

Graphical abstract

Keywords: Ramachandran plot, Molecular Dynamic Simulation, Protein structure, Variant of Uncertain Significance, Pathogenic, TP53
Abstract
The wide application of new DNA sequencing technologies is generating vast quantities of genetic variation data at unprecedented speed. Developing methodologies to decode the pathogenicity of the variants is imperatively demanding. We hypothesized that as deleterious variants may function through disturbing structural stability of their affected proteins, information from structural change caused by genetic variants can be used to identify the variants with deleterious effects. In order to measure the structural change for proteins with large size, we designed a method named RP-MDS composed of Ramachandran plot (RP) and Molecular Dynamics Simulation (MDS). Ramachandran plot captures the variant-caused secondary structural change, whereas MDS provides a quantitative measure for the variant-caused globular structural change. We tested the method using variants in TP53 DNA binding domain of 219 residues as the model. In total, RP-MDS identified 23 of 38 (60.5%) TP53 known Pathogenic variants and 17 of 42 (41%) TP53 VUS that caused significant changes of P53 structure. Our study demonstrates that RP-MDS method provides a powerful protein structure-based tool to screen deleterious genetic variants affecting large-size proteins.
1. Introduction
Clarification of the pathogenic impact of genetic variants is very challenging, as it relies on the combinational evidence derived from clinic, biostatistics, molecules and experiments [1], [2]. Recent application of new DNA sequencing technologies has drastically increased the power for genetic study, resulting in the accumulation of massive genetic variation data at population level. The vast quantity of accumulated variation data has far surpassed the capacity of the current annotation system [3]. The situation is well exemplified by the genetic variants collected from the cancer predisposition gene BRCA1 and BRCA2: 80% of the over 40,000 genetic variants identified from these two genes remain uncharacterized (https://brcaexchange.org/factsheet); of the characterized ones, over 30% of the BRCA1 and 40% of BRCA2 variants are classified as Variant of Uncertain Significance (VUS) due to the lack of functional evidence to determine their pathogenicity (https://www.ncbi.nlm.nih.gov/clinvar/). Therefore, developing new approaches to ease the challenge is urgently demanding.
Protein structure is stabilized by intramolecular interactions of hydrophobic, electrostatic, hydrogen bonding, and Van der Waals interactions. Depending on the position in a protein, a residue modified by a genetic variant can have no, mild, or severe influences on protein structure till inactivation of the affected protein [4], [5], [6], [7]. Therefore, we reasoned that protein structure could be used to identify the variants with deleterious effects [8]. Here, we defined deleterious variants as single amino acid substitution that causes overall structural deviation and impedes functionality. We also postulated that the structure-based methodologies should have these essential features: the targeted protein should have known protein structure in order to be used as the reference to judge the structural change caused by the variants, the methods should have high-throughput capacity, therefore, should be computational-based, in order to characterize a large number of variants simultaneously at low cost, and the results should be validatable by existing well-classified variants to confirm their reliablity.
Ramachandran plot is a graphical illustration for visualizing protein backbone energetic position in terms of torsion angles [9]. Ramachandran plot is one of the best theories in protein structure study with minimal discrepancy between experiments and simulations. The concept is based on the rigidity of the N-C peptide bond, in which the torsion angle ϕ and Ψ, representing and bonded atoms, are restricted by sterically unfavorable structure conformation due to collusion between non-bonded atoms. These physical limits are embedded with conformation information and deciphering the data provides essential insight for the protein structure. The essence of Ramachandran plot remains unchanged since it was developed but its reliability has been significantly improved in recent years [9], [10], [11]. Through capturing the distortion caused by genetic variants, we reasoned that the Ramachandran plot can be applied to study the influence of genetic variants on protein structure although this has not been tested in analyzing genetic variants [9], [10], [12], [13], [14]. Molecular Dynamics Simulation (MDS) is a computation-based atomistic simulation method. It analyzes physical movement of atoms and molecules after interacting for a fixed time period, and the trajectories are used to determine macroscopic thermodynamics properties of the targeted molecular structure. MDS has been widely used to analyze protein structural dynamics [15], [16], [17], and we also successfully applied MDS to characterize the genetic variants in BRCA1 BRCT domain [18]. Although MDS or Ramachandran plot alone provides an independent measure in protein structure, we reasoned that combination of Ramachandran Plot and MDS, we named it RP-MDS, could enhance the capacity of detecting the impact of genetic variants on protein structure.
TP53 is a tumor suppressor gene. It plays a key role in maintaining genome stability. Germline mutation in TP53 predisposes to a wide spectrum of early-onset cancers as exemplified by Li-Fraumeni syndrome [19], [20]. Three decades’ studies have identified over 1845 germline variants in TP53, 66% are located at the DNA binding domain (DBD) [21], [22], [23], [24], [25]. Despite extensive efforts made so far, over 60% of the germline variants in TP53 still remain as VUS due to the lack of functional evidence [24], [26]. In this study, we used TP53 germline variants as the model to test the use of RP-MDS method for genetic variant analysis. We observed that RP-MDS was able to identify 23 of the 38 known Pathogenic variants, and 17 of the 42 coding-change VUS, demonstrating that RP-MDS can effectively identify the deleterious genetic variants.
2. Materials and methods
2.1. Source of variants and modelling P53 mutant structure
We selected a total of 88 TP53 variants from ClinVar database, consisting of 38 Pathogenic, 8 Benign/Likely Benign, and 42 VUS variants (Supplementary Table 1). Single crystal DBD structure (native) of P53 were retrieved from the PDB database (PDB ID:2OCJ, at 2.05 Å) and the sequence numbering starts from 94 to 313 [27]. The structure was used as the template to build each P53 mutant structure using the UCSF Chimera software [28] and Modeller package [29]. The process was illustrated in Fig. 1.
Fig. 1.

Scheme of the study. Starting with the structures of native and mutant protein, each structure was submitted to GROMACS and simulated by MDS. The trajectories from MDS were utilized to create Ramachandran Plot and transformed into density plots by 2D Kernel Density Method. The average deviation of Benign, Likely Benign and native P53 was used as a trained data and compared with Pathogenic variants and VUS. Pathogenic variants were used to create criteria to classify VUS into deleterious and undefined groups.
2.2. Molecular dynamics simulations
Each mutant P53 DBD and wild-type P53 DBD structure was simulated using GROMACS molecular dynamics software, version 2020 [30]. A forcefield comparative simulation was performed between OPLS/AA and AMBER03, showing a comparable intramolecular number of Hydrogen bond (H bond) and Solvent-Accessible Surface Area (Supplementary Fig. S1). Thus, AMBER03 was chosen to model the protein complex. Zinc ion was described by a non-bonded model, which mimics the 4s4p3 vacant orbitals [31]. The protein structure was situated in the 10 × 10 × 10 nm simulation box, solvated with SPC/E water and neutralized with Cl– ions. The system was optimized with steep descent algorithm before 1 ns equilibration run at 298 K and 1 bar in the NPT ensemble using Berendsen thermostat and barostat. Forty ns production run was simulated for the system at 298 K and 1 bar in the NPT ensemble using V-rescale thermostat and Parrinello-Rahman barostat [32]. Verlet velocity algorithm was employed to integrate Newton’s equation of motion with a time step of 2 fs. Particle Mesh Ewald method was used to treat the long-range electrostatic interactions with the cut-off distance set at 1.0 nm. LINC algorithm was applied to constrain the hydrogen bond at equilibrium lengths and the trajectory frame of MD was saved every 15 ps [33].
2.3. Ramachandran plot analysis
Ramachandran plot for each mutant and native P53 was divided into various sub-regions following the established procedures [13]: α – helices [ϕ, ψ = (−63, −43)], β-strands [ϕ, ψ = (−130, 140)], PII – spirals [ϕ, ψ = (−45, +135)], γ′ – turns [ϕ, ψ = (−80, +80)], δ region [ϕ, ψ = (−63, −43)], and ε – region [ϕ, ψ = (+135, +135)]. The last 10 ns of the trajectory generated from MDS was utilized to create a Ramachandran plot. Each plot was transformed to density plot by Kernel density estimation using in-house python code with a grid dimension of 32 × 32 [34], [35].
The average density of the Benign, Likely Benign variants and wild type P53 was taken as a “trained data”, and standard deviation for each grid point was calculated. For each grid point, the Pathogenic variants were compared to the trained data. If the known variant was beyond the standard deviation, the grid point was marked as a significant “density deviation”. Subsequently, the percentage of the density-deviated grid points was calculated for each variant. The results were plotted against a lognormal distribution and passed the Anderson-Darling and Kolmogorov-Smirnov goodness of fit tests [36], [37]. Pathogenic variants generated a logarithmic mean of 3.452, with a scale sigma of 0.241, and upper and lower 95% boundaries at 3.529 and 3.376. (Table 1). Variants higher than 3.376 were set as the cut-off for deleterious, lower than 3.376 as ‘undefined’ [18].
Table 1.
Log-normal distributions for 38 pathogenic variant and goodness of fit test for the lognormal distribution.
| Μ, mean | σ, scale sigma | Lower 95% | Upper 95% | Goodness of Fit tests* | P-value | Decision at level (5%) | |
|---|---|---|---|---|---|---|---|
| Pathogenic | 3.452 | 0.241 | 3.376 | 3.526 | K-S test | 1 | Can't reject Lognormal |
| A-D test | 0.798 | Can't reject Lognormal |
*K-S:Kolmogorov-Smirnov test (36); A-D: Anderson-Darling test (37).
3. Results
3.1. Generating mutant protein structure
From the ClinVar database, we selected 88 variants including 38 Pathogenic, 8 Benign/Likely Benign (all available), and 42 coding-change VUS (Supplementary Table S1). These variants were located at 61 residue positions in P53 DBD region [21]: Y107, H115, S127, A129, M133, V143, D148, P151, P152, G154, V157, Y163, Q165, T170, V173, R175, C176, R181, G187, Q192, H193, I195, R202, R213, S215, V218, Y220, G226, C229, H233, N235, C238, C242, G244, G245, M246, R248, R249, L252, I254, I255, S260, N263, L264, L265, R267, V272, R273, A276, P278, G279, D281, R282, E285, L289, K292, G293, H296, G302, S303 and N310. For each changed residue, we built its P53 mutant structure and used MDS to measure the impact of the changed residue on protein structure. Fig. 2 shows typical examples of different impacts of the Pathogenic variant R175H and Benign variant N235S on P53 structure.
Fig. 2.

Examples of native P53 DBD structure and the variant-affected structure after 40 ns simulation. The well-determined Pathogenic variant R175H unbounded L1, L2 and L3 loops, in contrast to the intact core in the well-determined Benign variant N235S. Red colour shows the variants. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. MDS measurements
The dynamic effects of the variants on P53 DBD were investigated by MDS. The results showed that the RMSD of Cα atoms value was 0.313 ± 0.015 nm and Root Mean Square Fluctuation (RMSF) of the Cα atoms have a similar fluctuation at the residue Cα atoms 112–124, 178–190, 206–214, 221–232, 239–250, 289–301, which were consistent with the literature [15] which R, which used RMSD, RMSF, and H bond to analyze the impact of Pathogenic, Benign/Likely Benign and VUS variants on P53 structure [15].
Benign/Likely Benign mutants had average numbers of intramolecular H bond and RMSD of the protein backbone 135 ± 4.37 and 0.348 ± 0.045 nm, respectively (Supplementary Figs. S2a and S3a). Pathogenic mutants were separated into 2 regions: the lower bound regions occupied by Y163C, R175H, Y220C, G245D, G245S, R248Q, R273C, R282W, and the higher bound regions occupied by the remaining variants (Supplementary Figs. S2b and S3b) [21], with the average H bond in the lower and higher regions 98 ± 8.38 and 134 ± 4.66, respectively (Supplementary Fig. 2b). Similarly, RMSD showed two regions of displacement. VUS maintained the dynamics alike the native protein and Benign/Likely Benign mutants, with on average 133.8 and 0.335 nm for H bond and RMSD, except R249S with deviates of 103.9 and 0.668 nm for H bond and RMSD (Supplementary Figs. S2c and S3c). RMSF showed no significant structural deviation in comparison to Benign/Likely Benign (Supplementary Fig. S4c). The majority of variants (inclusive of Pathogenic, Benign/Likely Benign and VUS) fluctuated at 0.348 ± 0.045 nm and 135 ± 3.65 for RMSD and H bond. The results showed that the majority of P53 variants had similar structure dynamics for VUS. Using the cut-offs (H bond < 300, RMSD > 0.3 nm, RMSF > 0.25 nm, Gyration (Rg) > 1.7, SASA > 100 nm3) that successfully differentiated between deleterious and non-deleterious variants in BRCA1 BRCT domain [18], we observed that all except RMSF classified P53 variants into deleterious variants (Supplementary Table S2). In total, MDS (H Bond and RMSD only) was about to identify 8 Pathogenic variants with structural impact. Thus, MDS alone composed of H bond, RMSD, Gyration (Rg) and SASA were insufficient to differentiate a clear boundary between non-deleterious and deleterious VUS, reflecting the limited power of MDS for P53 DBD due likely to its much larger size (198 residues) than BRCA1 BRCT (95 residues) analyzed by MDS in our previous study.
3.3. Ramachandran plot
For each step of simulations, the torsion angle ϕ and Ψ for each residue were calculated and plotted (Fig. 3a) and converted into relative density graphs through Kernel density estimation (Fig. 3b). Native P53 had a high-density peak occupied at P-II region, β sheet region and α-helical region and a gentle peak at the δ′ (Fig. 3b). The bridge at γ′ indicated that there was a collectively strong NHi+2 to Oi backbone hydrogen bond. Notably, these deviations were unique to each variant. Analyzing the overall distribution of Pathogenic variants showed that the distribution of structural deviation lay at 28.0%, 38.2% and 33.4% for the 1st, 3rd quartiles, and the mean, respectively. These data were fitted against log-normal distribution and the bottom 95% tiles (3.376) were used to categorize deleterious regions. Deviation by pathogenic variants <95% tiles was considered as independent of conformation alteration, such as these associated with the disruption of protein-DNA or protein-protein interaction but not caused by P53 conformation change, therefore were excluded from further examination. Testing in the 38 Pathogenic variants showed that besides the 8 Pathogenic variants identified by MDS, Ramachandran plot was able to detect additional 15 variants with milder structural deviation that H bond, RMSD, and RMSF were unable to detect (Table 2).
Fig. 3.

Ramachandran plots for native p53. a) Ramachandran scatter plot for native P53. A torsional angle Ψ and ϕ were plotted for all residues. The fluctuation densities were concentrated at α helix (red), β strand (blue), γ (teal), δ (green), δ′ (purple) and PII strand (orange) regions. There was a minor fluctuation concentration at δ′ regions. The axis is represented at the top right side of the figure. b) 2D Kernel density plot for native P53 transformed from Ramachandran plot. Red to purple colours represent the degree of intensity from high to low. c) Ramachandran plots of native (black), Pathogenic (R175H, G245D, G245S, R248Q, R273C) and Benign (N235S) variants (Red). d) 2D Kernel density plot for the Pathogenic variants. The table shows the structural deviation from Ramachandran plot and the H bond for each variant. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
TP53 variants with deleterious effects on P53 structure.
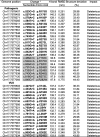 |
*The ones identified by H bond and RMSD highlighted in grey, by RP- MDS but not by H bond and RMSD in bold.
Fig. 3c, d exhibited examples of Ramachandra plots for the well-determined Pathogenic mutations. The Pathogenic variants had shifted from the native position (Fig. 3d) The polyproline-II (P-II) and β region peaks showed a noticeable modification in density implying a structural change. In particular, the torsional density for R175H, G245S and R248Q dissipated at β region and higher density at the P-II region, suggesting a weaken β strand formation and the strengthen P-II formation at the protein backbone. The substituted residue (red) fluctuated at a different torsional angle in comparison to the native residue (black) (Fig. 3c). This torsional angular change illustrated that the substituted residue interacted with a different part of the protein. Fig. 3c and d listed examples of the global torsional density deviation for the strong Pathogenic variants (R175H, G245D, G245S, R248Q, R273C), with the deviation of 43.8%, 38.7%, 49.4%, 43.6% and 46.3%, respectively, consistent with the results in H bond. Thus, Ramachandran plot provided a sensitive measure for the variants with deleterious effects on P53 structure.
3.4. Searching VUS with deleterious effects
Using the parameters determined from known Benign/Likely Benign and Pathogenic variants, we tested RP-MDS in 42 coding-change VUS variants to identify these with deleterious impact. Supplementary Fig. S5 illustrated the Ramachandran density plot for each of the 42 VUS variants. Under the deleterious >3.376 generated from known Pathogenic variants, Ramachandran plot identified 17 of 42 (41%) VUS (V143L, D148A, G154D, V157I, Q192R, V218G, C229Y, R249S, I254V, I255N, L264P, V272M, P278R, G293R, G293W, H296Y, G302E) causing apparent structural deviation. Thus, we classified these 17 VUS as deleterious variants (Table 2).
We tested the known 38 pathogenic variants, 8 Benign/Likely Benign variants and 42 VUS with missense 3D and SuSPect programs [38], [39]. Missense 3D was able to detect 13 pathogenic variants (R175G, R175H, C176Y, H193P, R213Q, Y220C, C238R, C242Y, G245D, G245S, G245V, L265P, R273P) and 8 VUS (C176W, G187D, S215R, V218G, L252P, I255N, P278R, G279R) with potential structural damage and all benign variants with no structural damage. SuSPect was able to detect all pathogenic variants, however, it failed to differentiate Benign/Likely Benign variants and classified all VUS as disease-associated variants. Here, our RP-MDS method was able to significantly increase the detection of deleterious variants.
4. Discussion
The conventional approaches for determining the pathogenicity of genetic variants rely on the evidence from experiment-based functional assays, biostatistics-based methods, evolution conservation-based algorithms, and clinical data. Previously, we developed an protein structure-based MDS approach to identify the deleterious variants and successfully applied it in classifying the variants in smaller-size BRCA1 BRCT domain. In this study, we further developed the MDS-based approach into RP-MDS approach in order to identify the deleterious variants in large-size functional domains. By using P53 DNA binding domain as the model, our study showed that RP-MDS fulfills our expectation. Data from our previous and current studies demonstrate that protein structure can be used for identifying the deleterious variants. This is particularly meaningful in current status of genetic variant annotation, considering the fact that protein structure for many disease genes have been well determined, lack of functional evidence for vast quantity of unclassified variants such as VUS, and advanced computational power allowing large-scale performance.
MDS is proficient to identify the deleterious variants with strong detrimental nature as demonstrated by the lower H bond and higher RMSD for the eight TP53 Pathogenic variants (Supplementary Fig. 2 and 3). For the Pathogenic variants with lesser severity, however, MDS showed inadequate sensitivity as their fluctuations are closer to the native P53. Similar situation was present in VUS that 41 of the 42 VUS showed no significant structural deviation (Supplementary Fig. 2c–4c). Our previous study demonstrated that MDS was sensitive in classifying the VUS in the BRCA1 BRCT domain [18]. The discrepancy is likely due to the size-difference between P53 DBD (213 residues) and BRCA1 BRCT domain (95 residues) that MDS has limited power to differentiate the structural changes in a larger protein structure. By using the information of backbone torsional angle from Ramachandran plot, this limitation is largely overcome allowing to expose the differences hid within the larger protein structure. This is well reflected by the increasingly identified TP53 structural-changing Pathogenic variants and VUS by RP-MDS.
The Ramachandran density plot demonstrated a dissimilar structural affinity for Pathogenic variants with the deleterious structural attitude in comparison to native P53. Fig. 3c showed the local residue torsional angle difference in comparison to the native structure and Fig. 3d showed that the Ramachandran density plot of its respective protein. P-II, α, β and δ′ regions (which are predominantly populated by folded proteins) were notably altered by the residue substitution. In this case, a dissimilar torsional angle in respect to the native residue inferred that the variant residue had positionally interacted with another part of the protein. Conversely, the substituted residue that fluctuated at a similar torsional angle had different interaction characteristics and consequentially affected the global structure. Thus, Ramachandran plot can effectively detect the deleterious characteristics of deleterious variants. Although Benign/Likely Benign variants had no noticeable disordered fluctuation, this information cannot be directly applied to assign the VUS variants with insignificant impact on the structure as non-deleterious one, as possibilities exist that these variants may still have deleterious effects through non-structural factors, e.g. post-translational modification, which may or may not disturb protein structure.
While many genetic variants can cause “loss of function” in the affected protein, it is often the case that many genetic variants can also cause “gain of function” consequence. The gain of function is often present in P53. For example, P53 R175, R248, and R273 are typical gain of function mutations, contributing to carcinogenesis [40], [41], [42], and P53 R249S is a typical conditional gain of function mutation [43], [44]. While the current structure-based approach used in our study can not distinguish between loss of function and gain of function mutations, it will be interesting to further explore the means to obtain the ability. These could include the selection of specific parameters, the use of known gain of function and loss of function p53 mutation as the control, or combination of structural changes with experimental methodologies.
Our study has limitations by the lack of Benign/Likely Benign variants in TP53 as the control. Comparing to a large number of Pathogenic and VUS variants, there were only 8 Benign/Likely Benign variants in ClinVar and International Agency for Research on Cancer (IARC) TP53 databases. This could relate with the nature of P53 that most of the variants could be deleterious because of its low thermodynamic and kinetic stability. In our analysis, we dessociated Zinc ion from P53. Polarization could be an attributing factor for the zinc ion dissociation, but the polarized force field is yet to be determined for P53. We cautiously disregarded to use the bonded model for the simulation as it artificially pull the L2 and L3 loops together., As such, it might become an artificial structure and the stability observed might not be real. Study showed that the zinc ion contributes to stability at 10 °C [45]. We reasoned that thermal contribution and entropy at 25 °C could easily delocalise zinc ion. Our reasoning is supported by the observation that “a significant fraction of p53 may exist in the zinc-free state under physiological conditions” [46], and zinc-free (apo) P53 is “both thermodynamically stable and kinetically accessible. Therefore, to understand how variants shift the balance of p53 conformational states, it is necessary to generate a complete thermodynamic model that includes parameters for the zinc-free form”. Thus we reasoned that choosing zinc-free structure can reflect better the impact of altered residues on the structure under physiological conditions . Lastly, our study excluded the interference by external organic molecule, i.e. DNA and other proteins, to avoid adverse impact on the speed of the simulation and increased complexity of the system.
In summary, the RP-MDS method provides a structural-based means to effectively identify deleterious variants in larger-size proteins. Its computational nature allows large-scale application for characterizing genetic variants in disease genes.
Funding
This work was funded by Macau Science and Technology Development Fund (085/2017/A2), Macau Science and Technology Development – Ministry of Science and Technology of People’s Republic of China fund (0077/2019/AMJ), grants from the University of Macau (SRG2017-00097-FHS, MYRG2019-00018-FHS), the Faculty of Health Sciences, University of Macau (Startup fund, FHSIG/SW/0007/2020P, FHS Innovation grant) (SMW). BT is the recipient of University of Macau Postdoctoral Fellowship Class A of the Macao Talent Program.
CRediT authorship contribution statement
Benjamin Tam: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing - original draft. Siddharth Sinha: Data curation, Resources, Software, Methodology, Writing - original draft. San Ming Wang: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Validation, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was performed at the high-performance computing cluster supported by Information and Communication Technology Office of the University of Macau.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.11.041.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary Fig. S1.

OPLS/AA and AMBER force field comparison. a) The number of intramolecular hydrogen bond in the system and b) the Solvent-Accessible Surface Area of the P53. Blue and Orange represent AMBER and OPLS/AA force field, respectively.
Supplementary Fig. S2.

Hydrogen bond (H bond) graph by 40 ns simulation. The plots were against native P53 (Black). a) 8 Benign/Likely Benign variants; b) 38 Pathogenic variants; and c) 42 VUS. The average H bond was 133 for native P53, 135 ± 4.37 for Benign/Likely Benign, and 135 ± 3.65 for VUS. Pathogenic mutants were separated into 2 regions, the higher and the lower region (Y163C, R175H, Y220C, G245D, G245S, R248Q, R273C, R282W) with 134 ± 4.66 and 98 ± 8.38 H bond, respectively. R249S (Gold) in VUS is identified to have deleterious structure and other VUS showed no significant deviation. The figure showed the H bond alone can only detect the variants with significant deleterious effects on protein structure.
Supplementary Fig. S3.

Root mean square deviation (RMSD) graph by 40 ns simulation. All were plotted against native P53 (Black). a) 8 Benign/Likely Benign variants; b) 38 Pathogenic variants; and c) 42 VUS. The average of RMSD is 0.320 nm for native P53, 0.348 for Benign/ Likely Benign and 0.327 nm for VUS. Pathogenic mutants were separated into 2 regions, the higher (Y163C, R175H, Y220C, G245D, G245S, R248Q, R273C, R282W) and the lower region with 0.672 ± 0.069 nm and 0.327 ± 0.050 nm, respectively. Only R249S in VUS was identified to have deleterious effects on protein structure, but not for other VUS. The figure showed that RMSD can only identify the variants with significant deleterious effects on protein structure.
Supplementary Fig. S4.

Root mean square fluctuation (RMSF) graph by 40 ns simulation. a) 8 Benign/Likely Benign variants; b) 38 Pathogenic variants; and c) 42 VUS. All were plotted against native P53 (Black). The Benign/Likely Benign and VUS variants show consistence RMSF in comparison to the native, whereas the region of large difference was between residue 286 – 307. RMSF for Pathogenic mutants has greater fluctuation in comparison to native P53. Only R249S was identified as deleterious and but not for other VUS, indicating that RMSF can only identify the variants with significant deleterious effects on protein structure.
Supplementary Fig. S5.

2D Kernel density plots for the 42 VUS (C176W, C229R, C229Y, D148A, D148Y, D281E, G154D, G154S, G187D, G226V, G279R, G293R, G293W, G302E, H115R, H233R, H296Y, I254V, I255N, K291N, K292R, L252P, L264P, M133I, N310D, N310K, P278R, Q192R, R181S, R249S, S127T, S215R, S260Y, S303G, S303N, T170M, T170R, V143L, V218G, V272M). The red to blue colors in the scale represent the degree of intensity from high to low. Each Ramachandran density plot provides visualization for the presence of structural deviations, the actual calculations were based on combined basic data from the variants of benign, likely benign, pathogenic, likely pathogenic and wild type. See detailed description in Materials and Methods.
References
- 1.He K.Y., Ge D., He M.M. Big data analytics for genomic medicine. Int J Mol Sci. 2017;18(2):412. doi: 10.3390/ijms18020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dankar F.K., Ptitsyn A., Dankar S.K. The development of large-scale de-identified biomedical databases in the age of genomics-principles and challenges. Hum Genomics. 2018;12(1) doi: 10.1186/s40246-018-0147-5. 19–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Federici G., Soddu S. Variants of uncertain significance in the era of high-throughput genome sequencing: a lesson from breast and ovary cancers. J Exp Clin Cancer Res. 2020;39(1) doi: 10.1186/s13046-020-01554-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho Y., Gorina S., Jeffrey P. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265(5170):346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 5.Joerger A.C., Ang H.C., Fersht A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci USA. 2006;103(41):15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eldar A., Rozenberg H., Diskin-Posner Y. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013;41(18):8748–8759. doi: 10.1093/nar/gkt630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joerger A.C., Fersht A.R. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. 2016;85(1):375–404. doi: 10.1146/annurev-biochem-060815-014710. [DOI] [PubMed] [Google Scholar]
- 8.Kumar S., Clarke D., Gerstein M.B. Leveraging protein dynamics to identify cancer mutational hotspots using 3D structures. Proc Natl Acad Sci USA. 2019;116(38):18962–18970. doi: 10.1073/pnas.1901156116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran G.N., Ramakrishnan C., Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7(1):95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 10.Vega M.C., Serrano L., Martínez J.C. Thermodynamic and structural characterization of Asn and Ala residues in the disallowed II′ region of the Ramachandran plot. Protein Sci. 2000;9(12):2322–2328. doi: 10.1110/ps.9.12.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wishart D.S., Nip A.M. Protein chemical shift analysis: a practical guide. Biochem Cell Biol. 1998;76(2-3):153–163. doi: 10.1139/bcb-76-2-3-153. [DOI] [PubMed] [Google Scholar]
- 12.Gromiha M.M., Oobatake M., Kono H., Uedaira H., Sarai A. Importance of mutant position in Ramachandran plot for predicting protein stability of surface mutations. Biopolymers. 2002;64(4):210–220. doi: 10.1002/bip.10125. [DOI] [PubMed] [Google Scholar]
- 13.Hollingsworth S.A., Karplus P.A. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol Concepts. 2010;1(3–4):271–283. doi: 10.1515/BMC.2010.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carugo O., Djinović-Carugo K. Half a century of Ramachandran plots. Acta Crystallogr D Biol Crystallogr. 2013;69(8):1333–1341. doi: 10.1107/S090744491301158X. [DOI] [PubMed] [Google Scholar]
- 15.Liu X., Tian W., Cheng J. Microsecond molecular dynamics simulations reveal the allosteric regulatory mechanism of p53 R249S mutation in p53-associated liver cancer. Comput Biol Chem. 2020;84:107194. doi: 10.1016/j.compbiolchem.2019.107194. [DOI] [PubMed] [Google Scholar]
- 16.Salsbury F.R., Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol. 2010;10(6):738–744. doi: 10.1016/j.coph.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pantelopulos G.A., Mukherjee S., Voelz V.A. Microsecond simulations of mdm2 and its complex with p53 yield insight into force field accuracy and conformational dynamics: microsecond Simulations of Mdm2. Proteins. 2015;83(9):1665–1676. doi: 10.1002/prot.24852. [DOI] [PubMed] [Google Scholar]
- 18.Sinha S., Wang S.M. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput Struct Biotechnol J. 2020;18:723–736. doi: 10.1016/j.csbj.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olivier M., Hollstein M., Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1) doi: 10.1101/cshperspect.a001008. a001008–a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li F.P., Fraumeni J.F., Jr., Mulvihill J.J. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48(18):5358–5362. [PubMed] [Google Scholar]
- 21.Freed-Pastor W.A., Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26(12):1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fortuno C., Cipponi A., Ballinger M.L. A quantitative model to predict pathogenicity of missense variants in the TP53 gene. Hum Mutat. 2019;40(6):788–800. doi: 10.1002/humu.23739. [DOI] [PubMed] [Google Scholar]
- 23.Demir Ö., Baronio R., Salehi F. Ensemble-based computational approach discriminates functional activity of p53 cancer and rescue mutants. PLoS Comput Biol. 2011;7(10) doi: 10.1371/journal.pcbi.1002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landrum M.J., Lee J.M., Riley G.R. . ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathe E., Olivier M., Kato S. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34(5):1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bittar C.M., Vieira I.A., Sabato C.S. TP53 variants of uncertain significance: increasing challenges in variant interpretation and genetic counseling. Fam Cancer. 2019;18(4):451–456. doi: 10.1007/s10689-019-00140-w. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y., Rosengarth A., Luecke H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr Sect D. 2007;63(3):276–281. doi: 10.1107/S0907444906048499. [DOI] [PubMed] [Google Scholar]
- 28.Pettersen E.F., Goddard T.D., Huang C.C. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Šali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 30.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1):43–56. [Google Scholar]
- 31.Stote R.H., Karplus M. Zinc binding in proteins and solution: a simple but accurate nonbonded representation. Proteins Struct Funct Bioinf. 1995;23(1):12–31. doi: 10.1002/prot.340230104. [DOI] [PubMed] [Google Scholar]
- 32.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. [Google Scholar]
- 33.Hess B., Bekker H., Berendsen H.J.C. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. [Google Scholar]
- 34.Parzen E. On estimation of a probability density function and mode. Ann Math Statist. 1962;33(3):1065–1076. [Google Scholar]
- 35.Rosenblatt M. Remarks on some nonparametric estimates of a density function. Ann Math Statist. 1956;27(3):832–837. [Google Scholar]
- 36.Massey F.J. The kolmogorov-smirnov test for goodness of fit. J Am Stat Assoc. 1951;46(253):68–78. [Google Scholar]
- 37.Anderson T.W., Darling D.A. Asymptotic theory of certain “goodness of fit” criteria based on stochastic processes. Ann Math Statist. 1952;23(2):193–212. [Google Scholar]
- 38.Ittisoponpisan S., Islam S.A., Khanna T. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J Mol Biol. 2019;431(11):2197–2212. doi: 10.1016/j.jmb.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yates C.M., Filippis I., Kelley L.A. SuSPect: enhanced prediction of single amino acid variant (SAV) phenotype using network features. J Mol Biol. 2014;426(14):2692–2701. doi: 10.1016/j.jmb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sigal A., Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000;60(24):6788–6793. [PubMed] [Google Scholar]
- 41.Hanel W., Marchenko N., Xu S. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;20(7):898–909. doi: 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller P.A.J., Vousden K.H. p53 mutations in cancer. Nat Cell Biol. 2013;15(1):2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 43.Fei Q., Shang K., Zhang J. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6(1):8651. doi: 10.1038/ncomms9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao P., Zeng S.X., Zhou X. Mutant p53 gains its function via c-Myc activation upon CDK4 phosphorylation at Serine 249 and consequent PIN1 binding. Mol Cell. 2017;68(6):1134–1146.e6. doi: 10.1016/j.molcel.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue Y., Wang S., Feng X. Effect of metal ion on the structural stability of tumour suppressor protein p53 DNA-binding domain. J Biochem. 2009;146(2):193–200. doi: 10.1093/jb/mvp055. [DOI] [PubMed] [Google Scholar]
- 46.Butler J.S., Loh S.N. Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry. 2003;42(8):2396–2403. doi: 10.1021/bi026635n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.