

Summary
We previously identified the N-quinoline-benzenesulfonamide (NQBS) scaffold as a potent inhibitor of nuclear factor-κB (NF-κB) translocation. Now, we report the structure-activity relationship of compounds with the NQBS scaffold in models of diffuse large B-cell lymphoma (DLBCL). We identified CU-O42, CU-O47, and CU-O75 as NQBS analogs with the most potent cytotoxic activity in DLBCL lines. Their anti-lymphoma effect was mediated by NF-κB sequestration to the cytoplasm of DLBCL cells. Internal Coordinates Mechanics analysis suggested direct binding between CU-O75 and IκBα/p50/p65 which leads to the stabilization of the NF-κB trimer. A whole cellular thermal shift assay confirmed direct binding of the NQBS to IκBα, an inhibitory component of the IκBα/p50/p65 trimer. Lymphoma cell line sequencing revealed CU-O75 induced downregulation of NF-κB-dependent genes and DeMAND analysis identified IκBα as one of the top protein targets for CU-O75. CU-O42 was potent in inhibiting tumor growth in two mouse models of aggressive lymphomas.
Subject Areas: Chemistry, Medical Biochemistry, Cancer
Graphical Abstract
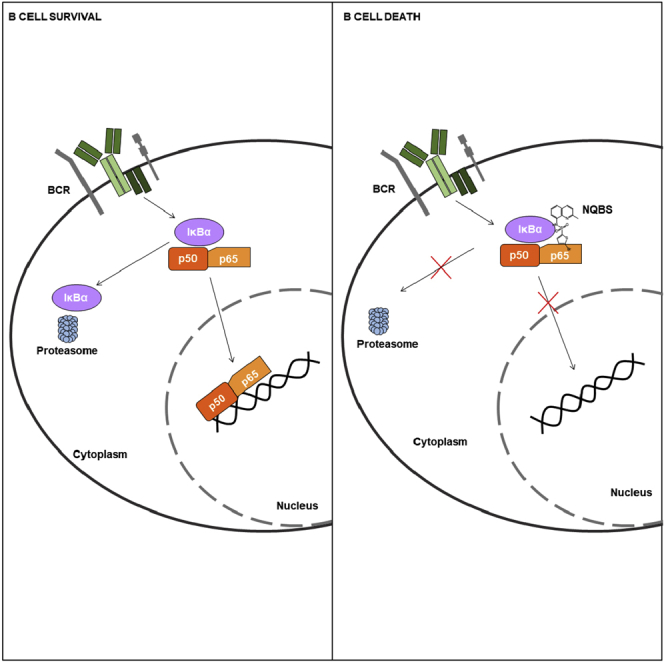
Highlights
-
•
NQBS inhibits NF-κB translocation to the nucleus by stabilizing IκKα-p50-p65 trimer
-
•
Exclusion of the NF-κB from the nucleus of the lymphoma cell leads to its rapid death
-
•
Preliminary in vivo data suggest NQBS to be efficacious and tolerable
Chemistry; Medical Biochemistry; Cancer
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma and represents an aggressive malignancy that is treated with combination immuno-chemotherapy. Large studies have shown that standard of care first-line therapy (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone - R-CHOP) results in long-term survival of approximately 50–60% of patients (Teras et al., 2016; Coiffier et al., 2002). For patients with refractory and relapsed disease, second-line salvage regimens with autologous stem cell transplant can provide a cure in up to 50% of patients. However, those patients with refractory disease and disease that relapses early after first-line treatment have particularly poor outcomes (Gisselbrecht et al., 2010). While immunotherapy-based approaches including bispecific T-cell engagers (Viardot et al., 2016) and chimeric antigen receptor T cells (CAR T) (Neelapu et al., 2017; Schuster et al., 2019) have shown impressive response rates in previously treated patients with DLBCL, continued relapses following these treatments necessitate the search for other novel treatment options with acceptable toxicity profiles, particularly in the elderly population (Coiffier et al., 2002).
DLBCL is a heterogeneous group of lymphomas defined by genetic and diagnostic markers which stratify patients into distinct prognostic groups. Historically, patients with activated B-cell (ABC) subtype of DLBCL have poorer outcomes compared to those with germinal center (GCB)-like DLBCL (Swerdlow et al., 2008; Alizadeh et al., 2000; Shaffer et al., 2012). The inherent resistance of the ABC DLBCL to standard treatment options is due, in part, to multiple genetic alterations involving proteins within the B-cell receptor (BCR), CD40L/CD40 and TLR/IL1R pathways, including CD79A and B, CARD11, MYD88, and A20, which result in constitutive nuclear factor-κB (NF-κB) activation (Pasqualucci, 2013; Davis et al., 2010; Ngo et al., 2010; Kato et al., 2009; Lenz et al., 2008). Addition of lenalidomide or ibrutinib to R-CHOP showed promising results in abrogating adverse outcomes associated with the ABC disease subtype (Nowakowski et al., 2015; Younes et al., 2014), and a recent report of a phase III study in non-GCB subtypes of DLBCL showed a modest benefit in adding ibrutinib to R-CHOP in patients younger than 60 years. Older patients treated with the same regimen experienced higher toxicity and further studies are needed (Younes et al., 2019).
NF-κB is one of the most ubiquitously implicated transcription factors in lymphoma development. NF-κB family consists of five proteins (p105/50, p100/p52, c-Rel, RelA/p65, and RelB) that regulate expression of >400 transcriptional target genes (Brasseres and Baldwin, 2006; Dutta et al., 2006; Luo et al., 2005). These encode for transcription factors, regulators of apoptosis, cell surface receptors, and early response genes, among others (Karin, 2006), that play a central role in the dysregulated growth and survival of most cancer cells.
Pathways leading to NF-κB activation are divided into the canonical (IκB dependent, via TNFα, IL-1, and LPS), non-canonical (IκB independent, via CD40/CD40L), and atypical (genotoxic stress, hypoxia, and reactive oxygen species) pathways, which all ultimately lead to the nuclear translocation of active NF-κB subunits (Ghosh and Baltimore, 1990; Ghosh et al., 1998; Oeckinghouse et al., 2011). Over-expression and constitutive activation of NF-κB is thought to be one of the central drivers in cancer biology including lymphomagenesis (Ghosh and Karin, 2002; Derudder et al., 2003; Xiao et al., 2001; Coope et al., 2002; Claudio et al., 2002; Dejardin et al., 2002). Identification of pharmacologic strategies to inhibit the activation of target NF-κB genes has thus become a key area of investigation (Herrington et al., 2015; Baud and Marin, 2009).
While over 700 inhibitors of the NF-κB signaling pathway have been experimentally identified (Gilmore and Herscovitch, 2006), very few of them have made it to the clinic. The Food and Drug Administration approved agents used in oncology and rheumatology, which act by modulating NF-κB indirectly, and include proteasome inhibitors [bortezomib (Kane et al., 2003) and carfilzomib (Herndon et al., 2013)], disease-modifying anti-rheumatic drugs (sulfasalazine and mesalamine), and anti-cytokine biologics [anakinra, infliximab, and adalimumab (Gilmore and Herscovitch, 2006)]. Hence, there is an urgent need for higher specificity, targeted therapeutic options to inhibit the NF-κB cascade.
High-throughput screening performed at the Columbia University and National Institutes of Health (NIH) identified small-molecule inhibitors of the NF-κB pathway: N-quinoline-benzenesulfonamides (NQBSs) (Xie et al., 2008), which were able to block nuclear translocation of NF-κB in human umbilical vein endothelial cells, were stimulated by TNFα. Here, we present data from structure-activity relationship (SAR) analyses, within preclinical models of DLBCL, that confirm the NF-κB-specific activity of NQBS analogs, leading to potent cytotoxic and pro-apoptotic effect in DLBCL models. This effect is mediated by direct binding of the NQBS analogs to IκBα, which stabilizes its interaction with NF-κB dimers and sequesters them in the cytoplasm.
Results
Chemical Synthesis and Lead Compound Identification
We performed SAR analysis on the previously identified NQBS structure aiming to identify derivatives with potent in vitro lymphoma cytotoxicity, while retaining their inherent NF-κB nuclear exclusion properties, an essential characteristic of the NQBS scaffold. All of the initially described compounds, as well as those that were additionally synthesized, were screened by an algorithm, validating three key aspects of an NF-κB-targeted compound: an ATP-based luminescence cell viability assay (anti-lymphoma effect), immunofluorescence microscopy (NF-κB subunit nuclear exclusion), and chemiluminescence-based electrophoretic mobility shift assay (EMSA) (inhibition of NF-κB subunit DNA binding) in 2 DLBCL cell lines (OCI-Ly1 – GCB DLBCL and OCI-Ly10 – ABC DLBCL). All of the derivatives tested in this manner are listed in Tables S1–S8. A general approach to NQBS synthesis is shown in Figure 1A. The chemical structures of three of the active compounds are shown in Figure 1B (CU-O42, CU-O47, and CU-O75), while the fourth structure represents the inactive analog molecule CU-O102, used in subsequent experiments as a negative control. Spectral data for these 4 compounds are presented in Figure S1.
Figure 1.

NQBS chemical structure
(A and B) (A) General synthetic pathways for the preparation of NQBS; (B) chemical structure of representative compounds (CU-O42, CU-O47, CU-O75, and CU-O102)
The effect of NQBS analogs in inducing cytotoxicity in lymphoma cell lines is shown via heatmap in Figure 2A. Figure S2 shows the growth inhibition IC50 values of CU-O75 across a spectrum of lymphoma lines including DLBCL and mantle cell lymphoma (MCL). CU-O75's IC50 values in these lines ranged between 0.75 and 1.5 μM. Neither of the active compounds exhibited clear differences in observed growth inhibition between the ABC and GCB DLBCL subtypes as shown in summary graph in Figure 2B. Similarly, lack of DLBCL subtype specificity was previously reported with another class of NF-κB-targeted compounds (IκB kinase [IKK] inhibitors) in the cell line models tested (Deng et al. 2015). NQBS-related compounds were potent inducers of apoptosis as shown in Figures 2C and 2D, and no clear difference was again observed in the induction of apoptosis between cell lines representing ABC and GCB DLBCL subtypes.
Figure 2.

Anti-lymphoma activity of NQBS
(A) Heatmap representing the activity of NQBS in inducing cell growth inhibition in 2 DLBCL lines (OCI-Ly1 and OCI-Ly10), measured by IC50 values using an ATP-based luminescence viability assay at 72 h. Representative compounds (CU-O42, CU-O47, CU-O75, and CU-O102) are highlighted. All of the experiments were performed in triplicates and repeated at least twice.
(B) NQBSs induce growth inhibition in lymphoma cell lines as measured by IC50 in an ATP-based luminescence viability assay at 24 h. Cell lines are color labeled as GCB DLBCL (red), ABC DLBCL (blue), and MCL (purple). All of the experiments were performed in triplicates and repeated at least twice.
(C) NQBSs induce apoptosis in lymphoma cell lines. PI and Yo-Pro flow cytometric analysis of CU-O42 effect in DLBCL cell lines at 24h. Cell lines are color labeled as GCB DLBCL (red) and ABC DLBCL (blue). Values represent means expressed as percentages compared with the untreated control; error bars represent SD. All of the experiments were performed in triplicates and repeated at least twice.
(D) Immunoblot analysis in OCI-Ly10 cell line treated with CU-O47 shows a cleavage of Caspase 8 and PARP, as well as a decrease in the level of XIAP and BID as markers of apoptosis induction.
Next, we performed a drug washout experiment in order to identify the duration of incubation needed for the biologic effect of NQBS-related compounds in DLBCL lines. OCI-Ly1 and OCI-Ly10 were incubated with CU-O47 and CU-O75 for 1 and 3 h, respectively (pulse exposure), after which the cells were washed and incubated in NQBS-free medium. Cell viability was analyzed at 24, 48, and 72 h, as shown in Figure S3. In the samples treated with 1h pulse, lymphoma growth inhibition was observed only at the highest concentration of CU-O47 and CU-O75, resulting in a significant right shift of the concentration:effect curve compared to continuously treated cells. Three-hour treatment with CU-O75 abrogated this difference proving that NQBSs yield their biologic effect on DLBCL cells in a remarkably rapid manner. Three-hour treatment with CU-O47 had a similar effect, displaying a concentration:effect curve approaching that of the continuous treatment group. Again, neither CU-O75 or CU-O47 exhibited selectivity for the ABC DLBCL.
NQBS-Mediated Inhibition of NF-κB Nuclear Translocation
While the initial discovery of NQBS-related compounds was made in a TNFα-stimulated human unbilical vein endothelial cell line (HUVEC) model, we sought to explore the effect of CU-O75, CU-O42, and CU-O47 in a cellular model with constitutive NF-κB activation, without TNFα stimulation. OCI-Ly10 is an ideal model as this cell line is derived from a patient with ABC DLBCL and has A20 and MYD88 mutations resulting in NF-κB overexpression (Ma et al., 2015). As shown in Figure 3A, untreated and TNFα-unstimulated OCI-Ly10 cells have p50 (red) present in the nucleus (top row, middle and right images). Treatment with CU-O42 for 6h results in p50 sequestration into the cytoplasm (third row, middle and right images). NQBS-related compounds do not affect nuclear localization of other transcription factors (e.g., Ku80 shown in green). Next, we wanted to analyze this effect on a quantitative protein level. We treated OCI-Ly10 cells with increasing concentrations of CU-O75 and analyzed the total nuclear levels of p50 and p65 proteins. Decrease in nuclear levels of both p50 and p65 was observed in both time and concentration-dependent manners (Figure 3B). We also used a EMSA to analyze the effect NQBSs have on direct NF-κB binding to DNA. As shown in Figure 3C, together with the observed decrease in the nuclear NF-κB, there was also decreased binding of NF-κB to DNA. Again, this CU-O75-mediated effect was both time and concentration dependent.
Figure 3.

Inhibition of NF-κB translocation
(A) Immunofluorescent microscopy analysis in OCI-Ly10 following a 6h incubation with CU-O42 shows p50 sequestration in the cytoplasm. This effect is NF-kB specific as it does not affect other transcription factors capable of translocating between the cytoplasm and the nucleus (e.g. Ku80)
(B) Immunoblotting analysis confirms decreased nuclear levels of p50 and p65 following CU-O75 treatment in OCI-Ly10 cells. A superfluous lane was cut out from the panels representing 3h and 6h treatment with CU-O75. This lane was an additional sample treated with TNFα. Since the effect of TNFα occurs quickly (within 5 min) and is lost by the 3h time point, this lane provided irrelevant information
(C) Chemiluminescence-based electrophoretic mobility shift assay (EMSA) analysis of the effect NQBSs have on direct NF-κB binding to DNA. There was a decreased binding of NF-κB to DNA following incubation of OCI-Ly10 cells with CU-O75. This effect was both time and concentration dependent. Three superfluous lanes were cut out from this image. As per manufacturer's instruction, we included 3 additional assay controls including: (1) a control without the lysate; (2) a control with excess unlabeled probe at 24h; and (3) untreated control. These additional lanes provided quality control but were irrelevant to the data.
(D) NQBS effect on nuclear levels of p50 and p65 in Jurkat cells following stimulation with TNFα. As early as 5 min after the addition of TNFα to the cell culture, there was a significant increase in nuclear levels of p50 and p65. A 4-h pre-incubation with CU-O47-inhibited TNFα-induced nuclear localization of p50 and p65. Incubation with either TNFα alone or both TNFα and CU-O75 did not have any effect on other transcription factors.
Finally, we wanted to validate the data that led to the NQBS discovery in a lymphoma model. For this, we used a Jurkat cell line, with no constitutive overactivation of NF-κB, similar to the HUVEC line requiring TNFα stimulation for NF-κB translocation to the nucleus. Figure 3D shows the effect on nuclear levels of p50 and p65 following stimulation with TNFα. As early as 5 min after the addition of TNFα to the cell culture, there was a significant increase in nuclear levels of p50 and p65. A 4-h pre-incubation with CU-O47 inhibited TNFα-induced nuclear localization of p50 and p65. Incubation with either TNFα alone or both TNFα and CU-O75 did not have any effect on other transcription factors (e.g., Ku80).
Mechanistic Elucidation of NQBS-Mediated NF-κB Inhibition
We then explored possible mechanism of NQBS-mediated inhibition of NF-κB. First, we wanted to rule out conventional mechanisms known to affect NF-κB subunit localization, as induced by proteasome and IKK inhibition. We investigated the activity of NQBS-related compounds on proteasome function. Graphs in Figure S4A represent the effect of NQBS on all three functions of the proteasome. While positive controls bortezomib and carfilzomib effectively inhibited the chymotrypsin-like activity of the proteasome, the NQBS-related compounds exerted minimal to no effect on chymotrypsin-like, trypsin-like, and caspase-like proteolytic activities of cellular proteasomes. This effectively proved that CU-O42, CU-O47, and CU-O75 were not proteasome inhibitors.
We also tested NBQS in the DISCOVERx kinome screen where CU-O42 and CU-O75 failed to inhibit any of the 403 kinases screened. Importantly, they did not inhibit any of the IKK family kinases. Furthermore, phosphorylated IκBα levels were not affected by treatment of OCI-Ly1, OCI-Ly10, or HBL-1 cells with CU-O42 (Figure S4B), confirming NQBS-related compounds did not act via IKK inhibition and were likely not protein kinase inhibitors.
Having proven that NQBSs do not act via established mechanisms, we sought to elucidate the novel mechanism by which NF-κB pathway inhibition may arise. For this, we focused on potential binding of these compounds to one or more members of the NF-κB protein family. To test this hypothesis, we used Internal Coordinate Mechanics software to identify potential binding sites between CU-O75 and NF-κB subunits and related proteins. Target proteins for this analysis included the subunits of the most prevalent NF-κB dimer in mammalian cells, p50 and p65, as well as IκBα, their inhibitory interacting partner.
ICM identified two putative CU-O75 docking sites on the p50-p65-IκBα trimer. As shown in Figure 4A, the first site is at the N terminus of IκBα and the second at the interface between the IκBα C terminus and the NF-κB dimer. Given the established contribution of IκBα′s C-terminus to the p50-p65-IκBα trimer's stability, we hypothesized that NBQS-related compounds may stabilize this trimeric interaction, thus preventing release and nuclear translocation of the dimer. To prove the direct interaction between NQBS and IκBα, we used the cellular thermal shift assay (CETSA), a recently developed assay to detect small-compound binding to proteins in whole-cell lysates, based on increased thermal stability of the target protein following direct small-compound protein interaction (Molina et al., 2013; Almqvist et al., 2016). Figure 4B shows the effect of the increasing temperatures on IκBα stability in OCI-Ly10 model treated with CU-O42 and CU-O75 compared to untreated cells. A temperature of 50.1°C led to full denaturation of IκBα in the control sample. A similar effect was observed when OCI-Ly10 cells were incubated with the inactive CU-O102 compound. Incubation with CU-O42 and CU-O75 led to significant increase in total IκBα at 50.1°C, consistent with increased, NQBS binding-mediated stability of the protein. Indeed, IκBα bands were easily detectable in samples collected at up to 55°C, translating into a thermal shift of 2.5–3.0°C (Figure 4C). Isothermal increase in the relative intensity observed in the CU-O75-treated sample compared to the vehicle control is shown in Figure 4D. This effect was not affected by translation inhibition as shown in Figure S4C. Despite the addition of cycloheximide to OCI-Ly10 cells, there was a significant increase in IκBα levels following 3- and 6-h treatments with NQBS (CU-O75 5μM), compared to dimethyl sulfoxide (DMSO) -treated cells at the same time intervals. Taken together, these data suggest that NQBS compounds inhibit NF-κB activity by stabilizing IκBα protein complexes, rather than by preventing IκBα degradation, thus increasing NF-κB sequestration in the cytoplasm.
Figure 4.

NQBS mechanism of action
(A) Internal Coordinate Mechanics (ICM) software analysis of potential binding sites between CU-O75 and NF-κB subunits. ICM identified two putative docking sites of CU-O75 on the p50-p65-IκBα trimer. First docking site is at the N terminus of IκBα and the second at the interface of C terminus of the IκBα with the p50-p65 dimer
(B) Whole cellular thermal shift assay (CETSA) for IκBα stability in OCI-Ly10 model treated with CU-O42 and CU-O75. While the temperature of 50.1°C led to full denaturation of IκBα in the control sample and in the sample treated with inactive CU-O102 compound, IκBα bands were readily detectable in samples treated with CU-O42 and CU-O75 at same temperatures
(C) Thermal shift of 2.5–3.0°C for IκBα protein mediated by CU-O42 and CU-O75 treatment of OCI-Ly10 cells
(D) Isothermal increase in the relative IκBα intensity observed in CU-O75-treated OCI-Ly10 cells compared to vehicle control.
Finally, we performed RNA sequencing from CU-O75-treated OCI-Ly10 cells and analyzed the changes in gene expression compared to the vehicle-treated controls. Of the total of 288 genes found to be significantly downregulated by CU-O75, 155 (54%) were NF-κB transcriptional targets (Figure 5A). Gene set enrichment analysis (GSEA) confirmed the observed downregulation of NF-κB-regulated genes following 12 h treatment with CU-O75 (p = 0.045, Figure 5B). We next used DeMAND (Woo Hoon et al., 2015), a proteome-wide algorithm, to elucidate the mechanism of action of small-molecule compounds, based on changes in mRNA expression. DeMAND integrates the change in dependency between each protein and its network-connected targets or interactors, in a time- and concentration-dependent series of small molecule perturbations, to identify the most directly affected proteins. In NQBS-treated OCI-Ly10 cells (IC20 of CU-O75 at 24 h), DeMAND analysis identified IκBα as a statistically significant candidate target of CU-O75 (p = 0.003), ranked in the top 100 proteins modified (Figure 5C).
Figure 5.

Gene expression changes following treatment with NQBS
(A) RNA sequencing from CU-O75-treated OCI-Ly10 demonstrates that of the total of 288 genes found to be significantly downregulated by CU-O75, 155 (54%) were NF-κB dependent
(B) Gene set enrichment analysis (GSEA) confirms downregulation of NF-κB controlled genes upon 12h treatment with CU-O75 (IC20) in OCI-Ly10
(C) DeMAND algorithm identifies IκBα as one of the top potential target proteins for CU-O75 (IC20).
Efficacy in In Vivo Models
To explore the anti-lymphoma effect of NQBS-related compounds in vivo, we chose 2 mouse lymphoma models. Figure 6A represents a double transgenic, CD19CherryLuciferase and λ-MYC mouse model that spontaneously develops aggressive B-cell lymphomas (Scotto et al., 2012). Once lymphoma development was demonstrable by luminescence analysis, we treated the animals (n = 3) with 4 daily intraperitoneal (i.p.) injections with CU-O42 at 10 mg/kg. CU-O42 rapidly reduced the luminescence signal measured in all regions of interest as shown graphically in Figure 6B. This effect was brief, and lymphomas grew rapidly following cessation of CU-O42 treatment.
Figure 6.

NQBS efficacy in vivo
(A) CU-O42 inhibits lymphoma growth in a double transgenic, CD19CherryLuciferase, and λ-MYC mouse model that spontaneously develops aggressive B-cell lymphomas; data for the representative animal shown.
(B) Graphic representation of CU-O42 tumor growth inhibition in double transgenic, CD19CherryLuciferase, and λ-MYC mouse model; data for the representative animal shown.
(C) DLBCL SCID (severe combined immunodeficiency) beige xenograft models with OCI-Ly1 (GC DLBCL) or HBL-1 (ABC DLBCL) demonstrating efficacy of CU-O42 in tumor growth inhibition (n = 10 for each of the treated groups)
(D) CU-O42-mediated tumor growth inhibition translated to statistically significant survival benefit in ABC DLBCL xenograft.
Next, we used two DLBCL SCID (severe combined immunodeficiency) beige xenograft models engrafting OCI-Ly1 (GCB DLBCL) or HBL-1 (ABC DLBCL). Since OCI-Ly10 does not form xenografts in SCID beige mice, we used HBL-1, another ABC cell line that forms xenografts. In this cell line, NQBS were able to prevent NF-κB translocation to the nucleus and cause lymphoma cell death similarly to OCI-Ly10. We treated both groups of mice (n = 10 for each cell line) with 10 mg/kg of CU-O42 for four consecutive days, administered i.p. on days 1 and 7. As shown in Figure 6C, CU-O42 was able to significantly inhibit lymphoma growth in mice with both ABC and GCB DLBCL xenografts compared to the DMSO-treated controls (n = 10 for each cell line). Interestingly, however, and potentially in accordance with the cell of origin hypothesis of our lymphoma xenografts, survival advantage was observed only in mice with HBL-1 lymphomas, an ABC DLBCL-derived line (Figure 6D).
Figure S5A shows that single i.p. and oral administration of CU-O75 achieved therapeutic plasma concentrations in mice that were equivalent to the concentrations in in vitro experiments observed to have biologic effect. I.p. administration of CU-O42 reached plasma levels that were higher than IC50 (631ng/mL). Additionally, mice treated with CU-O42 did not exhibit significant toxicity when observed for weight loss (Figure S5B) or myelosuppression (Figure S5C). All the animals from the xenograft experiment were sacrificed when the lymphomas surpassed a volume of 2000 mm3, and none was sacrificed due to observed toxicity of CU-O42.
Discussion
DLBCL is the most common aggressive lymphoma worldwide (Teras et al. 2016). While standard treatment with combination of immunotherapy and chemotherapy can achieve long-term remissions in a significant number of patients, approximately 40% of patients will eventually relapse (Coiffier et al., 2002). Mounting evidence suggests that morphologically identical disease, such as DLBCL, has a variety of genetic subtypes characterized by specific genetic lesions and oncogenic overexpression (Pasqualucci, 2013; Chapuy et al., 2018; Schmitz et al., 2018). It is, therefore, of utmost necessity to continue the search for novel treatment options for patients with DLBCL, with specific precision-medicine-based approaches, targeting drivers behind each of the lymphoma subtypes, such as NF-κB perturbation. This is particularly true in the elderly population where the efficacy of salvage therapy regimens is limited by its toxicity.
We are currently witnessing unprecedented developments in immunotherapeutic treatments for hematologic malignancies. Specifically, two recent approvals of CAR T-cell-based treatment options for patients with DLBCL include axicabtagene ciloleucel and tisagenlecleucel (Neelapu et al., 2017; Schuster et al., 2019). Additionally, PD1/PDL1 inhibitors have shown efficacy in DLBCL as well (Lesokhin et al., 2016). These are, however, treatments with significant side effects, particularly in the case of CAR T cells where cytokine release syndrome and neurotoxicity are common. Furthermore, the CAR T-based treatment approach is frequently used with the understanding that they are bridging therapies to allogeneic stem cell transplants which is itself treatment burdened by significant toxicities and transplant-related mortalities.
Given the advanced age of patients with DLBCL at diagnosis, a significant proportion may not be eligible for more aggressive immunotherapies such as CAR-T or stem cell transplants. Therefore, the development of additional therapeutic options with easy administration schedules and low-toxicity profiles, such as NQBS-related compounds, can satisfy the unmet need facing this large and growing DLBCL patient population (Teras et al., 2016; Coiffier et al., 2002). Some of them have already shown therapeutic potential as is the example of lenalidomide and rituximab in DLBCL (Wang et al., 2013).
In this manuscript, we focus on the preclinical development of a therapeutic approach, targeting one of the most important oncogenic pathways in DLBCL – NF-κB. NF-κB is responsible for uncontrolled division of DLBCL cells through activating mutations downstream of the BCR, CD40L/CD40, and TLR/IL1R pathways. These include CD79A and B, CARD11, MYD88, and A20 alterations, which result in constitutive activation of NF-κB (Davis et al., 2010). These genetic events lead to particularly adverse outcomes in patients with DLBCL whose disease carries the characteristics of activated B-cell type disease (Alizadeh et al., 2000). Multiple pro-inflammatory pathways converge to act through NF-κB as well, which suggests its selective inhibition may play a therapeutic role in a spectrum of non-malignant conditions, such as autoimmune diseases, including rheumatoid arthritis, inflammatory bowel disease, and psoriasis.
We have shown that NQBS-related compounds, in line with the high-throughput screening platform used for their identification, potently and rapidly inhibited NF-κB translocation to the nucleus of DLBCL cells. This effect was observed in multiple cell lines of both GCB and ABC origin and shown in multiple assays analyzing NF-κB subunit localization in the nucleus and its binding to DNA.
Mechanistically, these compounds act by directly binding to IκBα, increasing the cellular stability of the protein, as shown in our CETSA and cycloheximide pulse-chase assays. In turn, increased IκBα stability resulted in presumed greater binding to the p65/p50 dimer and sequestration of the complex to the cytoplasm of lymphoma cells. Consequently, the observed effect resulted in significant decrease in RNA expression of NF-κB-controlled target genes. Additionally, DeMAND analysis identified IκBα as a leading potential target of NQBS-related compounds as determined from the unique gene signature generated from lymphoma cells exposed to the drugs.
Finally, NQBS-related compounds potently inhibited lymphoma growth without significant toxicities observed in two in vivo models of DLBCL. Interestingly, and potentially in accordance with the cell of origin hypothesis of DLBCL, survival advantage was observed only in animals with ABC xenografts treated with NQBS, while NQBS-related compounds inhibited tumor growth of both GCB and ABC xenografts. It is important to note here that the number of animals used for this experiment (n = 10 for each group) is limited and that a larger number of animals could make the difference in survival of treated vs. untreated mice with OCI-Ly1 xenografts statistically significant. It is also noteworthy that while HBL-1 cell line engrafts sooner in experimental animals, eventually OCI-Ly1 overgrows HBL-1, so the lack of NQBS efficacy in GCB cell line with regards to overall survival benefit could be biased by the speed of cell line growth.
In conclusion, we present strong evidence that NQBS-related compounds potently inhibit NF-κB activity and translocation in preclinical models of DLBCL. In vivo efficacy and toxicity data provide encouraging evidence to support further drug development. Additional preclinical tests are being pursued to determine the suitability of these compounds for potential early phase clinical trials.
Limitations of the Study
These experiments in this manuscript are all performed in preclinical models of DLBCL, and further studies are needed to determine if NQBSs confer same mechanism of action that would be clinically meaningful. Main experiments in this study are performed using American Type Culture Collection (ATCC) -verified cell line models of DLBCL. Further work is need, particularly with primary DLBCL samples to confirm the findings reported.
Resource Availability
Lead Contact
Matko Kalac – matko.kalac@mssm.edu.
Materials Availability
Compounds reported in this study are protected by the US patent number 9,896,420.
Data and Code Availability
Microarray data are available in National Center for Biotechnology Information Gene Expression Omnibus database (accession number GSE161677).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Work presented in this publication was partly sponsored by the American Society of Clinical Oncology's Conquer Cancer Foundation through its Young Investigator Award, by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number TL1TR001875 to MK, and by the Center for Target Discovery and Development U01 CA217858 to AC, as well as by the S10 OD01235 and S10 OD021764 instrumentation grants to A.C. Work presented in this publication was partly sponsored by the National Institutes of Health through its S10 instrumentation program (grant ID 1S10OD018121) to D.W.L.
Author Contributions
M.K. designed and performed the experiments and wrote the manuscript, M.M. designed and performed the experiments and wrote the manuscript, A.R. synthesized the compounds and contributed to manuscript preparation, S.D. synthesized the compounds and contributed to manuscript preparation, L.S. designed and performed the experiments and contributed to manuscript preparation, M.M. designed and performed the experiments, M.B. and A.C. contributed in GSEA and DeMAND analysis, D.W.L. designed the experiments and contributed to the synthesis of the compounds, and O.A.O. designed the experiments and wrote the manuscript.
Declaration of Interests
A.C. is a founder, equity holder, consultant, and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc. N-quinolin-benzensulfonamides and related compounds' structures are submitted to the US Patent Office – US patent number 9,896,420. All remaining authors declare no conflicting interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101884.
Supplemental Information
References
- Alizadeh A.A., Eisen M.B., Davis R.E., Ma C., Lossos I.S., Rosenwald A., Boldrick J.C., Sabet H., Tran T., Yu X. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Almqvist H., Axelsson H., Jafari R., Dan C., Mateus A., Haraldsson M., Larsson A., Martinez Molina D., Artursson P., Lundbäck T., Nordlund P. CETSA screening identifies known and novel thymidylate synthase inhibitors and slow intracellular activation of 5-fluorouracil. Nat. Commun. 2016;7:11040. doi: 10.1038/ncomms11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V., Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasseres D.S., Baldwin A.S. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- Chapuy B., Stewart C., Dunford A.J., Kim J., Kamburov A., Redd R.A., Lawrence M.S., Roemer M.G.M., Li A.J., Ziepert M. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018;24:679–690. doi: 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E., Brown K., Park S., Wang H., Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat. Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- Coiffier B., Lepage E., Briere J., Herbrecht R., Tilly H., Bouabdallah R., Morel P., Van Den Neste E., Salles G., Gaulard P. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002;346:235–242. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- Coope H.J., Atkinson P.G., Huhse B., Belich M., Janzen J., Holman M.J., Klaus G.G., Johnston L.H., Ley S.C. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.E., Ngo V.N., Lenz G., Tolar P., Young R.M., Romesser P.B., Kohlhammer H., Lamy L., Zhao H., Yang Y. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C., Lipstein M., Rodriguez R., Serrano X.O., McIntosh C., Tsai W.Y., Wasmuth A.S., Jaken S., O'Connor O.A. The novel IKK2 inhibitor LY2409881 potently synergizes with histone deacetylase inhibitors in preclinical models of lymphoma through the downregulation of NF-κB. Clin. Cancer Res. 2015;21:134–145. doi: 10.1158/1078-0432.CCR-14-0384. [DOI] [PubMed] [Google Scholar]
- Dejardin E., Droin N.M., Delhase M., Haas E., Cao Y., Makris C., Li Z.W., Karin M., Ware C.F., Green D.R. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- Derudder E., Dejardin E., Pritchard L.L., Green D.R., Korner M., Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-α and lymphotoxin-β receptor activation. J. Biol. Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- Dutta J., Fan Y., Gupta N., Fan G., Gelinas C. Current insights into the regulation of programmed cell death by NF-κB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Baltimore D. Activation in vitro on NF-κB by its inhibitor IκB. Nature. 1990;344:679–682. doi: 10.1038/344678a0. [DOI] [PubMed] [Google Scholar]
- Ghosh S., May M.J., Kopp E.B. NF-κB and Rel proteins: evolutionary conserved mediators of immune responses. Annu. Rev. Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Karin M. Missing pieces in NF-κB puzzle. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Gilmore T.D., Herscovitch M. Inhibitors of NF-kB signaling:785 and counting. Oncogene. 2006;25:6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- Gisselbrecht C., Glass B., Mounier N., Singh Gill D., Linch D.C., Trneny M., Bosly A., Ketterer N., Shpilberg O., Hagberg H. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J. Clin. Oncol. 2010;28:4184–4190. doi: 10.1200/JCO.2010.28.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon T.M., Deisseroth A., Kaminskas E., Kane R.C., Koti K.M., Rothmann M.D., Habtemariam B., Bullock J., Bray J.D., Hawes J. US food and drug administration approval: carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013;19:4559–4563. doi: 10.1158/1078-0432.CCR-13-0755. [DOI] [PubMed] [Google Scholar]
- Herrington F.D., Carmody R.J., Goodyear C.S. Modulation of NF-kB signaling as a therapeutic target in autoimmunity. J. Biomol. Screen. 2015;21:223–242. doi: 10.1177/1087057115617456. [DOI] [PubMed] [Google Scholar]
- Kane R.C., Bross P.F., Farrell A.T., Pazdur R. Velcade: US FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Kato M., Sanada M., Kato I., Sato Y., Takita J., Takeuchi K., Niwa A., Chen Y., Nakazaki K., Nomoto J. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- Lenz G., Davis R.E., Ngo V.N., Lam L., George T.C., Wright G.W., Dave S.S., Zhao H., Xu W., Rosenwald A. Oncogenic CARD11 mutations in human diffuse large B-cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- Lesokhin A.M., Ansell S.M., Armand P., Scott E.C., Halwani A., Gutierrez M., Millenson M.M., Cohen A.D., Schuster S.J., Lebovic D. Nivolumab in patients with relapsed or refractory hemaologic malignancy: preliminary results of a phase Ib study. J. Clin. Oncol. 2016;34:2698–2704. doi: 10.1200/JCO.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.L., Kamata H., Karin M. The anti death machinery in IKK/NF-κB signaling. J. Clin. Immunol. 2005;25:541–550. doi: 10.1007/s10875-005-8217-6. [DOI] [PubMed] [Google Scholar]
- Ma J., Xing W., Coffey G., Dresser K., Lu K., Guo A., Raca G., Pandey A., Conley P., Yu H., Wang Y.L. Cerdulatinib, a novel dual SYK/JAK kinase inhibitor, has broad anti-tumor activity in both ABC and GCB types of diffuse large B cell lymphoma. Oncotarget. 2015;6:43881–43896. doi: 10.18632/oncotarget.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina D.M., Jafari R., Ignatushchenko M., Seki T., Larsson E.A., Dan C., Sreekumar L., Cao Y., Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;6141:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Eng. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo V.N., Young R.M., Schmitz R., Jhavar S., Xiao W., Lim K.H., Kohlhammer H., Xu W., Yang Y., Zhao H. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2010;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski G.S., LaPlant B., Macon W.R., Reeder C.B., Foran J.M., Nelson G.D., Thompson C.A., Rivera C.E., Inwards D.J., Micallef I.N. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J. Clin. Oncol. 2015;33:251–257. doi: 10.1200/JCO.2014.55.5714. [DOI] [PubMed] [Google Scholar]
- Oeckinghouse A., Hayden M.S., Ghosh S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L. The genetic basis of diffuse large B-cell lymphoma. Curr. Opin. Hematol. 2013;20:336–344. doi: 10.1097/MOH.0b013e3283623d7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R., Wright G.W., Huang D.W., Johnson C.A., Phelan J.D., Wang J.Q., Roulland S., Kasbekar M., Young R.M., Shaffer A.L. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Eng. J. Med. 2018;378:1396–1407. doi: 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- Scotto L., Kruithof-de Julio M., Paoluzzi L., Kalac M., Marchi E., Buitrago J.B., Amengual J., Shen M.M., O'Connor O.A. Development and characterization of a novel CD19CherryLuciferase (CD19CL) transgenic mouse for the preclinical study of B-cell lymphomas. Clin. Cancer Res. 2012;18:3803–3811. doi: 10.1158/1078-0432.CCR-11-2588. [DOI] [PubMed] [Google Scholar]
- Shaffer A.L., 3rd, Young R.M., Staudt L.M. Pathogenesis of human B cell lymphomas. Annu. Rev. Immunol. 2012;30:565–610. doi: 10.1146/annurev-immunol-020711-075027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H., Thiele J., Vardiman J.W. International Agency for Research on Cancer (IARC); 2008. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. [Google Scholar]
- Teras L.R., DeSantis C.E., Cerhan J.R., Morton L.M., Jemal A., Flowers C.R. US lymphoid malignancy statistics by world Health organization subtypes. CA Cancer J. Clin. 2016;66:443–459. doi: 10.3322/caac.21357. [DOI] [PubMed] [Google Scholar]
- Viardot A., Goebeler M.E., Hess G., Neumann S., Pfreundschuh M., Adrian N., Zettl F., Libicher M., Sayehli C., Stieglmaier J. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2016;127:1410–1416. doi: 10.1182/blood-2015-06-651380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Fowler N., Wagner-Bartak N., Feng L., Romaguera J., Neelapu S.S., Hagemeister F., Fanale M., Oki Y., Pro B. Oral lenalidomide with rituximab in relapsed or refractory diffuse large cell, follicular and transformed lymphoma: a phase II clinical trial. Leukemia. 2013;27:1902–1909. doi: 10.1038/leu.2013.95. [DOI] [PubMed] [Google Scholar]
- Woo J.H., Shimoni Y., Yang W.S., Subramaniam P., Iyer A., Nicoletti P., Rodríguez Martínez M., López G., Mattioli M., Realubit R. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162:441–451. doi: 10.1016/j.cell.2015.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G., Harhaj E.W., Sun S.C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Xie Y., Deng S., Thomas C.J., Liu Y., Zhang Y.Q., Rinderspacher A., Huang W., Gong G., Wyler M., Cayanis E. Identification of N-(quinolin-8-yl)benzenesulfonamides as agents capable of down-regulating NFκB activity within two separate high-throughput screens of NFκB activation. Bioorg. Med. Chem. Lett. 2008;18:329–335. doi: 10.1016/j.bmcl.2007.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A., Sehn L.H., Johnson P., Zinzani P.L., Hong X., Zhu J., Patti C., Belada D., Samoilova O., Suh C. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J. Clin. Oncol. 2019;37:1285–1295. doi: 10.1200/JCO.18.02403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A., Thieblemont C., Morschhauser F., Flinn I., Friedberg J.W., Amorim S., Hivert B., Westin J., Vermeulen J., Bandyopadhyay N. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol. 2014;15:1019. doi: 10.1016/S1470-2045(14)70311-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data are available in National Center for Biotechnology Information Gene Expression Omnibus database (accession number GSE161677).