

Abstract

Quantifying proteins based on peptide-coupled reporter ions is a multiplexed quantitative strategy in proteomics that alleviates the problem of ratio distortion caused by peptide cofragmentation, as commonly observed in other reporter-ion-based approaches, such as TMT and iTRAQ. Data-independent acquisition (DIA) is an attractive alternative to data-dependent acquisition (DDA) due to its better reproducibility. While multiplexed labeling is widely used in DDA, it is rarely used in DIA, presumably because current approaches lead to more complex MS2 spectra, severe ratio distortion, or to a reduction in quantification accuracy and precision. Herein, we present a versatile acetyl-alanine-glycine (Ac-AG) tag that conceals quantitative information in isobarically labeled peptides and reveals it upon tandem MS in the form of peptide-coupled reporter ions. Since the peptide-coupled reporter ion is precursor-specific while fragment ions of the peptide backbone originating from different labeling channels are identical, the Ac-AG tag is compatible with both DDA and DIA. By isolating the monoisotopic peak of the precursor ion in DDA, intensities of the peptide-coupled reporter ions represent the relative ratios between constituent samples, whereas in DIA, the ratio can be inferred after deconvoluting the peptide-coupled reporter ion isotopes. The proteome quantification capability of the Ac-AG tag was demonstrated by triplex labeling of a yeast proteome spiked with bovine serum albumin (BSA) over a 10-fold dynamic range. Within this complex proteomics background, BSA spiked at 1:5:10 ratios was detected at ratios of 1.00:4.87:10.13 in DDA and 1.16:5.20:9.64 in DIA.
Introduction
Advancements in liquid chromatography and mass spectrometry over the last decades have led to the maturation and widespread application of a variety of mass-spectrometry-based proteome quantification approaches. Label-free quantification is hampered by the effect of changing ionization efficiencies between LC–MS runs and the stochastic nature of precursor ion selection in the data-dependent acquisition (DDA) mode. Multiplex stable isotope labeling circumvents these issues and has been widely applied to label samples derived from different experimental conditions. Analysis of multiple samples labeled with distinct isotopes in a single LC–MS run leads to more reliable quantification. As reviewed by Arul and Robinson,1 isotope-label-based quantification methods can be classified into MS1 quantification, which is usually based on isotopic labeling resulting in different precursor masses of the same peptide derived from different samples, and MS2 quantification, which generally relies on isobaric labeling, resulting in identical precursor masses while quantifying based on fragment ions. Isobaric labeling strategies quantify constituent proteins of mixed samples by distinct fragment ions upon MS/MS without increasing the complexity of the MS1 spectra. These methods significantly improve the accuracy and precision as well as the throughput of quantitative proteomics in the DDA mode.2,3
Isobaric strategies can be further classified into three categories: (1) reporter-ion-based quantification (e.g., TMT2,4,5 and iTRAQ6,7); (2) peptide fragment ion based quantification, including isobaric peptide termini labeling (IPTL)-based approaches;8−10 (3) peptide-coupled reporter-ion-based quantification, like TMTc3,11 and EASI tag.12 Among these, the reporter-ion-based methods (TMT and iTRAQ) are most widely used because of their high multiplexing capacity and well-developed data-processing software. However, it has become increasingly apparent that reporter-ion-based quantification methods suffer from ratio distortion3,11,13,14 caused by peptide cofragmentation in which the produced reporter ions from different peptides are indistinguishable resulting in intensities that no longer reflect the ratios between the peptides and thus lead to inaccurate protein ratios. Various solutions to alleviate the ratio distortion problem have been described, such as narrowing the width of the precursor ion isolation window,3,11 gas-phase purification,14 and MultiNotch MS3,13,15 but these methods are not widely used as they rely on specific, state-of-the-art instrumentation, use scan routines that are difficult to optimize and control, or require MS3 scanning leading to a longer cycle time. Peptide fragment ion based quantification, an approach that is not affected by cofragmentation,8 has thus attracted attention: a growing number of IPTL-based approaches have been reported, such as Triplex-QITL,16 SMD-IPTL,10 and m-pIDL.17 In these methods, every labeling channel has a set of distinct fragment ions resulting in more precise quantification. However, the increased complexity18 of MS2 spectra requires custom-made data analysis tools, which has restricted widespread use. In contrast to the two methods mentioned above, quantifying proteins based on a precursor-specific, peptide-coupled reporter ion is not affected by peptide cofragmentation nor does it increase the complexity of the MS2 spectra. This approach was first proposed by Wühr et al.11 in 2012, but two limitations leading to unsatisfactory quantification accuracy and precision were pointed out: (1) the modest efficiency of forming peptide-coupled reporter ions and (2) the complicated isotope envelope of the peptide-coupled reporter ions. The improved TMTc+ approach3 applied a narrow precursor ion isolation window of 0.4 Th rather than the generally used 2 Th to select the monoisotopic peak, which significantly improved the quantification accuracy. The recently introduced EASI tag12 utilizes a sulfoxide-based tag to increase the formation efficiency of peptide-coupled reporter ions and applied an asymmetric isolation window to specifically isolate the monoisotopic peak to further simplify the isotope envelope of peptide-coupled reporter ions.
Besides the commonly applied DDA strategy, data-independent acquisition (DIA) approaches are becoming increasingly popular because they avoid the stochastic nature of DDA with its bias toward fragmenting higher intensity peptide ions and the resulting missing value problem between runs.19 In contrast to DDA, isolation of precursors for fragmentation in DIA does not rely on the peak intensities in MS1; instead, all precursors present within the isolation window will be fragmented and analyzed together. As a result, the problem of missing values of specific peptides between injections in a sample batch is largely avoided and reproducibility is improved. On the other hand, MS2 spectra are more complex as the fragment ions derived from multiple, coisolated precursor ions are present in the same spectrum, which makes analyzing DIA data more challenging than DDA data. It is worth noting that, fueled by improvements in the scan rate and resolution in mass spectrometry and the development of sophisticated data processing algorithms,19−21 the analysis of DIA data has been improved over the last decade, especially with respect to the reproducibility from run to run.20
However, DIA-based methods are not suitable for most current multiplexing approaches, which makes them inferior in terms of throughput to DDA-based methods, such as those based on TMT or iTRAQ, for which the reporter ions lose their quantification capability due to massive cofragmentation. For isotopic labeling and peptide fragment ion based isobaric labeling strategies, such as stable isotope labeling by amino acids in cell culture22,23 (SILAC) and IPTL, the peptides of different labeling channels have sets of distinct fragments ions, which multiplies the complexity of MS2 spectra with the number of labeled samples making identification of peptides more challenging. The recently published NeuCoDIA24 and MdFDIA25 methods apply neutron-encoding isotope labeling to achieve multiplexed DIA quantification, but they rely on ultrahigh resolution (resolution of MS2 >120k), which is not yet widespread and comes at the cost of a decreasing data acquisition rate. Thus, there is a need for a practical approach that can quantify multiplexed proteomics samples in DIA mode without compromising the data acquisition rate.
To establish a new peptide-coupled reporter-ion-based tag that avoids ratio distortion in DDA and improves the throughput of DIA-based methods without sacrificing the data acquisition rate, we propose a novel isobaric Ac-AG tag, which conceals quantitative information in isobarically labeled peptides and reveals it upon tandem MS in the form of peptide-coupled reporter ions. The ratios between labeling channels are simply reflected by the intensities of the peptide-coupled reporter ions in DDA mode, whereas the ratios can also be deduced from the peptide-coupled reporter ion isotope envelope in DIA mode after deconvolution. The Ac-AG tag is thus the first isobaric tag that can be applied for multiplexed quantitation in both DDA and DIA modes without having to resort to ultrahigh MS2 resolution.
Experimental Section
Details of chemicals and materials, the synthesis of triplex Ac-AG-tags (acetyl-alanine-glycine-p-nitrophenol, Ac-AG-PNP, see Figure 1C), reduction/alkylation and LysC digestion, selective N-terminal dimethylation of peptides, and LC–MS/MS analysis can be found in the Supporting Information.
Figure 1.

Schematic view of the Ac-AG approach. (A) Isobaric labeling steps; (B) LC–MS/MS of a mixture of triplex-labeled samples with DDA or DIA approaches; (C) functional design of the triplex-labeled Ac-AG-PNP tag (13C isotope locations are marked with asterisks).
Triplex Labeling of N-Terminally Dimethylated Peptides
5 μg of N-terminally dimethylated GTDWLANK, LysC peptides of BSA, or LysC peptides of yeast proteins was dissolved in 50 μL of 200 mM triethylammonium bicarbonate (TEAB) buffer of pH 8.5. Then, 4 μL of 50 mM Ac-A-13C2-G-PNP, 13C1-Ac-A-13C1-G-PNP, or 13C2-Ac-AG-PNP in DMF was added to the three peptide solutions, respectively. The reaction mixtures were shaken for 2 h at room temperature. To ensure complete labeling, 2 μL of the respective Ac-AG-PNP reagent was added again and incubated for 1 h more. Any esterification on the hydroxyl groups of Ser, Thr, or Tyr and excess PNP ester was hydrolyzed in the presence of 5% hydroxylamine hydrate at 55 °C for 5 min, and samples were then desalted by SPE using the STAGE (STop And Go Extraction) TIPS Desalting Procedure prior to LC–MS analysis.26 500 μL of 2% acetonitrile in water with 0.1% trifluoroacetic acid (TFA) was added to remove excess Ac-AG-COOH before eluting peptides from the STAGE tips with 60% acetonitrile in water with 0.1% TFA.
Database Searching and Quantification
LC–MS/MS raw files measured in DDA mode were analyzed with PEAKS_X+ (Bioinformatics Solutions Inc., Waterloo, Ontario, Canada) and searched against the Uniprot reference database of yeast (UP000002311, 6049 entries, downloaded on Jan. 20, 2020) into which the BSA entry (P02769) was inserted manually. LysC was selected as the enzyme, digestion mode was specific, and max missed cleavage sites was 0. A tolerance of 20 ppm for the precursor ion and 0.02 Da for the MS/MS fragment ions were applied. Carbamidomethylation (+57.02) on cysteine and dimethylation (+28.03) on the N-terminus were set as fixed modifications and oxidation (+15.99) on methionine as variable modification. For triplex labeling experiments, variable modifications on Lys were set as triplex isobaric Ac-AG tags with two 13C isotopes (+172.08). The results were filtered with a false discovery rate (FDR) of 0.5% for peptides. The filtered results were exported as a spectral library for DIA analysis using the default parameters of PEAKS_X+.
For identification of peptides in DIA, the raw data was searched against the prepared spectral library and the Uniprot reference database of yeast (UP000002311) complemented with BSA was selected as the PEAKS reference database. A tolerance of 20 ppm for the precursor ion and 0.02 Da for the MS/MS fragment ions were applied. The DIA results were also filtered with an FDR of 0.5% for peptides.
The PEAKS output of the peptides matched to proteins (protein-peptides.csv) and the peptide-spectrum matches (DB search psm.csv) were exported for both DDA and DIA searches. All the following steps were performed using in-house built Python scripts (available at https://github.com/tianxiaobo002/Ac-AG-tag_scripts_and_quantification_outputs). Only the unique peptides extracted from the protein-peptides.csv files were used for quantification. For every peptide spectrum match (PSM) that was derived from unique peptides in the DB search psm.csv files, the theoretical peptide-coupled reporter ion was calculated and grouped by scan number. The raw data was converted into a Python readable mgf file with RawConverter.27 All MS/MS spectra were extracted from the resulting mgf file and also grouped by scan number. Afterward, based on the scan number, the measured peak intensities in the mgf file were related to the corresponding theoretical peptide-coupled reporter ions, and the matched intensities were used to quantify peptides and ultimately proteins. The ratios of each PSM, peptide, and protein were calculated based on the peak intensities of the respective labeling channel. For quantification in DDA, the intensities of peptide-coupled reporter ions simply represent the relative quantification information. Examples of calculating ratios at the PSM, peptide, and protein levels can be found in Figure S1. Each peptide ratio was calculated from the three PSMs with the highest total peak intensity, and each protein ratio was calculated from the three peptides with the highest total peak intensity. For quantification in DIA, the only difference from the quantification in DDA is the correction of the 13C contribution in the isotope envelope of peptide-coupled reporter ions before calculating the ratios at the PSM level. Based on the peptide sequence derived from the PSM, the molecular formula is established, and from this, the native isotope envelope distribution of peptide-coupled reporter ions can be calculated and used to correct the 13C contribution. The corrected intensities were used for all the following calculations. Examples of correcting 13C contributions can be found in Figure S2.
The Python scripts and quantification outputs of triplex Ac-AG labeled BSA, yeast, and BSA-yeast samples at the PSM, peptide, and protein levels are available at https://github.com/tianxiaobo002/Ac-AG-tag_scripts_and_quantification_outputs. The raw mass spectrometry proteomics data of yeast and BSA-yeast samples have been deposited to the ProteomeXchange Consortium via the PRIDE28 partner repository with the dataset identifier PXD021187.
Results and Discussion
Design of the Ac-AG Tag and Labeling Strategy
During development of the SMD-IPTL10 method, we observed that the amide bond between the incorporated Ac-Ala and the side chain of the C-terminal Lys residue (produced by LysC protein digestion) fragments prior to the peptide backbone at lower stepped normalized collision energies (NCEs) generating a fragment ion covering the full sequence. We therefore assumed that an Ac-Ala-Gly tag on the side chain of Lys would have similar dissociation features and that the peptide bond at the C-terminal side of Gly would remain stable upon collision-induced dissociation (CID).29−33 Fragmentation between the Gly and Ac-Ala parts would thus generate a peptide-coupled reporter ion at low NCEs, and fragment ions of the peptide backbone would be generated at higher NCEs.12 In this way, the ions required for identification and quantification can be produced in the same MS2 spectrum by applying two NCEs.12 Multiple NCE values can be set in the Orbitrap acquisition software; when more than one value is set, the mass spectrometer will perform a stepwise fragmentation of the precursor ion, and all fragments created in the steps are collected and sent to the Orbitrap analyzer for detection in one scan. Since Ac-Ala does not have a good ionization site, it is expected to give rise to a neutral loss, and the peptide-coupled reporter ion with the attached Gly part from the Ac-AG tag will have the same charge state as the precursor ion.12
With different combinations of commercially available 13C-labeled acetic anhydride, alanine, and glycine, triplex isobaric Ac-AG tags were prepared as shown in Figure 1C and Scheme S1 (Supporting Information). The p-nitrophenol (PNP) ester was selected instead of the more widely used N-hydroxysuccinimide (NHS) ester because it has comparable reactivity with the epsilon amino group of Lys but a much better stability in the presence of water, which is important during purification of the synthesized Ac-AG-PNP tag. As derivatization of the C-terminal Lys with the Ac-AG tag might decrease the peptide ionization efficiency, the N-terminal amino groups of the peptides were dimethylated converting them into tertiary amines, which improves ionization.34−37 Dimethylation has also been reported to improve the completeness of the b-ion series.37 As shown in Figure 1A, the isobaric labeling method includes two steps: (1) selective N-terminal dimethylation38,39 and (2) labeling with the Ac-AG-PNP ester at the epsilon amino group of the C-terminal Lys residue. As there is no isotope label at the N-terminus and y-ions are expected to contain the full Ac-AG tag, all fragment ions of the peptide backbone originating from different labeling channels will have the same respective masses, which means that the complexity of MS2 spectra does not increase with the number of differentially labeled samples. Only the peptide-coupled reporter ions with different isotopic forms of Gly, produced after the loss of the complementarily labeled Ac-Ala part, will have different masses between labeling channels.
The differentially labeled samples are mixed prior to LC–MS/MS analysis, and the data acquisition can be performed in DDA or DIA mode, as shown in Figure 1B. In the analysis with DDA, a narrow precursor isolation window (e.g., 0.6 Th) allows isolation of the monoisotopic peak for fragmentation, and the resulting peptide-coupled reporter ions provide the quantitative readout with their respective intensities representing the relative ratios of the constituent peptides/proteins.3 In the case of DIA, the peptide-coupled reporter ions of different labeling channels form an isotope envelope due to the respective 13C contributions since the entire isotope envelope of the precursor is selected for fragmentation. The quantification information can be inferred from DIA data after deconvolution (see Figure S2 for details).
Derivatization with the Ac-AG Tag at the Peptide Level
To evaluate the efficiency of the labeling steps and investigate the influence of modification with the Ac-AG tag in combination with N-terminal dimethylation on ionization efficiency and charge state distribution, GTDWLANK was selected as the model peptide. FDWA was included as a reference peptide since it cannot react with the Ac-AG tag and will thus only show the effects on ionization efficiency and charge state distribution due to N-terminal dimethylation. By normalizing ionization efficiency with FDWA before and after labeling with the Ac-AG tag, the influence of the Ac-AG tag can be evaluated. The extracted ion chromatograms (XICs) of GTDWLANK, N-terminally dimethylated (N-dime)-GTDWLANK, and N-dime-GTDWLANK-GA-Ac were compared. As shown in Figure S3, GTDWLANK was efficiently converted to N-dime-GTDWLANK-GA-Ac with yields higher than 98% for both selective N-terminal dimethylation and labeling with the Ac-AG tag. We observed that the intensities of both model and reference peptides increased after N-terminal dimethylation and that there was a slight decline in the intensity of the model peptide upon labeling with the Ac-AG tag. However, the intensity was still higher than that of the original unmodified peptide. The charge state distribution of the model peptide before and after labeling with the Ac-AG tag showed that the doubly charged ion remains the most abundant (96.5%) species after modification (Table S1).
Optimizing the Fragmentation Energy for Ac-AG Labeled Peptides
The peptide-coupled reporter ion is used for quantification, and fragment ions of the peptide backbone are required for identification. Formation of both types of fragment ions in sufficient yields is thus a prerequisite for Ac-AG-tag-based quantification. Fragmentation of N-dime-GTDWLANK-GA-Ac was studied at different NCEs ranging from 18 to 32 (Figure S4). At an NCE of 18, the peptide-coupled reporter ion produced from the neutral loss of Ac-Ala was the dominant ion compared to the y- and b-ions from peptide backbone fragmentation (Figure 2A). As the NCE was increased stepwise from 18 to 28, the intensity of the peptide-coupled reporter ion gradually decreased and the ion was no longer detectable at an NCE of 28 (Figure S4). On the contrary, the intensities of ions from peptide backbone fragmentation increased from NCE 18 to 28 but dropped again when the NCE was increased to 32. Based on these results, an NCE of 18 was selected as the energy for producing the peptide-coupled reporter ion and an NCE of 28 for forming b- and y-ion fragments. Figure 2B shows the MS2 spectrum at a combined NCE of 18 and 28.
Figure 2.
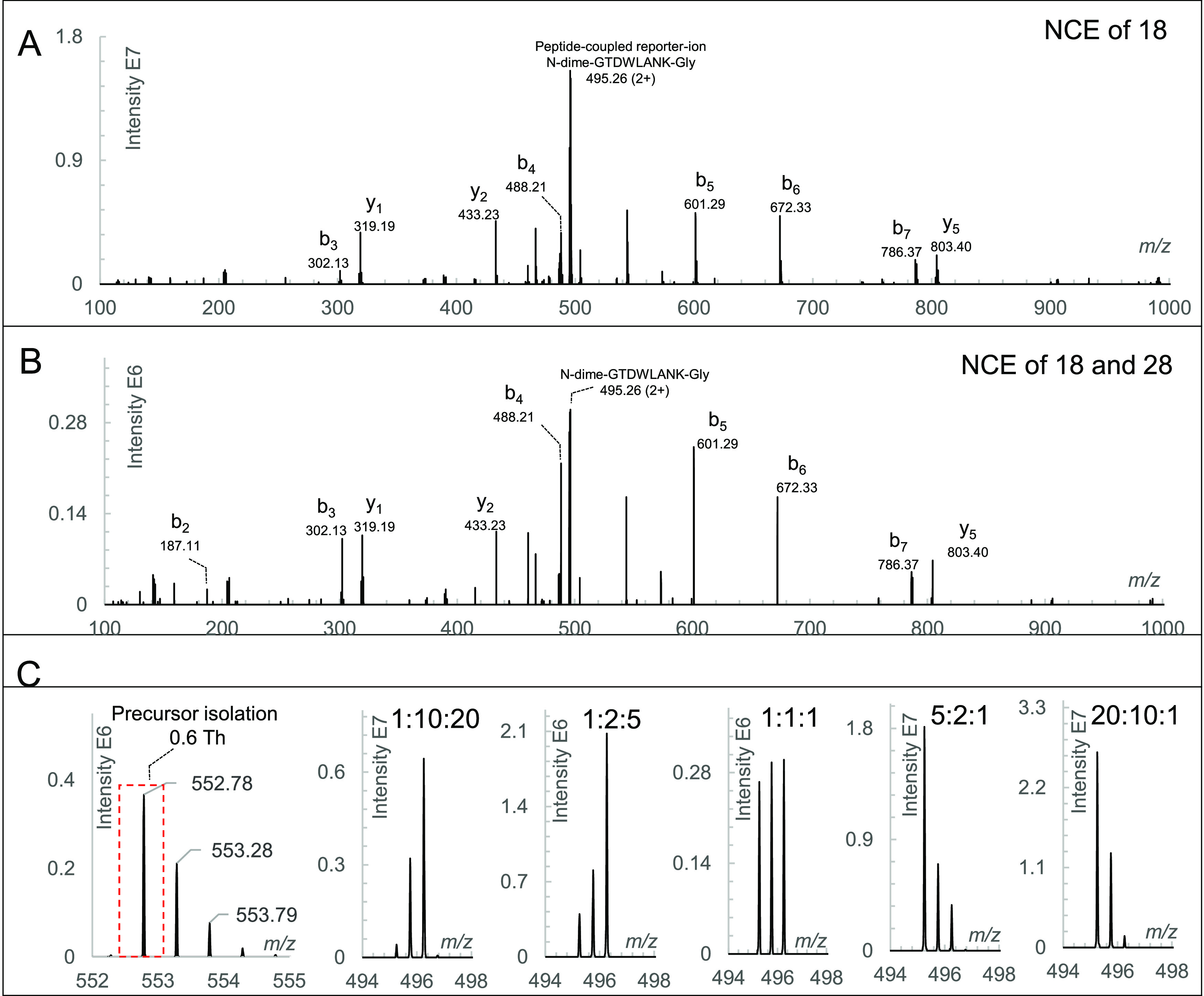
LC–MS/MS analysis of triplex-labeled GTDWLANK. (A) MS2 spectrum of triplex-labeled N-dime-GTDWLANK-GA-Ac with an NCE of 18 acquired in the parallel reaction monitoring mode; (B) MS2 spectrum of triplex-labeled N-dime-GTDWLANK-GA-Ac with a combined NCE of 18 and 28 acquired in the DDA mode; (C) precursor isolation with a window of 0.6 Th and peptide-coupled reporter ions in the DDA MS2 spectra at ratios of 1:10:20, 1:2:5, 1:1:1, 5:2:1, and 20:10:1.
The three labeled forms of N-dime-GTDWLANK-GA-Ac were first analyzed separately and found to have identical fragment ions of the peptide backbone but a distinct peptide-coupled reporter ion (Figure S5). To demonstrate the feasibility of the Ac-AG tag approach, triplex-labeled N-dime-GTDWLANK-GA-Ac peptides were mixed at ratios of 1:1:1, 1:2:5, 1:10:20, 5:2:1, and 20:10:1 followed by LC–MS/MS analysis. The measured ratios were 0.93:1.03:1.04, 0.98:1.98:5.04, 1.29:9.95:19.75, 5.05:1.94:1.02, and 20.15:9.69:1.16, respectively (Figure 2C).
DDA Analysis of Triplex-Labeled LysC Peptides of BSA
Triplex Ac-AG labeled N-terminally dimethylated LysC peptides of BSA were prepared, mixed at various ratios, and analyzed with LC–MS/MS in DDA mode with a narrow, 0.6 Th, precursor isolation window. To investigate the effect of different fragmentation energies on identification, NCEs of 24, 26, 28, and 30 were evaluated and the PSMs were counted to establish the optimal NCE for forming fragments of the peptide backbone. As shown in Figure S6, an NCE of 28 produced more PSMs than other NCEs, which is consistent with the result of N-dime-GTDWLANK-GA-Ac. As shown in Figure 3A, the log2-normalized ratios at the PSM level of the triplex-labeled BSA sample mixed at a ratio of 1:1:1 converge to zero for a more intense PSM. The corresponding peptide ratios were calculated based on the three PSMs with the highest total peak intensity. As shown in Figure 3B, the log2-normalized peptide ratios of the triplex-labeled BSA sample mixed at a ratio of 1:1:1 also converge to zero. Likewise, the protein ratio can be calculated from the three peptides with the highest total peak intensity. The ratio of the 1:1:1 mixed BSA sample was determined to be 0.93:1.07:1 at the protein level. Calculation details at the PSM, peptide, and protein levels can be found in Figure S1. Based on the medians of the log2-normalized peptide ratios, the 10:5:1 and 1:5:10 mixed triplex-labeled BSA samples were determined to be 9.51:5.24:1.24 and 1.16:5.03:9.78, as shown in Figure 3C,D, respectively.
Figure 3.

Analysis of Ac-AG triplex-tagged BSA-derived LysC peptides. (A) Log2-normalized ratio distribution at the PSM level at a mixing ratio of 1:1:1; (B) log2-normalized ratio distribution at the peptide level at a mixing ratio of 1:1:1; (C) log2-normalized ratio at the peptide level at a mixing ratio of 10:5:1; (D) log2-normalized ratio at the peptide level at a mixing ratio of 1:5:10. Expected values for log2-normalized mixing ratios are shown as dotted lines.
DDA Analysis of Triplex-Labeled LysC Peptides of the Yeast Proteome
To confirm the efficiency and reproducibility of the isobaric labeling steps and the quantification accuracy in complex samples, triplex-labeled LysC peptides of yeast proteins were prepared. An evaluation of the efficiency and reproducibility of selective N-terminal dimethylation on the proteome scale was reported in our previous work:40 for N-terminal dimethylation, more than 98% of LysC yeast peptides had a labeling yield exceeding 95%. To assess the labeling yield of the Ac-AG tag at the C-terminal Lys, Ac-AG was set as variable modification in the database search to simultaneously identify Ac-AG labeled peptides and peptides without the Ac-AG modification. Assuming that the peptides with and without modification have comparable ionization efficiencies (as shown in Figure S3), the Ac-AG labeling efficiency can be calculated, and more than 99% of all peptides had a labeling yield higher than 98%. Moreover, only 50 proteins were identified when no Ac-AG modification was selected, and most of them were identified by the C-terminal peptide of the protein. The charge state distributions of unlabeled LysC yeast peptides and N-dime-LysC peptides-GA-Ac were compared showing that the doubly charged peptide precursor ions remain most abundant after labeling with N-terminal dimethylation and the Ac-AG tag (Figure S7); the number of identified peptides remains comparable. Around 90% (1784 out of the 1996) of PSMs had peptide-coupled reporter ions with a complete isotope envelope, which can be used for quantification. Around 96% (9835 out of the 10,255) matched y-ions ions did not show the neutral loss due to fragmentation of the Ac-AG tag. The log2-normalized protein ratios of the triplex-labeled LysC yeast sample mixed at a ratio of 1:1:1 are shown in Figure 4A. Based on the medians of log2-normalized protein ratios (0.03, 0.01, and 0.01), the medians of measured ratios of the 1:1:1 mixed yeast sample were measured to be 1.02:1.00:1.00. The three channels have standard deviations of measured ratios of 0.12, 0.11, and 0.12, respectively.
Figure 4.

Analysis of Ac-AG tagged peptides from the LysC-digested yeast proteome. (A) Log2-normalized protein ratio distribution of triplex-labeled LysC yeast peptides mixed at a ratio of 1:1:1; (B) log2-normalized protein ratio distribution of triplex-labeled LysC yeast peptides mixed at a ratio of 1:2:5 combined with LysC BSA peptides mixed at a ratio of 1:5:10. Expected values for log2-normalized mixing ratios are shown as dotted lines.
Quantifying BSA in a Background of Yeast Proteins in DDA Mode
To examine the quantification capabilities and dynamic range of the Ac-AG tag in a complex background, a BSA-yeast proteomics sample consisting of triplex-labeled LysC-digested BSA mixed at 1:5:10 and triplex-labeled LysC yeast peptides mixed at 1:2:5 was prepared (Figure S8A) followed by LC–MS/MS analysis. In the BSA-yeast sample, 660 proteins were identified and 600 proteins were quantified by the peptide-coupled reporter ions. Around 60 identified proteins could not be quantified due to the low quality of PSMs lacking a complete isotope envelope of peptide-coupled reporter ions or incorrect peptide assignments. As shown in Figure 4B, the medians of log2-normalized protein ratios of the LysC yeast sample mixed at a ratio of 1:2:5 were determined to be 0.00, 1.05, and 2.30, which are close to the expected values of 0.00, 1.00, and 2.32, respectively. For the triplex-labeled BSA sample mixed at a ratio of 1:5:10, the measured ratio was determined to be 1.00:4.87:10.13.
Exploiting the Ac-AG Tag for Multiplex Quantification in the DIA Mode
The recently reported DIA multiplexed approaches, NeuCoDIA24 and MdFDIA25 incorporate isotopes by cell culture, while the mdDiLeu approach uses chemical labeling.41 These methods employ neutron encoding,42 which makes them rely on ultrahigh resolution (>120k) at the fragment ion level with, as a consequence, a reduced data acquisition rate. In contrast, the Ac-AG tag can be used to chemically label peptides derived from any type of sample, and the resulting peptide-coupled reporter ion can be properly differentiated with a resolution of 17.5k for the MS2 spectra.
In contrast to the DDA mode with a narrow precursor ion isolation window, wide isolation windows are used in DIA, meaning that the complete isotope envelope of precursor ions is selected for fragmentation. As a result, the isotope envelopes of peptide-coupled reporter ions of adjacent labeling channels overlap due to the contribution of heavier (13C) isotopes. An isolation window of 29 Th was applied with a 2 Th overlap between adjacent isolation windows to capture the entire isotope envelope of a given precursor ion in at least one isolation window. As shown in Figure 5A, the complete isotope envelope was observed for the peptide-coupled reporter ion of N-dime-GTDWLANK-GA-Ac (+2), as well as for different mixing ratios of triplex-labeled N-dime-GTDWLANK-GA-Ac. The intensities of the individual isotopic peaks of the isotope envelope of the peptide-coupled reporter ion contain the information that is needed for quantification. Different from DDA, it is necessary to deconvolve the overlapping isotopic envelopes of peptide-coupled reporter ions from the different labeling channels prior to using the corrected intensities of the monoisotopic peaks for ratio calculations at the PSM level. The peptide sequence, determined based on the PSM, is converted to the corresponding molecular formula of the peptide-coupled reporter ion allowing calculation of its native isotope envelope distribution. This distribution is subsequently used to correct the intensity of the respective monoisotopic peaks (see Figure S2 for further details). The corrected intensities of all monoisotopic peaks were used for the following ratio calculations at the PSM, peptide, and protein levels.
Figure 5.

Analysis of triplex-labeled N-dime-GTDWLANK-GA-Ac (A) and BSA-yeast in the DIA mode (B). (A) From left to right, the peptide-coupled reporter ion of a single channel of the precursor ion of N-dime-GTDWLANK-GA-Ac (+2) and the peptide-coupled reporter ions in the DIA MS2 spectra at ratios of 1:2:5, 5:2:1, 1:10:20, and 20:10:1; (B) analysis of the BSA-yeast sample in the DIA mode. Log2-normalized protein ratio distribution of triplex-labeled LysC yeast peptides mixed at a ratio of 5:2:1 combined with LysC BSA peptides mixed at a ratio of 1:5:10. Expected values for log2-normalized mixing ratios are shown as dotted lines.
To determine whether the DIA approach gives accurate quantitative results, we analyzed samples of triplex-labeled LysC peptides of BSA mixed at ratios of 1:1:1, 1:2:5, 1:10:20, 5:2:1, and 20:10:1 in DIA mode with a 29 Th isolation window. All the measured ratios were close to the original mixing ratios, as shown in Figure S9. We subsequently analyzed a BSA-yeast proteome sample consisting of triplex-labeled LysC-digested BSA mixed at a ratio of 1:5:10 and triplex-labeled LysC yeast peptides mixed at a ratio of 5:2:1 (Figure S8B). Using the spectral library established based on identification results from the previous DDA analysis, 450 proteins were identified and 416 proteins were quantified. As shown in Figure 5B, based on the medians (2.31, 0.97, and 0.12) of log2-normalized protein ratios, the medians of measured ratios of the 5:2:1 mixed yeast sample were determined to be 4.96:1.96:1.09. For the triplex-labeled BSA sample mixed at a ratio of 1:5:10, the measured ratio was determined to be 1.16:5.2:9.64.
Based on the aforementioned results, we believe that the Ac-AG tag is an attractive alternative of peptide-coupled reporter ion quantitation to TMTc3 and EASI tag.12 Although the Ac-AG tag exhibits good quantification capability across a 10-fold dynamic range in both DDA and DIA modes, there is still room for improvement: (1) the labeling capacity; even though the capacity can likely be increased with commercially available isotopically labeled amino acids, it compares unfavorably to the 16-plex that can be achieved with TMTpro,2 although TMTpro cannot be used in DIA mode; (2) the width of the precursor isolation window in DDA mode; the currently used isolation window of 0.6 Th is not small enough to clearly isolate the monoisotopic peak from a triply charged or quadruply charged precursor ion where the intervals between isotopic peaks are 0.33 and 0.25, respectively. So, for quantifying triply or quadruply charged peptides, isotope cluster deconvolution is also required. (3) While the Ac-AG tag exhibits good accuracy and precision across a 10-fold dynamic range in DIA mode, it may suffer from overlapping isotope envelopes of peptide-coupled reporter ions in cases where m/z values are very close to each other. Our results indicate that such situations did not occur frequently. Obtaining accurate peptide ratios in such cases depends on (a) increasing the resolution at the MS2 level at the expense of longer cycle times and (b) developing more advanced deconvolution algorithms that do not assume that the observed isotope envelopes are derived from single peptide-coupled reporter ions.
Conclusions
We describe an isobaric labeling approach based on the Ac-AG tag that maintains the advantages of existing peptide-coupled reporter ion-based quantification methods in DDA but also allows multiplexing in the DIA mode without sacrificing the rate of data acquisition and complicating MS2 spectra. While the labeling capacity is currently limited to triplex, it is potentially expandable with commercially available isotopically labeled amino acids. A combined NCE of 18 and 28 was used during data acquisition to generate both the peptide-coupled reporter ion for quantification and the peptide backbone fragment ions for identification in the same MS2 spectrum. The proteome quantification capabilities in DDA and DIA were demonstrated by triplex labeling of a yeast proteome spiked with BSA over a 10-fold dynamic range. The Ac-AG tag is easily prepared and makes multiplexed quantification in both DDA and DIA modes accessible to a wider community of proteome researchers.
Acknowledgments
We gratefully acknowledge the China Scholarship Council (CSC) for a Ph.D. fellowship to X.T. X.T. thanks Jos Hermans for helping with the LC–MS analyses. This work is partially funded by the Open Technology Programme of Toegepaste en Technische Wetenschappen (TTW) with project number 15230, which is financed by the Netherlands Organisation for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03858.
Chemicals and materials, synthesis of isobarically labeled acetyl-alanine-glycine-p-nitrophenol ester, reduction/alkylation and LysC digestion, selective N-terminal dimethylation of peptides, LC–MS/MS analysis, Figure S1. Examples of calculating ratios for PSMs, peptides, and proteins, Figure S2. Correcting the 13C contribution in the peptide-coupled reporter ion, Figure S3. Derivatization of GTDWLANK, Figure S4. MS2 spectra of triplex labeled N-dime-GTDWLANK-GA-Ac (mixed at 1:1:1) at various NCEs, Figure S5. Separate analysis of the three channels of triplex isobarically labeled N-dime-GTDWLANK-GA-Ac, Figure S6. Number of peptide spectral matches (PSMs) of N-dime-BSA peptides-GA-Ac at NCEs of 24, 26, 28, and 30, Figure S7. Charge state distributions of unlabeled and Ac-AG labeled LysC yeast peptides, Figure S8. Scheme of preparing the BSA-yeast proteomics sample, Figure S9. Triplex-labeled LysC BSA mixed at various ratios and analyzed in the DIA mode, Figure S10. LC–MS/MS of the triplex Ac-AG-PNP tag, Scheme S1. Synthesis approach for the triplex Ac-AG-PNP tags, Table S1. Charge state distribution of GTDWLANK before and after labeling with the Ac-AG tag (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Arul A. B.; Robinson R. A. S. Sample Multiplexing Strategies in Quantitative Proteomics. Anal. Chem. 2019, 91, 178–189. 10.1021/acs.analchem.8b05626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.; Wölmer N.; Koncarevic S.; Selzer S.; Böhm G.; Legner H.; Schmid P.; Kienle S.; Penning P.; Höhle C.; Berfelde A.; Martinez-Pinna R.; Farztdinov V.; Jung S.; Kuhn K.; Pike I. TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Anal. Chem. 2019, 91, 15941–15950. 10.1021/acs.analchem.9b04474. [DOI] [PubMed] [Google Scholar]
- Sonnett M.; Yeung E.; Wühr M. Accurate, Sensitive, and Precise Multiplexed Proteomics Using the Complement Reporter Ion Cluster. Anal. Chem. 2018, 90, 5032–5039. 10.1021/acs.analchem.7b04713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.; Schäfer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Neumann T.; Johnstone R.; Mohammed A. K.; Hamon C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Dayon L.; Hainard A.; Licker V.; Turck N.; Kuhn K.; Hochstrasser D. F.; Burkhard P. R.; Sanchez J. C. Relative Quantification of Proteins in Human Cerebrospinal Fluids by MS/MS Using 6-Plex Isobaric Tags. Anal. Chem. 2008, 80, 2921–2931. 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- Ross P. L.; Huang Y. N.; Marchese J. N.; Williamson B.; Parker K.; Hattan S.; Khainovski N.; Pillai S.; Dey S.; Daniels S.; Purkayastha S.; Juhasz P.; Martin S.; Bartlet-Jones M.; He F.; Jacobson A.; Pappin D. J. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Ow S. Y.; Cardona T.; Taton A.; Magnuson A.; Lindblad P.; Stensjö K.; Wright P. C. Quantitative Shotgun Proteomics of Enriched Heterocysts from Nostoc sp. PCC 7120 Using 8-Plex Isobaric Peptide Tags. J. Proteome Res. 2008, 7, 1615–1628. 10.1021/pr700604v. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Strozynski M.; Kozielski F.; Treumann A.; Thiede B. Isobaric Peptide Termini Labeling for MS/MS-Based Quantitative Proteomics. J. Proteome Res. 2009, 8, 4333–4341. 10.1021/pr900425n. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Arntzen M. O.; Strozynski M.; Treumann A.; Thiede B. Isobaric Peptide Termini Labeling Utilizing Site-Specific N-Terminal Succinylation. Anal. Chem. 2011, 83, 4775–4781. 10.1021/ac200229w. [DOI] [PubMed] [Google Scholar]
- Tian X.; de Vries M. P.; Visscher S. W. J.; Permentier H. P.; Bischoff R. Selective Maleylation-Directed Isobaric Peptide Termini Labeling for Accurate Proteome Quantification. Anal. Chem. 2020, 92, 7836–7844. 10.1021/acs.analchem.0c01059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wühr M.; Haas W.; McAlister G. C.; Peshkin L.; Rad R.; Kirschner M. W.; Gygi S. P. Accurate Multiplexed Proteomics at the MS2 Level Using the Complement Reporter Ion Cluster. Anal. Chem. 2012, 84, 9214–9221. 10.1021/ac301962s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S. V.; Meier F.; Wichmann C.; Cox J.; Mann M.; Meissner F. EASI-tag enables accurate multiplexed and interference-free MS2-based proteome quantification. Nat. Methods 2018, 15, 527–530. 10.1038/s41592-018-0037-8. [DOI] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger C. D.; Lee M. V.; Hebert A. S.; McAlister G. C.; Phanstiel D. H.; Westphall M. S.; Coon J. J. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 2011, 8, 933–935. 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Nusinow D. P.; Jedrychowski M. P.; Wuhr M.; Huttlin E. L.; Erickson B. K.; Rad R.; Haas W.; Gygi S. P. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem. 2014, 86, 7150–7158. 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Yin H.; Xie L.; Zhang Y.; Zhang L.; Yang P. Y.; Lu H. A novel triplex isobaric termini labeling quantitative approach for simultaneously supplying three quantitative sources. Anal. Chim. Acta 2018, 1001, 70–77. 10.1016/j.aca.2017.11.004. [DOI] [PubMed] [Google Scholar]
- Liu J.; Zhou Y.; Shan Y. C.; Zhao B.; Hu Y.; Sui Z. G.; Liang Z.; Zhang L.; Zhang Y. A Multiplex Fragment-Ion-Based Method for Accurate Proteome Quantification. Anal. Chem. 2019, 91, 3921–3928. 10.1021/acs.analchem.8b04806. [DOI] [PubMed] [Google Scholar]
- Xie L.-Q.; Zhang L.; Nie A.-Y.; Yan G.-Q.; Yao J.; Zhang Y.; Yang P.-Y.; Lu H.-J. ITMSQ: A software tool for N- and C-terminal fragment ion pairs based isobaric tandem mass spectrometry quantification. Proteomics 2015, 15, 3755–3764. 10.1002/pmic.201400513. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Navarro P.; Tate S.; Röst H.; Selevsek N.; Reiter L.; Bonner R.; Aebersold R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11, O111.016717. 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M.; Finney G.; Hoopmann M. R.; Merrihew G.; Toth M. J.; MacCoss M. J. Deconvolution of Mixture Spectra from Ion-Trap Data-Independent-Acquisition Tandem Mass Spectrometry. Anal. Chem. 2010, 82, 833–841. 10.1021/ac901801b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. C.; Gillet L. C.; Rosenberger G.; Röst H. L.; Vichalkovski A.; Gstaiger M.; Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat. Methods 2013, 10, 1246–1253. 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- Jiang H.; English A. M. Quantitative analysis of the yeast proteome by incorporation of isotopically labeled leucine. J. Proteome Res. 2002, 1, 345–350. 10.1021/pr025523f. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Minogue C. E.; Hebert A. S.; Rensvold J. W.; Westphall M. S.; Pagliarini D. J.; Coon J. J. Multiplexed Quantification for Data-Independent Acquisition. Anal. Chem. 2015, 87, 2570–2575. 10.1021/ac503593d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Y.; Zhang Y.; Zhang L.; Tao T.; Lu H. MdFDIA: A Mass Defect Based Four-Plex Data-Independent Acquisition Strategy for Proteome Quantification. Anal. Chem. 2017, 89, 10248–10255. 10.1021/acs.analchem.7b01635. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Ishihama Y.; Mann M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- He L.; Diedrich J.; Chu Y. Y.; Yates J. R. III Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal. Chem. 2015, 87, 11361–11367. 10.1021/acs.analchem.5b02721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yılmaz Ş.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2018, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A. R.; Jones J. L.; Somogyi A.; Wysocki V. H. Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. J. Am. Chem. Soc. 1996, 118, 8365–8374. 10.1021/ja9542193. [DOI] [Google Scholar]
- Mák M.; Mezö G.; Skribanek Z.; Hudecz F. Stability of Asp-Pro bond under high and low energy collision induced dissociation conditions in the immunodominant epitope region of Herpes simplex virion glycoprotein D. Rapid Commun. Mass Spectrom. 1998, 12, 837–842. . [DOI] [PubMed] [Google Scholar]
- Hogan J. M.; McLuckey S. A. Charge state dependent collision-induced dissociation of native and reduced porcine elastase. J. Mass Spectrom. 2003, 38, 245–256. 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Triscari J. M.; Pasa-Tolic L.; Anderson G. A.; Lipton M. S.; Smith R. D.; Wysocki V. H. Dissociation Behavior of Doubly-Charged Tryptic Peptides: Correlation of Gas-Phase Cleavage Abundance with Ramachandran Plots. J. Am. Chem. Soc. 2004, 126, 3034–3035. 10.1021/ja038041t. [DOI] [PubMed] [Google Scholar]
- Tiwary S.; Levy R.; Gutenbrunner P.; Salinas Soto F.; Palaniappan K. K.; Deming L.; Berndl M.; Brant A.; Cimermancic P.; Cox J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519–525. 10.1038/s41592-019-0427-6. [DOI] [PubMed] [Google Scholar]
- Hsu J. L.; Huang S. Y.; Chow N. H.; Chen S. H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75, 6843–6852. 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- Guo K.; Ji C.; Li L. Stable-Isotope Dimethylation Labeling Combined with LC–ESI MS for Quantification of Amine-Containing Metabolites in Biological Samples. Anal. Chem. 2007, 79, 8631–8638. 10.1021/ac0704356. [DOI] [PubMed] [Google Scholar]
- Cho K.-C.; Kang J. W.; Choi Y.; Kim T. W.; Kim K. P. Effects of peptide acetylation and dimethylation on electrospray ionization efficiency. J. Mass Spectrom. 2016, 51, 105–110. 10.1002/jms.3723. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Li L. De Novo Sequencing of Neuropeptides Using Reductive Isotopic Methylation and Investigation of ESI QTOF MS/MS Fragmentation Pattern of Neuropeptides with N-Terminal Dimethylation. Anal. Chem. 2005, 77, 7783–7795. 10.1021/ac051324e. [DOI] [PubMed] [Google Scholar]
- Qin H.; Wang F.; Zhang Y.; Hu Z.; Song C.; Wu R.; Ye M.; Zou H. Isobaric cross-sequence labeling of peptides by using site-selective N-terminus dimethylation. Chem. Commun. 2012, 48, 6265–6267. 10.1039/c2cc31705b. [DOI] [PubMed] [Google Scholar]
- Koehler C. J.; Arntzen M. O.; de Souza G. A.; Thiede B. An Approach for Triplex-Isobaric Peptide Termini Labeling (Triplex-IPTL). Anal. Chem. 2013, 85, 2478–2485. 10.1021/ac3035508. [DOI] [PubMed] [Google Scholar]
- Tian X.; de Vries M. P.; Permentier H. P.; Bischoff R. A Collision-Induced Dissociation Cleavable Isobaric Tag for Peptide Fragment Ion-Based Quantification in Proteomics. J. Proteome Res. 2020, 19, 3817–3824. 10.1021/acs.jproteome.0c00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X.; Frost D. C.; Yu Q.; Li M.; Gu T.-J.; Li L. Mass Defect-Based DiLeu Tagging for Multiplexed Data-Independent Acquisition. Anal. Chem. 2020, 92, 11119–11126. 10.1021/acs.analchem.0c01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert A. S.; Merrill A. E.; Bailey D. J.; Still A. J.; Westphall M. S.; Strieter E. R.; Pagliarini D. J.; Coon J. J. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 2013, 10, 332–334. 10.1038/nmeth.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.