

Abstract
Introduction
The NLRP3 inflammasome is closely related to diabetes and atherosclerosis. Recent studies suggest NIMA-related kinase 7 (NEK7) is necessary for NLRP3 inflammasome activation during potassium efflux. However, the expression of the NEK7/NLRP3 inflammasome pathway in diabetic lower extremity arterial disease (DLEAD) is unclear. The present study aimed to explore whether the NEK7/NLRP3 inflammasome pathway is involved in the pathogenesis of DLEAD.
Research design and methods
The serum levels of interleukin-1β (IL-1β) and IL-18 in the control group (n=39), diabetes without lower extremity artery diseases group (n=39) and DLEAD group (n=85) were measured. H&E and Von Kossa staining were used to observe the vasculature of amputated feet from patients with diabetic foot. Furthermore, immunohistochemical staining, immunofluorescence and western blot were used to detect the expression of NEK7 and the NLRP3 inflammasome.
Results
The serum IL-1β level in the DLEAD group was significantly increased compared with that in the control group and diabetes without lower extremity artery disease group. The serum IL-18 level was significantly higher in the DLEAD group and diabetes without lower extremity artery disease group than in the control group. H&E staining showed that the subintimal tissue of the arteries of patients with diabetic foot were highly thickened and exhibited irregular atherosclerotic plaques, and the arterial lumen was nearly occluded. Von Kossa staining showed dense brown-black calcium salt deposits in the vascular mesangium. Moreover, the expression of NEK7 and the NLRP3 inflammasome was significantly increased in the vascular cells of patients with diabetic foot, especially in vascular smooth muscle cells.
Conclusion
The NEK7/NLRP3 inflammasome pathway might be involved in the pathogenesis of DLEAD.
Keywords: atherosclerosis, diabetic foot, diabetes mellitus, type 2, inflammation
Significance of this study.
What is already known about this subject?
The nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome is closely related to diabetes and atherosclerosis.
Recent studies suggest that NIMA-related kinase 7 (NEK7) is necessary for NLRP3 inflammasome activation during potassium efflux.
The endogenous expression of the NEK7/NLRP3 inflammasome pathway in diabetic lower extremity arterial disease (DLEAD) is unclear.
What are the new findings?
The level of serum interleukin (IL)-1β and serum IL-18, which are downstream effector molecules of the NLRP3 inflammasome, increases in patients with DLEAD.
The expression of NEK7 and the NLRP3 inflammasome was significantly increased in the vascular cells of patients with diabetic foot, especially in vascular smooth muscle cells.
How might these results change the focus of research or clinical practice?
The NEK7/NLRP3 inflammasome pathway might be involved in the pathogenesis of DLEAD. The findings may provide new insight into the inflammation mechanism of diabetic macroangiopathy.
Introduction
Diabetic lower extremity arterial disease (DLEAD) is the main form of diabetic macroangiopathy (DMA), one of the chronic complications of diabetes and a specific type of atherosclerosis. As the atherosclerosis is secondary to diabetes, especially in type 2 diabetes, the vascular damage of diabetes appears earlier and more aggressive and serious than that in the non-diabetic population.1 DMA involves a wide range of arteries, including the coronary artery, cerebral artery, renal artery, peripheral artery and so on.2 However, routine clinical treatments including the reduction of blood glucose and blood pressure do not prevent the progression of DMA. Therefore, the incidence of DMA and its complications, including stroke, refractory heart failure and diabetic foot increases each year. The mortality of patients with diabetes mellitus (DM) is significantly increased, and their life quality is seriously affected. This phenomenon indicates an unknown pathogenic mechanism of DMA that needs to be explored.2
Activation of the NLRP3 inflammasome is closely related to metabolic inflammatory diseases such as diabetes, atherosclerosis, obesity and non-alcoholic fatty liver.3 The NLRP3 inflammasome belongs to a large family of cytoplasmic sensors, and it is produced by nucleotide-binding oligomerization domain-like receptors activated by endogenous substances (such as cholesterol crystals and urate crystals).3 It comprises NLRP3 scaffolds, apoptosis-associated speck-like proteins containing card (ASC) and pro-caspase-1. Activation of the NLRP3 inflammasome can trigger pro-caspase-1 to generate activated caspase-1 and cleave pro-IL-1β and pro-IL-18 to produce IL-1β and IL-18.4 Ferreira et al5 found that db/db mice exhibited vascular overexpression of caspase-1 and IL-1β, increased plasma IL-1β levels, activated caspase-1 in peritoneal macrophages and reduced acetylcholine (ACh) vasodilation, compared with db/m mice. Treatment of db/db mice with MCC950, a selective NLRP3 inhibitor, decreased plasma IL-1β and partly restored ACh vasodilation.5 Several studies showed that IL-1β activated by the NLRP3 inflammasome pathway and the elevated IL-18 levels in ApoE-/- mice could promote atherosclerotic plaque vulnerability.6 Moreover, mice lacking NLRP3 inflammasome components fed with a high-fat diet displayed improved insulin sensitivity and glucose tolerance.7 These studies indicated that the NLRP3 inflammasome contributes to diabetes-associated vascular dysfunction. Our previous study confirmed that activation of NLRP3 inflammasome mediates myocardial injury induced by lipopolysaccharide.8 Downregulation of the NLRP3 inflammsome can inhibit inflammation and alleviate myocardial injury and development of diabetes and its complications such as diabetic cardiomyopathy.8–10
Recent studies suggest that the NLRP3 inflammasome activation pathway might include the following: (1) metabolite or endotoxin-induced damage to the cell membrane or ATP and cell membrane receptor P2×7 binding to open potassium channels, causing potassium outflow and activation of the NLRP3 inflammasome; (2) oxidative stress caused by reactive oxygen species; (3) destruction of lysosomes and the release of cathepsin; and (4) induction of NLRP3 inflammasome activation by fatty acids, peptides and toxins through the microporous structure of the membrane.11 Among these NLRP3 inflammasome activation pathways, the potassium efflux mode which is mediated by NIMA-related kinase 7 (NEK7) plays a major role.11 NEK7 might aslo mediate NLRP3 polymerization and punctate ASC aggregation. NEK7 might be another component of the NLRP3 inflammasome.11 Inhibiting of NEK7 is expected to be an effective treatment for inhibition of NLRP3 inflammasome-related diseases.11 12 Ma et al13 found that the NEK7/NLRP3 inflammasome pathway was expressed at low levels in peripheral blood mononuclear cells of patients with systemic lupus erythematosus and that is negatively correlated with disease activity. At present, the endogenous expression of the NEK7/NLRP3 inflammasome pathway in DMA, especially in DLEAD is unclear and must be further elucidated.
Therefore, we observed the expression of NEK7 and the NLRP3 inflammasome in the arteries of amputated feet of patients with diabetic foot and measured the serum levels of IL-1β and IL-18, which are downstream effector molecules of the NLRP3 inflammasome, in patients with DLEAD, to examine the hypothesis that the NEK7/NLRP3 inflammasome pathway might participate in the progression of DLEAD and to provide a new scientific basis for the pathogenesis of DMA.
Materials and methods
Subjects
The study population consisted of 163 patients divided into a DLEAD group, a DM without lower extremity artery diseases group, and a control group (n=85, 39 and 39, respectively) with mean ages of 61.54±7.73, 53.82±7.71, and 57.51±10.99 years, respectively. Basic data were recorded for all subjects and included age, sex, height, weight, medical history, smoking history, drinking history, and body mass index (BMI). The diagnostic criteria for diabetes were as follows: (1) fasting blood glucose ≥7.0 mmol/L or (2) random blood glucose ≥11.1 mmol/L. Patients included in the DLEAD group consisted of those with lower extremity arteries with thickening of the intima-media and plaque formation assessed by ultrasonography. The exclusion criteria were thromboangiitis, Takayasu arteritis, acute lower extremity arterial embolism, and idiopathic arterial thrombosis. The criteria for inclusion in the control group were exclusion of hypertension, metabolic syndrome, DM and other cardiovascular diseases by history, physical examination, laboratory and ECG examination.
Up to 15 patients with type 2 diabetes with diabetic foot undergoing amputation surgery at the Department of Orthopedics, The Fourth Hospital of Harbin Medical University, (Harbin, China) were recruited into our study. The patients had no history of severe trauma or malignant tumour. The main causes of amputation were infection, severe ischemia and local gangrene. The general information, and past medical history including age, gender, and history of DM, hypertension, tobacco and alcohol use were recorded for all patients. Then a physical examination and biochemical tests were conducted, including fasting blood glucose, blood lipid and renal function. The upper limb arteries from five patients were collected after surgical removal following a traffic accident and used for control studies.
Data collection
Blood samples were collected in tubes containing disodium ethylenediaminetetraacetate dihydrate (Na2-EDTA) 1 mg/mL in the morning after an overnight fast. The serum was obtained by centrifugation at 3000 rpm for 10 min at 4°C, and the samples were stored at −80°C until analysis. The fasting serum glucose, lipid profile, serum creatinine, blood urea, serum uric acid, and cystain C levels were measured using an automatic analyzer (Cobas 8000, Roche, Switzerland). Serum levels of IL-1β and IL-18 were determined by ELISA measurements according to the manufacturer’s protocol using an IL-1β and an IL-18 ELISA kit (SEA563Hu and SEA064Hu, Cloud-Clone Corp, China).
Ultrasound
Ultrasound scans of the lower extremity arteries were performed with a GE Vivi E9 ultrasound system (USA) equipped with a 7.5-MHz linear array transducer by the same experienced sonographer. The patient was in a supine position with both legs exposed. The angle between sound beam direction and blood flow was <60°. The intima-media thickness (IMT) of the common femoral artery was measured by two-dimensional ultrasound. When the IMT was>0.15 cm, the atheromatous plaques were formed. Additionally, the plaque size, number, echo and the vascular diameter were also detected. The blood flow filling, stenosis and occlusion of the vascularture were observed by colour Doppler flow imaging. The degree of stenosis was calculated by dividing the residual diameter (N) by the vascular diameter at the end of the stenosis where the normal vascular diameter (D) was restored using the following formula : (1 -N/D)×100= degree of stenosis.14
H&E staining, immunohistochemistry and immunofluorescence technique
The arteries were isolated, fixed in 4% paraformaldehyde and then embedded in paraffin. The embedded arteries were cut into 5 mm sections, stained with H&E and then examined by light microscopy. Huawei P20 mobile phone was used to record images.
Formaldehyde-fixed paraffin sections were incubated with primary antibodies including anti-NEK7 (1:100, BioSS), anti-NLRP3 (1:100, CST), anti-ASC (1:100, Santa Cruz) and anti-caspase-1 (1:100, CST) antibodies overnight at 4℃ and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 hour at 37℃. Huawei P20 mobile phone was used to record images.
Additionally, the immunofluorescence technique was applied. After antigen retrieval, the slides were incubated with the primary antibodies overnight at 4°C. Next, an antimouse or antirabbit fluorescein isothicyanate (FITC) or tetramethyl rhodamine isothiocyanate (TRITC)-conjugated secondary antibody (1:100, Invitrogen) was added for 2 hours at 37°C. Then 4',6-diamidino-2-phenylindole (DAPI) was added at a dilution of 1:20 000 for 5 min, and the slides were cover-slipped with SlowFade Antifade (Invitrogen). The images were taken at 100 and 200 power magnification using an ix70 microscope and Magnafire V.1.1 software (both Olympus), and Image Pro-Plus software was used to generate colour composite images.
Von Kossa staining
Von Kossa staining was performed to examine calcification of the arteries according to the manufacturer’s instructions (Von Kossa staining kit, Solarbio, Beijing, China), and calcium staining of the arteries was analysed by light microscopy. Huawei P20 mobile phone was used to record images.
Western blot analysis
Protein extracts from arteries were homogenized in lysis buffer. The protein mixture was then placed in boiling water for 5 min and briefly centrifuged at low speed to collect denatured proteins. Protein samples were resolved on 6% (for NLRP3) or 12.5% (for NEK7, ASC, caspase-1, IL-1β, IL-18 and β-actin) Tris/glycine SDS-polyacrylamide gel in running buffer containing 25 mmol/L Tris, 192 mmol/L glycine, and 0.1% sodium dodecyl sulfate (SDS). The proteins were then transferred to a nitrocellulose membrane for 2 hours at 4°C. Then, the membranes were incubated with the primary antibodies including anti-β-actin (1:3000 diluted in TBS-T), anti-NLRP3 (1:1000), anti-NEK7 (1:500), anti-ASC (1:500), anti-caspase-1 (1:1000), anti-IL-1β (1:500), or anti-IL-18 (1:500) overnight at 4°C–8°C and then incubted with secondary antibodies (HRP-conjugated antigoat or antirabbit IgG) for 1 hour at room temperature. Protein expression was analysed using NIH image software and normalized to that of β-actin.
Statistical analysis
The data are presented as the mean±SD. For comparisons between two variables, the unpaired Student’s t-test was used. One-way ANOVA followed by the Newman-Keuls test was used to compare more than two groups. A value of p<0.05 was considered statistically significant.
Results
Clinical parameters of patients with DLEAD
This study enrolled 39 patients in the control group, 39 patients with diabetes without lower extremity arterial disease, and 85 patients with DLEAD, including 13 patients with a history of smoking, 9 with a history of drinking, and 65 with a history of hypertension. Age, sex, BMI, fasting blood glucose, serum cholesterol, triglycerides, high-density lipoprotein, low-density lipoprotein, blood urea nitrogen, creatinine, uric acid, and cystatin C were compared among the three groups of patients (table 1). In the DLEAD group, the patient age was older, and the blood urea nitrogen, creatinine and cystatin C were increased (p<0.05, table 1), and the BMI, fasting blood glucose, and serum cholesterol values were significantly increased (p<0.01, table 1) compared with the control group. Compared with the diabetes without lower extremity arterial disease group, the DLEAD group showed increased BMI, triglyceride, creatinine and cystatin C values (p<0.05, table 1) and a significantly older age (p<0.01, table 1).
Table 1.
Comparison of clinical parameters among the three groups of patients
| Control group (n=39) | DM without LEAD group (n=39) | DLEAD group (n=85) | |
| Age (year) | 57.51±10.99 | 53.82±7.71 | 61.54±7.73*† |
| Gender (male/female, n) | 19/20 | 21/18 | 55/29 |
| BMI, kg/m2 | 22.67±1.48 | 26.47±3.48** | 25.36±2.93**‡ |
| FSG, mmol/L | 5.18±0.36 | 8.88±3.54** | 9.46±7.72** |
| CHOL, mmol/L | 4.33±0.85 | 5.37±1.55** | 5.04±1.49** |
| TG, mmol/L | 1.42±0.77 | 3.15±3.30** | 2.14±2.04‡ |
| HDL, mmol/L | 1.07±0.18 | 1.18±0.19 | 1.17±0.24 |
| LDL, mmol/L | 2.66±0.78 | 3.15±1.03 | 2.99±0.90 |
| BUN, mmol/L | 5.23±1.13 | 6.18±1.92* | 6.24±1.93* |
| Cr, μmol/L | 68.01±11.85 | 68.43±19.85 | 79.26±22.59*‡ |
| UA, μmol/L | 305.9±69.12 | 291.6±64.71 | 307.80±88.90 |
| Cys C, mg/L | 0.75±0.18 | 0.85±0.28 | 1.05±0.46*‡ |
*p<0.05 vs control group, **p<0.01 vs control group
†p<0.01 vs DM without LEAD group
‡p<0.05 vs DM without LEAD group
BMI, body mass index; BUN, blood urea nitrogen; CHOL, cholesterol; Cr, creatinine; Cys C, cystatin C; DLEAD, diabetic with lower extremity artery disease group; DM without LEAD, diabetes mellitus without lower extremity artery disease group; FSG, fasting serum glucose; HDL, high-density lipoprotein; LDL, low density lipoprotein; TG, triglyceride; UA, uric acid.
Serum IL-1β and IL-18 in patients with DLEAD
The serum IL-1β levels in the control group, diabetes without lower extremity arterial disease group and DLEAD group were 38.56±13.35 pg/mL, 44.89±35.37 pg/mL, and 66.03±71.27 pg/mL, respectively. The serum IL-1β level in the DLEAD group was significantly higher than that in the control group (p<0.05, figure 1), which was significantly higher than that in the diabetes without lower extremity arterial disease group (p<0.05, figure 1). The serum IL-18 levels in the control group, diabetes without lower extremity arterial disease group and DLEAD group were 32.49±4.73 pg/mL, 42.81±11.60 pg/mL, and 41.17±13.88 pg/mL, respectively. Compared with the control group, patients with diabetes with lower extremity arterial disease showed significantly increased serum IL-18 levels (p<0.01, figure 1). Moreover, the serum IL-18 levels were significantly higher in the diabetes without lower extremity arterial disease group than in the control group (p<0.01, figure 1).
Figure 1.

The serum IL-1β and IL-18 levels in patients with DLEAD were increased. The data are expressed as the (mean±SD). Serum IL-1β levels and IL-18 levels were measured. Compared with the control group, *p<0.05, **p<0.01; compared with the DM without lead group, p<0.05. Con, control group (n=39); DM without lead, diabetes mellitus without lower extremity artery disease group (n=39); DLEAD, diabetic with lower extremity artery disease group (n=85); IL, interleukin.
Vascular ultrasound images and pathological images of diabetic foot patients
The anterior tibial arteries, one branch of lower extremity arteries from patients with diabetic foot were selected for vascular ultrasound, H&E staining and Von Kossa staining. Vascular ultrasound showed solid echogenic filling in the anterior tibial artery of patients with diabetic foot. Color Doppler flow imaging showed no blood flow signal in the artery. Therefore, the anterior tibial artery occlusion was diagnosed by ultrasound (figure 2A). H&E staining showed that the intimal tissue was highly thickened, and irregular atherosclerotic plaques had formed. The fibrous cap of the plaques exhibited a large amount of amorphous necrotic material, visible cholesterol crystals, and destruction of the vascular mesenteric fibres which were closed due to occlusion (figure 2B). Von Kossa staining of the vasculature revealed dense brown-black calcium salt particle deposits in the middle vascular layer (figure 2C), indicating the presence of atherosclerosis and vascular calcification in patients with diabetic lower extremity vascular disease.
Figure 2.

Ultrasound, H&E staining and Von Kossa staining of artery of patients with diabetic foot. The bar represents 200 µm. ATA, arteriae tibialis anterior; Con, control group (n=5); DLEAD, diabetic lower extremity arterial disease (n=12).
Expression of NEK7 and the NLRP3 inflammasome in the vasculature of patients with diabetic foot
Immunohistochemistry and western blot analysis revealed that NEK7 expression was significantly increased in the vascular tissues of patients with diabetic foot. Moreover, the expression of NLRP3 inflammasome molecules (NLRP3, ASC, caspase-1) was significantly increased (figures 3 and 4).
Figure 3.
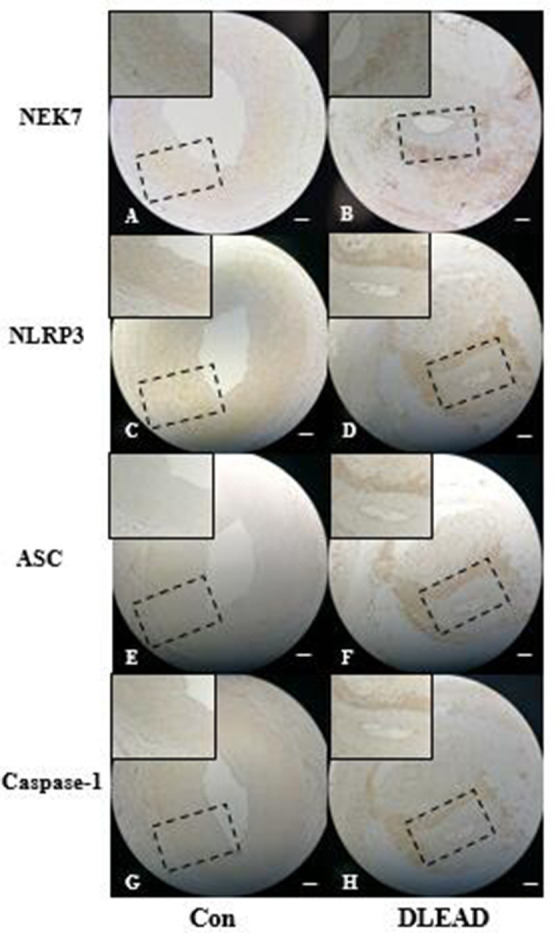
Immunohistochemical staining of the expression of NEK7 and the NLRP3 inflammasome in arteries of patients with diabetic foot. The bar represents 200 µm. ASC, apoptosis-associated speck-like proteins containing card; Con, control group (n=5); DLEAD, diabetic lower extremity arterial disease (n=12).
Figure 4.

Western blot showing NEK7 and the NLRP3 inflammasome protein expression in arteries of patients with diabetic foot. The bar represents 200 µm. ASC, apoptosis-associated speck-like proteins containing card; Con, control group; DLEAD, diabetic lower extremity arterial disease, n=4 at least in each group; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL, interleukin.
Expression of NEK7 and the NLRP3 inflammasome in vascular smooth muscle cells of patients with diabetic foot
Immunofluorescence detection of the NLRP3 inflammasome (NLRP3, ASC, and caspase-1) and its upstream mediator NEK7 in the vasculature of patients with diabetic foot showed that NEK7 and the NLRP3 inflammasome (NLRP3, ASC, and caspase-1) were significantly increased in vascular smooth muscle cells (figure 5).
Figure 5.
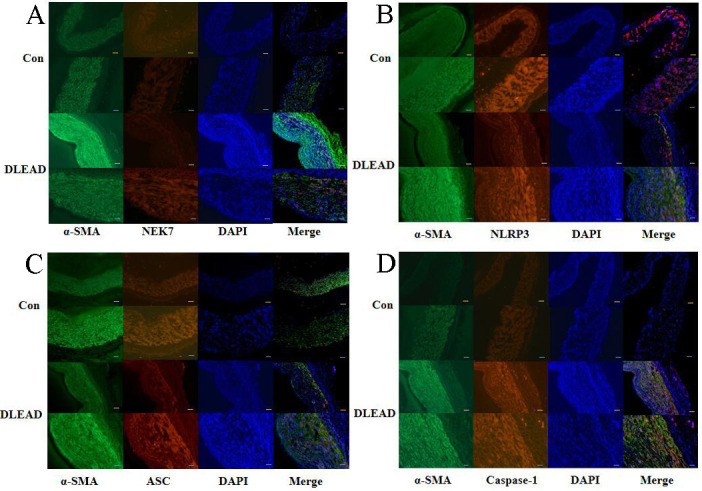
Immunofluorescence staining of NEK7, the NLRP3 inflammasome, and α-smooth muscle actin (α-SMA) in arteries of patients with DLEAD. The yellow bar represents 100 µm, and the white bar represents 200 µm. Con, control group; DAPI, 4',6-diamidino-2-phenylindole; DLEAD, diabetic lower extremity arterial disease; n=4 at least in each group.
Discussion
The key findings in the present study were as follows: (1) the levels of serum IL-1β and serum IL-18, which are downstream effector molecules of the NLRP3 inflammasome, are increased in DLEAD; (2) NEK7 and the NLRP3 inflammasome show significantly increased expression in the arteries of patients with diabetic foot; and (3) overexpression of the NEK7/NLRP3 inflammasome mainly occurs in vascular smooth muscle cells.
Excessive inflammation due to macrophages in the circulation, endothelial cells in the vascular intima, vascular smooth muscle cells in the media, fibroblasts in the vascular adventitia and perivascular fat can cause diabetic vascular injury. 1 15 The main mechanism of atherosclerosis secondary to diabetes is inflammation mediated by the NLRP3 inflammasome.15 Recent studies suggest that NEK7 is necessary for activation of the NLRP3 inflammasome during potassium efflux.11 When inflammasome activation occurs due to overnutrition or consumption of a Western diet, NEK7 interacts with NLRP3 to form the NLRP3 inflammasome.11 16 Moreover, inhibition of NEK7 can alleviate NLRP3 inflammasome-related diseases.11 12 However, at present, endogenous expression of the NEK7/NLRP3 inflammasome pathway in DMA, especially in DLEAD, is unclear and must be further elucidated.
DMA is a complex process of vascular injury and repair.1 In the present study, H&E and Von Kossa staining of the vasculature in patients with diabetic foot showed that DLEAD included atherosclerosis and extensive calcifications. Diabetes can accelerate the process of atherosclerosis and vascular calcification.17 These two pathological conditions will cause contraction and diastolic dysfunction of the arteries of the lower limbs, increase the stiffness and reduce the expandability of the blood vessels, affect the blood supply of the lower limbs, and participate in the occurrence of diabetic foot.18
When NLRP3 is activated, ASC recruits pro-caspase-1 to assemble an NLRP3 inflammasome.3 Subsequently, this inflammasome can trigger pro-caspase-1 to generate activated caspase-1 and then cleave pro-IL-1 β and pro-IL-18 to produce IL-1β and IL-18.4 IL-1β and IL-18 play a vital role in atherosclerosis and diabetes and are involved in plaque formation and are related to plaque stability.19 Inhibition of IL-1β expression can reduce the area of vascular plaque formation in ApoE-/- mice and improve blood glucose levels in patients with diabetes.19 IL-18 is an early marker of type 2 diabetes, and inhibition of IL-18-binding protein can alleviate endothelial dysfunction and diabetic atherosclerosis.15 20 Plasma IL-1β and IL-18 levels were significantly increased in carotid atherosclerosis patients.21 Moreover, the plasma IL-1β and IL-18 levels in the unstable plaque group were more elevated than those in the stable plaque group.21 In coronary atherosclerotic heart disease (CAD), the levels of IL-1β and IL-18 in human myeloid leukemia mononuclear cells (THP-1 cells) were distinctly higher than those in non-CAD subjects, and these levels were positively correlated with the level of NLRP3 inflammasome expression.22 In the early stages of atherosclerotic and type 2 diabetic plaque formation, mRNA expression and plasma levels of the NLRP3 inflammasome, IL-1β and IL-18 were significantly elevated in blood mononuclear cells of patients with type 2 diabetes with atherosclerosis.23 Consistently, our study found that the patients with DLEAD showed significantly increased serum levels of IL-1β and IL-18 compared with the control group, confirming the existence of a chronic inflammatory status in patients with DLEAD. Moreover, the serum IL-1β levels in the DLEAD group were higher than those in the diabetes without lower extremity artery disease group. Our results indicate that the inflammatory status in patients with DLEAD is more serious than that in patients with diabetes, and the serum IL-1β level might be a biomarker of DLEAD.
In this study, the expression of the NLRP3 inflammasome (NLRP3, ASC, caspase-1) was significantly increased in the arteries of patients with DLEAD, indicating that activation of the NLRP3 inflammasome may be involved in the pathological progress of DLEAD. In human carotid atherosclerotic plaques, the NLRP3 inflammasome (NLRP3, ASC and caspase-1) is overexpressed and is associated with plaque stability.21 23 24 Furthermore, inhibition of NLRP3 inflammasome activation can alleviate vascular smooth muscle cells calcification and decrease the IL-1β level.25 Thus, our findings suggest that the inflammatory state mediated by the NLRP3 inflammasome might trigger atherosclerosis and vascular calcification and aggravate diabetic vascular injury. However, what causes NLRP3 inflammasome activation in DLEAD?
Previous studies have suggested that activation of the NLRP3 inflammasome is complex and may involve a multifactorial, multipathway activation process.26 NEK7, which was recently discovered, is a key protein in the activation of the NLRP3 inflammasome in the potassium outflow mode.11 NEK7 acts as a potassium-specific receptor and can interact with NLRP3 inflammasome subunits in the case of potassium efflux.12 26 Furthermore, NEK7 can mediate NLRP3 polymerization, punctate ASC aggregation, and activate the NLRP3 inflammasome.26–28 Some scholars have suggested that NEK7 may be another component of the NLRP3 inflammasome. 11 28 Oridonin forms a covalent bond with cysteine 279 of NLRP3 in the NACHT domain (one of the domains of NLRP3) to block the interaction between NLRP3 and NEK7, thereby inhibiting NLRP3 inflammasome assembly and activation and preventing inflammation.12 The NEK7/NLRP3 complex may play a protective role in systemic lupus erythematosus as it has shown low expression levels in the peripheral blood mononuclear cells of affected patients.13 At present, few reports exist on the NEK7/NLRP3 inflammasome signalling pathway, especially with regard to diabetes and its complications. Therefore, we examined the occluded lower extremity arteries of patients with diabetes. Our finding showed that NEK7 expression was significantly increased in occluded lower extremity arteries. This finding indicated that the NEK7/NLRP3 inflammasome signalling pathway might be involved in diabetic vascular injury. Furthermore, immunofluorescence staining indicated that expression of the NEK7/NLRP3 inflammasome signalling pathway mainly occurred in vascular smooth muscle cells. These results suggest that activation of the NEK7/NLRP3 inflammasome in vascular smooth muscle cells might be the key mechanism of DLEAD. Therefore, inhibition of NEK7/NLRP3 inflammasome pathway activation in vascular smooth muscle cells might become a new target for delaying DMA.
The present study has some limitations. First, only a small number of patients with diabetes was included. Second, we could not explain in detail how hyperglycemia and advanced glycation end products activate the NEK7/NLRP3 inflammasome pathway in vascular smooth muscle cells and participate in the development of DLEAD. Further studies will focus on investigating the underlying mechanism of this process.
In summary, the present study demonstrated that the NEK7/NLRP3 inflammasome pathway might be involved in the pathogenesis of DLEAD (figure 6). The findings may provide new insight into the inflammation mechanism of DMA.
Figure 6.
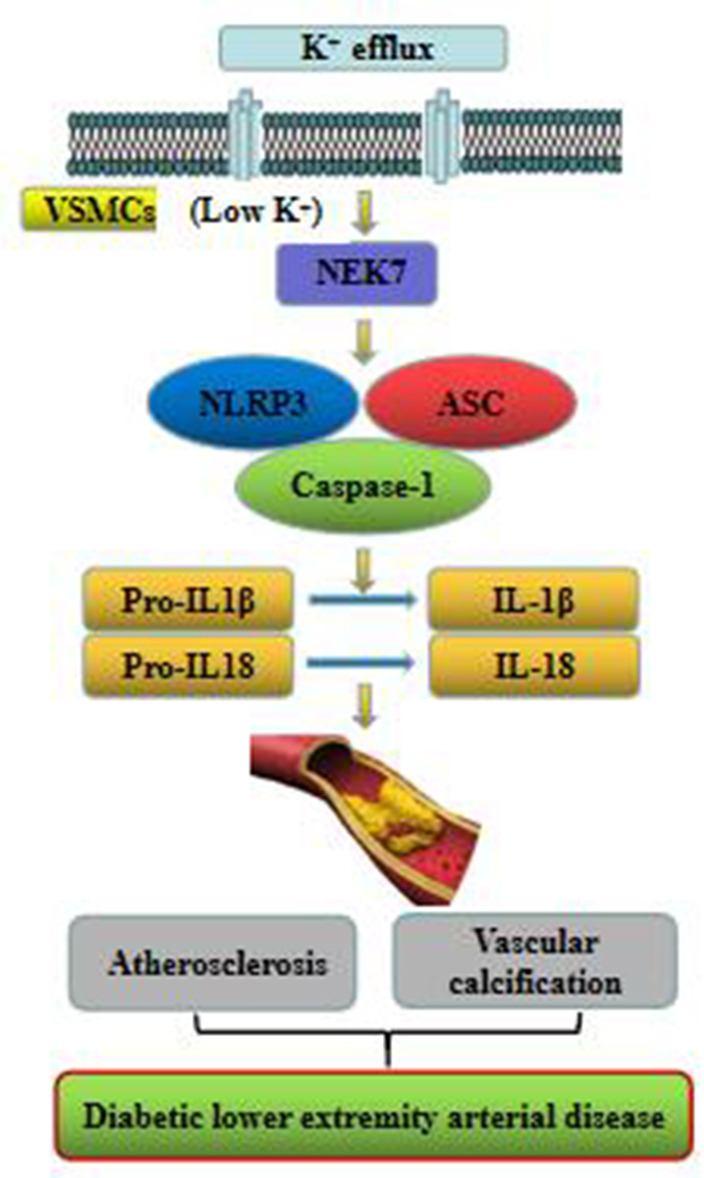
Expression of the NEK7/NLRP3 inflammasome pathway in patients with diabetic lower extremity arterial disease. ASC, apoptosis-associated speck-like proteins containing card; IL, interleukin; VSMCs, vascular smooth muscle cells.
Acknowledgments
The authors thank pathologists ShuJun Zhang and MingHan Zhu from the Pathology Department of the Fourth Hospital of Harbin Medical University for help with the stain of pathology.
Footnotes
HC and PW contributed equally.
Contributors: HC contributed to the study design, data collection, statistical analysis, manuscript writing and approval. PW contributed to data collection and approval. XD contributed to ultrasonography. BZ contributed to the WB analysis process and manuscript revision.
Funding: The present study was supported by ‘Chunhui plan’ Cooperative Scientific Research Project of the Ministry of Education of China (Grant no. HLJ2019021 to HC), Post-doctoral funding from the Government of Heilongjiang Province (Grant no. LBH215-178 to HC), the Health and Family Planning Commission of Heilongjiang Province (Grant no. 2016–119 to HC), the Cultivating Fund of the Fourth Affiliated Hospital of Harbin Medical University (Grant no. HYDSYPY201508 to HC), the Provincial Youth Science fund of Heilongjiang Province (Grant no. QC2016106 to BZ), the Heilongjiang Natural Science Foundation (Grant no.ZD201701 to XD), and the National Natural Science Foundation of China (Grant no. 81 271 646 to XD).
Competing interests: None declared.
Patient consent for publication: Not required.
Ethics approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Ethical approval for this study was obtained from the ethics committee of the Fourth Hospital of Harbin Medical University, Harbin, People's Republic of China (2019-SCILLSC-09).
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request. Some or all data, models, or code generated or used during the study are available from the corresponding author by request.
References
- 1.Madonna R, Pieragostino D, Balistreri CR, et al. . Diabetic macroangiopathy: pathogenetic insights and novel therapeutic approaches with focus on high glucose-mediated vascular damage. Vascul Pharmacol 2018;107:34 10.1016/j.vph.2018.01.009 [DOI] [PubMed] [Google Scholar]
- 2.Katakami N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J Atheroscler Thromb 2018;25:27–39. 10.5551/jat.RV17014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strowig T, Henao-Mejia J, Elinav E, et al. . Inflammasomes in health and disease. Nature 2012;481:278–86. 10.1038/nature10759 [DOI] [PubMed] [Google Scholar]
- 4.Xu C, Lu Z, Luo Y, et al. . Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat Commun 2018;9:4092. 10.1038/s41467-018-06522-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferreira NS, Bruder-Nascimento T, Pereira CA, et al. . Nlrp3 inflammasome and mineralocorticoid receptors are associated with vascular dysfunction in type 2 diabetes mellitus. Cells 2019;8:1595. 10.3390/cells8121595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fusco R, Siracusa R, Genovese T, et al. . Focus on the role of NLRP3 inflammasome in diseases. Int J Mol Sci 2020;21:4223 10.3390/ijms21124223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen H, Gris D, Lei Y, et al. . Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011;12:408–15. 10.1038/ni.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B, Liu Y, Sui Y-B, et al. . Cortistatin inhibits NLRP3 inflammasome activation of cardiac fibroblasts during sepsis. J Card Fail 2015;21:426–33. 10.1016/j.cardfail.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 9.Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018;61:21–8. 10.1007/s00125-017-4390-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birnbaum Y, Bajaj M, Qian J, et al. . Dipeptidyl peptidase-4 inhibition by saxagliptin prevents inflammation and renal injury by targeting the Nlrp3/ASC inflammasome. BMJ Open Diabetes Res Care 2016;4:e000227. 10.1136/bmjdrc-2016-000227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Y, Zeng MY, Yang D, et al. . Nek7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016;530:354–7. 10.1038/nature16959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He H, Jiang H, Chen Y, et al. . Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun 2018;9:2550. 10.1038/s41467-018-04947-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma ZZ, Sun HS, Lv JC, et al. . Expression and clinical significance of the NEK7-NLRP3 inflammasome signaling pathway in patients with systemic lupus erythematosus. J Inflamm 2018;15:16. 10.1186/s12950-018-0192-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tendera M, Aboyans V, Sievert H, et al. . Esc guidelines on the diagnosis and treatment of peripheral artery diseases: document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteries: the task force on the diagnosis and treatment of peripheral artery diseases of the European Society of cardiology (ESC). Eur Heart J 2011;32:2851–906. 10.1093/eurheartj/ehr211 [DOI] [PubMed] [Google Scholar]
- 15.An N, Gao Y, Si Z, et al. . Regulatory mechanisms of the NLRP3 inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Front Immunol 2019;10:1592. 10.3389/fimmu.2019.01592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christ A, Günther P, Lauterbach MAR, et al. . Western diet triggers NLRP3-Dependent innate immune reprogramming. Cell 2018;172:e14:162–75. 10.1016/j.cell.2017.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yahagi K, Kolodgie FD, Lutter C, et al. . Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus. Arterioscler Thromb Vasc Biol 2017;37:191–204. 10.1161/ATVBAHA.116.306256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mozos I, Malainer C, Horbańczuk J, et al. . Inflammatory markers for arterial stiffness in cardiovascular diseases. Front Immunol 2017;8:1058. 10.3389/fimmu.2017.01058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peiró C, Lorenzo Óscar, Carraro R, et al. . IL-1β Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front Pharmacol 2017;8:363. 10.3389/fphar.2017.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durpès M-C, Morin C, Paquin-Veillet J, et al. . PKC-β activation inhibits IL-18-binding protein causing endothelial dysfunction and diabetic atherosclerosis. Cardiovasc Res 2015;106:303–13. 10.1093/cvr/cvv107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi X, Xie W-L, Kong W-W, et al. . Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis 2015;24:2455–66. 10.1016/j.jstrokecerebrovasdis.2015.03.024 [DOI] [PubMed] [Google Scholar]
- 22.Satoh M, Tabuchi T, Itoh T, et al. . Nlrp3 inflammasome activation in coronary artery disease: results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin Sci 2014;126:233–41. 10.1042/CS20130043 [DOI] [PubMed] [Google Scholar]
- 23.Lee J, Wan J, Lee L, et al. . Study of the NLRP3 inflammasome component genes and downstream cytokines in patients with type 2 diabetes mellitus with carotid atherosclerosis. Lipids Health Dis 2017;16:217. 10.1186/s12944-017-0595-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paramel Varghese G, Folkersen L, Strawbridge RJ, et al. . Nlrp3 inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc 2016;5:e003031. 10.1161/JAHA.115.003031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen C, Yang X, Yan Z, et al. . Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol 2013;168:2242–7. 10.1016/j.ijcard.2013.01.211 [DOI] [PubMed] [Google Scholar]
- 26.Gong T, Yang Y, Jin T, et al. . Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol 2018;39:393–406. 10.1016/j.it.2018.01.009 [DOI] [PubMed] [Google Scholar]
- 27.Sharif H, Wang L, Wang WL, et al. . Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019;570:338–43. 10.1038/s41586-019-1295-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H, Wang Y, Li X, et al. . Nlrp3 activation and mitosis are mutually exclusive events coordinated by Nek7, a new inflammasome component. Nat Immunol 2016;17:250–8. 10.1038/ni.3333 [DOI] [PMC free article] [PubMed] [Google Scholar]