

Graphical abstract
Keywords: Microbiome, Benchmarking, Software development, Best practices, Metagenomics, Amplicon sequencing, Marker-gene sequencing
Abstract
Microbiomes are integral components of diverse ecosystems, and increasingly recognized for their roles in the health of humans, animals, plants, and other hosts. Given their complexity (both in composition and function), the effective study of microbiomes (microbiomics) relies on the development, optimization, and validation of computational methods for analyzing microbial datasets, such as from marker-gene (e.g., 16S rRNA gene) and metagenome data. This review describes best practices for benchmarking and implementing computational methods (and software) for studying microbiomes, with particular focus on unique characteristics of microbiomes and microbiomics data that should be taken into account when designing and testing microbiomics methods.
1. Introduction
“What we observe is not nature itself, but nature exposed to our method of questioning.”-Werner Heisenberg
Microbial communities have colonized and influence practically every ecosystem on the planet Earth, impacting environmental sciences [1], agriculture [2], [3], [4], and human health [5], [6], [7], [8], [9]. The host and ecosystem services provided by microbes are legion [10], [11], [12], [13]. It is no surprise that U.S. medical research spending on the human microbiome has reached US$1.7 billion in the past decade [7], [14], given the increasing discovery of microbial impacts on cancer [15], [16], obesity [17], [18], pharmacological effects [19], [20], infant health [21], [22], [23], [24], and susceptibility to disease [25]. Furthermore, we are becoming increasingly aware of the services microbes provide with regard to agriculture, waste-water treatment [26], [27], and climate change [28], [29].
The study of all facets of the microbiome [30], [31], e.g. microbial composition, diversity, and function as they interact with the biotic and abiotic features of the environment in which they live, is often referred to as the field of microbiome science [32], or microbiomics. Microbiomics has become a large interdisciplinary “multi-omic” field that incorporates classical microbiology, chemistry, metatranscriptomics, metaproteomics, metabolomics, culturomics, ecology, phylogenetics, systems biology, et cetera [33], [34]. The field of microbiome science is still a rapidly growing interdisciplinary field, the resulting data explosion [35] has led to a commensurate rise in novel analytical approaches to parse, curate, and analyze multi-omic data [36]. These myriad tools and datatypes make it increasingly difficult to interpret, compare, standardize, and benchmark the quality of the data and analytical methods in a consistent and meaningful way [31], [36], [37], [38]. These issues confound our ability to translate multi-omics research into clinical applications [39]. Precise computational techniques are needed to process, normalize, and analyze microbiomics datasets to support reproducible research into the role of microbiomes in health and the environment.
1.1. Marker-gene and metagenome sequencing
Prior to embarking on a microbiome survey, researchers must consider the intent of the study [40], [41], and determine what it is they want to investigate. These questions have important implications for logistics, cost, and biological inference, particularly in the area of translational science [42], [43], [44]. Measuring the composition of a microbiome (what taxa/species are present) is addressed most commonly by the use of amplicon-based / marker gene sequencing approaches to perform a microbial census [45], [46], across a variety of sample and treatment types. The functional potential of a microbial community can be inferred indirectly by marker-gene surveys [47], [48], [49], [50], or through direct observation of the functional genes and pathways by whole-metagenome sequencing surveys [51]. Measurements of functional activity in a microbiome can be derived through metabolomics [52], proteomics [53], [54], and transcriptomics [16] approaches. When applying and integrating several of these approaches, i.e. performing a “multi-omics” study, greater insights can be gained into community behavior and interactions with the host and/or environment [33], [55], [56], [57], [58], [59], [60], [61]. For the purposes of this review, we will focus on sequencing-based methods, though many of the same concerns (e.g., regarding sample and data characteristics, test data, and benchmarking approaches) will generally apply to other microbiomics techniques.
Conducting a microbial census via amplicon sequencing involves massively parallel sequencing of specific marker genes (such as 16S rRNA genes) or other DNA targets that are PCR-amplified directly from environmental DNA, and sequencing these pooled amplicons in parallel. These amplicon sequence reads can then be classified by comparison to a reference sequence database to identify their origin (e.g., the species or taxonomic group). The relatively low cost of this method enables sequencing of hundreds to thousands of samples simultaneously [62], making it useful for tracking microbial compositions in large surveys [63]. Additionally, the relatively low complexity of amplicon sequence data make it much more computationally tractable for a wide range of research applications [64], [65], [66]. Both short- [67] and long-read sequencing [68] can be used for taxonomic classification, but short reads are less reliable for species-level identification [67], [68], and both are insufficient for strain-level identification, though sequence variant information can be used to differentiate “phylotypes” at the sub-species level [69], [70].
Metagenome sequencing involves massively parallel sequencing of DNA fragments extracted directly from environmental samples [37], [51], [66], [71], [72], [73]. These sequence reads can then be computationally re-assembled into full or partial genomes [74], [75], [76] or taxonomically “binned” to identify their origin [9], [10]. Metagenome sequencing avoids the amplification and resolution biases that limit amplicon sequencing [51], [77], [78], [79], but still suffers from its own set of methodological and computational limitations and biases [37], [80], [81], [82], including issues with sequencing host or other non-target DNA [83], which must be carefully considered when designing an experiment. Furthermore, the significantly greater cost [84] and computational complexity [64], [65], [66] make metagenome sequencing more fiscally and technically challenging to work with, compared to amplicon sequencing approaches. “Shallow” metagenome sequencing allows taxonomic profiling at a comparable cost to amplicon sequencing [85], but lacks the coverage necessary for functional profiling or for genome re-assembly.
Amplicon and metagenome sequencing methods share similar properties that must be considered when designing and benchmarking computational methods to analyze these data. DNA extraction [83], storage [86], contamination [87], and other experimental biases [42], [72], [88], [89] can skew measurements from both methods [37], [80]. Sequencing errors are inherent to all modern sequencing chemistries/technologies [90], potentially introducing false-positive errors and skewing diversity estimates if uncorrected [69], [70], [91], [92]. Both approaches measure only the relative abundances of genes/taxa, unless additional steps are used to estimate their absolute abundances [93], [94], [95], [96], [97]. This means that most microbiome sequence datasets exhibit compositional properties that violate the assumptions of many conventional statistical approaches [98], [99]. Sequencing depth can vary across samples, leading to heated debate about appropriate normalization approaches for comparing samples [100], [101], [102]. Finally, the high ratio of genetic diversity to sample size (for most experiments and sample types) means that data typically exhibit high sparsity and a large component-to-sample ratio (p≫n). Addressing these issues remains an active area of research [103], [104].
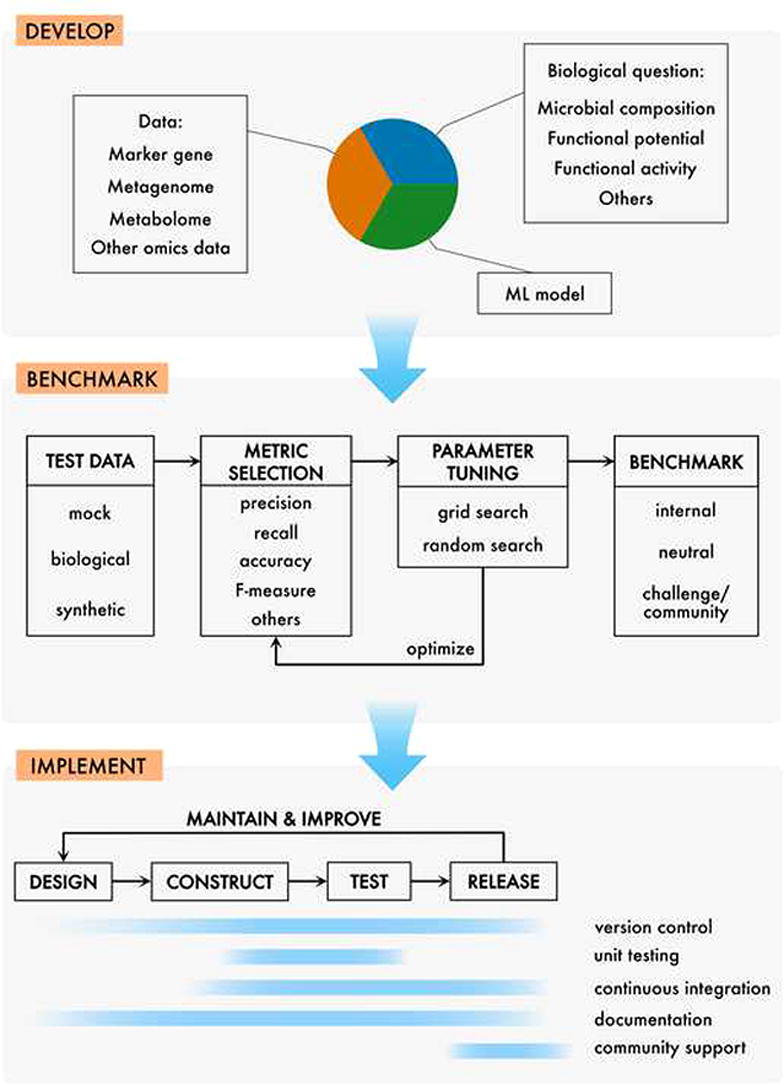
The goal of this review is to serve as a starting point for developing and benchmarking computational methods for microbiomics (Fig. 1). As such, a thorough review of existing methodology and benchmarks is out of scope. Nevertheless, to serve as a guide for future benchmarking endeavors, readers are referred to a selection of existing benchmarks for marker gene quality filtering [91], [105], normalization [100], [101], [102], taxonomic classification [67], clustering/denoising [90], [92], [106], [107], correlation detection [108], supervised learning [109], [110], overall workflows [111], [112], metagenome assembly [113], [114], [115], and metagenome taxonomic binning and classification [113], [116].
Fig. 1.

An overview of a microbiomics method development workflow. Typically, a method is developed to address one or more biological questions, e.g., concerning microbial composition (genetic or taxonomic), dynamics, or functional activity. Depending on the question, various data types can be used to feed a machine learning model or statistical test(s). Once developed, the method should be subject to a suite of benchmarks to assess its performance. A range of choices should be made here, as to what data to use, which performance metric to apply and what kind of benchmark to employ. Finally, to optimize accessibility for the research community, the method should be implemented into a software package/plugin, applying best practices for software development, including version control, testing and continuous integration, documentation and, finally, community support. Naturally, the three steps presented here may overlap in the development cycle, e.g. some part of benchmarking may be started already in the development phase. Software implementation also often starts early, with the first version of the working code. Generally, however, the transition from “develop” through “benchmark” to “implement” becomes natural as the project progresses.
2. Benchmarking
Benchmarks are essential to assess the performance of any computational method before releasing it to the public [117]. Methods for microbiome analysis are no exception, and benchmarks must be well designed to reflect the diverse operating conditions encountered in analysis of microbiomes. Benchmarking can be conducted to provide measures of absolute performance for a new method (e.g., runtime and memory requirements), as well as relative performance (runtime, accuracy, etc) of multiple methods. Other authors have reviewed fundamental aspects of bioinformatics methods benchmarking [117], [118], [119], [120], [121]; the goal of the current review is to amend that information with a focus on benchmarking practices that are of particular relevance to microbiomics. This section is primarily intended for microbiomics methods developers who wish to adopt best practices for development and benchmarking, but the fundamental goals of benchmarking are discussed below (see the section “Benchmarking Basics for Non-Developers”) for the benefit of microbiomics researchers and others who use computational methods (rather than develop them) and who would like more insight into the practical value of benchmarking.
“Internal” benchmarking (performed by the researchers developing a new method) is essential for any new method, particularly when this method is designed to compete against existing methods. Developers of new methods should incorporate multiple evaluation metrics and structure their tests to avoid the “self-assessment trap” when benchmarking [120]. “Neutral” benchmarks of multiple methods (performed by researchers not involved in the development of any of the methods under investigation) are important assessments that can provide unbiased evaluation of tools used by the research community [122], provided that all methods and tests are used properly. Benchmarking “challenges” and community benchmarks (performed by multiple teams of researchers who have developed competing methods) can be particularly valuable for large-scale relative benchmarks, ensuring that each method is used appropriately [111], [113]. The use of double blinding in benchmarking “challenges” can be particularly useful for evaluating the ability of different methods to generalize to unseen datasets, and avoiding implicit bias [120].
2.1. Test data
Selecting appropriate test data is a critical component of benchmarking. These data should reflect the use cases that a method is intended to address. In the case of microbiome analysis methods, this often includes performance for characterization of a diverse range of sample types or species (if the tool is intended for general use) to sufficiently sample the range of experimental conditions under which the method is designed to operate.
We differentiate between “mock”, “biological”, cross-validation, and simulated data. Mock data are real biological samples that have been created with a known property such as taxonomic composition, which can be used to test methodological accuracy. Biological data are distinct from mock data in that we may not know what the tested method should infer from the data. Biological data are typically not gathered primarily for the purpose of testing methods. Cross validation is more strictly a testing procedure than a type of data, but we use it in this context for data that is artificially split (usually multiple times) into mutually exclusive subsets. One subset is then used for fitting or training a model or technique, and one for validation or testing. Simulated data are data that are generated artificially, usually pseudo-randomly under a parametric model.
Each data type has strengths and weaknesses. Mock data can represent a gold standard but are often limited in availability due to the expense of generating them. Biological data represent real operating conditions and are a true test of a given method, but their use is limited by our knowledge of an objective truth. Simulated and cross-validated data are inexpensive to generate and flexible, allowing exhaustive testing of a method, but that also allows authors to overemphasise strengths or weaknesses of the method, intentionally or otherwise. Ultimately any reasonable approach to benchmarking needs to contain a balance of these data types.
2.1.1. Mock communities
“Mock” communities consist of mixtures of microbial cells with known composition (i.e., mixed at known ratios) and taxonomic identities (i.e., the marker genes or genomes of individual members have been sequenced) [123], [124]. These communities are then analyzed to profile their composition (e.g., with marker-gene or metagenome sequencing). Mock communities have seen widespread use in microbiome methods benchmarking [9], [76], [91], [92], [111], [125], [126], [127], because they provide known compositions for ground-truthing, but represent real experimentally derived data (incorporating the various technical, biological, and human errors inherent to microbiome profiling methods) and hence allow investigators to assess performance of diverse methods under real operating conditions. This latter attribute is a double-edged sword: testing under operating conditions (e.g., challenged by sequence errors) can be important for differentiating the performance of methods that otherwise perform well under simulated conditions [67], and complements cross-validation and simulation approaches, but can introduce other challenges, as discussed below.
The hybrid properties of mock communities make them useful for testing a diverse range of both wet-lab [111], [128], [129], [130], [131], [132], and computational methods [9], [91], [111], but also exposes them to limitations. Mock communities are laborious to create, as they must be physically generated from axenic cultures, and the marker-genes or genomes should be sequenced prior to use for testing sequencing-based methods. Hence, the cost of creating a mock community is a limiting constraint (though the costs pay off with repeated use), and inevitably these communities can only capture limited diversity. At best, they can realistically simulate only low-diversity sample types, such as some foods and other selective environments, and provide simplistic representations of more diverse communities such as gut microbiomes.
The introduction of technical, biological, and human errors during the creation of mock communities can also create significant challenges for their use in research. In the worst cases, significant human or technical errors could render a mock community unusable, e.g., if cross-contamination or sequencing errors skew results excessively. Even in the best cases, low to moderate levels of error are incorporated and it can be impossible to differentiate shortcomings of the methods being benchmarked from underlying technical errors [67], creating the false impression that a particular method is imperfect when, in fact, this is an inherent feature of the test data. For this reason, mock communities are best used to evaluate relative performance in the context of benchmarking, rather than the absolute accuracy of a specific method [67]. Similarly, mock communities have been long recommended as positive controls in microbiome sequencing experiments [91], and evaluating the relative performance of mock community accuracy across multiple sequencing runs (with identical methods) can be a useful method to detect potentially disruptive batch effects that can lead to spurious results [127].
Various resources exist for researchers interested in using mock communities for various purposes. We previously created mockrobiota (http://mockrobiota.caporasolab.us/) as a public resource for mock community datasets, allowing researchers to share and use mock community data generated under different experimental technological conditions (e.g., using different sequencing platforms, marker genes, and shotgun metagenomes), including the taxonomic composition of those mock communities [123]. Having a diverse collection of mock communities makes mockrobiota useful for benchmarking purposes, as methods can be tested on multiple communities and multiple sequencing platforms to avoid overfitting to single test datasets (discussed below). Physical mock communities can also be acquired from various government [133] and commercial sources including ATCC (https://www.lgcstandards-atcc.org/) and Zymo Research (https://www.zymoresearch.com/), enabling researchers to use these as positive controls in sequencing, or to analyze with specific technological platforms.
Several best practices should be considered when making mock communities in-house. Mock community members should be sequenced (ideally have their genomes sequenced) so that the actual expected sequence is known, rather than just the taxonomy. This is useful for benchmarking denoising methods and others for upholding sequence fidelity in sequence experiments [69]. Strain information and, ideally, persistent taxonomic identifiers should be used to label mock community members, so that taxonomic information can be updated in step with the tumultuous field of microbial systematics [134]. Typically, cellular DNA is used in mock communities but this can pose problems for multiple-copy marker genes, particularly when the precise copy number is unknown. Synthetic mock communities provide advantages for high-precision testing [135], though their potential applications are dependent on design methodology (e.g., synthetic amplicons are of limited use for shotgun metagenomics), limiting their usefulness for general method benchmarking. Creators of mock communities should provide detailed metadata on the construction and composition of mock communities to facilitate accurate re-use [123], and users of mock communities should carefully consider that information when re-using those mock communities (or data generated from mock communities) to avoid improper use.
2.1.2. Biological data
Objective test data for which the “correct” answer is known (e.g., simulated and mock community data), is essential for good methods benchmarking, but provides little or no insight into real operating conditions. Simulated and mock data are typically simplified and cannot represent the challenges encountered in real experiments, which are complicated by errors (human, sequencing, and other technical errors) and biological diversity that can be difficult or impossible to effectively simulate. Some methods that work well in simulated tests (“on paper”) unravel when faced with real data. Thus, testing on biological data is often beneficial to demonstrate method performance “in the field”, even if those samples do not provide objective measurements (e.g., known composition of a microbial community).
Biological testing typically involves selecting an appropriate set of samples to demonstrate performance under a range of conditions. For example, different sample types can be used to demonstrate effective operation of a method in highly diverse communities (e.g., soil, gut), moderately diverse (e.g., water), and relatively low-diversity communities (e.g., host surfaces) (Fig. 2). These tests can consist of analyzing published data, with the expectation that the new method will outperform the existing “gold standard”, or can evaluate unpublished samples, with the goal of demonstrating effective performance on real samples while acknowledging that the “correct” answer cannot be known.
Fig. 2.

Microbial diversity varies widely by sample type across the planet, as measured across 9787 samples from the Earth Microbiome Project [1]. A, Boxplots measuring the distribution of alpha diversity (as Shannon entropy) in each sample type (boxes show quartile values, diamonds indicate outlier values). B, Unweighted UniFrac principal coordinates analysis (PCoA) measures similarity between samples based on community-wide phylogenetic similarity. Samples are categorized by their “empo_3” sample type. Pre-computed data (Shannon diversity and PCoA coordinates) were collected from the published EMP study data at ftp://ftp.microbio.me/emp/.
In some cases, though, real samples are an effective ground truth, and can serve as primary test data in lieu of simulated or mock communities. This is often the case for supervised learning methods that are designed to predict sample class (e.g., disease state, sample type) or other objective measurements [109], [110], [136], [137]. In those cases, the true value can be objectively measured and validated, and hence biological samples provide known test cases. Hence, the microbiome machine learning literature usually uses real samples to test accuracy of supervised classification and regression methods [138].
Various resources exist for researchers who want to access existing biological data for methods benchmarking. Most marker-gene and metagenome data generated in published literature is (or should be!) deposited in public nucleotide sequence databases, following journal requirements and data standards such as MIMARKS [139], MISAG, MIMAG [74], and FAIR [140]. Hence, resources including NCBI-SRA [141], European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home), and Qiita [142] provide direct access to published sequence data and metadata that can be used for methods benchmarking of raw sequence data. Other databases provide access to processed datasets, e.g., observation matrices useful for benchmarking machine learning methods, including Qiita [142] and ML Repo [138].
2.1.3. Cross validation
Cross validation is a well-established technique from the field of machine learning [109], [110], [136], [137] that tests a method’s predictive power on unseen data. The general method is to partition the available samples into training and validation sets, train on the training set and test on the validation set, then repeat the process until all of the available data has been in a validation set. Many methods exist for generating the partitions but the common k-fold cross-validation technique partitions the data into k sets of equal size, then uses one of those k sets as the validation set for each iteration. Cross validation is applicable to a range of problems, but the archetypal example is that of training a supervised learning classifier, which in the context of a microbiomics experiment could include taxonomic classification of DNA sequences, sample classification, and metagenome taxonomic binning and classification. In these examples a database of samples with known classifications is used to “train” an algorithm to extrapolate those classifications for unseen samples. In the case of sample classification, the goal is to predict sample characteristics, e.g., sampling location [3] or demographic information [104], using microbiome data (e.g., sequence or species counts in each sample) as predictors. In the case of taxonomic classification (or DNA sequence annotation more generally), samples consist of single genetic sequences and classifications consist of taxonomic (or functional) annotations.
Cross validation techniques can be manipulated to test specific features. For example, for taxonomic classification, the training and validation sets can be chosen to emphasise the classifier’s performance on sequences that it should not be able to classify, because they come from taxa that were not present in the training set [67]. Generally, though, it is desirable that the data sets are chosen to reflect the method’s performance in realistic scenarios. Stratification of samples is a variance-reduction technique that attempts to ensure that classes are represented evenly across the folds in k-fold cross validation. As this technique reduces variance in the results between folds, it increases the statistical power of the test.
Of practical importance to supervised learning classification problems are the distribution of classifications among the samples. For instance, if cases of a rare pathology are as common as healthy samples in validation sets, then a method that errs toward false positives will overperform. When performing cross validation it is also important to eliminate any possible information leakage, that is inadvertently allowing information that is only supposed to be present in the training set to also be present in the validation set. For example, when testing the effect of imbalanced taxonomic distribution on taxonomic classifiers by cross validating over empirical taxonomic distributions, it is important to not use the empirical distributions of the samples in the validation set when training the classifiers [67], [143].
Automated tools now exist for cross validation of sample classification methods [110], [137] and taxonomic classification methods [144]. Excellent APIs also exist for cross validation and measurement of performance [145].
While many publications perform cross validation over a specific data set, the performance of a method is more strongly supported by cross validation across studies, for instance being able to use data from one study to draw correct conclusions about data from another study [104], [137], [143], [146].
2.1.4. Simulated data
Simulation can be used in some scenarios where cross validation is difficult, such as where a ground truth is not known about the data or where the method does not have a training or fitting step, such as parametric approaches or unsupervised learning. This includes techniques for data normalization and differential abundance testing [101], marker gene quality filtering, clustering, or denoising [70], overall workflows, taxonomic classification [147], diversity estimation [148], and metagenome assembly [149].
Simulated data offer a powerful degree of flexibility and control, and are inexpensive to generate compared to mock or biological data. This power is also its downfall, however, as results can be manipulated easily. It is a cliché to generate data under a parametric model that incorporates statistical features that a bioinformatic method was built to handle, and to then show how competing models fail to perform in the presence of those features, i.e. the algorithms under investigation are not independent [150]. Simulation must therefore be handled with care or used in conjunction with the other types of data mentioned above. Good simulation should not ignore important features of real data and conclusions should be drawn carefully. Two examples where simulation is particularly useful are for statistical power calculations [151], [152] or for objective goodness-of-fit calculations using parametric bootstraps [153]. In both cases the purpose of simulation is not to model the entire problem, but rather it is a tool for performing analytically intractable calculations.
Simulating the error inherent in genetic sequencing technology [154], [155], [156] is useful for benchmarking sequence denoising methods and metagenome assembly and binning methods [149], but is becoming less useful for other microbiomics methods (e.g., taxonomic classification of marker-gene sequences) as chemical and statistical techniques converge on single-nucleotide sequencing resolution [69], [70] and all of the variance becomes attributable to evolutionary history. Hence, cross validation is more common for the purpose of benchmarking supervised learning methods.
2.2. Parameter tuning
Parameter settings can critically impact method performance, and should be examined in many benchmarking studies. Developers should define default parameter settings for their methods ideally through the use of a well-designed benchmark, recommendations for which are given below. Method users should be aware, however, that default parameter settings do not absolve the user from responsibility in defining reasonable settings in their application. Likewise, developers of competing methods should not assume that default settings are a divine mandate, and that testing competing methods with default settings necessarily represent typical use cases, as discussed below. In general, parameters are exposed to end users when the setting is meant to be adjusted for different applications.
Parameter tuning allows method developers and users to evaluate the impact of parameter settings on method performance through benchmarking. This can either be performed via a complete “grid search” of all possible parameter values or a representative range, or through a “random search” of settings subset from the complete grid [157]. Machine-learning methods, and other computational methods with complex, interacting, and frequently non-intuitive or non-transparent parameters, can benefit from automated parameter tuning strategies, for example using Bayesian or evolutionary optimizers [158], [159], [160]. For most bioinformatics applications, however, the number of possible parameter permutations is limited and a complete or random grid search is sufficient, and can provide information about parameter behavior to end users [67].
When performing a benchmark, it is essential that neutrality is maintained regarding parameter tuning, to avoid introducing bias for any particular method. If any one method (such as a new method being developed) is tuned extensively, but other methods do not undergo any tuning, the comparison is unequal and potentially biased [117]. One strategy for maintaining neutrality is to use default settings for all methods being compared, following the rationale that this scenario represents usage by novice users who are less likely to manually adjust parameter settings, though this assumption may be overly simplistic for some benchmarks [117] or could even introduce bias, such as when default settings are intended for specific applications. Another strategy is to use the same parameter tuning methodology for all methods being compared. This is essential to avoid biases when parameter tuning is being conducted for any one method, such as a new method under development, and can be useful for evaluating the performance landscape across multiple parameter settings and applications [67]. Hence, parameter tuning is useful for both internal and neutral (independent) benchmarks. Full grid searches are not necessary for all benchmarks that perform tuning; selection of rationally defined settings can be sufficient, provided that the investigators are knowledgeable about each method, its parameter settings, and are careful to avoid introducing bias in this selection [117].
2.3. Performance metrics
The next step in benchmarking is to choose appropriate performance metrics. Ideally, multiple metrics should be chosen to avoid implicit bias by researchers testing their own methods [120], as well as by independent benchmarks. Optimizing a method to overperform in any one specific performance metric can come at the expense of other performance metrics, and hence overall method performance should be evaluated based on multiple metrics. Performance metrics are often specific to the benchmarking task, and an exhaustive review is out of the scope of this discussion, but we highlight some metrics as notable examples relevant for microbiomics.
Many methods can be evaluated using classification performance measures, as either direct or indirect evaluations of method performance (e.g., the performance of a DNA sequence clustering or assembly method can be evaluated by looking at its impact on predicted taxonomic composition). As an example of the detailed considerations that are necessary when choosing performance metrics, we cover classifier performance metrics in more detail below (Box 1). The extension of classifier metrics to cross validation in the regression case (where the predicted quantity is continuous, eg. [4], [161]) is straightforward. For instance, the default method for scoring regressors in scikit-learn is the coefficient of determination (or R2, not to be confused with other correlation coefficients) [145], which attempts to capture the tradeoff between bias and variation. Mean squared error and several other metrics are also widely used, depending on the application.
For evaluation of sequence clustering, denoising, or quality filtering approaches, DNA sequence “purity” can be a useful performance metric using both reference-based methods, e.g., dissimilarity between observed and expected sequences [69], or reference-independent methods, such as the Matthews correlation coefficient [162], [163]. The performance of various DNA sequence analysis methods and pipelines can also be compared via estimates of alpha diversity (within-community diversity) or beta diversity (between-community distance or dissimilarity) versus the expected diversity measurements [91], [111]. Similarly, the performance of alpha and beta diversity metrics can be evaluated by comparison to expected diversity measurements for simulated or mock communities [148], [164]. Metagenome profiling methods are commonly evaluated by many of these same approaches (e.g., alpha diversity, beta diversity, and classification metrics) [113]. Metagenome assemblers can be evaluated using a variety of reference-based approaches, e.g., by evaluating alignment to reference genomes or rRNA gene sequences [114].
Outside of metrics that measure performance of microbiomic methods, two other important considerations for many applications are runtime and memory usage. If it is not possible to process sufficient data in a reasonable time frame or on accessible hardware, the method might be limited in its application. For sample and taxonomic classification, these tasks have been made easier in recent years with the advent of denoising techniques [69], [70], resulting in roughly an order of magnitude fewer sequences to process at each step. This is countered, on the other hand, by expanding reference databases that can lead to considerable overhead and delays when training classifiers [144]. These parameters are usually easily monitored using standard system tools. Resource requirements and performance metrics are under-reported in the literature and should generally be reported when possible [119].
Box 1. A deep dive: measuring classifier performance.
In this section we cover several performance metrics that are applicable to methods benchmarking in general, but were, at least initially, developed for different types of classification tasks (e.g., feature classification, taxonomic classification, sample prediction).
There are several methods for measuring accuracy in cross validation that emphasize different properties of the classifiers. Precision, recall, and F-measure [165] are common metrics, but other accuracy metrics are applicable to specific benchmarking tasks such as sequence clustering [166] and metagenome assembly [114]. Descriptions of precision and recall are frequently written for binary classifiers, but for taxonomic or sample classification binary classification is the exception rather than the rule. For example, the current finest Greengenes [167] taxonomy contains 5,405 taxa, each of which is usually represented as a single class. Precision measures the fraction of classifier predictions that match the true class of a sample:
where Tp is the number of predictions that match the true class (also known as true positives) and Fp is the number of predictions that do not match the true class of a sample (false positives). Table 1 shows a hypothetical confusion matrix that counts the number of samples that belong to three orders of Bacteria and how a classifier might have classified them. In this example, precision for Lactobacillales would be 234/357 (the number of correct classifications over the corresponding row total).
Table 1.
A hypothetical confusion matrix that counts the number of samples that belong to three orders of Bacteria and how a classifier might have classified them.
| True Order |
|||||
|---|---|---|---|---|---|
| Lactobacillales | Pseudomonadales | Enterobacteriales | Total | ||
| Predicted Order | Lactobacillales | 234 | 34 | 89 | 357 |
| Pseudomonadales | 56 | 142 | 21 | 219 | |
| Enterobacteriales | 78 | 11 | 68 | 157 | |
| Total | 368 | 187 | 178 | ||
Recall is the fraction of samples for which the predictions match the true class::
where Fn is the number of samples for which the predictions did not match the true class (false negatives). In the example in Table 1, the recall for Lactobacillales would be 234/368 (the number of correct classification over the corresponding column total). F-measure is the harmonic mean of precision and recall:
As it is sometimes possible to trade precision for recall, F-measure provides a single number that rewards good precision and recall and penalizes poor performance in either.
So far we have considered precision, recall, and F-measure for a single class. There are several ways to generalize single-class measures to summarize the performance of a multiclass classifier. One method is microaveraging, where the number of correct classifications and the number of samples are summed over all classes before precision and recall are calculated. For Table 1, precision would be the sum of the diagonals divided by the sum of the row totals (444/733), recall would be the sum of the sum of the diagonals divided by the sum of the columns totals (444/733), and F-measure would be their harmonic mean (444/733). Clearly, for microaveraging, precision and recall are equal, so they are also equal to F-measure. There is a further complication for taxonomic classification that can make the microaveraged precision diverge from the microaveraged recall, which we will mention below.
The accuracy score is easier to understand and more descriptively named. It is the number or proportion of classifications that were correct. The accuracy score is also easier to understand in the multiclass scenario. For Table 1, accuracy is the sum of diagonals divided by the sum over the whole table (444/733), making it the same as the F-measure in this instance. However, the calculation of accuracy is not exactly the same as F-measure so where precision and recall differ, accuracy will not capture the trade-off between them, which again we will explore below.
There are important nuances that need to be considered when choosing measures of performance. An important (but seldom considered) consideration for microbiomics is how the measure incorporates imbalanced classes. Microaveraging, as described above, implicitly weights each sample as being equally important, which is rarely the case. Different samples (or classes, or sample types, or species, or genes) can have different importance because of how often they are observed in reality and because of their importance to the research question. For instance, the real distribution of taxa in biological samples is usually a long way from the distribution of taxa in standard databases [143] and the relative importance of false positives and false negatives must be carefully weighed, for instance in medical screening [168]. Therefore the calculations should be weighted. For instance, if Lactobacillales, Pseudomonadales, and Enterobacteriales comprise 47%, 28%, and 25% of the expected observations respectively, they could be weighted accordingly when assessing accuracy to avoid giving undue importance to rarer or less important observations.
For evaluation of taxonomic classification, there is another complication, which is that it is common for taxonomic classifiers to offer what is generally considered an advanced feature in the field of supervised learning classification. Taxonomic classifiers are usually configurable to abstain from classification at any taxonomic level if they are not sufficiently confident of the classification [67], [169]. This trick can increase precision without reducing recall, causing precision and recall to differ even with microaveraging. In such cases F-measure would also differ, and the change would not be reflected in standard accuracy calculations. Although precision, recall, F-measure, and accuracy can be computed using standard packages (e.g. scikit-learn [145]), modified precision, recall, and F-measure metrics have been used [67], [143] and implemented in specialized software packages [144] to appropriately score instances of incomplete taxonomic classification and misclassification.
2.4. Overfitting
Overfitting occurs when any method, model, or analysis is developed to fit a particular dataset too closely, reducing its ability to generalize to other datasets. This phenomenon occurs at the junction of data selection, parameter tuning, and metric selection, and hence its remedy relies on judicious benchmark design. Overfitting commonly occurs when too few data (or even a single dataset) are used for method optimization and evaluation. This leads to poor performance when that method is applied to other datasets. Hence, multiple datasets should be used for benchmarking whenever possible. Different data types or usage scenarios can also lead to different performance characteristics, so tests should be designed to capture the range of intended usage conditions. For example, we have shown that taxonomic classification algorithms display variable performance for different marker genes, and hence general parameter settings should be balanced to perform well for multiple marker genes [67].
Microbial diversity is another characteristic of microbiome studies that can impact method performance, and should be accounted for in benchmark design to avoid overfitting to particular sample types. Methods that are tuned using one sample type (such as stool) may generalize poorly to other sample types (such as soil). Historically, most mock communities used in the literature have been designed to simulate human stool [123], creating a blind spot if methods are optimized using these mock communities alone. The generation of mock communities for other samples types will provide researchers with opportunities to benchmark their methods with more diverse datasets. This is another motivation to pair mock community tests (which necessarily employ a limited range of microbial diversity) with simulated and real data for method evaluations.
2.5. Benchmarking resources
Methods benchmarking continues to be a major bottleneck for computational methods development. As the preceding sections have described, selecting appropriate test data, evaluation metrics, and competing methods can be a time-consuming and challenging process. Moreover, this selection process increases opportunities for self-assessment bias by methods developers [120], and leads to a proliferation of non-standardized benchmarking methodology, rendering meta-analyses of methods performance in the literature futile.
Benchmarking “challenges” provide partial relief, allowing researchers to perform (typically blinded) evaluation of methods using standard test data, and a centralized, independent evaluation process [111], [113]. Testing frameworks, containing standardized test data and evaluation metrics, provide another means for continuous, extensible methods evaluation, whereby new methods can be optimized and tested against pre-computed performance for existing methods, and new datasets can be added to extend the test cases [120]. LEMMI (Live Evaluation of computational Methods for Metagenome Investigation) [116] is an outstanding example of such a continuous testing framework for shotgun metagenomics taxonomic classifiers, featuring both a repository for pre-computed results, method containerization (to ensure long-term availability), standardized workflows and evaluation metrics, and a website for display of the latest results (https://lemmi.ezlab.org/). There is an ongoing need for testing frameworks for other marker-gene and metagenome analysis methods beyond taxonomic classification.
Other software packages provide useful functions to help streamline methods benchmarking. RESCRIPt (https://github.com/bokulich-lab/RESCRIPt) [144] features methods for reproducible generation, curation, and evaluation of nucleotide sequence reference databases. Both RESCRIPt and q2-quality-control (https://github.com/qiime2/q2-quality-control) feature different evaluation tools for measuring precision, recall, F-measure, and other accuracy metrics based on comparison of observed versus expected results, making these packages useful for high-level testing workflows. For evaluation of metagenome assemblies, MetaQUAST [114] contains several evaluation metrics and has been widely adopted including for community benchmarks [113].
2.6. Benchmarking basics for non-developers
This section is intended to give a general and concise overview of why benchmarking matters to microbiomics researchers, to provide methods users who are not involved in method development or testing greater insight into its practical value. Users of microbiomics methods should be aware that well-designed benchmarks are critical for both optimizing and validating the accuracy of these methods, and it is important (as a method user) to evaluate the quality of the benchmarks themselves, to assess whether the results can generalize to one’s own experimental conditions (e.g., sample type, experimental technologies, expected diversity).
Marker-gene and metagenome sequencing can exhibit technical variation from batch effects, DNA extraction, sample handling and processing differences, as well as differences in bioinformatics analysis pipelines [111]. Hence the methodology must be well controlled to deliver accurate and reproducible measurements. Best practices for method development (including software implementation and continuous testing) as well as benchmarking are paramount to optimizing individual methods, comparing methods to each other, and evaluating the performance of different methods across the range of conditions encountered in the laboratory (e.g., different sample types, data complexity, et cetera). Selection (or generation) of appropriate test data, methods, performance metrics, and parameter tuning are all critical steps in the creation of robust and unbiased benchmarks. The importance of these criteria during method development and benchmarking have been discussed in detail above, but for the purposes of method users we have compiled the following checklist to assist evaluating method performance in the literature:
-
1.
Test data typically should allow measurements of method accuracy. This should include a “ground truth” of some type, e.g., samples with known composition. This will normally consist of one or more mock communities or simulated datasets (as discussed above) [123], [133], [135]. In some cases, a ground truth may not be necessary, for example to measure the consistency among methods rather than their accuracy [170]
-
2.
Test data should be appropriate to the intended usage, and for the different use cases that a user can reasonably expect to encounter (if those different use cases can be expected to impact performance). This may include sample type, degrees of microbial diversity/complexity, or different targets (e.g., marker genes) [67], [78], [147], [171].
-
3.
Multiple datasets and types of data should be used for method testing and optimization. Method users should be aware that testing on a single dataset could lead to overfitting (as discussed above), and such an “optimized” method may not generalize well to other data. Benchmarks that test on multiple datasets and types of data demonstrate the variation in performance across multiple systems and technologies [67], [147], [171].
-
4.
Selecting methods for comparison to a new method or in a benchmarking study should be performed judiciously. Typically the methods used in this comparison should represent one or more “gold standards”, i.e. methods that are both popular in the field and demonstrated to have superior accuracy according to previous benchmarks [113], [171].
-
5.
Performance metrics should be selected to evaluate criteria that are important to users, e.g. scalability, accuracy, etc. Ideally, multiple relevant metrics should be used (as appropriate) to present the multi-dimensional performance of each method. As a method user, you should evaluate whether the methods selected reflect your usage scenario and technical constraints, or whether an incomplete assessment of performance is given. Major benchmarking challenges [113] and continuous benchmarks [116] give notable examples of the virtues of using a panel of performance metrics.
-
6.
All methods should be optimized, within reason, to provide a fair challenge in any large-scale benchmark. Using methods “off the shelf” (i.e., with default parameter settings) can often lead to suboptimal results, as these parameters are intended to be adjusted by a well-trained user, and a fair benchmark should evaluate the accuracy of each method (not just the new method(s)) by testing a range of parameter settings when appropriate. For example, we have shown that even older methods for taxonomy classification can perform well against newer methods when parameter optimization is performed [67].
Box 2. Benchmarking basics: example of taxonomy classification.
As an example of how to set up and evaluate a benchmarking study, we use the example of taxonomic classification of microbial DNA sequences (Fig. 3). In this imaginary example, a promising new method for taxonomy classification has been developed. To validate this new method, its performance is benchmarked against one or more other methods. The benchmarking process can be divided into the following stages:
-
1.
Data selection. Test data must be selected from appropriate sources that represent the intended uses, and also match the goals of the benchmark. Hence, test data selection goes hand-in-hand with test selection. In this example, both cross-validation and ground truthing are performed, and so three different data types are used: reference sequences (e.g., acquired using RESCRIPt [144]) to perform cross-validation; mock community data with known compositions (e.g., from mockrobiota [123]); and biological data (which ideally would represent multiple sample types).
-
2.
Methods selection. The goal of most benchmarking studies is to demonstrate the similarity or superiority of a new method to existing methods. Its performance can be tested against one or more “gold standard” methods for taxonomy classification, e.g., a naive Bayes classifier [67], [169]. Relevant parameters should be selected for each method, to perform parameter tuning.
-
3.
Metric selection. Appropriate metrics should be selected to evaluate key performance characteristics. In the case of taxonomy classification, accuracy will be crucial, but runtime and memory use are also important characteristics. In this example we use F-measure to measure accuracy (Fig. 3), but ideally multiple accuracy metrics should be used to evaluate multiple characteristics (e.g., precision and recall). For more details, see above (Box 1).
Fig. 3.

An example of a benchmarking workflow for development of a new taxonomic classification method. Test data can be retrieved from multiple sources to obtain: (1) reference sequences for cross validation or simulation (e.g., using RESCRIPt [144]); (2) mock community data and known compositions (e.g., from mockrobiota [123]); and (3) biological data, e.g., microbiome sequence data from Qiita [142]. Data can either be classified directly to evaluate results (e.g., for mock community data, for which the true composition is known), or split into k-folds for cross-validation where at each iteration (k-1) folds are used for model training (represented by grey boxes) and the last fold (green box) is used to evaluate model performance. In the case of taxonomic classification, classification accuracy can be scored using metrics like F-measure. Resource utilization is also recorded and compared to the “Gold Standard” method of choice. If either of the metrics is unsatisfactory, the model can be optimized (e.g., via a grid search of parameter settings) and the process is repeated. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Software implementation
Writing good software is an important step in providing accessible, rigorously tested computational methods to end users (in this case, microbiome researchers), as well as to facilitate community development of open-source research software. Other authors have reviewed best practices for developing research software [172], [173], [174], [175]. These same general guidelines (with few exceptions or additions!) apply to microbiomics software best practices, and should be read more deeply by those wishing to devote their lives (or some portion thereof) to developing robust, quality research software. We will reiterate here only a few of the main tenets of software development best practices, as a primer for those who are entering this field for the first time, and for the general edification of those readers interested in the life-cycle of computational methods development.
3.1. Existence != accessibility
Publishing a computational method does not mean that others will be able to use it in their own research. An accessible, maintained, well-documented, well-tested, and appropriately benchmarked software implementation should exist if the general scientific community is expected to utilize a method in their own research. Thorough documentation, intuitive interfaces, and good software development practices will make a method more accessible to users and contributors [176].
Given the significant amount of time and resources spent on developing and benchmarking a computational method, proper implementation is essential to provide useful (and usable) research tools. In the context of the global effort to advance microbiomics, the main goal in developing a research method should be reaching as wide of a user base as possible and that can only be achieved by making the developed methods publicly available to everyone but also, and more importantly, easily applicable. Furthermore, providing useful software implementations of computational methods supports research transparency and reproducibility in microbiomics [177].
3.2. Version control
The first and probably one of the most important aspects in writing software capable of generating reproducible results (a critical aspect of scientific research as well as method integrity!) is keeping track of exact versions of both the source code as well as any external packages/libraries used by that code. Even single version changes can have significant effects on the end results, often making software unusable or, in the worst case, simply incorrect. The widely accepted solution to this problem is to use a version control system (VCS) like Git (https://git-scm.com) or Subversion (https://subversion.apache.org). Version control systems enable developers to easily track every change to their codebase, compare code between versions and revert to previous versions should a problem be identified. Moreover, in the case of highly collaborative projects, where multiple developers work on the same files, a VCS makes it relatively easy to merge changes committed by multiple developers and resolve potential conflicts. In any VCS the developers can decide for themselves on which level of granularity they want to keep track of the changes, from committing only large, significant code changes to tracking minor changes. In the very least, it is recommended to occasionally archive copies of the code so that one can approximately follow the development flow and track down where potential bugs could have been introduced [172].
3.3. Data provenance tracking
In the spirit of encouraging reproducibility, it is advised to record all of the operations performed on the data as it passes through the developed workflow. Ideally, all of those steps should be automated and stored in a standardized way. What is run, why and with what parameters, what the input/output data were — these are some examples of details that should be tracked [178]. QIIME 2’s decentralized provenance tracking system is an outstanding example of how automated provenance tracking can be implemented [179]. All data types, parameters, and actions used in the course of a QIIME 2 workflow get recorded in data provenance stored in every QIIME 2 result file, such that the entire workflow used to obtain any file can be retraced to replicate that result. QIIME 2′s flexible, plugin-based architecture enables others to develop QIIME 2 plugins that can adopt this provenance system (as well as other features of QIIME 2), allowing developers to utilize these features in new or existing software packages.
Depending on the complexity of the analysis, simpler ways of keeping track of analysis steps may also be used. One of the easiest to implement is informing the user about what is being done at every step with the help of logging modules that are available for most, if not all, modern programming languages. In such a way, users can generate and store logs of their analyses that could, potentially, help them diagnose any issues, should a need arise. However, such an approach requires diligent record-keeping to ensure that workflow and environment details are stored with results.
3.4. Optimization vs. complexity
A common pitfall in software development is premature optimization and over-complication of code. Complication can hinder widespread use and adaptation by others, and hence it is a guiding principle to only make things as complicated as they need to be. Write simple, well-commented code that does things well and is understandable by other people, rather than devise a solution that is over-optimized but hard to follow due to its complexity [174].
3.5. Do not reinvent the wheel
Existing software libraries should be used whenever possible to streamline code, boost performance, and to benefit from community efforts. Regardless of the field of use, many intermediary actions, calculations and methods very likely have already been developed by others and should be used instead of writing code producing the same expected result. Whenever possible (or reasonable), one should rather make use of well-written and -maintained libraries and packages available in public repositories and keep track of their exact versions used (as already discussed above). This approach will streamline testing (provided it is well-tested in the third-party library; see also below), save development time, and through usage contribute to the integrity of community resources [175]. Exceptions may be made when these packages are poorly written, maintained, or tested; exhibit inferior performance; introduce licensing issues, dependency conflicts, or overcomplicated dependencies (e.g., installation issues); or for trivial actions.
3.6. Testing and continuous integration
No researcher would like to find out weeks after publishing their work that their results were incorrect due to a software mistake [180]. To ensure validity of the results obtained when using custom code, appropriate testing should be incorporated into the development cycle, preferably as early as possible. Appropriate unit tests (tests asserting that a specific method/piece of code functions as expected) should be written for every new component. The purpose is not only to validate that the expected results are obtained every time the code is run, but also that introduction of new code or alterations of the existing code do not change the expected output. In such cases, a failing test quickly informs the developer that breaking changes have been introduced and the code needs to be fixed.
Moreover, such unit tests should become a part of the so-called continuous integration (CI) process. The basic idea behind CI is that developers frequently commit their code to a shared repository where a set of automated checks will be run to ensure quality of the new code. Usually, code that does not pass selected criteria would not be allowed to be integrated with the existing codebase and the identified issues will need to be addressed first. There are many tools (freely available for the open-source community) that can be used to achieve that. Some examples are TravisCI (https://travis-ci.org/), GitHub Actions (https://github.com/features/actions) or CircleCI (https://circleci.com/).
Ideally, unit tests should not only assess whether the basic software functionality is maintained. They should test as many edge cases as possible and should anticipate the near-infinite number of ways that users can make mistakes: e.g., using input parameters that are out of range, data formats that are not supported, or call functions with incorrect arguments. Those cases should be tested and accounted for in the code. For developers, the payoff for writing comprehensive tests is less time supporting users who discover those cases for themselves.
Another beneficial common practice in writing tests is to turn bugs into unit tests as they are discovered. In such a way, one would design a test that fails given an existing bug and passes when the bug gets fixed (without changing the results of any other test) [174]. Even though writing code to test other code may seem somewhat superfluous at face value, the benefits of this practice rapidly outweigh the cost in effort, particularly (but not only) in collaborative projects [181].
3.7. Community support
Every piece of software has two human components: the developers and the users. Very often users would not be involved much in the development cycle. The developers would provide the users with the “final” version of the product and the users would perhaps report issues as they discover them but rarely (at least in the non-open-source community) are they directly involved in the development, unless the software development was commissioned by the users themselves. In the research community it should, however, be encouraged for the users (i.e., researchers) to get involved in the software development process as they understand best what the software should be able to achieve.
Open “issue” trackers and feature requests allow users (as well as developers) to report bugs, request new features, or describe potential improvements. This also allows other users (and developers) to track and contribute to solving or responding to these “issues”. Various issue trackers exist, a popular example being GitHub's (https://www.github.com) issue lists and project boards that can facilitate exchange of ideas between users and developers. Such exchange also fosters the idea of open science and helps the entire community to grow and collectively work towards the same goal rather than rely on a subset of people (developers) who would provide tools for a group of users. To make it as easy as possible for new contributors, any project should contain some form of contributing guidelines, which describe how to file a bug report or suggest a new feature, how to set up the environment and run the tests, how to structure a “pull request” (code contribution), et cetera. Additionally, whenever possible, it is a good practice to explicitly label issues to describe the content: for example, labeling (and ranking) bugs versus enhancements can support effective triage processes, and labeling “good first issues” will help new contributors identify issues that they can more easily solve [182].
For widely used research software, having a dedicated support forum or other online community can also be beneficial for connecting project developers and users. This also separates development tools (e.g., issue trackers) from user support discussions. Forums also encourage users to engage and support one another. Fostering greater user–developer connection through an online community or support forum can provide long-term benefits, e.g., through facilitating discussion of new features, beta-testing new features and documentation, and creating a venue for user-generated documentation and tutorials.
3.8. Documentation
Documentation is a critical component of all software projects. Software developers must resist the “paradox of documentation”, that those most knowledgeable about a project are the least in need of its documentation, and hence less motivated to write it [183]. Preparing good documentation can (and should!) occupy a significant amount of the total development time, as it provides crucial information to non-experienced users without the need to explore any of the code. Good documentation should include both descriptions of methods/components provided within the given software/tool, as well as examples and/or tutorials for the users to explore to familiarize themselves with the software in an accessible and pragmatic way. Ideally, a sample dataset is included to enable users to learn how to use the software (and recognize correct operation and outputs) without needing to provide their own data (e.g. in cases where they want to test the tool before getting the actual experimental data). Test data should typically consist of a small and low-complexity dataset, to reduce runtime/computational overhead, and to facilitate interpretation of results by new users, but supplying example datasets of varying size and complexity can be beneficial. If using a smaller-than-typical example dataset, the documentation should clarify this fact to prevent confusion among users who notice that runtime and computational resource use is substantially higher for their own (full-sized) dataset.
Another aspect to consider is documenting code for the developers themselves. Practices as simple as giving meaningful names to files/functions/variables (and following or establishing standardized conventions for naming these) can help new developers contribute to the project, and support community-driven development [175], [182]. Moreover, providing short descriptions of each method's purpose, required arguments, and return values in combination with any of the popular documentation-generating tools (e.g. Sphinx for Python, https://www.sphinx-doc.org) is an easy way to keep and maintain a “self-generating” documentation [174].
Community-driven documentation can be beneficial, particularly during the initial phase of a project, as users and contributors often know the documentation needs and gaps best, this greatly reduces the “paradox of documentation” [176], [183]. Community forums and issue trackers, as described above, can help facilitate contributing and beta-testing “unofficial” code before incorporating into the official documentation (which for many software projects is tied to the release cycle and hence cannot be updated as frequently or regularly). Involving the community in this way cultivates the sharing of knowledge and increases collaborative interaction through increasingly intrinsic means, the benefits of which cannot be overstated. Thus, we greatly encourage that “Documentation Sprints” coincide with “Code Sprints.”
4. Conclusions
Computational method development is a critical component that supports microbiome research, and care must be taken to create accurate, accessible tools. Rigorous benchmarks and robust software implementations (including comprehensive unit testing) are needed to support ongoing methodological advancements and to facilitate their use by end users (microbiome scientists). Continuous benchmarking and crowdsourced benchmarks [119] may enable more rapid methodological improvements, standardization, and balanced comparisons. Observing these best practices will lead to more transparent, reproducible research to support ongoing discoveries that illuminate the manifold roles of microbiomes in the health of the planet earth and its inhabitants.
Author statement
All authors contributed to writing and reviewing the final manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Thompson L.R., Sanders J.G., McDonald D., Amir A., Ladau J., Locey K.J. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551:457–463. doi: 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trivedi P., Leach J.E., Tringe S.G., Sa T., Singh B.K. Plant-microbiome interactions: from community assembly to plant health. Nat Rev Microbiol. 2020 doi: 10.1038/s41579-020-0412-1. [DOI] [PubMed] [Google Scholar]
- 3.Bokulich N.A., Thorngate J.H., Richardson P.M., Mills D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc Natl Acad Sci USA. 2014;111:E139–E148. doi: 10.1073/pnas.1317377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bokulich N.A., Collins T.S., Masarweh C., Allen G., Heymann H., Ebeler S.E. Associations among wine grape microbiome, metabolome, and fermentation behavior suggest microbial contribution to regional wine characteristics. MBio. 2016;7. doi: 10.1128/mBio.00631-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanson B.M., Weinstock G.M. The importance of the microbiome in epidemiologic research. Ann Epidemiol. 2016;26:301–305. doi: 10.1016/j.annepidem.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Foxman B., Martin E.T. Use of the microbiome in the practice of epidemiology: a primer on -omic technologies. Am J Epidemiol. 2015;182:1–8. doi: 10.1093/aje/kwv102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Proctor L. Priorities for the next 10 years of human microbiome research. Nature. 2019;569:623–625. doi: 10.1038/d41586-019-01654-0. [DOI] [PubMed] [Google Scholar]
- 8.Cullen C.M., Aneja K.K., Beyhan S., Cho C.E., Woloszynek S., Convertino M. Emerging priorities for microbiome research. Front Microbiol. 2020;11:136. doi: 10.3389/fmicb.2020.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert CLD, Qin J, Kunin V, Engelbrektson A, Ochman H, Hugenholtz P, et al. A framework for human microbiome research. Nature 2012;486:215–21. [DOI] [PMC free article] [PubMed]
- 10.Gonzalez A., King A., Robeson M.S., 2nd, Song S., Shade A., Metcalf J.L. Characterizing microbial communities through space and time. Curr Opin Biotechnol. 2012;23:431–436. doi: 10.1016/j.copbio.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hacquard S., Garrido-Oter R., Gonzalez A., Spaepen S., Ackermann G., Lebeis S. Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe. 2015;17:603–616. doi: 10.1016/j.chom.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 12.McKenney E.A., Koelle K., Dunn R.R., Yoder A.D. The ecosystem services of animal microbiomes. Mol Ecol. 2018;27:2164–2172. doi: 10.1111/mec.14532. [DOI] [PubMed] [Google Scholar]
- 13.Duar R.M., Henrick B.M., Casaburi G., Frese S.A. Integrating the ecosystem services framework to define dysbiosis of the breastfed infant gut: the role of B. infantis and human milk oligosaccharides. Front Nutr. 2020;7:33. doi: 10.3389/fnut.2020.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.NIH Human Microbiome Portfolio Analysis Team A review of 10 years of human microbiome research activities at the US National Institutes of Health, Fiscal Years 2007–2016. Microbiome. 2019;7:31. doi: 10.1186/s40168-019-0620-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.García-Castillo V., Sanhueza E., McNerney E., Onate S.A., García A. Microbiota dysbiosis: a new piece in the understanding of the carcinogenesis puzzle. J Med Microbiol. 2016;65:1347–1362. doi: 10.1099/jmm.0.000371. [DOI] [PubMed] [Google Scholar]
- 16.Poore G.D., Kopylova E., Zhu Q., Carpenter C., Fraraccio S., Wandro S. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020 doi: 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massier L., Chakaroun R., Tabei S., Crane A., Didt K.D., Fallmann J. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut. 2020 doi: 10.1136/gutjnl-2019-320118. [DOI] [PubMed] [Google Scholar]
- 18.Piccolo B.D., Graham J.L., Stanhope K.L., Nookaew I., Mecer K.E., Chintapalli S.V. Diabetes-associated alterations in the cecal microbiome and metabolome are independent of diet or environment in the UC Davis type 2-diabetes mellitus rat model. Am J Physiol-Endocrinol Metabolism. 2018;8:214. doi: 10.1152/ajpendo.00203.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pryor R., Martinez-Martinez D., Quintaneiro L., Cabreiro F. The role of the microbiome in drug response. Annu Rev Pharmacol Toxicol. 2019 doi: 10.1146/annurev-pharmtox-010919-023612. [DOI] [PubMed] [Google Scholar]
- 20.Saad R., Rizkallah M.R., Aziz R.K. Gut Pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog. 2012;4:16. doi: 10.1186/1757-4749-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferretti P., Pasolli E., Tett A., Asnicar F., Gorfer V., Fedi S. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe. 2018;24 doi: 10.1016/j.chom.2018.06.005. 133–45.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milani C., Duranti S., Bottacini F., Casey E., Turroni F., Mahony J. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev. 2017:81. doi: 10.1128/MMBR.00036-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominguez-Bello M.G., De Jesus-Laboy K.M., Shen N., Cox L.M., Amir A., Gonzalez A. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med. 2016;22:250–253. doi: 10.1038/nm.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bokulich N.A., Chung J., Battaglia T., Henderson N., Jay M., Li H. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med. 2016 doi: 10.1126/scitranslmed.aad7121. 8:343ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martino C., Kellman B.P., Sandoval D.R., Clausen T.M., Marotz C.A., Song S.J. Bacterial modification of the host glycosaminoglycan heparan sulfate modulates SARS-CoV-2 infectivity. Microbiology. 2020 doi: 10.1101/2020.08/17.238444. [DOI] [Google Scholar]
- 26.Riva V., Riva F., Vergani L., Crotti E., Borin S., Mapelli F. Microbial assisted phytodepuration for water reclamation: Environmental benefits and threats. Chemosphere. 2020;241 doi: 10.1016/j.chemosphere.2019.124843. [DOI] [PubMed] [Google Scholar]
- 27.de Celis M., Belda I., Ortiz-Álvarez R., Arregui L., Marquina D., Serrano S. Tuning up microbiome analysis to monitor WWTPs’ biological reactors functioning. Sci Rep. 2020;10:4079. doi: 10.1038/s41598-020-61092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez R., Durán P. Natural holobiome engineering by using native extreme microbiome to counteract the climate change effects. Front Bioeng Biotechnol. 2020;8:568. doi: 10.3389/fbioe.2020.00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee A., Cornejo J., Bandopadhyay R. Emergent climate change impact throughout the world: call for “Microbiome Conservation” before it’s too late. Biodivers Conserv. 2020;29:345–348. doi: 10.1007/s10531-019-01886-6. [DOI] [Google Scholar]
- 30.Marchesi J.R., Ravel J. The vocabulary of microbiome research: a proposal. Microbiome. 2015;3:31. doi: 10.1186/s40168-015-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berg G., Rybakova D., Fischer D., Cernava T., Vergès M.-C.C., Charles T. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8:103. doi: 10.1186/s40168-020-00875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shetty S.A., Lahti L. Microbiome data science. J Biosci. 2019;44 doi: 10.1007/s12038-019-9930-2. [DOI] [PubMed] [Google Scholar]
- 33.Misra B.B., Langefeld C.D., Olivier M., Cox L.A. Integrated omics: tools, advances, and future approaches. J Mol Endocrinol. 2018 doi: 10.1530/JME-18-0055. [DOI] [PubMed] [Google Scholar]
- 34.Jansson J.K., Hofmockel K.S. The soil microbiome-from metagenomics to metaphenomics. Curr Opin Microbiol. 2018;43:162–168. doi: 10.1016/j.mib.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Stephens Z.D., Lee S.Y., Faghri F., Campbell R.H., Zhai C., Efron M.J. Big data: astronomical or genomical? PLoS Biol. 2015;13 doi: 10.1371/journal.pbio.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kyrpides N.C., Eloe-Fadrosh E.A., Ivanova N.N. Microbiome data science: understanding our microbial planet. Trends Microbiol. 2016;24:425–427. doi: 10.1016/j.tim.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Nayfach S., Pollard K.S. Toward accurate and quantitative comparative metagenomics. Cell. 2016;166:1103–1116. doi: 10.1016/j.cell.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nayfach S., Bradley P.H., Wyman S.K., Laurent T.J., Williams A., Eisen J.A. Automated and accurate estimation of gene family abundance from shotgun metagenomes. PLoS Comput Biol. 2015;11 doi: 10.1371/journal.pcbi.1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitsios G.D., Morowitz M.J., Dickson R.P., Huffnagle G.B., McVerry B.J., Morris A. Dysbiosis in the intensive care unit: Microbiome science coming to the bedside. J Crit Care. 2017;38:84–91. doi: 10.1016/j.jcrc.2016.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young V.B. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ. 2017;356 doi: 10.1136/bmj.j831. [DOI] [PubMed] [Google Scholar]
- 41.Prosser J.I. Putting science back into microbial ecology: a question of approach. Philos Trans R Soc Lond B Biol Sci. 2020;375:20190240. doi: 10.1098/rstb.2019.0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allaband C., McDonald D., Vázquez-Baeza Y., Minich J.J., Tripathi A., Brenner D.A. Microbiome 101: studying, analyzing, and interpreting gut microbiome data for clinicians. Clin Gastroenterol Hepatol. 2019;17:218–230. doi: 10.1016/j.cgh.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staley C., Kaiser T., Khoruts A. Clinician guide to microbiome testing. Dig Dis Sci. 2018;63:3167–3177. doi: 10.1007/s10620-018-5299-6. [DOI] [PubMed] [Google Scholar]
- 44.Tyler A.D., Smith M.I., Silverberg M.S. Analyzing the human microbiome: a “how to” guide for physicians. Am J Gastroenterol. 2014;109:983. doi: 10.1038/ajg.2014.73. [DOI] [PubMed] [Google Scholar]
- 45.Schloss P.D., Handelsman J. Status of the microbial census. Microbiol Mol Biol Rev. 2004;68:686–691. doi: 10.1128/MMBR.68.4.686-691.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Louca S., Mazel F., Doebeli M., Parfrey L.W. A census-based estimate of Earth’s bacterial and archaeal diversity. PLoS Biol. 2019;17 doi: 10.1371/journal.pbio.3000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Douglas G.M., Maffei V.J., Zaneveld J.R., Yurgel S.N., Brown J.R., Taylor C.M. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020 doi: 10.1038/s41587-020-0548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jun S.-R., Robeson M.S., Hauser L.J., Schadt C.W., Gorin A.A. PanFP: pangenome-based functional profiles for microbial communities. BMC Res Notes. 2015;8:479. doi: 10.1186/s13104-015-1462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wemheuer F., Taylor J.A., Daniel R., Johnston E., Meinicke P., Thomas T. Tax4Fun2: a R-based tool for the rapid prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene marker gene sequences. Bioinformatics. 2018;490037 doi: 10.1101/490037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narayan N.R., Weinmaier T., Laserna-Mendieta E.J., Claesson M.J., Shanahan F., Dabbagh K. Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genomics. 2020;21:56. doi: 10.1186/s12864-019-6427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharpton T.J. An introduction to the analysis of shotgun metagenomic data. Front Plant Sci. 2014;5:209. doi: 10.3389/fpls.2014.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quinn R.A., Melnik A.V., Vrbanac A., Fu T., Patras K.A., Christy M.P. Global chemical effects of the microbiome include new bile-acid conjugations. Nature. 2020 doi: 10.1038/s41586-020-2047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin H., He Q.-Y., Shi L., Sleeman M., Baker M.S., Nice E.C. Proteomics and the microbiome: pitfalls and potential. Expert Rev Proteomics. 2019;16:501–511. doi: 10.1080/14789450.2018.1523724. [DOI] [PubMed] [Google Scholar]
- 54.Long S., Yang Y., Shen C., Wang Y., Deng A., Qin Q. Metaproteomics characterizes human gut microbiome function in colorectal cancer. NPJ Biofilms Microbiomes. 2020;6:14. doi: 10.1038/s41522-020-0123-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinu F.R., Beale D.J., Paten A.M., Kouremenos K., Swarup S., Schirra H.J. Systems biology and multi-omics integration: viewpoints from the metabolomics research community. Metabolites. 2019:9. doi: 10.3390/metabo9040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hasin Y., Seldin M., Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:83. doi: 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Issa Isaac N., Philippe D., Nicholas A., Raoult D., Eric C. Metaproteomics of the human gut microbiota: challenges and contributions to other OMICS. Clin Mass Spectrometry. 2019;14:18–30. doi: 10.1016/j.clinms.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang S., Chaudhary K., Garmire L.X. More is better: recent progress in multi-omics data integration methods. Front Genet. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Q., Wang K., Wu W., Giannoulatou E., Ho J.W.K., Li L. Host and microbiome multi-omics integration: applications and methodologies. Biophys Rev. 2019;11:55–65. doi: 10.1007/s12551-018-0491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu C., Zhou F., Ren J., Li X., Jiang Y., Ma S. A selective review of multi-level omics data integration using variable selection. High Throughput. 2019:8. doi: 10.3390/ht8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graw S., Chappell K., Washam C.L., Gies A., Bird J., Robeson M.S. Multi-omics data integration considerations and study design for biological systems and disease. Molecular-Omics. 2020 doi: 10.1039/d0mo00041h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Minich JJ, Humphrey G, Benitez RAS, Sanders J, Swafford A, Allen EE, et al. High-Throughput Miniaturized 16S rRNA Amplicon Library Preparation Reduces Costs while Preserving Microbiome Integrity. mSystems 2018;3:557. [DOI] [PMC free article] [PubMed]
- 63.Poretsky R., Rodriguez-R L.M., Luo C., Tsementzi D., Konstantinidis K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0093827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sims D., Sudbery I., Ilott N.E., Heger A., Ponting C.P. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15:121–132. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 65.Luo C., Rodriguez-R L.M., Konstantinidis K.T. Chapter twenty-three – a user’s guide to quantitative and comparative analysis of metagenomic datasets. In: DeLong E.F., editor. Vol. 531. Academic Press; 2013. pp. 525–547. (Methods in enzymology). [DOI] [PubMed] [Google Scholar]
- 66.Ranjan R., Rani A., Metwally A., McGee H.S., Perkins D.L. Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun. 2016;469:967–977. doi: 10.1016/j.bbrc.2015.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bokulich N.A., Kaehler B.D., Rideout J.R., Dillon M., Bolyen E., Knight R. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson J.S., Spakowicz D.J., Hong B.-Y., Petersen L.M., Demkowicz P., Chen L. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10:5029. doi: 10.1038/s41467-019-13036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J.A., Holmes S.P. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017;2. doi: 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed]
- 71.Liu Y.-X., Qin Y., Chen T., Lu M., Qian X., Guo X. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell. 2020 doi: 10.1007/s13238-020-00724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quince C., Walker A.W., Simpson J.T., Loman N.J., Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35:833–844. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- 73.Jovel J., Patterson J., Wang W., Hotte N., O’Keefe S., Mitchel T. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front Microbiol. 2016;7:459. doi: 10.3389/fmicb.2016.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bowers R.M., Kyrpides N.C., Stepanauskas R., Harmon-Smith M., Doud D., Reddy T.B.K. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–731. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grieb A., Bowers R.M., Oggerin M., Goudeau D., Lee J., Malmstrom R.R. A pipeline for targeted metagenomics of environmental bacteria. Microbiome. 2020;8:21. doi: 10.1186/s40168-020-0790-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singer E., Andreopoulos B., Bowers R.M., Lee J., Deshpande S., Chiniquy J. Next generation sequencing data of a defined microbial mock community. Sci Data. 2016;3 doi: 10.1038/sdata.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schloss P.D. The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Z., DeSantis T.Z., Andersen G.L., Knight R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucl Acids Res. 2008;36 doi: 10.1093/nar/gkn491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soergel D.A.W., Dey N., Knight R., Brenner S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012 doi: 10.1038/ismej.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McLaren MR, Willis AD, Callahan BJ. Consistent and correctable bias in metagenomic sequencing experiments. Elife 2019;8. doi: 10.7554/eLife.46923. [DOI] [PMC free article] [PubMed]
- 81.R Marcelino V, Holmes EC, Sorrell TC. The use of taxon-specific reference databases compromises metagenomic classification. BMC Genomics 2020;21:184. [DOI] [PMC free article] [PubMed]
- 82.Manor O., Borenstein E. MUSiCC: a marker genes based framework for metagenomic normalization and accurate profiling of gene abundances in the microbiome. Genome Biol. 2015;16:53. doi: 10.1186/s13059-015-0610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bjerre R.D., Hugerth L.W., Boulund F., Seifert M., Johansen J.D., Engstrand L. Effects of sampling strategy and DNA extraction on human skin microbiome investigations. Sci Rep. 2019;9:17287. doi: 10.1038/s41598-019-53599-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kuczynski J., Lauber C.L., Walters W.A., Parfrey L.W., Clemente J.C., Gevers D. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2011;13:47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, et al. Evaluating the Information Content of Shallow Shotgun Metagenomics. mSystems 2018;3. https://doi.org/10.1128/mSystems.00069-18. [DOI] [PMC free article] [PubMed]
- 86.Song S.J., Amir A., Metcalf J.L., Amato K.R., Xu Z.Z., Humphrey G. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSystems. 2016;1:e00021–e116. doi: 10.1128/mSystems.00021-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Salter S.J., Cox M.J., Turek E.M., Calus S.T., Cookson W.O., Moffatt M.F. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watson E.-J., Giles J., Scherer B.L., Blatchford P. Human faecal collection methods demonstrate a bias in microbiome composition by cell wall structure. Sci Rep. 2019;9:16831. doi: 10.1038/s41598-019-53183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Knight R., Vrbanac A., Taylor B.C., Aksenov A., Callewaert C., Debelius J. Best practices for analysing microbiomes. Nat Rev Microbiol. 2018;16:410–422. doi: 10.1038/s41579-018-0029-9. [DOI] [PubMed] [Google Scholar]
- 90.Nearing J.T., Douglas G.M., Comeau A.M., Langille M.G.I. Denoising the denoisers: an independent evaluation of microbiome sequence error-correction approaches. PeerJ. 2018;6 doi: 10.7717/peerj.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bokulich N.A., Subramanian S., Faith J.J., Gevers D., Gordon J.I., Knight R. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huse S.M., Mark Welch D.B., Morrison H.G., Sogin M.L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palmer J.M., Jusino M.A., Banik M.T., Lindner D.L. Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. PeerJ. 2018;6 doi: 10.7717/peerj.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barlow J.T., Bogatyrev S.R., Ismagilov R.F. A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities. Nat Commun. 2020;11:2590. doi: 10.1038/s41467-020-16224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tkacz A., Hortala M., Poole P.S. Absolute quantitation of microbiota abundance in environmental samples. Microbiome. 2018;6:110. doi: 10.1186/s40168-018-0491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jian C., Luukkonen P., Yki-Järvinen H., Salonen A., Korpela K. Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLoS ONE. 2020;15 doi: 10.1371/journal.pone.0227285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rao C., Coyte K.Z., Bainter W., Geha R.S., Martin C.R. Multi-kingdom quantitation reveals distinct ecological drivers of predictable early-life microbiome assembly. bioRxiv. 2020 [Google Scholar]
- 98.Gloor G.B., Macklaim J.M., Pawlowsky-Glahn V., Egozcue J.J. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aitchison J. The statistical analysis of compositional data 1986. https://doi.org/10.1007/978-94-009-4109-0.
- 100.Thorsen J., Brejnrod A., Mortensen M., Rasmussen M.A., Stokholm J., Al-Soud W.A. Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome. 2016;4:62. doi: 10.1186/s40168-016-0208-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weiss S., Xu Z.Z., Peddada S., Amir A., Bittinger K., Gonzalez A. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:59. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McMurdie P.J., Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10 doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li H. Microbiome, metagenomics, and high-dimensional compositional data analysis. Annu Rev Stat Appl. 2015;2:73–94. [Google Scholar]
- 104.Martino C., Shenhav L., Marotz C.A., Armstrong G., McDonald D., Vázquez-Baeza Y. Context-aware dimensionality reduction deconvolutes gut microbial community dynamics. Nat Biotechnol. 2020:1–4. doi: 10.1038/s41587-020-0660-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schloss P.D., Gevers D., Westcott S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schloss P.D., Westcott S.L. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2011;77:3219–3226. doi: 10.1128/AEM.02810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rideout J.R., He Y., Navas-Molina J.A., Walters W.A., Ursell L.K., Gibbons S.M. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ. 2014;2 doi: 10.7717/peerj.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weiss S., Van Treuren W., Lozupone C., Faust K., Friedman J., Deng Y. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016;10:1669–1681. doi: 10.1038/ismej.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Knights D., Costello E.K., Knight R. Supervised classification of human microbiota. FEMS Microbiol Rev. 2011;35:343–359. doi: 10.1111/j.1574-6976.2010.00251.x. [DOI] [PubMed] [Google Scholar]
- 110.Bokulich N.A., Dillon M.R., Bolyen E., Kaehler B.D., Huttley G.A., Caporaso J.G. q2-sample-classifier: machine-learning tools for microbiome classification and regression. J Open Res Softw. 2018;3 doi: 10.21105/joss.00934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sinha R., Abu-Ali G., Vogtmann E., Fodor A.A., Ren B., Amir A. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol. 2017;486:207–1086. doi: 10.1038/nbt.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Straub D, Blackwell N, Fuentes AL, Peltzer A, Nahnsen S, Kleindienst S. Interpretations of microbial community studies are biased by the selected 16S rRNA gene amplicon sequencing pipeline 2019:2019.12.17.880468. doi: 10.1101/2019.12.17.880468. [DOI] [PMC free article] [PubMed]
- 113.Sczyrba A., Hofmann P., Belmann P., Koslicki D., Janssen S., Dröge J. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat Methods. 2017;14:1063–1071. doi: 10.1038/nmeth.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mikheenko A., Saveliev V., Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2016;32:1088–1090. doi: 10.1093/bioinformatics/btv697. [DOI] [PubMed] [Google Scholar]
- 115.Latorre-Pérez A., Villalba-Bermell P., Pascual J., Vilanova C. Assembly methods for nanopore-based metagenomic sequencing: a comparative study. Sci Rep. 2020;10:13588. doi: 10.1038/s41598-020-70491-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seppey M., Manni M., Zdobnov E.M. LEMMI: a continuous benchmarking platform for metagenomics classifiers. Genome Res. 2020;30:1208–1216. doi: 10.1101/gr.260398.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Weber L.M., Saelens W., Cannoodt R., Soneson C., Hapfelmeier A., Gardner P.P. Essential guidelines for computational method benchmarking. Genome Biol. 2019;20:125. doi: 10.1186/s13059-019-1738-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boulesteix A.-L. Ten simple rules for reducing overoptimistic reporting in methodological computational research. PLoS Comput Biol. 2015;11 doi: 10.1371/journal.pcbi.1004191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mangul S., Martin L.S., Hill B.L., Lam A.K.-M., Distler M.G., Zelikovsky A. Systematic benchmarking of omics computational tools. Nat Commun. 2019;10:1393. doi: 10.1038/s41467-019-09406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Norel R., Rice J.J., Stolovitzky G. The self-assessment trap: can we all be better than average? Mol Syst Biol. 2011;7:537. doi: 10.1038/msb.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jelizarow M., Guillemot V., Tenenhaus A., Strimmer K., Boulesteix A.-L. Over-optimism in bioinformatics: an illustration. Bioinformatics. 2010;26:1990–1998. doi: 10.1093/bioinformatics/btq323. [DOI] [PubMed] [Google Scholar]
- 122.Boulesteix A.-L., Lauer S., Eugster M.J.A. A plea for neutral comparison studies in computational sciences. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0061562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bokulich NA, Rideout JR, Mercurio WG, Shiffer A, Wolfe B, Maurice CF, et al. mockrobiota: a Public Resource for Microbiome Bioinformatics Benchmarking. mSystems 2016;1. doi: 10.1128/mSystems.00062-16. [DOI] [PMC free article] [PubMed]
- 124.Highlander S. Mock community analysis. In: Nelson KE, editor. Encyclopedia of Metagenomics, vol. 10, New York, NY: Springer New York; 2013, p. 1–7.
- 125.Huse SM, Huber J a., Morrison HG, Sogin ML, Mark Welch DB. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 2007;8:R143. [DOI] [PMC free article] [PubMed]
- 126.Bokulich N.A., Mills D.A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl Environ Microbiol. 2013;79:2519–2526. doi: 10.1128/AEM.03870-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yeh Y-C, Needham DM, Sieradzki ET, Fuhrman JA. Taxon Disappearance from Microbiome Analysis Reinforces the Value of Mock Communities as a Standard in Every Sequencing Run. mSystems 2018;3. doi: 10.1128/mSystems.00023-18. [DOI] [PMC free article] [PubMed]
- 128.Cichocki N., Hübschmann T., Schattenberg F., Kerckhof F.-M., Overmann J., Müller S. Bacterial mock communities as standards for reproducible cytometric microbiome analysis. Nat Protoc. 2020;15:2788–2812. doi: 10.1038/s41596-020-0362-0. [DOI] [PubMed] [Google Scholar]
- 129.Fouhy F., Clooney A.G., Stanton C., Claesson M.J., Cotter P.D. 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. 2016;16:1–13. doi: 10.1186/s12866-016-0738-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abusleme L., Hong B.-Y., Dupuy A.K., Strausbaugh L.D., Diaz P.I. Influence of DNA extraction on oral microbial profiles obtained via 16S rRNA gene sequencing. J Oral Microbiol. 2014;6. doi: 10.3402/jom.v6.23990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Taylor D.L., Walters W.A., Lennon N.J., Bochicchio J., Krohn A., Caporaso J.G. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for illumina amplicon sequencing. Appl Environ Microbiol. 2016;82:7217–7226. doi: 10.1128/AEM.02576-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gohl D.M., Vangay P., Garbe J., MacLean A., Hauge A., Becker A. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol. 2016;34:942–949. doi: 10.1038/nbt.3601. [DOI] [PubMed] [Google Scholar]
- 133.Amos G.C.A., Logan A., Anwar S., Fritzsche M., Mate R., Bleazard T. Developing standards for the microbiome field. Microbiome. 2020;8:98. doi: 10.1186/s40168-020-00856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schoch C.L., Ciufo S., Domrachev M., Hotton C.L., Kannan S., Khovanskaya R. NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database. 2020:2020. doi: 10.1093/database/baaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hardwick S.A., Chen W.Y., Wong T., Kanakamedala B.S., Deveson I.W., Ongley S.E. Synthetic microbe communities provide internal reference standards for metagenome sequencing and analysis. Nat Commun. 2018;9:3096. doi: 10.1038/s41467-018-05555-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhou Y.-H., Gallins P. A review and tutorial of machine learning methods for microbiome host trait prediction. Front Genet. 2019;10:579. doi: 10.3389/fgene.2019.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pasolli E., Truong D.T., Malik F., Waldron L., Segata N. Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput Biol. 2016;12 doi: 10.1371/journal.pcbi.1004977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vangay P., Hillmann B.M., Knights D. Microbiome Learning Repo (ML Repo): A public repository of microbiome regression and classification tasks. GigaScience. 2019:8. doi: 10.1093/gigascience/giz042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yilmaz P., Kottmann R., Field D., Knight R., Cole J.R., Amaral-Zettler L. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol. 2011;29:415–420. doi: 10.1038/nbt.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wilkinson M.D., Dumontier M., Aalbersberg I.J.J., Appleton G., Axton M., Baak A. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data. 2016;3 doi: 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kodama Y, on behalf of the International Nucleotide Sequence Database Collaboration, Shumway M, Leinonen R. The sequence read archive: explosive growth of sequencing data. Nucleic Acids Res 2011;40:D54–6. [DOI] [PMC free article] [PubMed]
- 142.Gonzalez A., Navas-Molina J.A., Kosciolek T., McDonald D., Vázquez-Baeza Y., Ackermann G. Qiita: rapid, web-enabled microbiome meta-analysis. Nat Methods. 2018;15:796–798. doi: 10.1038/s41592-018-0141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kaehler B.D., Bokulich N.A., McDonald D., Knight R., Caporaso J.G., Huttley G.A. Species abundance information improves sequence taxonomy classification accuracy. Nat Commun. 2019;10:4643. doi: 10.1038/s41467-019-12669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Robeson MS, O'Rourke DR, Kaehler BD, Ziemski M, Dillon MR, Foster JT, Bokulich NA. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv 2020.10.05.326504; 10.1101/2020.10.05.326504. [DOI] [PMC free article] [PubMed]
- 145.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O. Scikit-learn: machine learning in Python. J Machine Learning Res. 2011;12:2825–2830. [Google Scholar]
- 146.Thomas A.M., Manghi P., Asnicar F., Pasolli E., Armanini F., Zolfo M. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med. 2019;25:667–678. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Almeida A., Mitchell A.L., Tarkowska A., Finn R.D. Benchmarking taxonomic assignments based on 16S rRNA gene profiling of the microbiota from commonly sampled environments. GigaScience. 2018:7. doi: 10.1093/gigascience/giy054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Willis A.D., Martin B.D. Estimating diversity in networked ecological communities. Biostatistics. 2020 doi: 10.1093/biostatistics/kxaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fritz A, Hofmann P, Majda S, Dahms E, Dröge J, Fiedler J, et al. CAMISIM: Simulating metagenomes and microbial communities n.d. doi: 10.1101/300970. [DOI] [PMC free article] [PubMed]
- 150.Aniba M.R., Poch O., Thompson J.D. Issues in bioinformatics benchmarking: the case study of multiple sequence alignment. Nucl Acids Res. 2010;38:7353–7363. doi: 10.1093/nar/gkq625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kelly B.J., Gross R., Bittinger K., Sherrill-Mix S., Lewis J.D., Collman R.G. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics. 2015;31:2461–2468. doi: 10.1093/bioinformatics/btv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Debelius J., Song S.J., Vazquez-Baeza Y., Xu Z.Z., Gonzalez A., Knight R. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol. 2016;17:217. doi: 10.1186/s13059-016-1086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Goldman N. Statistical tests of models of DNA substitution. J Mol Evol. 1993;36:182–198. doi: 10.1007/bf00166252. [DOI] [PubMed] [Google Scholar]
- 154.Huang W., Li L., Myers J.R., Marth G.T. ART: a next-generation sequencing read simulator. Bioinformatics. 2012;28:593–594. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ono Y., Asai K., Hamada M. PBSIM: PacBio reads simulator–toward accurate genome assembly. Bioinformatics. 2013;29:119–121. doi: 10.1093/bioinformatics/bts649. [DOI] [PubMed] [Google Scholar]
- 156.Yang C., Chu J., Warren R.L., Birol I. NanoSim: nanopore sequence read simulator based on statistical characterization. GigaScience. 2017;6:1–6. doi: 10.1093/gigascience/gix010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bergstra J., Bengio Y. Random search for hyper-parameter optimization. J Mach Learn Res. 2012;13:281–305. [Google Scholar]
- 158.Luo G. A review of automatic selection methods for machine learning algorithms and hyper-parameter values. Network Modeling Analysis in Health Informatics and Bioinformatics. 2016;5:1–16. [Google Scholar]
- 159.Nguyen V. 2019 IEEE second international conference on artificial intelligence and knowledge engineering (AIKE) 2019. Bayesian Optimization for Accelerating Hyper-Parameter Tuning. [DOI] [Google Scholar]
- 160.Bochinski E., Senst T., Sikora T. 2017 IEEE international conference on image processing (ICIP) 2017. Hyper-parameter optimization for convolutional neural network committees based on evolutionary algorithms. [DOI] [Google Scholar]
- 161.Hermans S.M., Buckley H.L., Case B.S., Curran-Cournane F., Taylor M., Lear G. Using soil bacterial communities to predict physico-chemical variables and soil quality. Microbiome. 2020;8:79. doi: 10.1186/s40168-020-00858-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochimica et Biophysica Acta (BBA) - Protein Structure 1975;405:442–51. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed]
- 163.Schloss PD. Application of a Database-Independent Approach To Assess the Quality of Operational Taxonomic Unit Picking Methods. mSystems 2016;1. doi: 10.1128/mSystems.00027-16. [DOI] [PMC free article] [PubMed]
- 164.Willis A.D. Rarefaction, alpha diversity, and statistics. Front Microbiol. 2019;10:2407. doi: 10.3389/fmicb.2019.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Manning C.D., Raghavan P., Schütze H. Cambridge University Press; 2008. Introduction to information retrieval. [Google Scholar]
- 166.Westcott SL, Schloss PD. OptiClust, an Improved Method for Assigning Amplicon-Based Sequence Data to Operational Taxonomic Units. mSphere 2017;2. doi: 10.1128/mSphereDirect.00073-17. [DOI] [PMC free article] [PubMed]
- 167.McDonald D., Price M.N., Goodrich J., Nawrocki E.P., DeSantis T.Z., Probst A. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Maxim L.D., Daniel Maxim L., Niebo R., Utell M.J. Screening tests: a review with examples. Inhalation Toxicol. 2014;26:811–828. doi: 10.3109/08958378.2014.955932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Glassman S.I., Martiny J.B.H. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. mSphere. 2018;3:1–5. doi: 10.1128/mSphere.00148-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lu J., Salzberg S.L. Ultrafast and accurate 16S rRNA microbial community analysis using Kraken 2. Microbiome. 2020;8:124. doi: 10.1186/s40168-020-00900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Noble W.S. A quick guide to organizing computational biology projects. PLoS Comput Biol. 2009;5 doi: 10.1371/journal.pcbi.1000424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sandve G.K., Nekrutenko A., Taylor J., Hovig E. Ten simple rules for reproducible computational research. PLoS Comput Biol. 2013;9 doi: 10.1371/journal.pcbi.1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wilson G., Aruliah D.A., Brown C.T., Chue Hong N.P., Davis M., Guy R.T. Best practices for scientific computing. PLoS Biol. 2014;12 doi: 10.1371/journal.pbio.1001745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Baxter S.M., Day S.W., Fetrow J.S., Reisinger S.J. Scientific software development is not an oxymoron. PLoS Comput Biol. 2006;2 doi: 10.1371/journal.pcbi.0020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kim Y.-M., Poline J.-B., Dumas G. Experimenting with reproducibility: a case study of robustness in bioinformatics. GigaScience. 2018:7. doi: 10.1093/gigascience/giy077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schloss P.D. Identifying and overcoming threats to reproducibility, replicability, robustness, and generalizability in microbiome research. MBio. 2018;9 doi: 10.1128/mBio.00525-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wilson G, Bryan J, Cranston K, Kitzes J, Nederbragt L, Teal TK. Good Enough Practices in Scientific Computing 2016. [DOI] [PMC free article] [PubMed]
- 179.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Miller G. Scientific publishing. A scientist’s nightmare: software problem leads to five retractions. Science. 2006;314:1856–1857. doi: 10.1126/science.314.5807.1856. [DOI] [PubMed] [Google Scholar]
- 181.Kane D.W., Hohman M.M., Cerami E.G., McCormick M.W., Kuhlmman K.F., Byrd J.A. Agile methods in biomedical software development: a multi-site experience report. BMC Bioinf. 2006;7:273. doi: 10.1186/1471-2105-7-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Steinmacher I., Graciotto Silva M.A., Gerosa M.A., Redmiles D.F. A systematic literature review on the barriers faced by newcomers to open source software projects. Inf Softw Technol. 2015;59:67–85. [Google Scholar]
- 183.Geiger R.S., Varoquaux N., Mazel-Cabasse C., Holdgraf C. The types, roles, and practices of documentation in data analytics open source software libraries: a collaborative ethnography of documentation work. Comput Support Coop Work. 2018;27:767–802. [Google Scholar]