

Abstract
N6-methyladenosine (m6A) is the most commonly occurring internal RNA modification to be found in eukaryotic mRNA and serves an important role in various physiological events. AlkB homolog 5 RNA demethylase (ALKBH5), an m6A demethylase, belongs to the AlkB family of dioxygenases and has been shown to specifically demethylate m6A in RNA, which is associated with a variety of tumors. However, its function in bladder cancer remains largely unclear. In the present study, we found that the expression of ALKBH5 was downregulated in bladder cancer tissues and cell lines. Low expression of ALKBH5 was correlated with the worse prognosis of bladder cancer patients. Furthermore, functional assays revealed that knockdown of ALKBH5 promoted bladder cancer cell proliferation, migration, invasion, and decreased cisplatin chemosensitivity in the 5637 and T24 bladder cancer cell lines in vivo and in vitro, whereas ALKBH5 overexpression led to the opposite results. Finally, ALKBH5 inhibited the progression and sensitized bladder cancer cells to cisplatin through a casein kinase 2 (CK2)α-mediated glycolysis pathway in an m6A-dependent manner. Taken together, these findings might provide fresh insights into bladder cancer therapy.
Keywords: ALKBH5, bladder cancer, m6A, protein kinase CK2α, cisplatin, glycolysis
Graphical Abstract
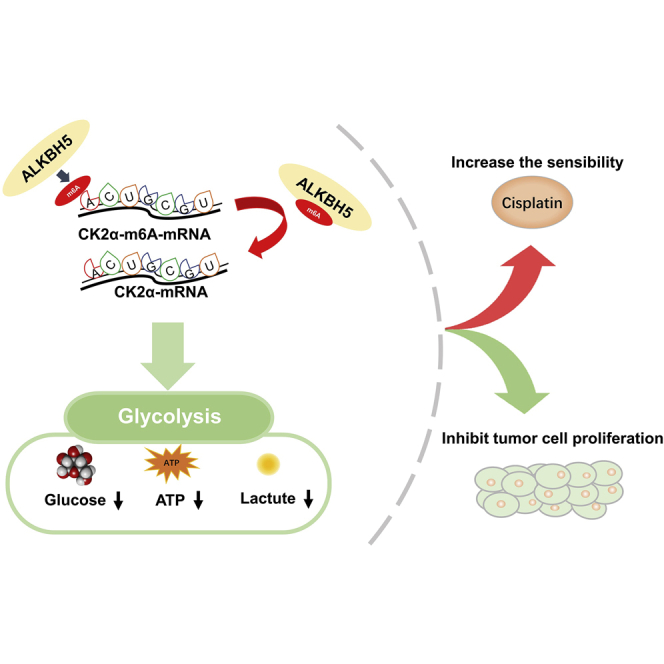
Yu et al. show that m6A demethylase ALKBH5 was correlated with the prognosis of bladder cancer patients, inhibited bladder cancer cell proliferation, and sensitized bladder cancer cells to cisplatin in vitro and in vivo by CK2α-mediated glycolysis in an m6A-dependent manner, which might provide fresh insights into bladder cancer therapy.
Introduction
Bladder cancer is the fourth most common malignancy in men, with ~81,190 new cases and 17,240 cases of mortality estimated during 2018 in the United States.1 Treatment is effected mainly through surgical intervention and chemotherapy.2 Cisplatin-based chemotherapy has been the first-class treatment of bladder cancer for many years, which has been proven to increase the 5-year overall survival of bladder cancer patients. However, the overall response rate of patients to chemotherapy for bladder cancer is only around 20%–40%. The sensitivity to cisplatin varies considerably among patients. In recent years, although significant progress has been made in terms of immunotherapy and targeted therapies for other types of tumors,2,3 no effective therapeutic target for bladder cancer has been identified to date. Resistance to the chemotherapy is still a major problem in terms of bladder cancer treatment. Therefore, a detailed understanding of the pathogenesis of bladder cancer is essential for identifying novel and effective therapeutic targets.
N6-methyladenosine (m6A) is the most common internal modification to be found in the mRNAs of most eukaryotes.4 A variety of tRNAs, rRNAs, long noncoding RNAs, and mRNA methylation events have been identified and shown to regulate RNA splicing,5, 6, 7 stability,8, 9, 10, 11, 12, 13 and translation.14, 15, 16 This mechanism involves m6A “writer,” “eraser,” and “reader” proteins that function by catalyzing, removing, or recognizing m6A-modified sites, thereby altering important biological processes accordingly. Recently, multiple studies have reported that m6A regulatory proteins fulfill important roles in the initiation and progression of different types of cancer, including breast cancer,17,18 glioblastoma,19 hepatocellular carcinoma,20,21 renal cell carcinoma,22 bladder cancer,23, 24, 25 and so on.26,27 Yang et al.23 first reported that methyltransferase-like 3 (METTL3) played an oncogenic role in bladder cancer. Cheng et al.24 confirmed that METTL3 promoted bladder cancer progression via the AFF4/nuclear factor κB (NF-κB)/Myc signaling network. Based on previous work in our laboratory, Han et al.25 reported that METTL3 promoted tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in an m6A-dependent manner. However, other m6A regulatory proteins have yet to be studied in bladder cancer.
AlkB homolog 5 RNA demethylase (ALKBH5), an m6A demethylase, belongs to the AlkB family of dioxygenases and has been shown to play different roles in different types of tumor. ALKBH5 may promote cancer by mediating the m6A demethylation of mRNA in glioblastoma,28 breast cancer,17 and ovarian cancer.29 In breast cancer, ALKBH5 was shown to mediate the m6A demethylation of homeobox transcription factor Nanog (NANOG) mRNA, thereby inducing the breast cancer stem cell phenotype.17 However, in pancreatic cancer, ALKBH5 was shown to inhibit tumor development by mediating the m6A demethylation of long noncoding RNA.30 These studies confirmed that ALKBH5 may play different roles in various types of cancer. In bladder cancer, Yang et al.23 demonstrated that ALKBH5 exerted opposite roles to those of METTL3. Cheng et al.24 also reported that ALKBH5 was downregulated in bladder cancer tissues compared with normal tissues. However, the mechanism and role of ALKBH5 in bladder cancer have yet to be elucidated.
Tumor cells prefer to consume glucose to produce lactate, even under oxygen-rich conditions. This phenomenon is referred to as the “Warburg effect” or “aerobic glycolysis.”31 The high glycolytic flux depends on glycolysis-associated genes, such as SRC-3, glucose transporter type 1 (GLUT1), GLUT3, lactic dehydrogenase A (LDHA), LDHB, hexokinase 1 (HK1), HK2, pyruvate kinase type M (PKM), and hypoxia-inducible factor 1-alpha (HIF-1α), resulting in production of pyruvate, alanine, and lactate. Previous studies reported that the protein kinase, casein kinase 2 (CK2), augments the Warburg effect, leading to increased proliferation, migration, and invasion of cancer cells.32,33 CK2 is a conserved, ubiquitously expressed protein serine/threonine kinase.34 CK2α is one of the essential catalytic subunits of the holoenzyme.35 Recently, high expression levels of CK2α were found in breast cancer,36 prostate cancer,37 lung cancer,38,39 head and neck cancer,40 colorectal cancer,41 gastric cancer,42 renal cell carcinoma,43 and bladder cancer,44 which closely associated with the development of tumors. A previous study from our research demonstrated that CK2α promoted bladder cancer cell survival via the glucose metabolic pathway.44 In addition, CK2 was shown to affect the cisplatin drug sensitivity in gastric cancer,45 non-small cell lung,46 and cholangiocarcinoma.47 Inhibitors of CK2 were subsequently shown to promote cancer cell apoptosis by increasing the sensitivity of cisplatin chemotherapy.46, 47, 48
In the present study, the roles and associated mechanisms of ALKBH5 in bladder cancer were investigated. These experiments revealed the following: that (1) ALKBH5 expression was downregulated in bladder cancer tissues, and low expression levels of ALKBH5 in bladder cancer was associated with a poor prognosis; (2) ALKBH5 inhibited bladder cancer cell proliferation in vitro and in vivo via CK2α-mediated glycolysis; (3) knockdown of ALKBH5 decreased cisplatin-induced apoptosis; and (4) ALKBH5 may have a suppressive role in bladder cancer by affecting the stability of CK2α mRNA in an m6A-dependent manner. To the best of our knowledge, this is the first comprehensive study to have identified that ALKBH5 may affect tumor progression by regulating m6A modification in bladder cancer, and the results obtained could provide fresh insights into the development of novel bladder cancer therapies. Therefore, ALKBH5 may act as a novel diagnosis predictor for patients with bladder cancer.
Results
ALKBH5 Was Significantly Downregulated in Human Bladder Cancer Tissues and Associated with Bladder Cancer Patient Prognosis
First, quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) and western blot results from 28 paired bladder cancer tissues demonstrated that ALKBH5 was significantly downregulated in bladder cancer tissues compared with normal tissues in mRNA (Figure 1A) and protein (Figure 1B) levels. ALKBH5 was also downregulated in five bladder cancer cell lines compared with SVHUC-1 cells (human ureteral epithelial immortalized cell line), selected as a normal bladder epithelial cell line (Figures 1C and 1D). Moreover, ALKBH5 was also found to be significantly downregulated in bladder cancer tissue in the GEO database (Figure S1A). Next, immunohistochemistry (IHC) analysis of tissue microarray (TMA) was performed to further explore the relationship between the expression of ALKBH5 and clinicopathological features of the patients (Figure 1E). Subsequent investigations revealed that the expression of ALKBH5 was associated with histological grade and tumor-lymph node-metastasis (TNM) stage (Table 1). The high-expression group experienced a lower grade and TNM stage. Furthermore, Kaplan-Meier survival curves revealed that patients with low levels of ALKBH5 expression had a worse prognosis and poorer overall survival rate compared with those with high ALKBH5 expression (Figure 1F). Therefore, we speculated that ALKBH5 acted as a suppressor in bladder cancer.
Figure 1.

ALKBH5 Was Downregulated in Bladder Cancer Tissues and Cell Lines and Served as a Prognostic Factor in Bladder Cancer
(A) Relative expression of ALKBH5 mRNA in the 28 pairs of bladder cancer tissues and matched adjacent normal tissues quantified by qRT-PCR. ALKBH5 was downregulated in bladder cancer tissues compared with that in adjacent normal tissues (p < 0.01). (B) The expression of ALKBH5 protein in 8 pairs of bladder cancer tissues (T) and adjacent normal tissues (N) by western blot were shown. (C and D) Relative expression of ALKBH5 in bladder cancer cell lines and immortalized normal bladder epithelial cell line SVHUC-1 by qRT-PCR and western blot. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (E) IHC analysis of ALKBH5 in bladder cancer tissue at 200× magnification. (F) Kaplan-Meier survival curves of overall survival in 161 bladder cancer patients based on ALKBH5 by IHC staining. The log-rank test was used to compare differences between two groups (p = 0.036).
Table 1.
Association of ALKBH5 Expression with Clinicopathologic Characteristics of Bladder Cancer Patients
| Parameters | Number of Cases | ALKBH5 Expression |
p Value | |
|---|---|---|---|---|
| Low (%) | High (%) | |||
| Age (Years) | ||||
| 40 | 30 | 10 | 0.487 |
|
| ≥60 | 121 | 97 | 24 | |
| Gender | ||||
| Male | 124 | 101 | 23 | 0.144 |
| Female | 37 | 26 | 11 | |
| Histological Grade | ||||
| Low | 108 | 80 | 28 | 0.033a |
| High | 53 | 47 | 6 | |
| TNM Stage | ||||
| Ta, T1 | 95 | 69 | 26 | 0.020a |
| T2–T4 | 66 | 58 | 8 | |
| Lymph Node Metastasis | ||||
| Negative | 152 | 122 | 30 | 0.179 |
| Positive | 9 | 5 | 4 | |
| CK2α | ||||
| Negative | 43 | 28 | 15 | 0.010a |
| Positive | 118 | 99 | 19 | |
Statistically significant.
GEO database (https://www.ncbi.nlm.nih.gov/geo/; GEO: GSE13507) analysis also showed that methyltransferase-like 14 (METTL14) and Wilms tumor 1-associating protein (WTAP) were downregulated in bladder cancer tissues (Figure S1A). However, the mRNA levels of METTL14 and WTAP were not statistically different between tumor tissues and adjacent normal tissues by qRT-PCR (Figure S1B). Moreover, fat mass and obesity-associated protein (FTO) was no statistical difference between tumor tissues and adjacent normal tissues by GEO database analysis and qRT-PCR (Figure S1).
ALKBH5 Inhibited Bladder Cancer Cell Proliferation and Migration In Vitro
To verify the function of ALKBH5 in vitro, T24 and 5637 cells were stably transfected with knockdown lentivirus, overexpression lentivirus, or control lentivirus. The expression levels of ALKBH5 were confirmed by qRT-PCR analysis and western blotting (Figure S2).
Cell counting kit-8 (CCK-8) assays indicated that ALKBH5 knockdown significantly increased cell proliferation (Figure 2A), whereas ALKBH5 overexpression decreased cell proliferation (Figure 2B). In colony-formation assays, knockdown of ALKBH5 increased colony formation (Figure 2C), whereas the colony-formation rate was reduced in ALKBH5-overexpressed cells (Figure 2D). Additionally, cell cycle analysis revealed that ALKBH5 knockdown decreased the percentage of bladder cancer cells at the G1 phase, whereas ALKBH5 overexpression had the opposite effect (Figures 2E and 2F). ALKBH5 knockdown was also observed to promote the cell-invasive and -migratory capabilities in the 5637 and T24 cell lines, whereas ALKBH5 overexpression had the opposite effect (Figure S3).
Figure 2.

ALKBH5 Inhibited Bladder Cancer Cell Proliferation In Vitro
(A and B) Cell proliferation assessed by CCK-8 assays. Knockdown of ALKBH5 promoted cell proliferation in 5637 and T24 cells. Overexpression of ALKBH5 inhibited cell proliferation in 5637 and T24 cells. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (C and D) Colony-formation assay showed that knockdown of ALKBH5 significantly increased the cloning number of 5637 and T24 cells compared with control group, whereas ALKBH5 overexpression significantly decreased the cloning number of 5637 and T24 cells. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (E and F) Cell cycle analyzed by flow cytometry. Histogram indicates the percentage of cells in G0/G1, S, and G2/M. Data represent the mean ± SD from three independent experiments, ∗p < 0.05.
Taken together, these results showed that ALKBH5 could be involved in bladder cancer proliferation, migration, and invasion, which acted as a tumor suppressor.
ALKBH5 Increased Sensitivity to Cisplatin In Vivo and In Vitro
CCK-8 and flow cytometry assays were used to investigate the effects of ALKBH5 on sensitivity to cisplatin. The CCK-8 assay results revealed that the viability of the cells was inhibited with an increasing concentration of cisplatin (Figures 3A and 3B). Knockdown of ALKBH5 dramatically decreased the suppression rate of cisplatin in 5637 and T24 cells compared with that in the negative control (NC) group. The half-maximal inhibitory concentration (IC50) results showed that ALKBH5 knockdown led to an increase in the IC50 value, and the sensitivity to cisplatin was subsequently decreased (Figure 3A), whereas ALKBH5 overexpression elicited the opposite effect (Figure 3B). Furthermore, it was revealed that the apoptotic rate of bladder cancer cells was significantly increased following treatment with cisplatin for 24 or 48 h. Additionally, knockdown of ALKBH5 decreased the apoptotic rate induced by cisplatin in 5637 and T24 cells, whereas overexpression of ALKBH5 increased the rate of apoptosis (Figures 3C–3E). In vivo, T24 cells transfected with ALKBH5 knockdown or the control group were injected subcutaneously into nude mice to investigate whether knockdown of ALKBH5 expression affected sensitivity to cisplatin on the bladder cancer cells (Figures 3F and 3G). Knockdown of ALKBH5 increased bladder cancer tumor size and weight in mice (Figures 3H and 3I). After cisplatin chemotherapy, the size and weight of tumor were deceased compared with treatment with the normal saline (Figures 3H and 3I). In the cisplatin chemotherapy groups, the size and volume of ALKBH5 knockdown group were larger than those of the short hairpin (sh)NC group (Figures 3G and 3I). Taken together, these results were consistent with the change of drug sensitivity and demonstrated that ALKBH5 was able to increase the cisplatin chemosensitivity of bladder cancer cells.
Figure 3.

ALKBH5 Increased Sensitivity to Cisplatin In Vivo and In Vitro
(A) Knockdown of ALKBH5 expression decreased the suppression rate of cisplatin and increased IC50 to cisplatin in 5637 and T24 cells by CCK-8. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (B) Overexpression of ALKBH5 expression increased the rate of cisplatin and decreased IC50 to cisplatin in 5637 and T24 cells by CCK-8. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (C–E) The percentage of apoptotic cells was increased after treatment with cisplatin for 48 h. Knockdown of ALKBH5 expression decreased the rate of cisplatin-induced apoptosis compared with control cells in 5637 and T24 cells, whereas overexpression of ALKBH5 increased the rate of cisplatin-induced apoptosis. Data represent the mean ± SD from three independent experiments. Student’s t test with two biological independent replicates was used to determine statistical significance, ∗p < 0.05. (F and G) Subcutaneous xenograft tumor model with ALKBH5 knockdown (shALKBH5) or control cells (shNC). Cisplatin or normal saline was injected intraperitoneally starting from day 7 of tumor inoculation. The red arrows indicate the location of the tumor. (H) Tumor weight was measured after 3 weeks from transplanting. Data represent the mean ± SD, ∗p < 0.05. (I) Tumor volume was measured every 3 days from transplanting. Data represent the mean ± SD, ∗p < 0.05.
ALKBH5 Correlated with CK2α Expression in Bladder Cancer Tissues and Cell Lines
Subsequently, The Cancer Genome Atlas (TCGA) database was used to search for potential mRNAs, which were regulated by ALKBH5. The analysis results revealed that ALKBH5 was associated with CK2α expression (Figure S4). Western blot assays confirmed that ALKBH5 knockdown dramatically upregulated the expression of CK2α and the glycolysis-associated proteins, GLUT, HK1, LDHA, LDHB, and PKM (Figure 4A), whereas ALKBH5 overexpression had the opposite effects in 5637 and T24 cells (Figure 4B). qRT-PCR assays confirmed that ALKBH5 knockdown upregulated the expression of CK2α and the glycolysis-associated mRNAs, GLUT, HK1, and LDHA (Figure S5A), whereas ALKBH5 overexpression had the opposite effects in 5637 and T24 cells (Figure S5B). Glucose uptake, lactate detection, and ATP detection assays revealed that ALKBH5 knockdown was able to significantly promote glucose utilization (Figure 4C) and lactate production (Figure 4D) and increase intracellular ATP levels (Figure 4E). However, ALKBH5 overexpression reduced the level of glycolysis in 5637 and T24 cells (Figures 4E–4G).
Figure 4.

ALKBH5 Was Correlated with CK2α Expression in Bladder Cancer Tissues and Cell Lines
(A and B) Western blot analysis of CK2α, GLUT, HK1, LDHA, LDHB, PKM, and GAPDH in 5637 and T24 cells with ALKBH5 knockdown or overexpression. (C–E) Knockdown of ALKBH5 expression increased uptake of glucose, lactate production, and the intracellular ATP level within 24 h. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (F–H) Overexpression of ALKBH5 expression suppressed the uptake of glucose, lactate production, and the intracellular ATP level. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (I) CK2α was expressed higher in bladder cancer tissues than in adjacent normal tissues by qRT-PCR (n = 28, p < 0.001). (J and K) A negative correlation between the expression of CK2α and ALKBH5 was shown in bladder cancer tissues by qRT-PCR and IHC. Scale bars, 100 μm.
Subsequently, the expression levels of ALKBH5, CK2α, and glycolysis-related genes were investigated in patient tissues. qRT-PCR assays revealed that the expression of CK2α was upregulated in bladder cancer tissues (Figure 4I) and negatively associated with ALKBH5 (Figure 4J). We also found that glycolysis-related genes GLUT, LDHA, and LDHB were negatively associated with ALKBH5 (Figure S5C). IHC analysis showed that the expression of ALKBH5 and CK2α predominantly occurred in the nucleus and cytoplasm (Figure 4K). A significant negative association of the expression of CK2α with ALKBH5 was also identified based on the paraffin-embedded tissues from the TMA analysis (Table 1). Taken together, these data strongly suggested that ALKBH5 negatively regulated CK2α expression and influenced glycolysis in bladder cancer tissues and cell lines.
ALKBH5 Reduced the Stability of the CK2α mRNA in an m6A-Dependent Manner
First, immunofluorescence assays revealed that ALKBH5 was widely distributed in the nucleus and cytoplasm (Figure 5A), which were consistent with the results of IHC. Subsequently, the ALKBH5 knockdown or overexpression cells were treated with actinomycin D (Act D). ALKBH5 knockdown was found to significantly increase the relative half-life of the CK2α transcript (Figure 5B). However, the half-life of the CK2αtranscript was decreased by overexpression of ALKBH5 in the 5637 and T24 cells (Figure 5C). These data suggested that ALKBH5 could reduce the stability of CK2α mRNA.
Figure 5.

ALKBH5 Modulated the Stability of the CK2α mRNA in an m6A-Dependent Manner
(A) Immunofluorescence staining of 5637 and T24 cells revealed that ALKBH5 was widely distributed in the nucleus and cytoplasm. (B) Knockdown of ALKBH5 expression increased the half-life of the CK2α transcript. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (C) Overexpression of ALKBH5 expression shortened the half-life of the CK2α transcript. Data represent the mean ± SD from three independent experiments, ∗p < 0.05. (D) RIP assay demonstrated that anti-ALKBH5 antibody was able to significantly enrich the level of CK2α mRNA compared with the anti-IgG antibody by qRT-PCR. The β-actin transcript was used as control. (E) The effect of ALKBH5 on a wild-type pLenti-UTR-Luc vector or a mutant pLenti-UTR-Luc vector was measured by luciferase reporter assays in 5637 and T24 cells. (F) The level of m6A in bladder cancer tissues was higher than that in adjacent normal tissues detected by the m6A RNA methylation quantification kit (∗p < 0.05). (G) m6A dot blot assays of 5637 and T24 cells with knockdown or overexpression of ALKBH5; methylene blue (MB) stain as loading control. The methylation of RNA decreased significantly after ALKBH5 overexpression, whereas it increased significantly after ALKBH5 knockdown. (H) The detection of CK2α m6A modification level by immunoprecipitation of m6A-modified mRNA in control or ALKBH5-overexpressed cells followed by qRT-PCR. Data represent the mean ± SD from three independent experiments, ∗p < 0.05
Subsequently, the RNA immunoprecipitation (RIP) assay demonstrated that the anti-ALKBH5 antibody was able to significantly enrich the level of CK2α mRNA compared with the anti-immunoglobulin G (IgG) antibody (Figure 5D). As a NC, the β-actin transcript was not detectable in the ALKBH5 and IgG immunocomplexes. These results strongly suggested that ALKBH5 was able to bind to the CK2α transcript physically. In particular, it was possible to hypothesize that ALKBH5 may bind to CK2α mRNA in vitro and reduce the stability of the CK2α transcript.
Furthermore, to investigate whether the CK2α 3′ untranslated region (UTR) was required for ALKBH5 in order to reduce the levels of CK2α expression, a dual-luciferase assay was performed using pLenti-UTR-Luc reporters that carried CK2α3′ UTR or an empty vector in 5637 and T24 ALKBH5-overexpressed cells and control cells. The results showed that overexpression of ALKBH5 decreased the luciferase activity of the CK2α 3′ UTR reporter vector but did not exert any effect on the empty vector (Figure 5E). Taken together, these data indicated that ALKBH5 could bind to the CK2α 3′ UTR.
Finally, with the use of the m6A RNA methylation quantification kit, we found the level of m6A in tumor tissues was significantly higher compared with that in adjacent normal tissues (Figure 5F). The results of the dot blot showed that knockdown of ALKBH5 led to an increase in the m6A level, and the overexpression of ALKBH5 reduced the level of m6A in bladder cancer cells (Figure 5G). m6A RIP (MeRIP) assays showed that overexpression of ALKBH5 decreased the m6A levels of the CK2α mRNAs in 5637 and T24 cells (Figure 5H). Together, all of these results indicated that ALKBH5 reduced the stability of CK2α mRNAs in an m6A-dependent manner.
CK2α Interference Decreased the Cell Proliferation and Increased the Extent of Apoptosis Induced by ALKBH5 in Bladder Cancer Cells
To assess the cell proliferation of CK2α as it interacts with ALKBH5 in bladder cancer, CK2α small interference (si)RNA (siCK2α) or a control (scramble [SCR]) were transfected in ALKBH5 knockdown or control cells. The results of the CCK-8 assay showed that CK2α knockdown led to a significant decrease in the cell growth rate. In addition, CK2α knockdown totally reversed the proliferation induced by ALKBH5 in bladder cancer cells (Figure 6A). The colony-formation assays also produced similar results (Figure 6B). Through the quantitative analysis of glycolysis, it was identified that siCK2α decreased the glucose uptake, lactic acid production, and ATP production of bladder cancer cells within 24 h (Figures 6C–6E), which suggested that ALKBH5 promoted cell proliferation through a CK2α-dependent glycolytic pathway. Furthermore, two selective inhibitors of CK2α, CX-4945 and 4,5,6,7-tetrabromobenzotriazole (TBB), were used for treatment in ALKBH5 knockdown and control cells. We found the optical density (OD) value fold (shALKBH5/shNC) and the cell numbers fold (shALKBH5/shNC) were decreased, as determined by the CCK-8 assays (Figure S6A) and colony-formation assays (Figure S6B). Additionally, treatment with CX-4945 led to an increase the apoptotic rate induced by knockdown of ALKBH5 expression in 5637 and T24 cells (Figure S7). These results showed that the ability of cell proliferation was decreased, and the apoptosis rate was increased, when the CK2α function was inhibited in bladder cancer cells.
Figure 6.

CK2α Interference Decreased the Promotion of Cell Proliferation Induced by ALKBH5 in Bladder Cancer Cells
(A) CCK-8 assays were used to measure the effect of siCK2α or SCR on 5637 and T24 cells with ALKBH5 knockdown. siCK2α could partly reduce cell growth induced by knockdown of ALKBH5 in 5637 and T24 cells. (B) Colony-formation assays showed that siCK2α could rescue the cell growth promoted by knockdown of ALKBH5 in 5637 and T24 cells. (C–E) siCK2α decreased glucose uptake, lactic acid production, and ATP production of bladder cancer cells within 24 h. Data represent the mean ± SD from three independent experiments, ∗p < 0.05
With the consideration of all of these results together, it was possible to verify that CK2α interference decreased cell proliferation, and increased the apoptotic rate induced by ALKBH5 in bladder cancer cells.
ALKBH5 Inhibited Tumorigenesis In Vivo
To further verify the effects of ALKBH5 on tumor growth in vivo, a subcutaneous xenograft tumor model was established. The tumors injected with ALKBH5 overexpression cells grew more slowly compared with those injected with control (NC) cells (Figure 7A). Overexpression of ALKBH5 inhibited bladder cancer tumor size and weight in mice (Figures 7B and 7C). Western blot, qRT-PCR, and IHC analyses showed that the expression of CK2α was significantly decreased in the ALKBH5 overexpression groups (Figures 7D and 7E). Furthermore, IHC analysis showed that expression of the cell proliferation marker, Ki67, was significantly decreased in the ALKBH5 overexpression groups. In summary, these results demonstrated that overexpression of ALKBH5 was able to inhibit tumorigenesis in vivo.
Figure 7.

ALKBH5 Inhibited Tumorigenesis In Vivo
(A) Subcutaneous xenograft tumor model with ALKBH5 overexpression (ALKBH5) or control cells (NC). (B) Tumor volume was measured at the indicated weeks after transplanting. Data represent the mean ± SD, ∗p < 0.05. (C) Tumor weight was measured after 3 weeks from transplanting. Data represent the mean ± SD, ∗p < 0.05. (D) Western blot and qRT-PCR analysis of CK2α and ALKBH5 expression in the tumor model. Data represent the mean ± SD, ∗p < 0.05. (E) IHC analysis showed that the expression of CK2α and cell proliferation marker Ki67 decreased in ALKBH5. Data represent the mean ± SD from three independent experiments, ∗p < 0.05 (F) Working model: ALKBH5 inhibited cell proliferation and sensitized bladder cancer cells to cisplatin by m6A-CK2α-mediated glycolysis.
Discussion
As a main component in the m6A eraser, ALKBH5 was reported to be downregulated in bladder cancer samples.24 In the present study, we further found that the downexpression of ALKBH5 was associated with TNM stage, histological grade, and poor prognosis of bladder cancer patients. We confirmed that ALKBH5 inhibited bladder cancer cell proliferation and sensitized cancer cells to cisplatin by CK2α-mediated glycolysis in an m6A-dependent manner. To the best of the authors’ knowledge, this is the first comprehensive study to have shown that ALKBH5 affected tumorigenesis by regulating the modification of m6A in bladder cancer.
Recent studies have also demonstrated that ALKBH5 acts as a suppressor in colon49 and pancreatic cancer.30 In bladder cancer, Yang et al.23 demonstrated that ALKBH5 exerted opposite roles to those of METTL3. Cheng et al.24 also reported ALKBH5 was downregulated in bladder cancer tissues compared with normal tissues. In the present study, we first verified that the level of ALKBH5 was significantly downregulated in bladder cancer tissues and cell lines, especially in T24 and 5637 cell lines. Furthermore, we found that ALKBH5 expression was associated with the histological grade and TNM stage in bladder cancer patients. In addition, bladder cancer patients with high expression levels of ALKBH5 experienced an improved prognosis, suggesting that ALKBH5 may be a prognostic indicator for bladder cancer patients. As ALKBH5 acting as a suppressor in bladder cancer, we selected T24 and 5637 cell lines with lower ALKBH5 expression for our follow-up functional experiments. We found that overexpression of ALKBH5 significantly inhibited the proliferation, migration, and invasion of T24 and 5637 cells. Similarly, in vivo experiments showed that overexpression of ALKBH5 in the T24 cells led to the development of smaller tumors in nude mice compared with the control group. These results confirmed that ALKBH5 played a suppressive role in bladder cancer.
Zhang et al.50 reported that ALKBH5 was associated with CK2α expression. In our present study, we confirmed that ALKBH5 and CK2α were significantly negatively correlated in bladder cancer tissues by western blotting, qRT-PCR, and IHC analyses. Furthermore, ALKBH5 was shown to significantly alter the level of CK2α and influence the glucose metabolic pathway. In our previous study, Zhang et al.44 reported that CK2α affected the glycolysis pathway. Furthermore, we found that knockdown of ALKBH5 expression significantly promoted glucose utilization and lactate production and increased intracellular ATP levels, whereas overexpression of ALKBH5 had the opposite effect. Glucose uptake, lactate, and ATP production were reduced by interfering with the level of CK2α in bladder cancer cells after knockdown of ALKBH5 expression, indicating the key dominance of ALKBH5 and CK2α in the whole process. These results confirmed that ALKBH5 was associated with CK2α expression in bladder cancer tissues and cell lines and could affect bladder cancer via CK2α-mediated glycolysis. The inhibitors of CK2α have been found to suppress tumor growth44,51,52 in acute and chronic leukemias,53 pancreatic cancer,54 and bladder cancer.44 In our study, we found that inhibitors of CK2α, CX-4945, or TBB55,56 could rescue the cell proliferation, glucose utilization, lactate production, and intracellular ATP levels and also the apoptotic rate induced by ALKBH5 knockdown in bladder cancer cells. CK2α siRNA assay revealed the identical results. All of these results suggested that ALKBH5 could inhibit cell proliferation via CK2α.
As a molecule that specifically demethylated m6A in single-stranded RNA,57 ALKBH5 was found to regulate the targeted genes by affecting mRNA stability. Zhang et al.28 reported that ALKBH5 mediated the m6A demethylation of FOXM1 mRNA, leading to enhanced FOXM1 expression in glioblastoma. Li et al.58 reported that ALKBH5 was able to control trophoblast invasion at the maternal-fetal interface by regulating the stability of CYR61 mRNA. Zhu et al.59 also reported that ALKBH5 reduced the TIMP3 mRNA stability depending on m6A demethylation modification in non-small cell lung cancer, which was consistent with the results presented in our study. Our study had shown that in bladder cancer cells, ALKBH5 could specifically recognize the m6A sites of the 3′ UTR in CK2α mRNA, which decreased the stability of CK2α mRNA and eventually led to a decrease in the protein level of CK2α. Dot blot and MeRIP assays showed that ALKBH5 regulated CK2α in an m6A-dependent manner. According to a previous study, we speculated that since the fate of m6A-modified mRNAs was also determined by the readers, the difference in the abundance, RNA affinity, and cumulative binding of m6A readers may lead to divergent results.60 The dual and opposing regulation of m6A modifications and the stability of mRNAs indicated that interference with the interaction between m6A and mRNA may be worthy of exploiting it therapeutically.
Taketo et al.26 reported that METTL3-depleted cells, with changing the level of m6A, showed higher sensitivity to anticancer reagents, such as gemcitabine, 5-fluorouracil, and cisplatin in pancreatic cancer. Several other studies have also shown that CK2α was able to affect the cisplatin drug sensitivity of gastric cancer,45 non-small cell lung cancer,46 and cholangiocarcinoma.47 The inhibitors of CK2α were able to promote cancer cell apoptosis by increasing the sensitivity of cisplatin chemotherapy.46, 47, 48 In our study, we found that ALKBH5 affected the sensitivity of cisplatin in vitro and in vivo through m6A-CK2α-mediated glycolysis, which may provide a potential novel therapeutic target for the treatment of bladder cancer. At present, m6A inhibitors have been developed for advancing traditional and regenerative medicine, particularly inhibitors of FTO, including meclofenamic acid (MA)/MA2, FB23/FB23-2, MO-I-500, and so on. MA/MA2, identified as a highly selective inhibitor of FTO, could suppress glioma tumorigenesis and prolong the lifespan in mice.61 FB23/FB23-2, a high selectivity toward FTO, suppressed proliferation and promoted the differentiation/apoptosis in acute myelocytic leukemia cells by binding to FTO directly.62 In melanoma, the combination of FTO inhibition and anti-PD-1 blockers may reduce resistance to immunotherapy.63 These reports indicate that FTO inhibitors, alone or in combination with standard therapeutic agents, provide the immense therapeutic potential to cancers, especially those with high FTO expression.64 Recently, several regulators, which are upstream of m6A proteins, could alter the m6A level via targeting m6A proteins, shedding light on the development of new therapies for cancers. SPI1 inhibits the development of malignant hematopoietic cells via regulating METTL14.65 However, no specific RNA modification inhibitors of ALKBH5 were found in the present. We hope more selective and efficacious drugs targeting m6A-related factors will be developed and explored for the treatment of a wide range of cancers.
Conclusions
Taken together, our results showed that ALKBH5 was downregulated in bladder cancer and associated with the prognosis of bladder cancer patients. Moreover, ALKBH5 significantly inhibited bladder cancer cell proliferation and sensitized bladder cancer cells to cisplatin in vitro and in vivo by CK2α-mediated glycolysis in an m6A-dependent manner, which might provide fresh insights into bladder cancer therapy.
Materials and Methods
Tissue Samples
All of the bladder cancer tumors and adjacent normal tissue samples were gathered from patients who were diagnosed with bladder cancer and who had undergone surgery in The First Affiliated Hospital of Nanjing Medical University between 2013 and 2016. Tissue samples were frozen in liquid nitrogen and immediately stored at −80°C prior to RNA extraction. All patients signed the informed consent form prior to surgery to grant permission that their clinical materials may be used for research purposes. This study was also approved by the Ethics Committee of The First Affiliated Hospital of Nanjing Medical University.
Cell Culture
The bladder cancer cell lines (T24, J82, UMUC3, 5637, and 253J) and the human ureteral epithelial immortalized cell line (SVHUC-1 cell) were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM (Gibco, Thermo Fisher Scientific, USA) containing 10% fetal bovine serum (FBS; Biological Industries, Israel) and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific, USA). All cell lines were cultured at 37°C in a humidified incubator containing 5% CO2.
TMA and IHC
TMA was constructed from 161 cases of formalin-fixed, paraffin-embedded bladder cancer tissues. IHC was performed on TMA to evaluate the level of ALKBH5 protein expression. TMA was treated with xylene and 100% ethanol, followed by decreasing concentrations of ethanol. After antigen retrieval, IHC was performed on TMA by blocking and staining with antibodies against ALKBH5 (1:1,000 dilution; Novus Biologicals, USA) and CK2α (1:1,000; Millipore Sigma, USA), respectively, followed by secondary antibody incubation and the standard avidin biotinylated peroxidase complex method. Hematoxylin was used for counterstaining, and images were captured using a microscope (Nikon, Japan). Standard staining protocols were used.49 Stained tissues were scored for staining intensity (SI) and the percentage of positive cells (PP). SI was scored on a scale of 0–3 (0, negative staining; 1, weak staining; 2, moderate staining; 3, strong staining), and PP was scored according to five categories: 0 (0% positive cells), 1 (≤10%), 2 (11%–50%), 3 (51%–80%), or 4 (>80%). The total score was calculated by multiplying the SI and PP scores, ranging from 0 to 12. Two pathologists who were blinded to the clinical parameters provided the respective scores. Different scores were divided into low-staining (0–7) and high-staining (8–12) groups.
Cell Transfection
Lentiviruses constructed for ALKBH5 knockdown or overexpression were obtained from Obio Technology (China). Cells were plated in 6-well dishes until 40% confluence was reached and infected with ALKBH5 overexpression lentivirus (termed ALKBH5), a NC, ALKBH5 knockdown lentivirus (termed shALKBH5-1 and shALKBH5-2), and SCR control (termed shNC) in 5637 and T24 bladder cancer cells. Pools of stably transduced cells were generated by selection using puromycin (2.5 μg/mL) for 2 weeks.
CK2α siRNA and NC plasmid were obtained from Genepharm Biotech (China). Transfections were performed using the Invitrogen Lipofectamine 2000 kit (Thermo Fisher Scientific, USA), according to the manufacturer’s instructions.
RNA Isolation and qRT-PCR
Total RNA was extracted from clinical samples or cultured cell lines using Invitrogen TRIzol reagent (Thermo Fisher Scientific, USA) and subsequently reverse transcribed into cDNA using Primescript RT reagent (Takara Bio, Japan), according to the manufacturer’s instructions. For mRNA analysis, qRT-PCR was performed with SYBR Premix Ex Taq Reagent (Takara Bio, Japan) using the StepOne Plus Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific, USA). The primers used for qRT-PCR were listed in Table S1. Fold changes in mRNA expression were calculated using the 2−ΔΔCt method and normalized against β-actin with ABI Step One software version 2.1.
RNA Stability
5637 and T24 cells transfected with the control lentivirus, ALKBH5 overexpression lentivirus, or ALKBH5 knockdown lentivirus were treated with 2 μg/mL Act D for 0, 2, 4, 6, 8, or 10 h. Total RNAs were harvested and then subjected to qRT-PCR analysis. The level of CK2α transcript was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) control, and the relative half-life of CK2α was calculated.
Western Blot Analysis
After the trypsinization of the cells, the protein was extracted using radioimmunoprecipitation assay (RIPA) buffer (Beyotime Institute of Biotechnology, China) containing protease inhibitors (Thermo Fisher Scientific, USA) and quantified using a bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of Biotechnology, China). Total protein was separated using SDS-PAGE (10% gels) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore Sigma, USA). After blocking with 5% nonfat milk at room temperature for 2 h, the membranes were immunostained with primary antibodies against primary antibodies ALKBH5 (1:1,000; Novusbio), GLUT1 (1:1,000; Epitomics, USA), LDHA (1:1,000; Epitomics, USA), LDHB (1:1,000; Epitomics, USA), PKM (1:1,000; Epitomics, USA), or HK1 (1:1,000; Cell Signaling Technology, USA) at 4°C overnight. After washing three times in Tris-buffered saline (TBS) containing Tween 20 (TBST), the membranes were incubated with a secondary antibody at room temperature for 1.5 h. Band signals were detected using a chemiluminescence system (Bio-Rad Laboratories, USA) and analyzed using Image Lab Software. The protein levels were normalized to GAPDH (1:1,000; Cell Signaling Technology, USA).
Cell Proliferation and Colony-Formation Assays
Pretreated cells were counted and seeded into a 96-well plate at a density of 2 × 103 (T24) cells/well or 4 × 103 (5637) cells/well. Cell proliferation was measured after 24, 48, 72, and 96 h using the CCK-8 assay (Dojindo Molecular Technologies, Japan). The absorbance was measured at 490 nm with a microplate reader following incubation at 37°C for 1 h, according to the manufacturer’s instructions.
For the colony-formation assay, pretreated cells were seeded into 6-well plates (800 cells/well). The cells were incubated for 1 or 2 weeks. Colonies were fixed in paraformaldehyde for 30 min, washed with PBS, and stained with 0.1% crystal violet. For the inhibitor experiments, inhibitor CX-4945 or TBB (MedChemExpress, USA) was added into the wells at the concentration of 5 or 100 μM, respectively, after cell attachment.
Cell Cycle Assay
Cells (1 × 106) were collected, washed with 2 mL PBS twice, and fixed with 75% pre-cold ethanol for 24 h at −20°C. After washing further with PBS twice and staining with propidium iodide (PI; BD Biosciences, USA) at room temperature for 30 min, cells were assessed by flow cytometry (Becton Dickinson, USA), and the cell cycle was analyzed using Cell Quest Modfit software version 5.0.
Transwell Cell Migration and Invasion Assay
Transfected cells were seeded into the upper chambers with serum-free medium, which was coated with or without Matrigel (BD Biosciences, USA) for the invasion and migration assays. Medium containing 10% FBS was added to the bottom chamber. After incubation at 37°C for every 12 h, the cells were fixed in methanol for 30 min and stained with 0.1% crystal violet for 30 min. Migratory and invasive cells were counted on a microscope in five randomly selected fields, and all of the experiments were repeated three times.
Glucose Uptake, Lactate Production, and ATP Detection
The glucose assay, lactic acid assay, and ATP assay kits (Jiancheng, China) were used to determine glucose uptake, lactate production, and ATP production, respectively, in bladder cancer cells, according to the manufacturer’s instructions.
IC50 Determination
The transfected cells were trypsinized and seeded into a 96-well plate at a density of 5,000 cells per well. Three parallel wells were set up, and the plate was incubated overnight in an incubator. Subsequently, the transfected cells were treated with 32, 16, 8, 4, 2, or 1 μM cisplatin (Tokyo Chemical Industry, Japan) for 24 h. Cell viability was measured by the CCK-8 method, according to the manufacturer’s instructions. IC50 values were calculated according to the probit regression model.66 All experiments were independently performed thrice. Suppression rate of cisplatin is 1 − (OD value of x μM/OD value of 0 μM) × 100%.
Apoptosis Assay
Following treatment with cisplatin (Tokyo Chemical Industry, Japan) for 24 and 48 h, the cells were digested with trypsin and stained with Annexin V-allophycocyanin (APC) and -PI (Fcmacs, China) for 30 min at room temperature. Finally, the cells were assessed by flow cytometry (Becton Dickinson, USA).
Xenograft Experiments In Vivo
Animal studies were approved by the Animal Research Ethics Committee of Nanjing Medical University. The BALB/c nude mice (4–6 weeks old) were obtained from the Model Animal Research Center of Nanjing Medical University. T24 cells stably transfected with the ALKBH5 overexpression lentivirus (ALKBH5) or control lentivirus (NC) were injected subcutaneously into the flank of each mouse. Tumor growth was monitored every 3 days by measuring the width (W) and length (L) with calipers, and the volume (V) of the tumor was calculated using the formula V = (W2 × L)/2. At 3 weeks after injection, the mice were euthanized, and tumors were removed, weighed, fixed, and embedded for IHC.
The BALB/c nude mice (4 weeks old) were obtained from the Model Animal Research Center of Nanjing Medical University. T24 cells stably transfected with the ALKBH5 knockdown lentivirus (shALKBH5) or control lentivirus (shNC) were injected subcutaneously into the flank of each mouse. Mice in the cisplatin group were injected intraperitoneally with cisplatin (2.5 mg/kg body weight, twice every week) starting from day 7 of tumor inoculation. Mice in the control group were injected intraperitoneally with equal volume of normal saline at the same time. Tumor growth was monitored every 3 days by measuring the width and length with calipers, and the volume of the tumor was calculated using the formula V = (W2 × L)/2.
Determination of the m6A Level
Total RNA from patient samples was isolated using Invitrogen TRIzol (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions, and treated with deoxyribonuclease I (Sigma-Aldrich; Merck KGaA, USA). RNA quality was analyzed using NanoDrop technology (Thermo Fisher Scientific, USA). The m6A RNA methylation quantification kit (Epigentek, USA) was used to measure the m6A content in the total RNAs. Briefly, 200 ng RNAs were coated on assay wells. Capture antibody solution and detection antibody solution were then added to assay wells separately, following the manufacturer’s instructions. The m6A levels were quantified colorimetrically by reading the absorbance of each well at a wavelength of 450 nm, and then calculations were performed based on the standard curve.
m6A Dot Blot Assay
The poly(A)+ RNAs were first denatured by heating at 65°C for 5 min and transferred onto a nitrocellulose membrane (Amersham; GE Healthcare, USA) with a Bio-Dot apparatus (Bio-Rad Laboratories, USA). The membranes were then UV crosslinked, blocked, incubated with m6A antibody (1:1,000; Abcam, USA) overnight at 4°C, and subsequently incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:3,000; Proteintech Group, USA). Finally, the membranes were visualized using the chemiluminescence system (Bio-Rad Laboratories, USA). The membrane stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2) was used to ensure consistency among the different groups.
Dual-Luciferase Reporter Assay
The adenine residue embedded within the consensus sequence located closest to the translation termination codon in the CK2α 3′ UTR was mutated (5′-AAACU-3′ to 5′-AAUCU-3′). Cells were cotransfected with plasmids containing the 3′ UTR of wild or mutant fragments from CK2α using Invitrogen Lipofectamine 3000 (Thermo Fisher Scientific, USA), according to the manufacturer’s protocol. At 48 h after transfection, firefly and Renilla luciferase activities were measured consecutively using the dual-luciferase reporter assay system (Promega, USA). Finally, the ratios of luminescence from firefly to Renilla luciferase were calculated.
RIP Assay
RIPA assay was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation kit (Millipore Sigma, USA), according to the manufacturer’s instructions. Briefly, bladder cancer cells were lysed with RIPA lysis buffer. Cell lysates were immunoprecipitated with anti-ALKBH5 antibody or nonimmunized IgG at 4°C overnight. After purification of the RNA, RT-PCR and qRT-PCR were performed to measure the levels of CK2α transcript in the ALKBH5 or IgG immunocomplexes.
MeRIP-qRT-PCR Assay
For the m6A RNA binding experiments, the RNAs of bladder cancer cells stably transfected with either the lentiviral overexpression or control ALKBH5 vector were isolated and treated with RNase I (Sigma-Aldrich, USA). RNAs were fragmented by sonication for 10 s on an ice-water mixture. Immunoprecipitations were performed using an anti-m6A antibody (1:1,000; Abcam, USA) previously bound to magnetic Life Technologies Dynabeads (Thermo Fisher Scientific, USA) in the RIPA buffer (Magna RIP RNA-Binding Protein Immunoprecipitation Kit; Millipore Sigma, USA) and incubated with DNA-free fragmented RNAs. Beads were then treated with proteinase K (20 mg/mL) for 1.5 h at 42°C. Subsequently, RNAs were extracted using phenol/chloroform/isoamyl alcohol and subjected to qRT-PCR using primers for CK2α and normalized to input.
Acknowledgments
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Research Ethics Committee of The First Affiliated Hospital of Nanjing Medical University and with the 1964 Helsinki declaration and its later amendments. All subjects have written, informed consent, and all written, informed consent to participate in the study was obtained from bladder cancer patients for samples to be collected from them. All data generated or analyzed during this study are included either in this article or in the Supplemental Information. This work was supported by the National Natural Science Foundation of China (grant numbers 82073306, 82072832, 81772711, and 81602235); Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD); Provincial Initiative Program for Excellency Disciplines of Jiangsu Province (grant number BE2016791); “333” Project of Jiangsu Province (LGY2016002 and 2018055); and Jiangsu Province’s Key Provincial Talents Program (ZDRCA2016006).
Author Contributions
Q.L. and H. Yang conceived of the study and carried out its design. H. Yu, X.Y., J.T., and S.S. performed the experiments. J.H., Z.Z., and J.L. collected clinical samples. H. Yu, X.Y., and J.T. analyzed the data and wrote the paper. Q.L. and H. Yang revised the paper. All authors read and approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.10.031.
Contributor Information
Qiang Lu, Email: doctorlvqiang@sina.com.
Haiwei Yang, Email: haiweiyang@njmu.edu.cn.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Felsenstein K.M., Theodorescu D. Precision medicine for urothelial bladder cancer: update on tumour genomics and immunotherapy. Nat. Rev. Urol. 2018;15:92–111. doi: 10.1038/nrurol.2017.179. [DOI] [PubMed] [Google Scholar]
- 3.Ribas A., Tumeh P.C. The future of cancer therapy: selecting patients likely to respond to PD1/L1 blockade. Clin. Cancer Res. 2014;20:4982–4984. doi: 10.1158/1078-0432.CCR-14-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yue Y., Liu J., He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343–1355. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao W., Adhikari S., Dahal U., Chen Y.S., Hao Y.J., Sun B.F., Sun H.Y., Li A., Ping X.L., Lai W.Y. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 7.Ping X.L., Sun B.F., Wang L., Xiao W., Yang X., Wang W.J., Adhikari S., Shi Y., Lv Y., Chen Y.S. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A.A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y.S. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz S., Mumbach M.R., Jovanovic M., Wang T., Maciag K., Bushkin G.G., Mertins P., Ter-Ovanesyan D., Habib N., Cacchiarelli D. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertero A., Brown S., Madrigal P., Osnato A., Ortmann D., Yiangou L., Kadiwala J., Hubner N.C., de Los Mozos I.R., Sadée C. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature. 2018;555:256–259. doi: 10.1038/nature25784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen C.A., Shyu A.B. Emerging Themes in Regulation of Global mRNA Turnover in cis. Trends Biochem. Sci. 2017;42:16–27. doi: 10.1016/j.tibs.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang C., Klukovich R., Peng H., Wang Z., Yu T., Zhang Y., Zheng H., Klungland A., Yan W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA. 2018;115:E325–E333. doi: 10.1073/pnas.1717794115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi H., Wang X., Lu Z., Zhao B.S., Ma H., Hsu P.J., Liu C., He C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–328. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O., Pestova T.V., Qian S.B., Jaffrey S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C., Samanta D., Lu H., Bullen J.W., Zhang H., Chen I., He X., Semenza G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA. 2016;113:E2047–E2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu Y., Lin Z., Wan A., Chen H., Liang H., Sun L., Wang Y., Li X., Xiong X.F., Wei B. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer. 2019;18:46. doi: 10.1186/s12943-019-1004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visvanathan A., Patil V., Arora A., Hegde A.S., Arivazhagan A., Santosh V., Somasundaram K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–533. doi: 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y., Peng C., Chen J., Chen D., Yang B., He B., Hu W., Zhang Y., Liu H., Dai L. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol. Cancer. 2019;18:127. doi: 10.1186/s12943-019-1053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M., Wei L., Law C.T., Tsang F.H., Shen J., Cheng C.L., Tsang L.H., Ho D.W., Chiu D.K., Lee J.M. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 22.Li X., Tang J., Huang W., Wang F., Li P., Qin C., Qin Z., Zou Q., Wei J., Hua L. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8:96103–96116. doi: 10.18632/oncotarget.21726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang F., Jin H., Que B., Chao Y., Zhang H., Ying X., Zhou Z., Yuan Z., Su J., Wu B. Dynamic m(6)A mRNA methylation reveals the role of METTL3-m(6)A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. 2019;38:4755–4772. doi: 10.1038/s41388-019-0755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng M., Sheng L., Gao Q., Xiong Q., Zhang H., Wu M., Liang Y., Zhu F., Zhang Y., Zhang X. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. 2019;38:3667–3680. doi: 10.1038/s41388-019-0683-z. [DOI] [PubMed] [Google Scholar]
- 25.Han J., Wang J.Z., Yang X., Yu H., Zhou R., Lu H.C., Yuan W.B., Lu J.C., Zhou Z.J., Lu Q. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol. Cancer. 2019;18:110. doi: 10.1186/s12943-019-1036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taketo K., Konno M., Asai A., Koseki J., Toratani M., Satoh T., Doki Y., Mori M., Ishii H., Ogawa K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018;52:621–629. doi: 10.3892/ijo.2017.4219. [DOI] [PubMed] [Google Scholar]
- 27.Li Z., Weng H., Su R., Weng X., Zuo Z., Li C., Huang H., Nachtergaele S., Dong L., Hu C. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z., Chen Y., Sulman E.P., Xie K., Bögler O. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu H., Gan X., Jiang X., Diao S., Wu H., Hu J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J. Exp. Clin. Cancer Res. 2019;38:163. doi: 10.1186/s13046-019-1159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Y., Hu H., Wang Y., Yuan H., Lu Z., Wu P., Liu D., Tian L., Yin J., Jiang K., Miao Y. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1 Methylation. Cell. Physiol. Biochem. 2018;48:838–846. doi: 10.1159/000491915. [DOI] [PubMed] [Google Scholar]
- 31.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 32.Yang K.M., Kim K. Protein kinase CK2 modulation of pyruvate kinase M isoforms augments the Warburg effect in cancer cells. J. Cell. Biochem. 2018;119:8501–8510. doi: 10.1002/jcb.27078. [DOI] [PubMed] [Google Scholar]
- 33.Im D.K., Cheong H., Lee J.S., Oh M.K., Yang K.M. Protein kinase CK2-dependent aerobic glycolysis-induced lactate dehydrogenase A enhances the migration and invasion of cancer cells. Sci. Rep. 2019;9:5337. doi: 10.1038/s41598-019-41852-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burnett G., Kennedy E.P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954;211:969–980. [PubMed] [Google Scholar]
- 35.Litchfield D.W. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romieu-Mourez R., Landesman-Bollag E., Seldin D.C., Traish A.M., Mercurio F., Sonenshein G.E. Roles of IKK kinases and protein kinase CK2 in activation of nuclear factor-kappaB in breast cancer. Cancer Res. 2001;61:3810–3818. [PubMed] [Google Scholar]
- 37.Laramas M., Pasquier D., Filhol O., Ringeisen F., Descotes J.L., Cochet C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2alpha) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer. 2007;43:928–934. doi: 10.1016/j.ejca.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 38.Daya-Makin M., Sanghera J.S., Mogentale T.L., Lipp M., Parchomchuk J., Hogg J.C., Pelech S.L. Activation of a tumor-associated protein kinase (p40TAK) and casein kinase 2 in human squamous cell carcinomas and adenocarcinomas of the lung. Cancer Res. 1994;54:2262–2268. [PubMed] [Google Scholar]
- 39.So K.S., Rho J.K., Choi Y.J., Kim S.Y., Choi C.M., Chun Y.J., Lee J.C. AKT/mTOR down-regulation by CX-4945, a CK2 inhibitor, promotes apoptosis in chemorefractory non-small cell lung cancer cells. Anticancer Res. 2015;35:1537–1542. [PubMed] [Google Scholar]
- 40.Faust R.A., Gapany M., Tristani P., Davis A., Adams G.L., Ahmed K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett. 1996;101:31–35. doi: 10.1016/0304-3835(96)04110-9. [DOI] [PubMed] [Google Scholar]
- 41.Lin K.Y., Tai C., Hsu J.C., Li C.F., Fang C.L., Lai H.C., Hseu Y.C., Lin Y.F., Uen Y.H. Overexpression of nuclear protein kinase CK2 α catalytic subunit (CK2α) as a poor prognosticator in human colorectal cancer. PLoS ONE. 2011;6:e17193. doi: 10.1371/journal.pone.0017193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin K.Y., Fang C.L., Chen Y., Li C.F., Chen S.H., Kuo C.Y., Tai C., Uen Y.H. Overexpression of nuclear protein kinase CK2 Beta subunit and prognosis in human gastric carcinoma. Ann. Surg. Oncol. 2010;17:1695–1702. doi: 10.1245/s10434-010-0911-9. [DOI] [PubMed] [Google Scholar]
- 43.Stalter G., Siemer S., Becht E., Ziegler M., Remberger K., Issinger O.G. Asymmetric expression of protein kinase CK2 subunits in human kidney tumors. Biochem. Biophys. Res. Commun. 1994;202:141–147. doi: 10.1006/bbrc.1994.1904. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X., Yang X., Yang C., Li P., Yuan W., Deng X., Cheng Y., Li P., Yang H., Tao J., Lu Q. Targeting protein kinase CK2 suppresses bladder cancer cell survival via the glucose metabolic pathway. Oncotarget. 2016;7:87361–87372. doi: 10.18632/oncotarget.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu W., Chen Q., Wang Q., Sun Y., Wang S., Li A., Xu S., Røe O.D., Wang M., Zhang R. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014;5:e1551. doi: 10.1038/cddis.2014.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang B., Yao J., Li B., Shao G., Cui Y. Inhibition of protein kinase CK2 sensitizes non-small cell lung cancer cells to cisplatin via upregulation of PML. Mol. Cell. Biochem. 2017;436:87–97. doi: 10.1007/s11010-017-3081-2. [DOI] [PubMed] [Google Scholar]
- 47.Zakharia K., Miyabe K., Wang Y., Wu D., Moser C.D., Borad M.J., Roberts L.R. Preclinical In Vitro and In Vivo Evidence of an Antitumor Effect of CX-4945, a Casein Kinase II Inhibitor, in Cholangiocarcinoma. Transl. Oncol. 2019;12:143–153. doi: 10.1016/j.tranon.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen F., Huang X., Wu M., Gou S., Hu W. A CK2-targeted Pt(IV) prodrug to disrupt DNA damage response. Cancer Lett. 2017;385:168–178. doi: 10.1016/j.canlet.2016.10.026. [DOI] [PubMed] [Google Scholar]
- 49.Jin H., Ying X., Que B., Wang X., Chao Y., Zhang H., Yuan Z., Qi D., Lin S., Min W. N6-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207. doi: 10.1016/j.ebiom.2019.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang M., Han G., Wang C., Cheng K., Li R., Liu H., Wei X., Ye M., Zou H. A bead-based approach for large-scale identification of in vitro kinase substrates. Proteomics. 2011;11:4632–4637. doi: 10.1002/pmic.201100339. [DOI] [PubMed] [Google Scholar]
- 51.Buontempo F., McCubrey J.A., Orsini E., Ruzzene M., Cappellini A., Lonetti A., Evangelisti C., Chiarini F., Evangelisti C., Barata J.T., Martelli A.M. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia. 2018;32:1–10. doi: 10.1038/leu.2017.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rabalski A.J., Gyenis L., Litchfield D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. 2016;22:2840–2847. doi: 10.1158/1078-0432.CCR-15-1314. [DOI] [PubMed] [Google Scholar]
- 53.Siddiqui-Jain A., Drygin D., Streiner N., Chua P., Pierre F., O’Brien S.E., Bliesath J., Omori M., Huser N., Ho C. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–10298. doi: 10.1158/0008-5472.CAN-10-1893. [DOI] [PubMed] [Google Scholar]
- 54.Hwang D.W., So K.S., Kim S.C., Park K.M., Lee Y.J., Kim S.W., Choi C.M., Rho J.K., Choi Y.J., Lee J.C. Autophagy Induced by CX-4945, a Casein Kinase 2 Inhibitor, Enhances Apoptosis in Pancreatic Cancer Cell Lines. Pancreas. 2017;46:575–581. doi: 10.1097/MPA.0000000000000780. [DOI] [PubMed] [Google Scholar]
- 55.Zhao T., Jia H., Li L., Zhang G., Zhao M., Cheng Q., Zheng J., Li D. Inhibition of CK2 enhances UV-triggered apoptotic cell death in lung cancer cell lines. Oncol. Rep. 2013;30:377–384. doi: 10.3892/or.2013.2407. [DOI] [PubMed] [Google Scholar]
- 56.Gray G.K., McFarland B.C., Rowse A.L., Gibson S.A., Benveniste E.N. Therapeutic CK2 inhibition attenuates diverse prosurvival signaling cascades and decreases cell viability in human breast cancer cells. Oncotarget. 2014;5:6484–6496. doi: 10.18632/oncotarget.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen L., Song C.X., He C., Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu. Rev. Biochem. 2014;83:585–614. doi: 10.1146/annurev-biochem-060713-035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X.C., Jin F., Wang B.Y., Yin X.J., Hong W., Tian F.J. The m6A demethylase ALKBH5 controls trophoblast invasion at the maternal-fetal interface by regulating the stability of CYR61 mRNA. Theranostics. 2019;9:3853–3865. doi: 10.7150/thno.31868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu Z., Qian Q., Zhao X., Ma L., Chen P. N6-methyladenosine ALKBH5 promotes non-small cell lung cancer progress by regulating TIMP3 stability. Gene. 2020;731:144348. doi: 10.1016/j.gene.2020.144348. [DOI] [PubMed] [Google Scholar]
- 60.Zhou Z., Lv J., Yu H., Han J., Yang X., Feng D., Wu Q., Yuan B., Lu Q., Yang H. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol. Cancer. 2020;19:104. doi: 10.1186/s12943-020-01216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G., Sun G., Lu Z., Huang Y., Yang C.G. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Y., Su R., Sheng Y., Dong L., Dong Z., Xu H., Ni T., Zhang Z.S., Zhang T., Li C. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35:677–691.e10. doi: 10.1016/j.ccell.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang S., Wei J., Cui Y.H., Park G., Shah P., Deng Y., Aplin A.E., Lu Z., Hwang S., He C., He Y.Y. m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019;10:2782. doi: 10.1038/s41467-019-10669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selberg S., Blokhina D., Aatonen M., Koivisto P., Siltanen A., Mervaala E., Kankuri E., Karelson M. Discovery of Small Molecules that Activate RNA Methylation through Cooperative Binding to the METTL3-14-WTAP Complex Active Site. Cell Rep. 2019;26:3762–3771.e5. doi: 10.1016/j.celrep.2019.02.100. [DOI] [PubMed] [Google Scholar]
- 65.Weng H., Huang H., Wu H., Qin X., Zhao B.S., Dong L., Shi H., Skibbe J., Shen C., Hu C. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell. 2018;22:191–205.e9. doi: 10.1016/j.stem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tao J., Lu Q., Wu D., Li P., Xu B., Qing W., Wang M., Zhang Z., Zhang W. microRNA-21 modulates cell proliferation and sensitivity to doxorubicin in bladder cancer cells. Oncol. Rep. 2011;25:1721–1729. doi: 10.3892/or.2011.1245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.