

Abstract
Optimization efforts were devoted to discover novel PDE10A inhibitors in order to improve solubility and pharmacokinetics properties for a long-term therapy against pulmonary arterial hypertension (PAH) starting from the previously synthesized inhibitor A. As a result, a potent and highly selective PDE10A inhibitor, 14·3HCl (half maximal inhibitory concentration, IC50 = 2.8 nmol/L and >3500-fold selectivity) exhibiting desirable solubility and metabolic stability with a remarkable bioavailability of 50% was identified with the aid of efficient methods of binding free energy predictions. Animal PAH studies showed that the improvement offered by 14·3HCl [2.5 mg/kg, oral administration (p.o.)] was comparable to tadalafil (5.0 mg/kg, p.o.), verifying the feasibility of PDE10A inhibitors for the anti-PAH treatment. The crystal structure of the PDE10A−14 complex illustrates their binding pattern, which provided a guideline for rational design of highly selective PDE10A inhibitors.
KEY WORDS: Phosphodiesterase 10A, Inhibitor, Benzimidazole derivatives, Crystal structure, Metabolic stability, Bioavailability, Pulmonary arterial hypertension
Graphical abstract
A potent and highly selective PDE10A inhibitor, 14·3HCl (IC50 = 2.8 nmol/L and >3500-folds selectivity) with a remarkable bioavailability of 50% was obtained to verify the feasibility for the anti-PAH treatment. The crystal structure of PDE10A–14 complex illustrated the binding pattern, providing a guideline for rational design of highly selective PDE10A inhibitors.

1. Introduction
Pulmonary arterial hypertension (PAH) is a rare but frequently fatal disease marked by particularly severe and progressive form of chronic pulmonary hypertension that frequently culminates in right ventricular heart-failure and premature death1. Although significant advances in understanding of the pathobiology of PAH have given rise to the development of several clinical therapeutics against this disease2, an unmet medical need still exists because of significant morbidity and mortality as well as inadequate improvement for life quality of PAH patients3,4. Nowadays, clinical drugs for PAH therapy include endothelin receptor antagonists (bosentan), prostacyclin analogs (treprostinil), soluble guanylate cyclase (sGC) stimulator (riociguat) and PDE5 inhibitors (sildenafil and tadalafil) as well as their combination. However, the majority of U.S. Food and Drug Administration (FDA) proved anti-PAH drugs still fall “short of the mark” in reversing or halting the progression of this disease in the long run on account of the pathobiologic complexity relative to the various signaling pathway5. In addition, PDE5 inhibitors have been limited in clinical use because of the side effects from low PDE subtype selectivity and rapid metabolization such as sildenafil6,7. Therefore, a novel PDE5 inhibitor (TPN171) with fewer side effects and an excellent pharmacokinetic profile has being investigated in a phase II clinical trial for the treatment of PAH8.
In the treatment of PAH, there is a growing body of evidence showing that nitric oxide (NO)-driven 3′,5′-cyclic guanosine monophosphate (cGMP) and prostacyclin (PGI2)-driven 3′,5′-cyclic adenosine monophosphate (cAMP) signaling pathways involve the up-regulation of the secondary messenger level and play a vital role in regulating vasomotor tone and vascular smooth muscle proliferation, thereby preventing pulmonary vascular from remodeling2. With this in mind, it is reasonable to hypothesize that the pharmacological dual regulation of both cAMP and cGMP levels may provide a synergistic effect for PAH treatment. Recently, PDE10 as a promising target for the therapy of PAH has been validated on the pathobiology9. PDE10 as a dual-substrate enzyme is capable of accommodating and degrading cAMP with a Km value of 56 nmol/L and cGMP with a Km value of 4.4 μmol/L, respectively10. PDE10 expression is detected not only in very high levels in human central nervous system (CNS) tissues, but also in the peripheral organs such as thyroid, testis, kidney, and lung11, highlighting that PDE10 is of interest as an appealing drug target for a broad range of diseases including CNS disorders12, lower urinary tract symptoms (LUTS)13, lung/colorectal cancer14,15 and PAH9,16.
However, there is still limited study based on PDE10 in terms of pharmacological intervention as an effective anti-PAH therapeutic approach. On the positive side, our recent exciting result has demonstrated that a highly selective PDE10A inhibitor A (or original 2b16, IC50 = 0.44 nmol/L, Scheme 1) served as a specific targeting chemical probe to exert significant pharmacodynamic effect on PAH treatment. Nevertheless, inhibitor A could not be dosed orally in mice due to its poor aqueous solubility and fast metabolism (rat in vivo blood terminal half-life t1/2 = 0.58 h). Due to the fact that PAH is a chronic and progressive disease, it is of paramount importance to achieve oral administration during daily therapy. Therefore, a novel PDE10 inhibitor should be identified with more potential drug-like characteristics for PAH therapy including superior aqueous solubility and metabolic stability as well as pharmacokinetic properties. Herein, we reported our effort on further structural optimization of compound A aiming to improve metabolic stability and the pharmacokinetics properties in order to further validate PDE10 inhibition as a dependable drug target for PAH treatment by oral administration. As a result, compound 14 was designed and identified to show potent inhibition with an IC50 of 2.8 nmol/L as well as selectivity of 3500-fold against other PDE subtypes. Furthermore, the pharmacokinetic properties of 14·3HCl have been remarkably improved with an oral bioavailability up to ∼50%, thus enabling in vivo studies by oral intake. Importantly, 14·3HCl significantly lowered the arterial pressure in PAH rats at a dosage of 2.5 mg/kg (p.o.). These findings further support the potential of PDE10 inhibition as an effective target for the anti-PAH therapy.
Scheme 1.

Structure optimized design of novel PDE10 inhibitors aiming to improve solubility and pharmacokinetics properties.
2. Results and discussion
2.1. Optimization of novel PDE10A inhibitors to improve metabolic stability and aqueous solubility
According to earlier structure–activity relationship studies and the X-ray crystal structure of the PDE10A−A complex16, the substituted benzimidazole ring of A formed an H-bond interaction with Y693 in the selectivity pocket Q217,18, contributing to its high binding affinity to PDE10A as well as higher selectivity over other PDEs. Therefore, this scaffold is preferably retained in our structure-based design for novel congeneric inhibitors. Although the 1,8-naphthalimide moiety of A acted as an H-bond donor with the conserved residue Q726, this large rigid aromatic scaffold tended to aggregate via π−π stacking interactions, leading to a detrimental effect on aqueous solubility. It proved challenging to modify this structural element to improve aqueous solubility without reducing its inhibitory potency. Introduction of a hydrophilic group on to a non-critical binding site of the compound and extending out into a solvent-exposed region could be a favorable optimization approach.
To test the above conjecture, molecular docking and molecular dynamics (MD) simulations based on the crystal complex of the PDE10A−A complex were first performed to preliminarily assess binding mode before the preparation of individual compounds (Supporting Information Fig. S1 and Table S1). The result of MD evaluations of novel compounds based on a benzimidazole moiety as an effective selective pocket binding group (SPBG) was available to estimate the binding affinity toward PDE1016. As a result, most of our newly designed compounds (Scheme 2) exhibited high potency inhibition towards PDE10A with IC50 values less than 100 nmol/L in the bioassay in vitro, while validating a similar binding affinity trend predicted among these compounds except individual differences. Our approach of structure-based design made the utmost of binding free energy predictions to greatly reduce synthetic loads and costs, thereby accelerating the time-consuming hit-to-lead process. As our initial efforts focused on shrinking the rigid aromatic scaffold of 1,8-naphthalimide, isoquinolin-1(2H)-one and quinoline were utilized as relatively small scaffold to afford compounds 1 and 2 with IC50 values of ∼100 and 9.1 nmol/L, respectively. The higher inhibitory potency of compound 2 was divinable due to remarkable binding free energies ∼6 kcal/mol lower than that of 1 (Supporting Information Table S1). The high potent inhibition of compound 2 inspired us to work on further optimization on structural design (Scheme 2). To our surprise, compound 4 with a vinyl linker gave better inhibitory potency (IC50 = 2.7 nmol/L) compared to compound 2 with an ethyl linker. It was probably due to the fact that conformationally restricted internal C C bridge reduced entropy compensation upon the ligand binding toward PDE10A19. More importantly, compound 4 exhibited a marked improvement in metabolic stability (rat liver microsomes, RLM t1/2 = 17.4 min) as compared to compound 2 (RLM t1/2 = 0.87 min, Supporting Information Table S2), which was attributed to limited conformations resulting in being more resistant to binding to drug metabolizing enzymes20. Higher stability and excellent binding affinity of compound 4 prompted us to carry out an in-depth investigation towards the effect of substitution on benzimidazole moiety. Unfortunately, none of the investigated compounds, 3–9 gained an advantage over compound 4 on evaluation of inhibitory potency against PDE10A (Table 1).
Scheme 2.

The general synthetic route summarized for the preparation of the compounds 1–14. Reagents and conditions: (a) methyl acrylate, NaOCH3, methanol, reflux, 4 h; (b) methyl acrylate, TEA, (PPh3)2PdCl2, N,N-dimethylformamide, Ar, 120 °C, overnight; (c) Pd/C, H2, ethanol, room temperature, overnight; (d) 6 mol/L NaOH, ethanol, room temperature, overnight; (e) i) 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), CH2Cl2, overnight; ii) acetic acid, 90 °C, overnight.
Table 1.
Inhibitory activities of compounds 1–14 against PDE10A.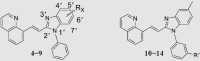
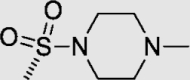
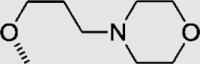
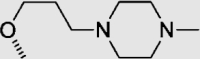
To investigate for in vivo efficacy, water solubility of the inhibitors had to be improved for oral administration. According to a similar molecular in silico docking method16 of PDE10A with compound 4 (Supporting Information Fig. S2), the binding mode indicated that the aryl ring at the N-1′ position of benzimidazole moiety could further be structurally optimized, especially with substituents on the meta-/para-position. This N-1′ aryl moiety seems to be far from the key protein surroundings and extend out into a solvent-exposed surface region. Meanwhile, the binding free energies of compounds 10–14 with values lower than −38 kcal/mol were found to speculate that their inhibitory activities against PDE10A could be better or comparable to that of compound 4. As a result, all designed compounds, 10–14 with different hydrophilic groups showed nanomolar or subnanomolar inhibitory activities against PDE10A with IC50 values lower than 2.8 nmol/L. Interestingly, the solubility of compounds substituted with the water-soluble morpholine and N-methyl piperazine were notably enhanced in the hydrochloride form, resulting in up to 11.9 and 4.7 mg/mL for 13·HCl and 14·3HCl solubility in deionized (D.I.) water (pH = 7.0), respectively. The C C bridge between two aromatic rings within 14·3HCl was insensitive to react with cysteine in aqueous phase (Supporting Information Fig. S3), indicating low probability formation of a covalent adduct in vivo21.
2.2. Synthesis of compounds 1−14
The synthetic route for the newly developed compounds is outlined in Scheme 2. The key carboxyl intermediates M-1, M-4, and M-5 were treated with different arylamines in the presence of HATU22, followed by acid-catalyzed ring-forming condensation reaction to afford final products 1–12, 13·3HCl and 14·3HCl in reasonable yields. All the new compounds were fully characterized by 1H NMR, 13C NMR spectroscopy and HR-MS, and found to fully agree with their proposed structures. To remove possible false hits, a pan-assay interfering compound substructures (PAINS) screening was performed by using the online program PAINS-Remover (http://cbligand.org/PAINS)23. All the synthesized compounds passed this test to minimize the risk of poor drug development choices, because starting hit compounds with problematic structures were alerted as PAINS on the account of being probably protein-reactive in vivo.
2.3. Structure−activity relationships
The inhibitory effects of compounds on PDEs activities were investigated by the radioisotope procedure with 3H-cGMP/3H-cAMP while papaverine served as a positive control16. Most of newly synthesized compounds exhibited high binding affinity toward PDE10A with IC50 values < 100 nmol/L. The introduction of the C C bond instead of the C–C single bond afforded a 3-fold enhancement of PDE10A inhibition in compound 4 relative to compound 2. Comparison of 3 with 4 showed that removal of the phenyl ring at the N-1′ position of benzimidazole moiety caused significantly decrease in inhibitory activity. As shown in Table 1, among compounds 4–9, methyl-substitution only at 5′-position of the benzimidazole moiety gave rise to the pronounced inhibition; on the other hand, there was no improvement of inhibitory potency observed for 5′-fluoro analogue 6, signifying the important hydrophobic effect of the methyl group. It was important to find that newly designed compounds 11–14 with different hydrophilic groups on the meta-position of the phenyl ring at the N-1′ position of benzimidazole moiety still afforded nanomolar or subnanomolar inhibitory activities against PDE10A with IC50 values lower than 2.8 nmol/L, highlighting that our rational molecular docking approach played a crucial role in guiding the structure-based design for PDE10A inhibitors.
2.4. Remarkable pharmacokinetic profile of 14·3HCl
On the basis of their remarkable in vitro parameters, compounds 10–12, 13·3HCl and 14·3HCl were progressed for evaluation of preliminary metabolic stability in a liver microsome assay. As shown in Table 2, compound 14·3HCl showed favorable metabolic stability with RLM t1/2 of 25 min, significantly more stable than other PDE10A inhibitors in Table 1, being regarded as a preferable lead to enable subsequent pharmacokinetic (PK) assessments.
Table 2.
Metabolic stability of 10–12, 13·HCl and 14·3HCl in rat liver microsomes.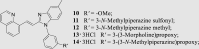
| Compd. | t1/2 (min)a | CLint [mL/(min·g·protein)]b |
|---|---|---|
| 10 | 2.1 | 999.4 |
| 11 | 1.5 | 1390 |
| 12 | 11 | 198.1 |
| 13·3HCl | 16 | 127.7 |
| 14·3HCl | 25 | 85.1 |
t1/2: elimination half-life.
CLint: intrinsic body clearance.
The PK profile of compound 14·3HCl in rats (approved by the Institutional Animal Care and Use Committee, Sun Yat-sen University, Guangzhou, China) is shown in detail in Table 3. To our delight, the PK (intravenous administration/i.v.) profile of compound 14·3HCl showed significant improvement with relatively low clearance (CL = 1398 ± 217 mL/h/kg) and higher plasma exposure (the mean area under the plasma concentration–time curve, AUC(0−∞) = 1819 ± 297 h·ng/mL) at a dose of 2.5 mg/kg, compared to A16 (CL = 2337 ± 470 mL/h/kg and AUC(0−∞) = 1099 ± 215 h·ng/mL). It is noteworthy that the 14·3HCl had good bioavailability of 50%, almost 20-fold higher than A (Supporting Information Table S3), highlighting a successful structural optimization strategy by means of conformational restriction and introduction of the N-methyl piperazine side chain24.
Table 3.
Pharmacokinetic profile of compound 14·3HCl in Sprague–Dawley rats.
| Route | t1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUC(0−t) (h·ng/mL) | AUC(0−∞) (h·ng/mL) | MRT(0−t) (h) | CL (mL/h/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|
| i.v.a | 6.7 ± 1.5 | 0.4 ± 0.4 | 230 ± 50 | 1666 ± 184 | 1819 ± 297 | 6.3 ± 0.8 | 1398 ± 217 | – |
| p.o.b | 5.2 ± 0.9 | 6.0 ± 2.0 | 272 ± 25 | 3310 ± 812 | 3551 ± 1008 | 8.4 ± 0.7 | – | 50 ± 12 |
AUC: area under the curve. MRT: mean residence time. CL: clearance. −Not applicable.
i.v. = intravenous administration, dose = 2.5 mg/kg.
p.o. = oral administration, dose = 10 mg/kg.
2.5. Crystal structure of PDE10A with 14
In view of the promising potency against PDE10A, the cocrystal profile of the PDE10−14 complex was pursued to reveal the binding motif. As illustrated in Fig. 1A, the quinoline group of 14 inserted in the hydrophobic clamp to form an H-bond with the invariant Q726 and had multiple π–π stacking interactions with the hydrophobic clamp consisting of F729 and I692/F696, which mainly contributed to its high affinity with PDE10 A10,16. As expected, the benzimidazole moiety of 14 was sandwiched by the unique selectivity pocket Q217,18 of PDE10A and formed another strong H-bond with Y693, accounting for not only its high inhibitory potency but remarkable selectivity over other PDEs. Furthermore, the water-solubilizing N-methylpiperazine group extended to the solvent region in a chair conformation and displayed no detrimental effect on the binding motif in agreement with our predicted mode. The 5′-subtituted methyl group on the benzimidazole ring also exhibited the optimal interaction orientation fitting well within the selectivity pocket Q2 of PDE10A. Additionally, the site-directed mutagenetic PDE10A (449–770) were designed to obtain the Y693A/Q726A/F729A mutant, respectively. As a result, the inhibitory potency of the compound 14 against these mutations were significantly decreased by 13-, 30- and 58-fold, respectively, in the comparison with that of the wild type (Supporting Information Fig. S5), which added weight to the highlight of the key residues for inhibitor binding like Q726, F729 and Y693.
Figure 1.

Crystal structure of the PDE10-14 (PDB code: 7BPI). (A) Surface presentation. The benzimidazole moiety and N-methylpiperazine group of 14 were involved in Q2 pocket and solvent region, respectively. (B) Ribbon model of 14 binding in the active site of PDE10A. The red and magentas dotted lines represent H-bonds and multiple π−π stacking interactions, respectively.
2.6. Excellent selectivity of compound 14·3HCl over other PDE isoforms
As seen in Supporting Information Table S5, the structure-based sequence alignment of the substrate-binding pocket shows dramatic variation of amino acids across PDE families10, which determines not only the shape and size of the binding pocket but also the binding affinity of different inhibitors. A selective binding pocket composed of adjacent amino acid residues of PDE10A has been demonstrated, which exclusively involves I692, Y693 and G725, respectively. The benzimidazole group of 14·3HCl well occupied this narrow selectivity pocket17,18, and showed a strong H-bond interaction with the specific Y693, accounting for the excellent selectivity over other PDEs. The selectivity profile of compound 14·3HCl against other PDE isoforms is summarized in Table 4. Compound 14·3HCl with the IC50 value of 2.8 nmol/L against PDE10A2 exhibited weak inhibition (IC50 > 10 μmol/L) towards the majority of the representative PDE subtypes including PDE1B, PDE2A, PDE3A, PDE4D2, PDE5A1, PDE7A1, PDE8A1 and PDE9A2, thereby showing more than 3500-fold selectivity across other PDE families. Apparently, the remarkable selectivity of compound 14·3HCl over other PDE subtypes implied potential for PAH treatment with minimal interference from other PDEs as well as fewer side effects anticipated, supporting further pharmacodynamics assessment studies for 14·3HCl.
Table 4.
Selectivity of 14·3HCl across PDE families.
| PDE | IC50 (μmon/L) | Selectivitya |
|---|---|---|
| PDE10A2 (449–770) | 2.8 × 10−3 | − |
| PDE1B (10–487) | >10 | >3500 |
| PDE2A (580–919) | >10 | >3500 |
| PDE3A (679–1087) | >10 | >3500 |
| PDE4D2 (86–413) | >10 | >3500 |
| PDE5A1 (535–860) | >10 | >3500 |
| PDE7A1 (130–482) | >10 | >3500 |
| PDE8A1 (480–820) | >10 | >3500 |
| PDE9A2 (181–506) | >10 | >3500 |
Selectivity fold = IC50(PDEs)/IC50(PDE10A2).
2.7. Pharmacological toxicity
After an oral administration, there was no obvious acute toxicity found for 14·3HCl in the course of evaluation for pharmacological safety using the same protocol previously reported16,25,26, in which this inhibitor was well tolerated up to a dose of 1.0 g/kg. Additionally, 14·3HCl showed a moderate affinity towards human ether-à-go-go-related gene (hERG) potassium channel with an IC50 value of 4.5 μmol/L (Supporting Information Fig. S4), corresponding to 1600-fold selectivity for PDE10A IC50 (2.8 nmol/L) in vitro, indicating a good safety margin of 36-fold between hERG IC50 and plasma peak concentration (Cmax, calculated (Calcd.) as ∼124.6 nmol/L at an oral dosage of 2.5 mg/g at 6 h in Supporting Information Table S4). As a result, compound 14·3HCl showed practical usability and potential to be used as a drug candidate for in vivo pharmacodynamic studies by means of oral delivery.
2.8. Notable therapeutic effects against PAH in rats
Subsequently, pharmacodynamic effects of 14·3HCl against PAH were evaluated in a monocrotaline (MCT)-induced PAH rat model16,25,26 (rat source: Beijing Vital River Laboratory Animal Technology Co., Ltd.; approved by the Institutional Animal Care and Use Committee, Sun Yat-sen University, Guangzhou, China). The typical symptoms of PAH were observed after 21 consecutive days of MCT injection (60 mg/kg, once), as shown by an enhancement of the mean pulmonary artery pressure (mPAP), right ventricle hypertrophy index (RVHI%), and wall thickness percentage (WT%) of the model group in comparison with the control group (Fig. 2A−C). Treatment with 14·3HCl or tadalafil decreased all the above symptoms of the rats in contrast to that of the model (no treatment) group, especially for mPAP. For RVHI% and WT%, similar trends were also observed. Oral administration of 14·3HCl at a dose of 2.5 mg/kg approximately provided a plasma concentration of Calcd. ∼84 nmol/L at 10 h (equivalent to 336 nmol/L at an oral dose of 10 mg/kg in Supporting Information Table S4), which indicated 30-fold higher than the IC50 (2.8 nmol/L) in vitro. It resulted in significantly ameliorating these PAH medical indicators in rats comparable to that of tadalafil at a dose of 5.0 mg/kg (p.o.), further indicating the feasibility of PDE10A inhibitors for the anti-PAH treatment.
Figure 2.

Effects of compound 14·3HCl and tadalafil on pulmonary arterial rat model in vivo. (A) Mean pulmonary artery pressure (mPAP) of different groups. (B) Right ventricle hypertrophy index (RVHI%) of different groups. (C) Wall thickness percentage (WT%) of different groups. (D) Body weight of different groups. (E) Representative images for each group with hematoxylin and eosin straining. Data shown represent the mean ± SD (n = 6–10 animals per group). ####P<0.0001 and ###P<0.001 vs. the control group; ∗P<0.05, ∗∗P<0.01 and ∗∗∗P<0.001 vs. the model group. Scale bar, 5 μm.
3. Conclusions
Given the previously-reported research findings of PDE10 inhibition as a potential anti-PAH target, there is a continuing interest in the discovery of novel PDE10A inhibitors with favorable druglike characteristics for a long-term therapy. Through optimization of the pharmacokinetic profile of the lead compounds via the structure-based design/optimization strategy using scaffold hopping and conformational restriction with the aid of efficient methods of binding free energy predictions, a novel, potent and highly selective PDE10A inhibitor 14 with IC50 = 2.8 nmol/L for PDE10A and more than 3500-fold selectivity over other PDEs was identified.
The crystal structure of the PDE10A−14 complex illustrates the binding motif, which provided a guideline for rational design of highly selective PDE10A inhibitors. More importantly, 14·3HCl was found to have favorable drug-like properties including high solubility (S = 4.7 mg/mL in D.I. water), remarkable in vivo metabolic stability (t1/2 = 5.2 h), low risk for hERG K+ channel block, and remarkable oral bioavailability (F = 50%). Furthermore, animal studies ascertained the therapeutic potential of PDE10 inhibition for the treatment of PAH by oral administration, in which 14·3HCl significantly improved typical disease medical indicators in the rat model. In short, our findings clearly demonstrate that PDE10 inhibition is a promising potential therapeutic target, which opens a vista for discovery of potent PDE10A inhibitors for treatment of PAH.
4. Experimental
All the reagents were purchased from commercial suppliers (Aldrich®, Shanghai, China; BIDE®, Shanghai, China; or Innochem® Beijing, China), which are used directly without further purification. Chemical HG/T2354-92 silica gel (200–300 mesh, Haiyang®, Qingdao, China) was used for chromatography. 1H NMR and 13C NMR spectra were recorded at room temperature on a Bruker AVANCE III 400 and 500 instrument (Bruker®, Zürich, Switzerland) with tetramethylsilane (TMS) as an internal standard. The following abbreviations are used: s (singlet), d (doublet), dd (two doublets), t (triplet), q (quartet), and m (multiplet). Coupling constants were reported in Hz. High-resolution mass spectra were recorded on a Shimadzu LC‒MS−IT-TOF mass spectrometer (Shimadzu®, Kyoto, Japan). The accurate number of HCl in the molecular structure of 14·3HCl was deduced by an ion chromatography (IC) method with the aid of Dionex ICS-900 (Thermo Fisher Scientific®, Sunnyvale, USA). The purity of compounds was determined by reverse-phase high-performance liquid chromatography (HPLC) analysis confirming to be over 95%. HPLC instrument: Shimadzu LC-20AT (Shimadzu®, column: Hypersil BDS C18, 5.0 μm, 150 mm × 4.6 mm (elite); detector: SPD-20A UV–Vis detector, UV detection at 254 nm; elution, methanol/H2O (100%–90%, v/v); column temperature: 25 °C; and flow rate = 0.8–1.0 mL/min.
4.1. General procedures for synthesis of compounds 4−14
To the solution of M-4 (1.0 mmol) in dichloromethane (DCM, 10 mL) was added HATU (760 mg, 3 mmol), DIPEA (516 mg, 4.0 mmol), and appropriate amine (1.0 mmol) with stirring at room temperature for 12 h. After completion of reaction, the mixture was extracted with ethyl acetate (EA, 3 × 50 mL). The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated in vacuo to afford crude intermediate directly used in the next step without purification. To the residue above was added acetic acid (10 mL) which was stirred at 90 °C overnight. Then the reaction mixture was alkalized with NaOH to pH 9–10, and extracted with EA (3 × 50 mL). The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (DCM/methanol, 10:1) to afford products 4–14 in two steps.
4.1.1. (E)-8-(2-(1-(3-Methoxyphenyl)-5-methyl-1H-benzo[d]imidazole-2-yl)vinyl)quinoline (10)
130 mg, yield 69.0%. 1H NMR (400 MHz, dimethyl sulfoxide-d6/DMSO-d6) δ 9.05 (d, J = 16.3 Hz, 1H), 8.98 (dd, J = 4.1, 1.8 Hz, 1H), 8.41 (dd, J = 8.3, 1.7 Hz, 1H), 8.04 (d, J = 7.1 Hz, 1H), 7.98 (d, J = 8.1 Hz, 1H), 7.62 (dt, J = 11.2, 5.0 Hz, 3H), 7.56 (s, 1H), 7.36 (d, J = 16.2 Hz, 1H), 7.18 (m, 4H), 7.08 (dd, J = 8.2, 1.1 Hz, 1H), 3.85 (s, 3H), 2.46 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 160.82, 150.99, 150.69, 145.67, 143.71, 137.10, 136.76, 134.82, 133.77, 132.62, 132.53, 131.35, 129.56, 128.70, 127.24, 127.08, 125.01, 122.35, 119.90, 119.14, 116.58, 115.29, 113.32, 110.39, 56.06, 21.72. HR-MS (ESI-TOF) m/z [M+H]+ Calcd. for C26H21N3O 392.1757, Found 392.1750.
4.1.2. (E)-8-(2-(5-Methyl-1-(3-((4-methylpiperazin-1-yl)sulfonyl)phenyl)-1H-benzo[d]imidazole-2-yl)vinyl) quinoline (11)
52 mg, yield 48.4%. 1H NMR (400 MHz, DMSO-d6) δ 8.98 (m, 2H), 8.41 (dd, J = 8.3, 1.7 Hz, 1H), 8.08 (d, J = 7.2 Hz, 1H), 8.01 (m, 4H), 7.88 (s, 1H), 7.60 (m, 3H), 7.37 (d, J = 16.2 Hz, 1H), 7.13 (m, 2H), 2.98 (s, 4H), 2.47 (s, 3H), 2.27 (s, 4H), 2.11 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 151.14, 150.80, 145.65, 137.19, 137.10, 136.62, 134.68, 133.61, 133.13, 132.90, 132.66, 132.03, 129.69, 128.70, 128.29, 127.34, 126.94, 126.74, 125.30, 122.35, 119.35, 116.49, 110.02, 53.88, 46.26, 45.70, 21.70. HR-MS (ESI-TOF) m/z [M+H]+ Calcd. for C30H29N5O2S 524.2115, Found 524.2108.
4.1.3. (E)-8-(2-(5-Methyl-1-(3-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-benzo[d]imidazole-2-yl)vinyl) quinoline (12)
30 mg, yield 40.2%. 1H NMR (400 MHz, CDCl3) δ 8.99 (d, J = 16.2 Hz, 1H), 8.91 (dd, J = 4.1, 1.7 Hz, 1H), 8.15 (dd, J = 8.3, 1.7 Hz, 1H), 7.86 (d, J = 7.2 Hz, 1H), 7.78 (d, J = 7.3 Hz, 1H), 7.68 (s, 1H), 7.57 (t, J = 7.7 Hz, 1H), 7.51 (t, J = 7.7 Hz, 2H), 7.45 (dd, J = 11.1, 6.8 Hz, 3H), 7.41 (d, J = 8.0 Hz, 1H), 7.09 (m, 2H), 3.66 (s, 2H), 2.61 (s, 4H), 2.53 (s, 3H), 2.50 (s, 4H), 2.29 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 151.20, 149.87, 146.21, 143.60, 140.26, 136.20, 136.14, 134.82, 134.61, 133.73, 132.84, 129.71, 129.07, 128.61, 128.50, 127.80, 127.34, 126.32, 126.10, 124.60, 121.45, 119.38, 117.38, 109.62, 61.88, 54.52, 51.93, 45.11, 21.67. HR-MS (ESI-TOF) m/z [M+H]+ Calcd. for C31H31N5 474.2652, Found 474.2634.
4.1.4. (E)-4-(3-(3-(5-Methyl-2-(2-(quinolin-8-yl)vinyl)-1H-benzo[d]imidazole-1-yl)phenoxy)propyl)morpholine hydrochloride (13·3HCl)
30 mg, yield 57.5%. 1H NMR (400 MHz, MeOD-d4) δ 9.14 (m, 2H), 8.89 (d, J = 8.0 Hz, 1H), 8.34 (s, 1H), 8.30 (d, J = 7.6 Hz, 1H), 7.95 (dd, J = 7.8, 4.5 Hz, 1H), 7.88 (s, 1H), 7.77 (s, 1H), 7.73 (t, J = 7.4 Hz, 1H), 7.44 (m, 6H), 4.28 (s, 2H), 4.06 (d, J = 12.6 Hz, 2H), 3.86 (t, J = 12.0 Hz, 2H), 3.57 (d, J = 11.3 Hz, 2H), 3.43 (s, 2H), 3.20 (s, 2H), 2.62 (s, 3H), 2.35 (s, 2H). 13C NMR (126 MHz, MeOD-d4) δ 160.13, 147.51, 147.40, 144.30, 138.83, 138.41, 133.05, 132.53, 131.87, 131.69, 131.43, 130.84, 129.46, 128.71, 128.65, 122.34, 119.69, 117.57, 113.63, 113.24, 112.99, 112.34, 65.46, 63.67, 54.66, 52.00, 23.38, 20.35. HR-MS (ESI-TOF) m/z [M+H]+ Calcd. for C32H32N4O2 505.2598, Found 505.2591.
4.1.5. (E)-8-(2-(5-Methyl-1-(3-(3-(4-methylpiperazin-1-yl)propoxy)phenyl)-1H-benzo[d]imidazole-2-yl)vinyl)quinoline hydrochloride (14·3HCl)
14, 2.0 g, yield 42.6%. To the solution of 14 (1.0 g 1.9 mmol) in DCM (10 mL) was added hydrochloric acid in 1,4-dioxane (4 mol/L, 3.8 mmol) with stirring at room temperature until multiple solid separated out. Then the yellow solid was filtered to obtain the product of 14·3HCl (951 mg, yield 85.8%). 1H NMR (400 MHz, MeOD-d4) δ 9.12 (d, J = 16.3 Hz, 1H), 8.99 (d, J = 3.1 Hz, 1H), 8.47 (d, J = 8.1 Hz, 1H), 8.17 (d, J = 6.2 Hz, 1H), 8.11 (d, J = 7.9 Hz, 1H), 7.68 (m, 5H), 7.41 (m, 5H), 4.29 (s, 2H), 3.76 (s, 8H), 3.54 (s, 2H), 3.03 (s, 3H), 2.61 (s, 3H), 2.39 (s, 2H). 13C NMR (126 MHz, MeOD-d4) δ 160.12, 149.96, 148.24, 144.68, 140.72, 138.50, 138.11, 133.23, 131.81, 131.61, 131.41, 131.02, 130.93, 130.19, 128.87, 128.31, 126.78, 122.02, 119.72, 117.49, 113.62, 113.08, 112.20, 110.47, 65.27, 23.67, 20.35. HR-MS (ESI-TOF) m/z [M+H]+ Calcd. for C33H35N5O 518.2914, Found 518.2895. Cl% Calcd. 16.9%, Found 16.2% by IC.
4.2. In vitro enzymatic activity assay
The catalytic domains of PDE1B (10–487), PDE2A (580–919), PDE3A (679–1087), PDE4D2 (86–413), PDE5A1 (535–860), PDE7A1 (130–482), PDE8A1 (480–820), PDE9A2 (181–506), and PDE10A (449–770) were purified using a similar protocol in our previously published protocol16,27,28. 3H-cGMP was the substrate for the biological test against PDE1B, PDE2A and PDE5A while 3H-cAMP for those of PDE3A, PDE4D, PDE7A, PDE8A, and PDE10A. Firstly, 3H-cGMP/3H-cAMP was diluted with the assay buffer (20–50 mmol/L Tris-HCl (pH 8.0), 10 mmol/L MgCl2, and 1 mmol/L DTT) to 20,000–30,000 cpm per assay. The mixture was then performed at 25 °C for 15 min and terminated by the addition of 0.2 mol/L ZnSO4. 0.2 mol/L Ba(OH)2 was added with a precipitate formed. The unreacted 3H-cGMP/3H-cAMP was left in the supernatant. The radioactivity in the supernatant was measured in 2.5 mL of Ultima Gold liquid scintillation cocktail using a PerkinElmer 2910 liquid scintillation counter (PerkinElmer® Tri-carb2910TR, Waltham, USA). For the measurement of IC50 of compounds, eight different concentrations were used, and each measurement was at least repeated three times.
5. Associated content
The atomic coordinates and structure factors have been deposited into the RCSB Protein Data Bank with accession number 7BPI.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21708052, 21877134, 81602955, and 81703341), Science Foundation of Guangzhou City (201904020023, China), Fundamental Research Funds for the Central Universities (Nos. 19ykpy126 and 19ykpy123, China), China Postdoctoral Science Foundation (Nos. 2019M663325 and 2019M663326), Guangdong Province Higher Vocational Colleges & Schools Pearl River Scholar Funded Scheme (2016, China). We appreciate Prof. Hengming Ke at the University of North Carolina, Chapel Hill, for his valuable expertise in the process of molecular cloning, expression, purification, crystal structure, and bioassay of PDEs.
Author contributions
Yuncong Yang conducted methodology and validation. Sirui Zhang investigated in validation. Qian Zhou investigated in methodology. Chen Zhang investigated in methodology and software. Yuqi Gao conducted validation and formal analysis. Hao Wang investigated the study. Zhe Li investigated in software and validation. Deyan Wu and Yinuo Wu validated the data. Yi-You Huang conducted methodology and validation, and writing-reviewed the manuscript. Lei Guo investigated the study, writing-reviewed and edited the manuscript. Hai-Bin Luo supervised the whole study, writing-reviewed and edited the manuscript. All the authors approved the final version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.04.003.
Contributor Information
Yi-You Huang, Email: huangyy287@mail.sysu.edu.cn.
Lei Guo, Email: guolei7@mail.sysu.edu.cn.
Hai-Bin Luo, Email: luohb77@mail.sysu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Xiong P.Y., Potus F., Chan W., Archer S.L. Models and molecular mechanisms of world health organization group 2 to 4 pulmonary hypertension. Hypertension. 2018;71:34–55. doi: 10.1161/HYPERTENSIONAHA.117.08824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M., Lau Edmund M.T., Montani D., Jaïs X., Sitbon O., Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130:2189–2208. doi: 10.1161/CIRCULATIONAHA.114.006974. [DOI] [PubMed] [Google Scholar]
- 3.Benza R.L., Miller D.P., Barst R.J., Badesch D.B., Frost A.E., McGoon M.D. An evaluation of long-term sur-vival from time of diagnosis in pulmonary arterial hypertension from the REVEAL registry. Chest. 2012;142:448–456. doi: 10.1378/chest.11-1460. [DOI] [PubMed] [Google Scholar]
- 4.Humbert M., Sitbon O., Chaouat A., Bertocchi M., Habib G., Gressin V., et al. Survival in patients with idi-opathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 5.Benza R.L., Park M.H., Keogh A., Girgis R.E. Management of pulmonary arterial hypertension with a focus on combination therapies. J Heart Lung Transplant. 2007;26:437–446. doi: 10.1016/j.healun.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 6.Gbekor E., Betheil S., Fawcett L., Mount N., Phillips S. Selectivity of sildenafil and other phosphodiester-ase type 5 (PDE5) inhibitors against all human phosphodiesterase families. Eur Urol Suppl. 2002;1:63. [Google Scholar]
- 7.Mirone V., Fusco F., Rossi A., Sicuteri R., Montorsi F. Tadalafil and vardenafil vs sildenafil: a review of patient-preference studies. BJU Int. 2009;103:1212–1217. doi: 10.1111/j.1464-410X.2008.08267.x. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z., Jiang X., Zhang X., Tian G., Yang R., Wu J., et al. Pharmacokinetics-driven optimization of 4(3H)-pyrimidinones as phosphodiesterase type 5 inhibitors leading to TPN171, a clinical candidate for the treatment of pulmonary arterial hypertension. J Med Chem. 2019;62:4979–4990. doi: 10.1021/acs.jmedchem.9b00123. [DOI] [PubMed] [Google Scholar]
- 9.Tian X., Vroom C., Ghofrani H.A., Weissmann N., Bieniek E., Grimminger F., et al. Phosphodiesterase 10A upregulation contributes to pulmonary vascular remodeling. PLoS One. 2011;6 doi: 10.1371/journal.pone.0018136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H., Liu Y., Hou J., Zheng M., Robinson H., Ke H. Structural insight into substrate specificity of phosphodiesterase 10. Proc Natl Acad Sci U S A. 2007;104:5782–5787. doi: 10.1073/pnas.0700279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakics V., Karran E.H., Boess F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology. 2010;59:367–374. doi: 10.1016/j.neuropharm.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Geerts H., Spiros A., Roberts P. Phosphodiesterase 10 inhibitors in clinical development for CNS disor-ders. Expert Rev Neurother. 2017;17:553–560. doi: 10.1080/14737175.2017.1268531. [DOI] [PubMed] [Google Scholar]
- 13.Hennenberg M., Schott M., Kan A., Keller P., Tamalunas A., Ciotkowska A., et al. Inhibition of adrenergic and non-adrenergic smooth muscle contraction in the human prostate by the phosphodiesterase 10-selective inhibitor TC-E 5005. Prostate. 2016;76:1364–1374. doi: 10.1002/pros.23208. [DOI] [PubMed] [Google Scholar]
- 14.Li N., Lee K., Xi Y., Zhu B., Gary B.D., Ramírez-Alcántara V., et al. Phosphodiesterase 10A: a novel target for selective inhibition of colon tumor cell growth and β-catenin-dependent TCF transcriptional activity. Oncogene. 2014;34:1499–1509. doi: 10.1038/onc.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu B., Lee K., Canzoneri J., Ramirez-Alcantara V., Sigler S., Gary B., et al. Abstract 4372: phos-phodiesterase 10A inhibition suppresses lung tumor cell growth by activating PKG to inhibit ras and Wnt signaling. Cancer Res. 2015;75:4372. [Google Scholar]
- 16.Huang Y.Y., Yu Y.F., Zhang C., Chen Y., Zhou Q., Li Z., et al. Validation of phosphodiesterase-10 as a novel target for pulmonary arterial hypertension via highly selective and subnanomolar inhibitors. J Med Chem. 2019;62:3707–3721. doi: 10.1021/acs.jmedchem.9b00224. [DOI] [PubMed] [Google Scholar]
- 17.Verhoest P.R., Chapin D.S., Corman M., Fonseca K., Harms J.F., Hou X., et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J Med Chem. 2009;52:5188–5196. doi: 10.1021/jm900521k. [DOI] [PubMed] [Google Scholar]
- 18.Jansen C., Kooistra A.J., Kanev G.K., Leurs R., de Esch Ijp, de Graaf C. PDEStrIAn: a phosphodiesterase structure and ligand interaction annotated database as a tool for structure-based drug design. J Med Chem. 2016;59:7029–7065. doi: 10.1021/acs.jmedchem.5b01813. [DOI] [PubMed] [Google Scholar]
- 19.Williams D.H., Stephens E., O'Brien D.P., Zhou M. Understanding noncovalent interactions: ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes. Angew Chem Int Ed Engl. 2004;43:6596–6616. doi: 10.1002/anie.200300644. [DOI] [PubMed] [Google Scholar]
- 20.Varma M.V.S., Obach R.S., Rotter C., Miller H.R., Chang G., Steyn S.J., et al. Physicochemical space for op-timum oral bioavailability: contribution of human intestinal absorption and first-pass elimination. J Med Chem. 2010;53:1098–1108. doi: 10.1021/jm901371v. [DOI] [PubMed] [Google Scholar]
- 21.Niu W., Guo L., Li Y., Shuang S., Dong C., Wong M.S. Highly selective two-photon fluorescent probe for ratiometric sensing and imaging cysteine in mitochondria. Anal Chem. 2016;88:1908–1914. doi: 10.1021/acs.analchem.5b04329. [DOI] [PubMed] [Google Scholar]
- 22.Matharu D.S., Flaherty D.P., Simpson D.S., Schroeder C.E., Chung D., Yan D., et al. Optimization of potent and selective quinazolinediones: inhibitors of respiratory syncytial virus that block RNA-dependent RNA−polymerase complex activity. J Med Chem. 2014;57:10314–10328. doi: 10.1021/jm500902x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baell J.B., Holloway G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z., Tang W. Drug metabolism in drug discovery and development. Acta Pharm Sin B. 2018;8:721–732. doi: 10.1016/j.apsb.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schermuly R.T., Kreisselmeier K.P., Ghofrani H.A., Yilmaz H., Butrous G., Ermert L., et al. Chronic sildena-fil treatment inhibits monocrotaline-induced pulmonary hypertension in rats. Am J Respir Crit Care Med. 2004;169:39–45. doi: 10.1164/rccm.200302-282OC. [DOI] [PubMed] [Google Scholar]
- 26.Wu D., Zhang T., Chen Y., Huang Y., Geng H., Yu Y., et al. Discovery and optimization of chromeno[2,3-c]pyrrol-9(2H)-ones as novel selective and orally bioavailable phosphodiesterase 5 inhibitors for the treatment of pulmonary arterial hypertension. J Med Chem. 2017;60:6622–6637. doi: 10.1021/acs.jmedchem.7b00523. [DOI] [PubMed] [Google Scholar]
- 27.Wu X.N., Huang Y.D., Li J.X., Yu Y.F., Qian Z., Zhang C., et al. Structure-based design, synthesis, and biological evaluation of novel pyrimidinone derivatives as PDE9 inhibitors. Acta Pharm Sin B. 2018;8:615–628. doi: 10.1016/j.apsb.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu D., Huang Y., Chen Y., Huang Y.Y., Geng H., Zhang T., et al. Optimization of chromeno[2,3-c]pyrrol-9(2H)-ones as highly potent, selective, and orally bioavailable PDE5 inhibitors: structure–activity relationship, X-ray crystal structure, and pharmacodynamic effect on pulmonary arterial hypertension. J Med Chem. 2018;61:8468–8473. doi: 10.1021/acs.jmedchem.8b01209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.