

Abstract
Background:
Previous studies have shown that α7 nicotinic acetylcholine receptor (nAChR) has a critical role in the regulation of pain sensitivity and neuroinflammation. However, pharmacological effects of α7 nAChR activation in the hippocampus on neuroinflammatory mechanisms associated with allodynia and hyperalgesia remain unknown. We have determined the effects of 3a,4,5,9b-tetrahydro-4-(1-naphthalenyl)-3H-cyclopentan[c]quinoline-8-sulfonamide (TQS), an α7 nAChR positive allosteric modulator, on lipopolysaccharide (LPS)-induced allodynia and hyperalgesia in mice. We also evaluated the effects of TQS on immunoreactivity of microglial marker ionized-calcium binding adaptor molecule 1 (Iba-1), phospho-nuclear factor-κB (p-NF-κB p65), tumor necrosis factor-alpha (TNF-α), and norepinephrine (NE) level.
Methods:
Mice were treated with (0.25, 1 or 4 mg/kg, ip) followed by LPS (1 mg/kg, ip) administration. Allodynia and hyperalgesia were determined using von Frey filaments and hot plate respectively. Immunoreactivity of Iba-1, p-NF-κB p65, and TNF-α, were measured in the hippocampus using immunofluorescence assay. Hippocampal NE level was evaluated using high performance liquid chromatography.
Results:
LPS administration resulted in allodynia and hyperalgesia in mice after six h. Systemic administration of TQS prevented LPS-induced allodynia and hyperalgesia. TQS pretreatment significantly decreased the immunoreactivity of Iba-1, p-NF-κB, and TNF-α in CA1 and DG regions of the hippocampus. In addition, TQS reversed LPS-induced NE reduction in the hippocampus.
Conclusions:
Taken together, our results suggest that TQS prevented LPS-induced allodynia and hyperalgesia, upregulation of TNF-α expression and NE level reduction involving microglial α7 nAChR in part in the hippocampus. Therefore, these findings highlight the important effects of α7 nAChR allosteric modulator against symptoms of inflammatory pain.
Keywords: Nicotinic receptor, Pain, Microglia, TNF-α, Hippocampus
Introduction
Inflammatory pain is characterized by painful sensation due to non-noxious stimuli (allodynia) and increased response to painful sensation (hyperalgesia) [1–3]. Several factors mediate pain sensitivity linked to inflammation such as immune cells, enzymes, chemokines, cytokines, and neurotransmitters [4]. Currently, the conventional pain relieving drugs, including anticonvulsants, antidepressants, opioids, and sodium channel blockers have limited pharmacological efficacy associated with numerous central nervous system (CNS) related adverse effects in managing inflammatory pain [5]. Thus, it is necessary to explore novel CNS targets for the effective treatment of inflammatory pain.
Emerging evidence suggests that α7 nicotinic acetylcholine receptors (nAChRs) have an important role in modulating the pain and inflammation [6–9]. The α7 nAChR exhibits orthosteric and allosteric binding sites that are targeted by diverse ligands [10]. Although different ligands acting at α7 nAChR show anti-inflammatory and antinociceptive effects [7,8,11,12], the receptor could undergo desensitization when it responds to high concentration of the ligands acting onto orthosteric site. However, the ligands binding onto allosteric site such as positive allosteric modulators (PAMs) exhibit a greater action compared to compounds acting on orthosteric site [10,13]. Therefore, PAMs are considered an alternative pharmacological tool that enhances the activity of α7 nAChRs. Beside neuronal α7 nAChRs, immune cells also express α7 nAChRs that have reported to have anti-inflammatory features [9]. Moreover, microglia, well known immune cells, widely distributed in the CNS is implicated in inflammatory pain [14]. The α7 nAChRs expressed by microglia were suggested to modulate microglial activation and inflammation [9,12,15–17]. However, anti-inflammatory effects of the ligands activating α7 nAChRs associated with antinociceptive effects were shown primarily in the spinal cord in rodent models of chronic or inflammatory pain [7,8,11]. Since the pain processing and maintenance occurs in the brain [18], the ani-inflammatory properties of α7 nAChR PAMs in the specific brain region during pain-like symptoms associated with inflammation are not well understood.
Preclinical and clinical studies have revealed that the hippocampus, a limbic brain region, plays a critical role in processing and maintenance of pain signals [19–22]. Previous reports have indicated that sustained elevated level of tumor necrosis factor-alpha (TNF-α), a proinflammatory cytokine produced primarily by microglia, in the hippocampus induces inflammatory pain-like symptoms in animal models [14,23–25]. Furthermore, intracerebroventricular injection of recombinant TNF-α induces inflammatory pain-like symptoms [24,26,27]. Thus, increasing TNF-α results in pain hypersensitivity [28] and the TNF-α was suggested to impact the adrenergic system by decreasing norepinephrine (NE) level in the hippocampus as another contributor for developing pain associated with inflammation [4].
The purpose of present study was to investigate the effects of 3a,4,5,9b-tetrahydro-4-(1-naphthalenyl)-3H-cyclopentan[c]quinoline-8-sulfonamide (TQS), α7 nAChR positive allosteric modulator (PAM), in mice treated with lipopolysaccharide (LPS). This animal model was selected because systemic administration of LPS leads to an elevation in nociception and inflammatory process in the CNS [29–31]. We hypothesized that the changes in allodynia and hyperalgesia would be correlated with changes in relevant inflammatory markers such as ionized calcium-binding adapter molecule 1 (Iba-1, a microglial maker), phosphor-nuclear factor-κB (p-NF-κB p65), and TNF-α that interrupts NE neurotransmission in the hippocampus. Therefore, the effects of TQS on Iba-1, NF-κB, TNF-α and NE in the hippocampus were evaluated during LPS administration.
Materials and methods
Animals
Male C57BL/6 J mice obtained from Jackson Laboratory (Bar Harbor, ME, USA) were housed four per cage (29 × 18 × 12 cm). Standard laboratory conditions were followed for animals (relative humidity 50–60%, 22 ± 2 °C) with a 12-h light/dark cycle and water and food ad libitum. Mice (10–12 weeks old) were undergone to behavioral experiments between 09.00 and 16.00 h in a blind manner regarding to drug treatment. All methods and experiments were following the National Institutes of Health guidelines for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee at South Dakota State University. Good Laboratory Practice was performed based on ARRIVE guidelines. Limited animals suffering was totally attempted.
Drugs and chemicals
LPS isolated from Escherichia coli(serotype 055:B5; Sigma-Aldrich, St. Louis, MO, USA) and methyllycaconitine (MLA; Sigma-Aldrich) were dissolved in physiological saline (0.9% sodium chloride). TQS (Tocris Bioscience, Ellisville, MO, USA) was reconstituted in physiological saline containing 1% dimethyl sulfoxide (DMSO) and 0.5% tween 80. All chemicals were prepared freshlyand administered intraperitoneally.
Experimental protocol
In experiment 1, mice were randomized into six groups: three control groups [vehicle and three groups treated with different doses of TQS (0.25, 1 or 4 mg/kg)] and four LPS groups [vehicle + LPS and three groups treated with the same previous doses of TQS + LPS]. In experiment 2, mice were randomized into five groups: control, MLA (3 mg/kg), LPS, TQS (4 mg/kg) + LPS, and MLA + TQS (4 mg/kg) + LPS. Tactile allodynia and thermal hyperalgesia showed highest peak six h after intraperitoneal administration of LPS (1 mg/kg) as described previously [16,29]. TQS was given 30 min before LPS administration. Animals received MLA 10 min before TQS injection. Equal volume of vehicle (saline + 1% DMSO + 0.5% tween 80) was administered to control mice. The doses for TQS and MLA were selected based on previous reports [16,32]. The mice were sacrificed and, their brain tissues collected six h after LPS administration.
Tactile allodynia
Tactile allodynia was evaluated by measuring 50% paw withdrawal threshold six h after LPS administration as described previously [16] using series of calibrated von Frey filaments (Stoelting, Inc., Wood Dale, IL, USA) and wire mesh boxes (Stoelting, Inc., Wood Dale, IL, USA). Briefly, different filaments of various forces starting with 0.16 g buckling weight were applied vertically on plantar surface of hind paw. Hind paw lifting was considered as a positive score, and the next filament was then applied with light force for second value. If paw withdrawal did not occur within 5 s for second time, the next greater force filament was then applied. The resulting series of positive/negative measurements were used to calculate paw withdrawal threshold.
Thermal hyperalgesia
Thermal hyperalgesia was evaluated by measuring the latency time six h after LPS administration as described previously [16] using analgesia meter (IITC Life Science Inc., Woodland Hills, CA, USA). Briefly, latency time was quantified using heat source to induce thermoreceptors exhibited by foot. Each animal was exposed to hot plate maintained at 54.0 ± 0.1 °C. Thirty s was chosen as an endpoint to avoid tissue harm.
Immunofluorescence assay
Immunofluorescence assay was conducted as described previously [15]. Briefly, 14 μM coronal hippocampal tissue slices were washed with phosphate-buffered saline (PBS). The tissues were retrieved using 0.01 Mcitrate buffer (pH 6.0) heated at 90 °C in a water bath for 10 min. Hippocampal tissue sections were then blocked and incubated with primary antibody against Iba-1 (Wako, Osaka, Japan), p-NF-κB p65 (Cell Signaling Technology, Danvers, MA, USA), and TNF-α (Santa Cruz Biotech, Dallas, TX, USA) overnight at 4 °C. Secondary antibody labeled with fluorescein isothiocyanate (FITC) (Santa Cruz Biotechnology, Dallas, TX, USA) was applied on the sections. Mounting medium containing 4’,6’-diamidino-2-phenylindole (DAPI) was used to mount tissue on slides. Laser scanning confocal microscope (Olympus Fluoview FV1200) was used to determine immunofluorescence. Integrated density of protein immunoreactivity was performed using Image J software (NIH, Bethesda, MD, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Hippocampi were dissected from 1-mm coronal sections using Allen Brain Atlas and mouse brain stereotaxic coordinates and stored at −80 °C until further analysis. Total RNA was extracted from hippocampal tissue using trizole reagent (Invitrogen, Carlsbad, CA, USA) based on manufacturer’s instructions. The cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA) and Master Cycler Personal (Eppendorf, Hauooauge, NY, USA) as described previously [17]. Real-time PCR was conducted for resulting cDNA. Primers sequences were purchased from Integrated DNA Technologies (Coralville, IA, USA) as shown in Table 1. The cycle threshold (Ct) value was measured for each gene to calculate relative expression using delta-delta Ct method.
Table 1.
Sequence of primers used in current investigation in qRT-PCR.
| Gene | Primer sequence (5’−3’) |
| GAPDH | GTGGAGTCATACTGGAACATGTAG (forward) |
| AATGGTGAAGGTCGGTGTG (reverse) | |
| TNF-α | TCTTTGAGATCCATGCCGTTG (forward) |
| AATGGTGAAGGTCGGTGTG (reverse) |
Enzyme-linked immunosorbent assay (ELISA)
Hippocampal tissue samples were stored at −80 °C until further analysis. The tissues were then homogenized in tris buffer saline (1X) containing protease inhibitor mix (complete, Mini, Roche, Indianapolis, IN, USA) and 1% triton X-100. The brain tissue homogenate solutions were centrifuged at 17000×g for 30 min at 4 °C. The collected supernatants were stored at −80 °C until analysis. Quantification of hippocampal TNF-α was assessed by ELISA kit (eBioscience, San Diego, CA, USA), according to manufacturer’s instructions.
NE assay
NE assay was performed as described previously with minor modification [33]. Briefly, hippocampal tissue samples were stored at −80 °C until further analysis. During assay, hippocampi were homogenized in 0.1 N perchloric acid (1:5 as g/ml) and then centrifuged (14,000 ×g) for 30 min at 4 °C. Supernatants were collected and injected (20 μl) onto a high performance liquid chromatography (HPLC) system. The system consisted of electrochemical detection (ED) unit (ESA Inc., Chelmsford, MA, USA), a solvent delivery module, a Coulochem III detector equipped with a 5011A analytical cell and 5020 guard cell, C-18 analytical column (BetaBasic-18 column, 150 × 3 mm, Thermo Hypersil-Keystone, PA, USA), and ESA chromatography data system (EZ Chrom Elite, Chelmsford, MA, USA). 5011A analytical cell and 5020 guard cell (set at +350 mV) were used with setting the detector at 100 nA for both electrodes (electrode 1: −150 mV, electrode 2: +220 mV). The solvent mobile phase (75 mM NaH2PO4, 25 μM EDTA, 1.7 mM 1-octane sulfonic acid, 100 μL/L triethylamine, 100 mL/L of acetonitrile and the pH adjusted to 3.0) was used with 0.15 mL/min flow rate. The amount of NE in hippocampi was calculated based on obtained peaks from data system and the standard solutions (1–100 ng/mL). The data were expressed as NE ng/mg (tissue weight).
Statistical analysis
All behavioral data except MLA experiments were analyzed using two-way ANOVA with consideration LPS vs. control as a factor interacting with the factor of treatments. One-way ANOVA was used for all other data. For multiple comparisons, Tukey’s post hoc test was conducted with consideration p < 0.05 to be significant between the groups. GraphPad Prism (GraphPad Inc., San Diego, CA, USA) was used to analyze data expressed as mean ± SEM.
Results
Effects of TQS on LPS-induced allodynia
To examine the effect of TQS on tactile allodynia induced by LPS in mice, von Frey filament test was performed. Two-way ANOVA indicated that TQS pretreatment significantly prevented LPS-induced decreased paw withdrawal threshold (Fig. 1A; F3,37 = 4.275; p < 0.05). LPS treated group exhibited significant reduction (p < 0.0001) in paw withdrawal threshold as compared to control animals indicating the presence of substantial tactile allodynia. Moreover, TQS (1 or 4 mg/kg) significantly reversed the LPS effects by increasing paw withdrawal threshold indicating substantial antiallodynic effects. TQS-treated groups at basal level did not exhibit significant differences for paw withdrawal threshold compared to control group.
Fig. 1.
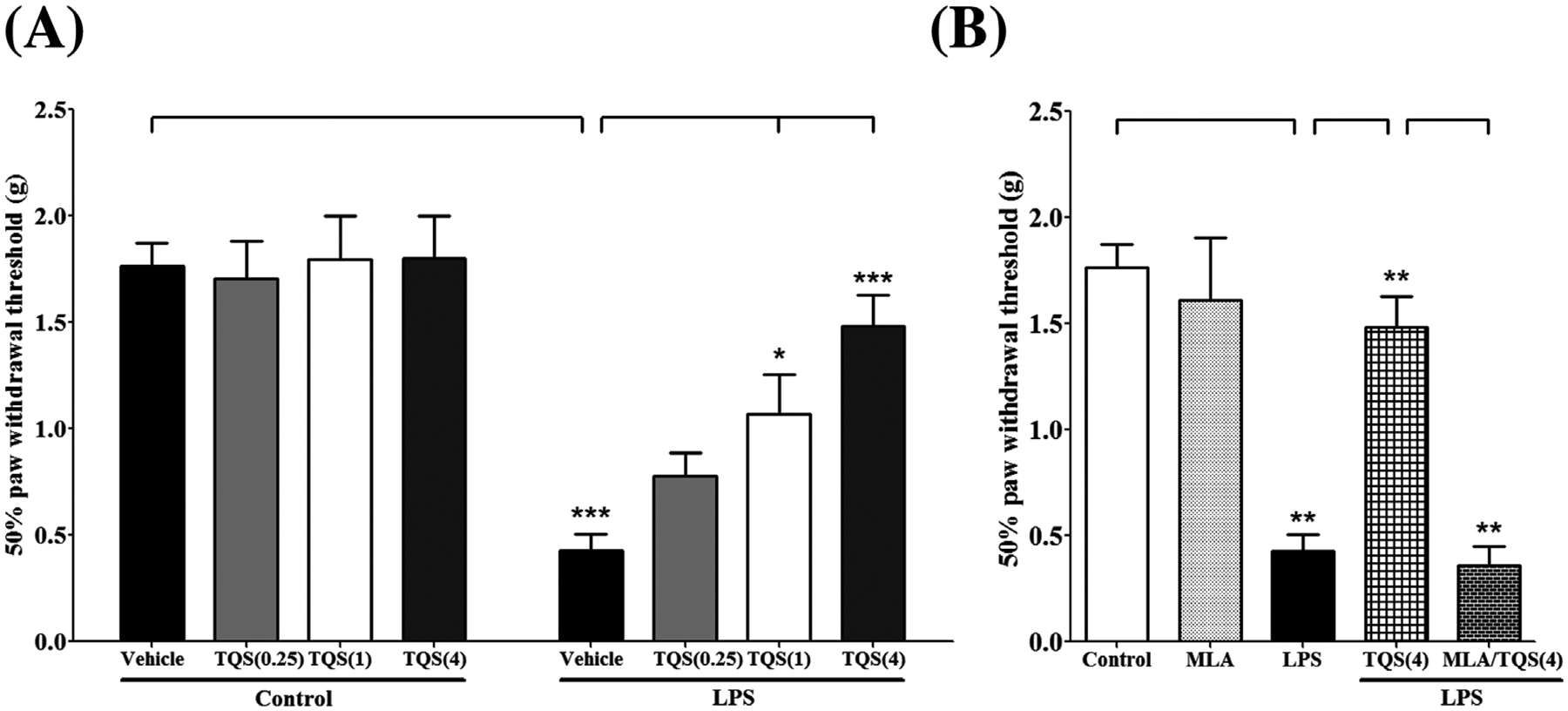
Assessment of antinociceptive-like effects of TQS using von Frey filaments in mice. (A) Effects of TQS on paw withdrawal threshold during LPS-induced allodynia. (B) Effects of MLA on antiallodynic response of TQS. Data are expressed as mean ± SEM, n = 4–8/group. *p < 0.05; **p < 0.001; ***p < 0.0001.
To determine the involvement of α7 nAChR in antiallodynic effects of TQS in the LPS model, the α7-selective antagonist MLA was injected in animals along with TQS (4 mg/kg) to determine if it would block TQS action. One-way ANOVA followed by post hoc test for multiple comparisons indicated that MLA (3 mg/kg) pretreatment significantly restricted (p < 0.01) antiallodynic effects of TQS by decreasing paw withdrawal threshold (Fig. 1B; F4,28 = 23.26; p < 0.001). However, mice did not show a significant change for paw withdrawal threshold when treated with MLA alone compared to control.
Effects of TQS on LPS-induced hyperalgesia
To evaluate whether TQS can reduce hyperalgesia induced by LPS in mice, hot plate tests were performed. Two-way ANOVA indicated that TQS pretreatment significantly prevented LPS-induced decreased latency time for paw withdrawal (Fig. 2A; F3,32 = 5.680; p < 0.01). LPS treated group exhibited significant reduction (p < 0.01) in latency time as compared to control animals indicating the presence of substantial thermal hyperalgesia. Moreover, TQS (4 mg/kg) significantly (p < 0.05) reversed the LPS effects by increasing latency time indicating substantial antihyperalgesic effects. TQS treated groups at basal level did not exhibit significant difference latency time compared to control group.
Fig. 2.
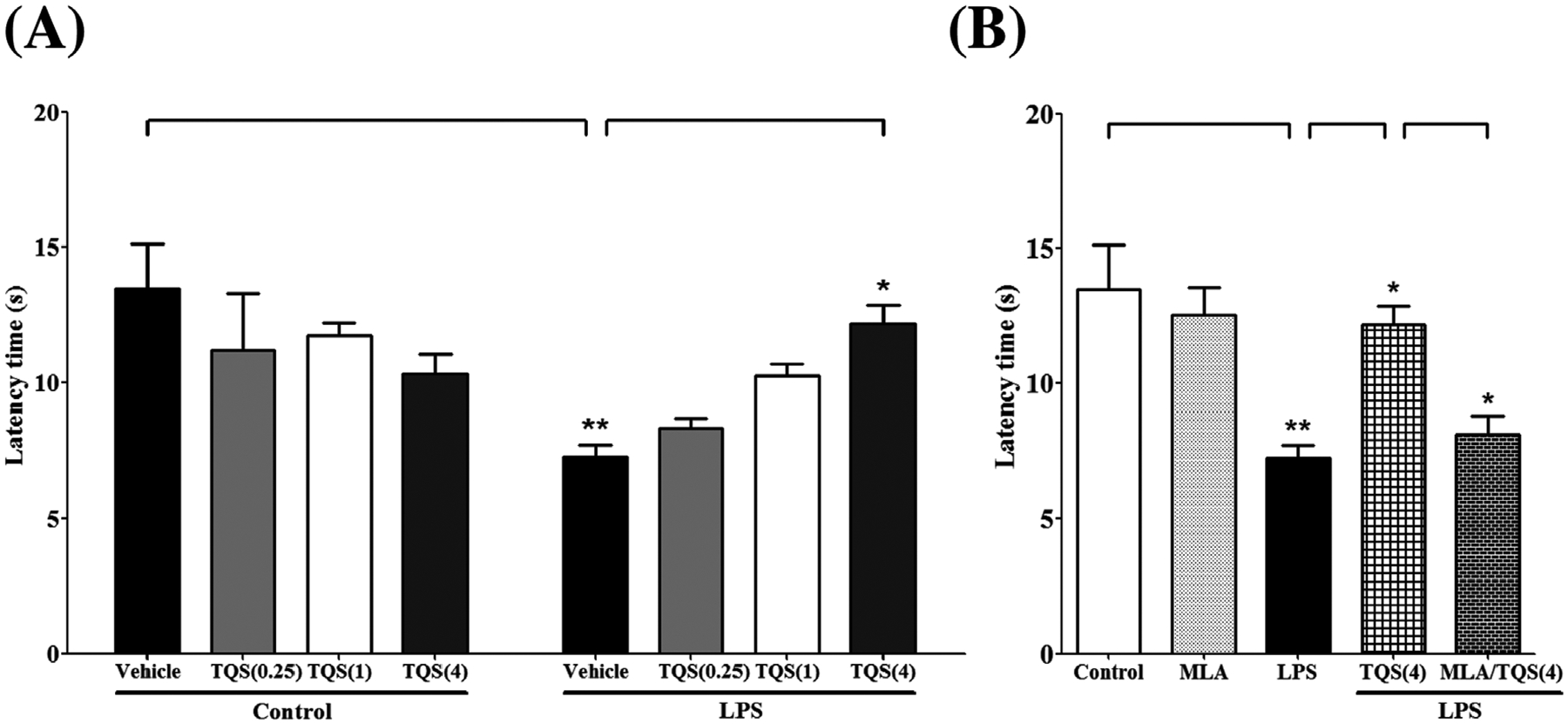
Assessment of antinociceptive-like effects of TQS using hot plate in mice. (A) Effects of TQS on latency time during LPS-induced hyperalgesia. (B) Effects of MLA on antihyperalgesic response of TQS. Data are expressed as mean ± SEM, n = 4–6/group. *p < 0.05; **p < 0.01.
MLA was tested in mice to examine the involvement of α7 nAChR in antihyperalgesic effects of TQS during LPS-induced hyperalgesia. MLA was injected 10 min before TQS (4 mg/kg) administration. One-way ANOVA followed by post hoctest for multiple comparisons indicated that MLA (3 mg/kg) pretreatment significantly blocked (p < 0.05) antihyperalgesic effects of TQS by decreasing latency time (Fig. 2B; F4,24 = 8.380; p < 0.001). However, mice did not show a significant change for latency time when treated with MLA alone compared to control.
Effects of TQS on Iba-1 immunoreactivity in CA1 and DG in the hippocampus
To examine the effects of TQS on LPS-induced microglial activation in the hippocampus, Iba-1 immunoreactivity was determined in CA1 and DG regions of the hippocampus. One-way ANOVA showed that TQS significantly affected Iba-1 immunoreactivity in CA1 (Fig. 3A; F2,9 = 18.34; p < 0.001) and DG (Fig. 3B; F2,11 = 14.93; p < 0.001) during LPS-induced allodynia and hyperalgesia. Post hoc test for multiple comparisons showed that LPS (1 mg/kg) significantly increased the immunoreactivity of Iba-1 in CA1 (p < 0.05) and DG (p < 0.001). Moreover, TQS (4 mg/kg) significantly (p < 0.05) reduced the Iba-1 immunoreactivity in both regions of the hippocampus.
Fig. 3.

Effects of TQS on Iba-1 immunoreactivity during LPS-induced allodynia and hyperalgesia in CA1 (A) and DG (B) regions of the hippocampus. The Iba-1 expression was quantified using immunofluorescence assay. Representative images show the immunofluorescence in CA1 (C) and DG (D) region. Magnification 20 X, Scale bar = 50 um. Data are expressed as mean ± SEM, n = 4–5/group; *p < 0.05, **p < 0.001.
Effects of TQS on p-NF-κB p65 immunoreactivity in CA1 and DG in the hippocampus
To assess the effects of TQS on LPS-induced NF-κB activation in the hippocampus, p-NF-κB p65 immunoreactivity was determined in CA1 and DG regions of the hippocampus. One-way ANOVA showed that TQS significantly affected p-NF-κB p65 immunoreactivity in CA1 (Fig. 4A; F2,10 = 10.95; p < 0.01) and DG (Fig. 4B; F2,10 = 9.567; p < 0.01) during LPS-induced allodynia and hyperalgesia. Post hoc test for multiple comparisons showed that LPS (1 mg/kg) significantly increased the immunoreactivity of p-NF-κB p65 in CA1 (p < 0.05) and DG (p < 0.01). Furthermore, TQS (4 mg/kg) significantly reduced the p-NF-κB p65 immunoreactivity in CA1 (p < 0.01) and DG (p < 0.05) regions.
Fig. 4.
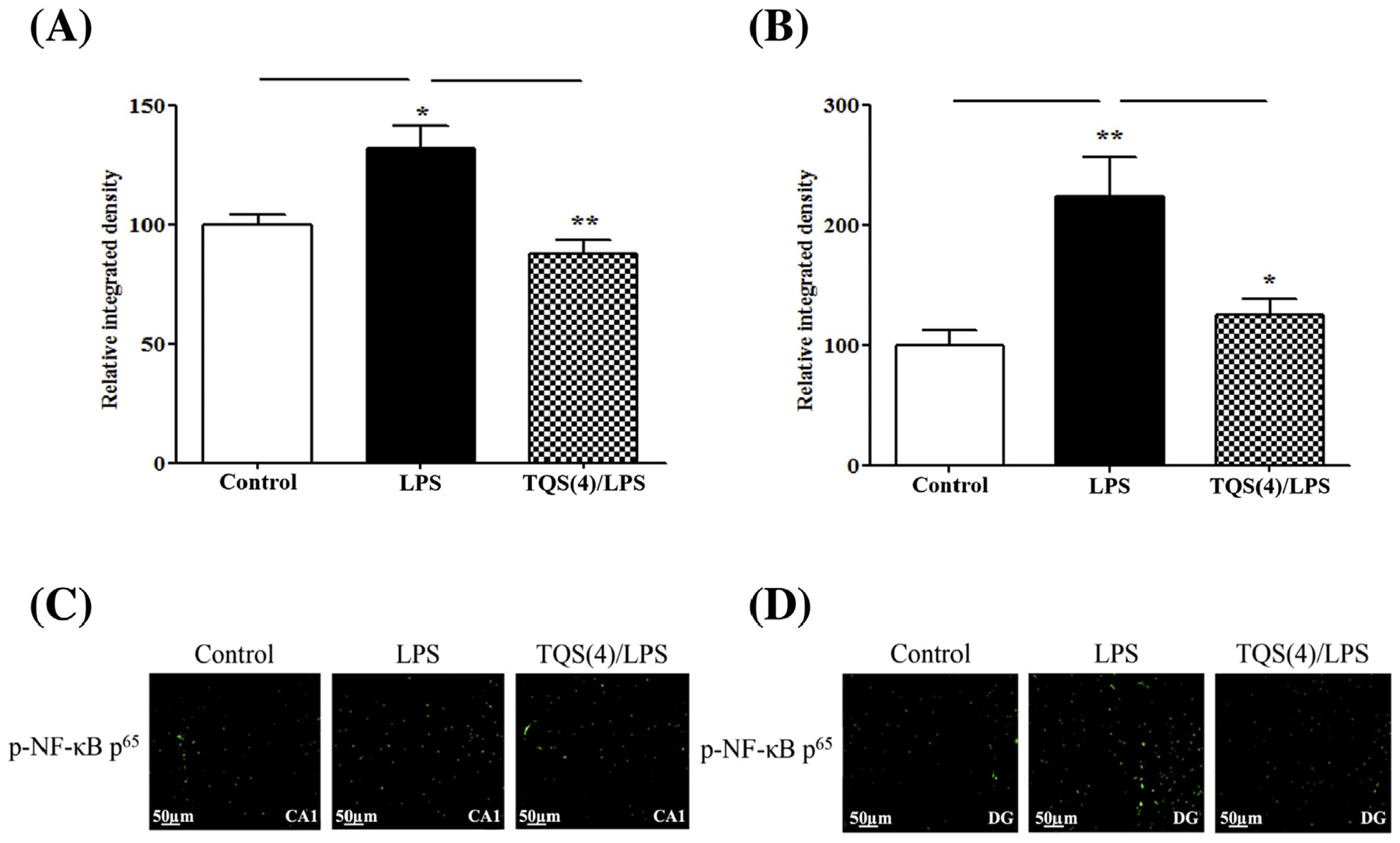
Effects of TQS on p-NF-κB p65 immunoreactivity during LPS-induced allodynia and hyperalgesia in CA1 (A) and DG (B) regions of the hippocampus. The p-NF-κB p65 expression was quantified using immunofluorescence assay. Representative images show the immunofluorescence analysis in CA1 (C) and DG (D) regions. Magnification 20 X, Scale bar = 50 um. Data are expressed as mean ± SEM, n = 4/group. *p < 0.05; **p < 0.01.
Effects of TQS on levels of TNF-α and NE in the hippocampus
We quantified the effects of TQS on mRNA and protein of TNF-α in the hippocampus using qRT-PCR and ELISA, respectively. One-way ANOVA showed that TQS significantly decreased the expression of TNF-α mRNA (Fig. 5A; F3,12 = 15.09; p < 0.0001) and protein (Fig. 5B; F3,21 = 43.45; p < 0.0001). Multiple comparisons of means showed that LPS (1 mg/kg) significantly increased the expression of TNF-α mRNA (p < 0.01) and protein (p < 0.001). Furthermore, TQS (4 mg/kg) significantly decreased TNF-α mRNA (p < 0.05) and protein (p < 0.001) expressions during LPS-induced allodynia and hyperalgesia. MLA clearly (p < 0.001) blocked the TQS effects by increasing the expression of TNF-α mRNA in the hippocampus.
Fig. 5.

Effects of TQS on levels of TNF-α and NE in the hippocampus during LPS-induced allodynia and hyperalgesia in mice. The TNF-α mRNA expression (A) and protein level (B) were quantified using qRT-PCR and ELISA, respectively. (C) The NE level was measured by HPLC-EC. Data are expressed as mean ± SEM, n = 4–7/group. *p < 0.05; **p < 0.01; ***p < 0.001.
In addition, one-way ANOVA revealed that TQS significantly affected LSP-induced reduction of hippocampal NE levels (Fig. 5C; F3,20 = 5.943; p < 0.001). While multiple comparisons of means revealed that LPS (1 mg/kg) significantly (p < 0.05) decreased the level of NE in the hippocampus, TQS (4 mg/kg) significantly (p < 0.01) reversed the LPS-induced reduction in NE level corresponding to its reduction in allodynia and hyperalgesia.
Effects of TQS on TNF-α immunoreactivity in CA1 and DG in the hippocampus
To evaluate the effects of TQS on the expression of TNF-α in CA1 and DG during antiallodynic and antihyperalgesic conditions, we quantified TNF-α integrated density. One-way ANOVA showed that TQS significantly affected TNF-α immunoreactivity in CA1 (Fig. 6A; F2,15 = 22.52; p < 0.0001) and DG (Fig. 6B; F2,15 = 28.55; p < 0.0001) during LPS-induced allodynia and hyperalgesia. Post hoc test for multiple comparisons showed that LPS (1 mg/kg) significantly (p < 0.001) increased the immunoreactivity of TNF-α in CA1 and DG. Moreover, TQS (4 mg/kg) significantly (p < 0.001) reduced the TNF-α immunoreactivity in both regions of the hippocampus.
Fig. 6.

Effects of TQS on TNF-α immunoreactivity during LPS-induced allodynia and hyperalgesia in CA1 (A) and DG (B) regions of the hippocampus. The TNF-α expression was quantified using immunofluorescence assay. Representative images show the immunofluorescence in CA1 (C) and DG (D) regions. Magnification 20 X, Scale bar = 50 um. Data are expressed as mean ± SEM, n = 5–7/group. *p < 0.001.
Discussion
In the present study, TQS pretreatment prevented LPS-induced allodynia and hyperalgesia. In addition, TQS altered the LPS-induced elevated immunoreactivity of Iba-1, p-NF-κB p65, and TNF-α in both of DG and CA1 and enhanced NE level reduced by the effects of LPS in the hippocampus. The TQS effects were reversed by MLA, an α7 nAChR antagonist. Taken together, our results indicate that TQS reduces nociceptive sensitivity associated with relevant inflammatory markers and increasing NE level involving activation α7 nAChR in the hippocampus.
Previous studies have indicated the involvement of α7 nAChR in regulating pain hypersensitivity in animal models. For example, α7 nAChR knockout mice exhibited an increase in pain-related responses or resisted for pharmacological treatment with α7 nAChR agonist to decrease pain-related responses [8,34]. Consistent with our data, several studies have shown that inflammatory and neuropathic pain-related symptoms in animal models can be reduced by selective α7 nAChR agonists such as TC-7020, choline, and PNU282987 [8,35,36]. However, desensitization is a major issue that could limit α7 nAChR efficiency when full α7 nAChR agonists are used [37]. Thus, α7 nAChR type II PAMs like TQS [10,13] can modulate desensitization and enhance the receptor activity against pain-related symptoms [7,34,38]. Since TQS is an α7 nAChR PAM type II, our results indicate that TQS can inhibit LPS-induced allodynia and hyperalgesia as mentioned above. In addition, MLA reverses the antiallodynic and antihyperalgesic effects produced by TQS, confirming the notion that α7 nAChRs are involved in these pharmacological effects. Interestingly, TQS treatment alone cannot enhance tactile allodynia and thermal hyperalgesia at basal level. We propose that LPS may cause interruption for cholinergic tone that could be enhanced by TQS.
It should be noted that although the effects of α7 Type II PAMS on α7 receptors in neurons and heterologous expression systems are associated with increased in channel activation [10], the α7 receptors in immune cells that mediate the cholinergic anti-inflammatory pathway appear to function differently. Signals mediated by α7 receptors in these cells are more likely to be metabotropic which are consistent with the effects we have observed on the regulation of intracellular signaling pathways [9]. As modulators of the α7 conformational states, PAMs and silent agonists such as NS6740 appear to be effective activators of these pathways, even in the absence of ion channel currents [6,39].
During neuroinflammation, NF-κB is involved in inflammatory signaling pathway that results in TNF-α production primarily from microglia [40]. We have shown that TQS inhibits the activation of microglia and NF-κB in the hippocampus [16,17]. In the present study, we examined TQS effects on these inflammatory markers in CA1 and DG regions of the hippocampus. Previous reports have indicated that CA1 and DG of the hippocampus play an important role in mediating responses to painful stimuli [25,41]. Our findings support the evidence that both of CA1 and DG show upregulation of the neuroinflammatory signaling during tactile allodynia and thermal hyperalgesia. The results indicate that LPS leads to activation of microglia and NF-κB in these hippocampal regions by increasing immunoreactivity of Iba-1 and p-NF-κB p65 which are reversed by TQS.
Our results indicate that TQS markedly decreases the LPS-induced expression of TNF-α in the hippocampus. This suggests that TQS-mediated actions against LPS-induced allodynia and hyperalgesia may be mediated by decreasing TNF-α expression in the hippocampus. Furthermore, MLA reversed TQS-induced decrease TNF-α mRNA expression in the hippocampus, indicating a decrease in TNF-α expression which is correlated with TQS effects on α7 nAChR. Moreover, TQS decreased the LPS-stimulated immunoreactivity of TNF-α in both DG and CA1 regions of the hippocampus. In addition, our results have shown that LPS causes a decrease in NE level during pain hypersensitivity. This might be correlated with increased TNF-α as described earlier. Taken together, these results suggest that TQS mediate its antiallodynic and antihyperalgesic effects at least in part involving increased release of NE and low level of TNF-α in the hippocampus. However, additional studies are required to investigate the effects of TQS in other brain regions for multiple reasons. First, the nociceptive pathway extends from the periphery to several CNS regions such as spinal cord, medulla, pons, midbrain, thalamus, and hypothalamus [42]. Second, systemic LPS administration may induce inflammatory mediators in the previously mentioned regions.
The mechanisms involved regarding NE release are regulated by the stimulation of α2-adrenoceptors by NE at presynaptic locations [43]. Cytokines and neurotransmitters are major factors that play a key role in the mediation of bidirectional communication between neurons and immune systems, thus maintaining homeostasis. The abnormal production of one or both of these mediators greatly affects the signaling associated with other mediator resulting pathogenesis [31]. This decrease in NE is associated with dysregulation of G-protein coupling with presynaptic α2-adrenoceptor mediated by TNF-α [18,42,44,45]. Our findings indicate that LPS resulted in increased TNF-α expression and reduced NE level indicating dysregulation in α2-adrenoceptors. Since TQS pretreatment resulted in decreased the expression of TNF-α and increased the level of NE in the hippocampus, these effects may indicate that α2-adrenoceptors return to their normal function as reported by other studies [46].
In summary, our findings indicate that TQS prevents LPS-induced allodynia and hyperalgesia, upregulation of TNF-α expression and reduction of NE level in the hippocampus (Fig. 7). Therefore, these findings highlight the important effects of α7 nAChR allosteric modulator against some symptoms of inflammatory pain. It is noteworthy to mention that the studies reported here obtained from male mice, future studies may be required using female mice to determine any gender differences.
Fig. 7.
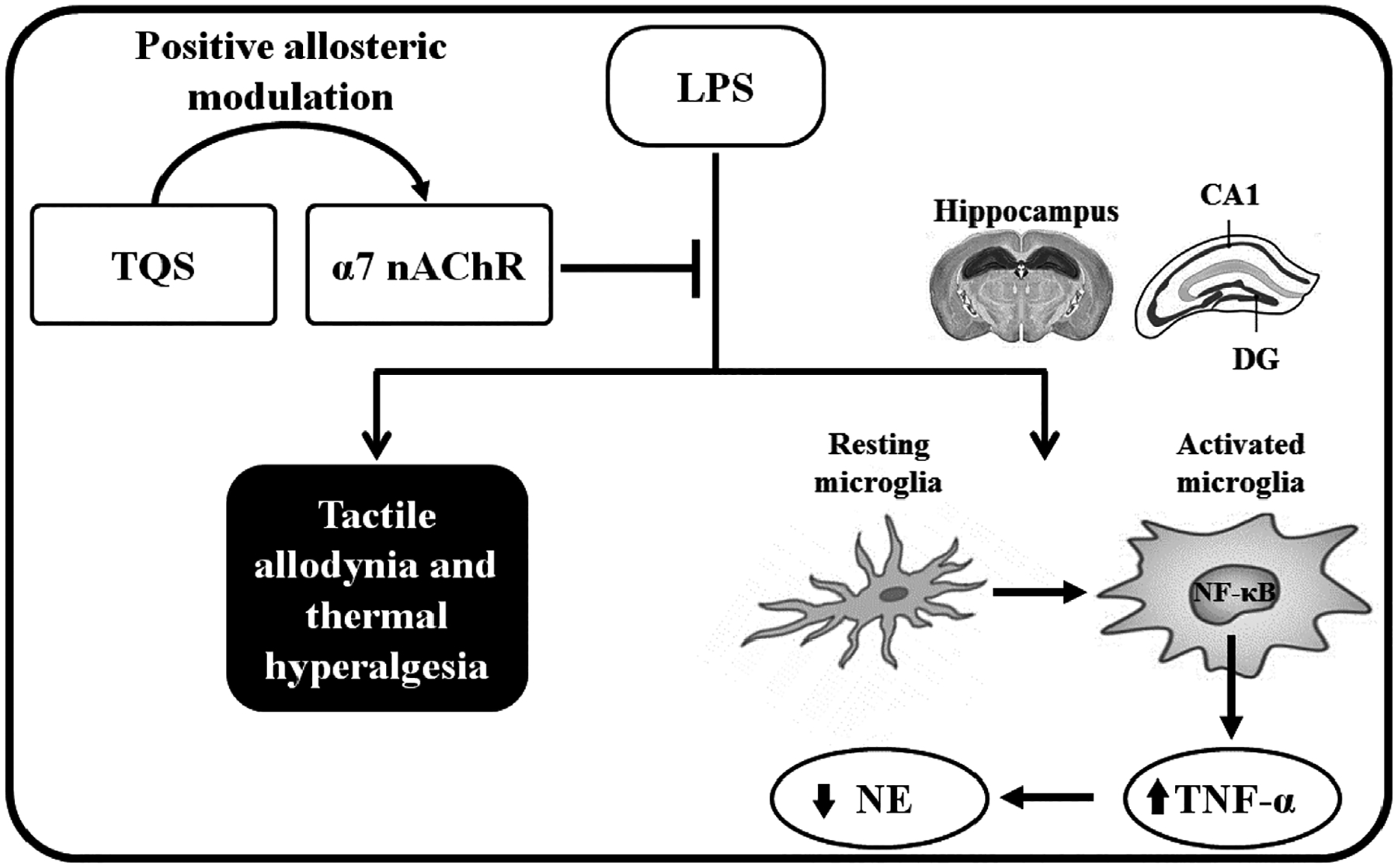
A schematic showing the proposed mechanisms of TQS on LPS-induced allodynia and hyperalgesia. Briefly, TQS binds at allosteric site of α7 nAChR in the presence of endogenous acetylcholine that binds at orthosteric site of α7 nAChR [10] in the hippocampus. These interactions lead to α7 nAChR activation that inhibits LPS-induced microglial activation and nuclear factor-kappa-B (NF-κB), a transcription factor for tumor necrosis factor-alpha (TNF-α) [16,17]. The reduction in NF-κB expression leads to a decrease in TNF-α production from microglia. Since TNF-α has a role in the reduction of neuronal norepinephrine (NE) level [23], low production of TNF-α by the effects of TQS could enhance NE amount. These effects may be linked for decreasing mechanical allodynia and thermal hyperalgesia at least in part.
Acknowledgments
This study was supported by Fulbright Foundation, USA and SDSU Research Foundation (SR). RLP is supported by NIH grant GM 57481.
Abbreviations:
- CNS
central nervous system
- DG
dentate gyrus
- Iba-1
ionized calcium-binding adapter molecule 1
- LPS
lipopolysaccharide
- MLA
methyllycaconitine
- nAChRs
nicotinic acetylcholine receptors
- NE
norepinephrine
- PAM
positive allosteric modulator
- p-NF-κB
phosphor-nuclear factor-κB
- TNF-α
tumor necrosis factor-alpha
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- [1].Wang F, Xu S, Shen X, Guo X, Peng Y, Yang J. Spinal macrophage migration inhibitory factor is a major contributor to rodent neuropathic pain-like hypersensitivity. Anesthesiology 2011;114(3):643–59. [DOI] [PubMed] [Google Scholar]
- [2].Zhuo M Neuronal mechanism for neuropathic pain. Mol Pain 2007;3:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Harden RN. Chronic neuropathic pain. Mechanisms, diagnosis, and treatment. Neurologist 2005;11(2):111–22. [DOI] [PubMed] [Google Scholar]
- [4].Fasick V, Spengler RN, Samankan S, Nader ND, Ignatowski TA. The hippocampus and TNF: common links between chronic pain and depression. Neurosci Biobehav Rev 2015;53:139–59. [DOI] [PubMed] [Google Scholar]
- [5].Yaksh TL, Woller SA, Ramachandran R, Sorkin LS. The search for novel analgesics: targets and mechanisms. F1000Prime Rep 2015;7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Papke RL, Bagdas D, Kulkarni AR, Gould T, AlSharari SD, Thakur GA, et al. The analgesic-like properties of the alpha7 nAChR silent agonist NS6740 is associated with non-conducting conformations of the receptor. Neuropharmacology 2015;91:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Freitas K, Negus SS, Carroll FI, Damaj MI. In vivo pharmacological interactions between a type II positive allosteric modulator of alpha7 nicotinic ACh receptors and nicotinic agonists in a murine tonic pain model. Br J Pharmacol 2013;169(3):567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Donvito G, Bagdas D, Toma W, Rahimpour E, Jackson A, Meade JA, et al. The interaction between alpha 7 nicotinic acetylcholine receptor and nuclear peroxisome proliferator-activated receptor-alpha represents a new antinociceptive signaling pathway in mice. Exp Neurol 2017;295:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 2007;151(7):915–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Williams DK, Wang J, Papke RL. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem Pharmacol 2011;82(8):915–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sun R, Zhang W, Bo J, Zhang Z, Lei Y, Huo W, et al. Spinal activation of alpha7-nicotinic acetylcholine receptor attenuates posttraumatic stress disorder-related chronic pain via suppression of glial activation. Neuroscience 2017;344:243–54. [DOI] [PubMed] [Google Scholar]
- [12].Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 2004;89(2):337–43. [DOI] [PubMed] [Google Scholar]
- [13].Gronlien JH,Hakerud M,Ween H,Thorin-Hagene K,Briggs CA,Gopalakrishnan M, et al. Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol 2007;72(3):715–24. [DOI] [PubMed] [Google Scholar]
- [14].Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron 2018;100(6):1292–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Alzarea S, Rahman S. Effects of alpha-7 nicotinic allosteric modulator PNU 120596 on depressive-like behavior after lipopolysaccharide administration in mice. Prog Neuropsychopharmacol Biol Psychiatry 2018;86:218–28. [DOI] [PubMed] [Google Scholar]
- [16].Abbas M, Rahman S. Effects of alpha-7 nicotinic acetylcholine receptor positive allosteric modulator on lipopolysaccharide-induced neuroinflammatory pain in mice. Eur J Pharmacol 2016;783:85–91. [DOI] [PubMed] [Google Scholar]
- [17].Abbas M, Alzarea S, Papke RL, Rahman S. The alpha7 nicotinic acetylcholine receptor positive allosteric modulator attenuates lipopolysaccharide-induced activation of hippocampal IkappaB and CD11b gene expression in mice. Drug Discov Ther 2017;11(4):206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Millan MJ. Descending control of pain. Prog Neurobiol 2002;66(6):355–474. [DOI] [PubMed] [Google Scholar]
- [19].Zimmerman ME, Pan JW, Hetherington HP, Lipton ML, Baigi K, Lipton RB. Hippocampal correlates of pain in healthy elderly adults: a pilot study. Neurology 2009;73(19):1567–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schweinhardt P, Lee M, Tracey I. Imaging pain in patients: is it meaningful? Curr Opin Neurol 2006;19(4):392–400. [DOI] [PubMed] [Google Scholar]
- [21].Duric V, McCarson KE. Persistent pain produces stress-like alterations in hippocampal neurogenesis and gene expression. J Pain 2006;7(8):544–55. [DOI] [PubMed] [Google Scholar]
- [22].Bingel U, Quante M, Knab R, Bromm B, Weiller C, Buchel C. Subcortical structures involved in pain processing: evidence from single-trial fMRI. Pain 2002;99(1–2):313–21. [DOI] [PubMed] [Google Scholar]
- [23].Covey WC, Ignatowski TA, Knight PR, Spengler RN. Brain-derived TNFalpha: involvement in neuroplastic changes implicated in the conscious perception of persistent pain. Brain Res 2000;859(1):113–22. [DOI] [PubMed] [Google Scholar]
- [24].Ignatowski TA, Covey WC, Knight PR, Severin CM, Nickola TJ, Spengler RN. Brain-derived TNFalpha mediates neuropathic pain. Brain Res 1999;841(1–2):70–7. [DOI] [PubMed] [Google Scholar]
- [25].Martuscello RT, Spengler RN, Bonoiu AC, Davidson BA, Helinski J, Ding H, et al. Increasing TNF levels solely in the rat hippocampus produces persistent pain-like symptoms. Pain 2012;153(9):1871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Covey WC, Ignatowski TA, Renauld AE, Knight PR, Nader ND, Spengler RN. Expression of neuron-associated tumor necrosis factor alpha in the brain is increased during persistent pain. Reg Anesth Pain Med 2002;27(4):357–66. [DOI] [PubMed] [Google Scholar]
- [27].Oka T, Wakugawa Y, Hosoi M, Oka K, Hori T. Intracerebroventricular injection of tumor necrosis factor-alpha induces thermal hyperalgesia in rats. Neuroimmunomodulation 1996;3(2–3):135–40. [DOI] [PubMed] [Google Scholar]
- [28].Shubayev VI, Myers RR. Upregulation and interaction of TNFalpha and gelatinases A and B in painful peripheral nerve injury. Brain Res 2000;855(1):83–9. [DOI] [PubMed] [Google Scholar]
- [29].Yoon SY, Patel D, Dougherty PM. Minocycline blocks lipopolysaccharide induced hyperalgesia by suppression of microglia but not astrocytes. Neuroscience 2012;221:214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Maier SF, Wiertelak EP, Martin D, Watkins LR. Interleukin-1 mediates the behavioral hyperalgesia produced by lithium chloride and endotoxin. Brain Res 1993;623(2):321–4. [DOI] [PubMed] [Google Scholar]
- [31].Dantzer R Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev 2018;98(1):477–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Callahan PM, Hutchings EJ, Kille NJ, Chapman JM, Terry [86_TD$DIFF][60_TD$DIFF]Jr. AV. Positive allosteric modulator of alpha7 nicotinic-acetylcholine receptors, PNU-120596 augments the effects of donepezil on learning and memory in aged rodents and non-human primates. Neuropharmacology 2013;67:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Roni MA, Rahman S. Antidepressant-like effects of lobeline in mice: behavioral, neurochemical, and neuroendocrine evidence. Prog Neuropsychopharmacol Biol Psychiatry 2013;41:44–51. [DOI] [PubMed] [Google Scholar]
- [34].Alsharari SD, Freitas K, Damaj MI. Functional role of alpha7 nicotinic receptor in chronic neuropathic and inflammatory pain: studies in transgenic mice. Biochem Pharmacol 2013;86(8):1201–7. [DOI] [PubMed] [Google Scholar]
- [35].Loram LC, Taylor FR, Strand KA, Maier SF, Speake JD, Jordan KG, et al. Systemic administration of an alpha-7 nicotinic acetylcholine agonist reverses neuropathic pain in male Sprague Dawley rats. J Pain 2012;13(12):1162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rowley TJ, McKinstry A, Greenidge E, Smith W, Flood P. Antinociceptive and anti-inflammatory effects of choline in a mouse model of postoperative pain. Br J Anaesth 2010;105(2):201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Papke RL, Kem WR, Soti F, Lopez-Hernandez GY, Horenstein NA. Activation and desensitization of nicotinic alpha7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther 2009;329(2):791–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Munro G, Hansen R, Erichsen H, Timmermann D, Christensen J, Hansen H. The alpha7 nicotinic ACh receptor agonist compound B and positive allosteric modulator PNU-120596 both alleviate inflammatory hyperalgesia and cytokine release in the rat. Br J Pharmacol 2012;167(2):421–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bagdas D, Wilkerson JL, Kulkarni A, Toma W, AlSharari S, Gul Z, et al. The alpha7 nicotinic receptor dual allosteric agonist and positive allosteric modulator GAT107 reverses nociception in mouse models of inflammatory and neuropathic pain. Br J Pharmacol 2016;173(16):2506–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014;81(5):1009–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Soleimannejad E, Naghdi N, Semnanian S, Fathollahi Y, Kazemnejad A. Antinociceptive effect of intra-hippocampal CA1 and dentate gyrus injection of MK801 and AP5 in the formalin test in adult male rats. Eur J Pharmacol 2007;562(1–2):39–46. [DOI] [PubMed] [Google Scholar]
- [42].Romanelli P, Esposito V. The functional anatomy of neuropathic pain. Neurosurg Clin N Am 2004;15(3):257–68. [DOI] [PubMed] [Google Scholar]
- [43].Dixon WR, Mosimann WF, Weiner N. The role of presynatpic feedback mechanisms in regulation of norepinephrine release by nerve stimulation. J Pharmacol Exp Ther 1979;209(2):196–204. [PubMed] [Google Scholar]
- [44].Yaksh TL. Pharmacology of spinal adrenergic systems which modulate spinal nociceptive processing. Pharmacol Biochem Behav 1985;22(5):845–58. [DOI] [PubMed] [Google Scholar]
- [45].Ignatowski TA, Spengler RN. Tumor necrosis factor-alpha: presynaptic sensitivity is modified after antidepressant drug administration. Brain Res 1994;665(2):293–9. [DOI] [PubMed] [Google Scholar]
- [46].Ignatowski TA, Sud R, Reynolds JL, Knight PR, Spengler RN. The dissipation of neuropathic pain paradoxically involves the presence of tumor necrosis factor-alpha (TNF). Neuropharmacology 2005;48(3):448–60. [DOI] [PubMed] [Google Scholar]