Y, Luo Z et al Autophagy mediates avian influenza H5N1 pseudotyped particle‐induced lung inflammation through NF‐κB and p38 MAPK signaling pathways. Am J Physiol Lung Cell Mol Physiol 2014; 306:L183–L195. [DOI] [PubMed] [Google Scholar]
- 50. Casanova JE. Bacterial autophagy: offense and defense at the host‐pathogen interface. Cell Mol Gastroenterol Hepatol 2017; 4:237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK‐dependent, NF‐κB‐independent control of autophagic gene expression. Oncogene 2011; 30:1727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang S, Qiang L, Sample A, Shah P, He YY. NF‐κB signaling activation induced by chloroquine requires autophagosome, p62 protein, and c‐Jun N‐terminal kinase (JNK) signaling and promotes tumor cell resistance. J Biol Chem 2017; 292:3379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De Faveri F, Chvanov M, Voronina S et al LAP‐like non‐canonical autophagy and evolution of endocytic vacuoles in pancreatic acinar cells. Autophagy 2019:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Quantification of TNF‐α and IL‐1β in BMDMs stimulated with proteinase K‐digested lipids. The Seeded BMDMs were either stimulated with 120μg/mL of purified lipids or proteinase K‐digested lipids (0·1 mg/ml proteinase K for 1 h at 37°C followed by 100°C for 15 min to inactivate the enzyme) for 24 h. The cell culture supernatants were collected and the concentrations of TNF‐α and IL‐1β were determined by ELISA. *P < 0·05 denotes significant difference between the compared groups.
Fig. S2. Nuclease digestion failed to decrease M. pneumoniae lipids‐induced TNF‐α and IL‐1β production in mouse macrophages. BMDMs were seeded on 24‐well plates and were stimulated with 120 μg/mL of lipids or nuclease‐pretreated lipids (80 pμg/mL of RNase and 100 μg/mL DNase for 4h at 37°C) for 24 h. The cell culture supernatants were collected and the concentrations of TNF‐α and IL‐1β were determined by ELISA. *P < 0·05 denotes significant difference between the compared groups.
Fig. S3. Lipase abrogates M. pneumoniae lipids‐induced TNF‐α and IL‐1β production. BMDMs were cultured in 24‐well plates and incubated with 120μg/mL of M. pneumoniae lipids or lipase digested‐lipids (0·1 mg/mL lipoprotein lipase for 6h at 37 °C) for 24 h. The amounts of TNF‐α and IL‐1β in the culture medium were determined by ELISA. *P < 0·05 denotes significant difference between the compared groups.
Data Availability Statement
The data that support the findings of this study are available in the Supporting information of this article.