

Abstract
Melatonin (MLT) is a well-known pineal hormone possessed with remarkable biological activities. However, its low oral bioavailability and high first-pass metabolism rate are important pharmacokinetics problems. Therefore, 5 MLT derivatives (1-5) were designed and synthesised in our group to solve these problems. In this work, in silico analysis of all synthetic derivatives for pharmacokinetic and drug-likeness parameters were predicted by SwissADME software. The results revealed that all derivatives (1-5) met the requirements for ideal oral bioavailability and CNS drugs. The molecular docking showed that the acetyl-MLT derivative (1) and the un-substitution at N1-position derivative 5 would be substrates of CYP1A2, while the lipophilic substituted N1-position derivatives 2-4 could not be metabolised by CYP1A2. Moreover, all N-amide derivatives (1-4) were hydrolysed and released less than 2.33% MLT after 4-hour incubation in 80% human plasma. It seemed that these derivatives preferred to behave like drugs rather than prodrugs of MLT. These findings confirmed that the addition of bulky groups at the N1-position of the MLT core could prolong the half-life, increase drug absorption and penetrate the blood brain barrier into the CNS.
Keywords: Melatonin, N-amide derivative, metabolism, ADME, molecular docking
Introduction
Melatonin (N-acetyl-5-methoxytryptamine; MLT) is a hormone secreted primarily from the pineal gland in the brain. In humans, MLT is biosynthesised from tryptophan via a 4 step pathway. Firstly, hydroxylation at the 5-position of the indole ring is catalysed by tryptophan 5-hydroxylase and carboxylation subsequently occurs to generate serotonin. After that, the amine group of serotonin is acetylated and the hydroxyl group is methylated to obtain MLT.1 MLT is a pleiotropic molecule that can play a key role in a variety of important physiological functions or in pathological conditions like cancer, inflammation and neurodegeneration.2-4 MLT crosses cell membranes and acts through non-receptor mediated mechanisms as a scavenger for reactive oxygen species and reactive nitrogen species, either by directly reducing the concentration of highly reactive hydroxyl radicals or by stimulating the antioxidative enzymes superoxide dismutase and glutathione peroxidase.2,3 Moreover, MLT could efficiently protect neuronal cells from neurotoxins such as amyloid-β (Aβ) via antioxidant and anti-amyloid properties.5 In clinical trials, the toxicity of melatonin is remarkably low, and no serious side effects have been reported.4,6
Many studies have examined the pharmacokinetics of MLT and its bioavailability. The pharmacokinetic studies found that exogenously administrated MLT displays poor bioavailability (3%-56%) as a consequence of its extensive hepatic first-pass metabolism. The half-life of exogenous MLT is between 12 and 48 minutes. Cytochrome P450 enzyme CYP1A2 plays a major role in the metabolism of MLT.7 This hepatic enzyme converts MLT to 6-hydroxymelatonin, which is subsequently bound to sulphate and glucuronide and excreted in the urine.8,9 Our group has designed the derivatives of MLT with the aim to overcome the pharmacokinetic problems of MLT (Figure 1). Lipophilic derivatives of MLT demonstrated a remarkable antioxidant activity and have been proposed to be neuroprotective agents.10,11 In this study, we investigated the MLT release profile of MLT derivatives to identify whether these compounds behaved like drugs or prodrugs. In addition, we employed computational methods to investigate the nature of the interaction between the derivatives and the major metabolising enzyme CYP1A2 and to determine their physicochemical parameters and the drug-likeness of the compounds.
Figure 1.

Structure of all compounds in this study.
Materials and Methods
In silico pharmacokinetics prediction
The pharmacokinetic properties of all compounds were predicted by an online accessible web tool: SwissADME program of the Molecular Modelling Group of the Swiss Institute of Bioinformatics,12 which is publicly available at http://www.swissadme.ch. The software computed various pharmacokinetic properties and descriptors: the octanol/water partitioning coefficient, aqueous solubility, brain/blood permeability, and human gastrointestinal absorption (HIA) capability. Percentage of oral absorption (%ABS) was calculated according to the method of Zhao et al13 using the following equation:
where TPSA was topological polar surface area
Metabolism prediction
To further investigate the binding mode of MLT derivatives, molecular docking calculations were performed using Autodock VINA.14 The crystal structure of human microsomal P450 1A2 (CYP1A2) in complex with alpha-naphthoflavone (PDB code: 2HI4, retrieved from the RCSB Protein Data Bank) was used as the receptor.15 The bound inhibitor and water molecules were removed and only the coordinates of the protein complexed with heme molecules were kept. The 3D structures of ligands were built and their geometry optimisations (HF/3-21G) were conducted utilising the Gaussian03 program.16 The AutoDockTool (ADT) version 1.5.6 was used to prepare all the docking input files.17 All hydrogen atoms were added to the enzyme. The Kollman united atom and Gasteiger-Marsili charges were assigned for protein and ligand, respectively.18 All of the compounds were docked into the active site of CYP1A2 with the centre of 1.50, 22.90 and 17.50 along x, y and z axis, respectively. A box size of 30 × 30 × 30 Å with a grid spacing of 1 Å was set up. During the docking calculation protocol, the protein was treated as a rigid body while rotation and translation of ligands were allowed. For each complex, the predicted binding free and mode of binding for the selection of the docking pose were based on information from previous work.19 To avoid the methylation process, the distance between the site of metabolism (SOM) of the ligand and the Fe(II) of heme should not be less than ca. 8 Å or the SOM of the ligand should point away from the heme ion. Molecular representation was prepared using Chimera.20
Determination of the hydrolysis percentages of compounds
All the synthesised compounds were preliminarily evaluated for their ability to release MLT using an 80% human plasma assay by a method slightly modified from previous reports.21,22 Briefly, a solution of 5 mM of each MLT derivative was prepared in acetonitrile (1 mL) and added to 9 mL of 80% human plasma in phosphate buffered solution (PBS) pH 7.4. This solution was incubated and gently shaken at 37 ± 0.5°C in a water bath. An aliquot of 400 µL was withdrawn from each tube after 0.5, 1, 2, 3 and 4 hours. The solution was added to cold acetonitrile (400 µL) and placed in an ice bath in order to stop any further hydrolysis. The mixture was centrifuged at 15 000 ×g and 4°C for 10 minutes. The clear supernatant (500 µL) obtained from each tube was filtered and analysed by HPLC. Chromatographic conditions were a stationary phase Venusil® C18 Plus (4.6 × 250 mm) column, 5 μm particle size (Bonna-Agela Technologies, China) at room temperature. The mobile phase consisted of acetonitrile and water (50:50) with a flow rate 1.0 mL/min. The excitation and emission wavelengths of fluorescence detection were set at 286 nm and 346 nm, respectively. Compound concentrations and percentages in the mixture were quantified by peak area calculation and analysis. Human plasma in this study was obtained from the blood bank of Srinagarind Hospital, Khon Kaen University. The protocol for the hydrolysis study of MLT derivatives was approved by the Khon Kaen University Ethics Committee for Human Research (HE621275).
Results and Discussion
ADME properties and drug-likeness
The physicochemical properties and drug-likenesses of the derivatives were calculated by the SwissADME program (Table 1). The derivatives presented n-octanol and water partition coefficient (cLogP) values in the range 1.99 to 4.17 (cLogP < 5), the number of hydrogen bond acceptors and donors in the derivatives were in the acceptable range (HBA < 10 and HBD < 5), and the molecular weight of all compounds was less than 500, which indicated that the derivatives met all criteria of Lipinski’s rule.23 Topological polar surface areas (TPSA) were found in the range of 51.04 to 60.33 Å2 and the number of rotatable bonds were less than 10, which accorded to Veber’s rule.24 Water solubility (cLogS) values ranged between –4.99 and –2.38, indicating soluble to moderately soluble, and the percentage oral absorption (%ABS) values of all compounds ranged from 88% to 90%, indicating that these derivatives would have good membrane permeability.
Table 1.
In-silico ADME properties of MLT, 5-MT and compounds 1-5.
| Compounds | MW | cLogP | cLogS | TPSA | NORTB | HBA | HBD | %ABS | Lipinski’s violation |
|---|---|---|---|---|---|---|---|---|---|
| MLT | 232.28 | 1.86 | –2.34 | 54.12 | 5 | 2 | 2 | 90.33 | 0 |
| 5-MT | 190.24 | 1.68 | –2.44 | 51.04 | 3 | 2 | 2 | 91.39 | 0 |
| 1 | 274.32 | 1.99 | –2.38 | 60.33 | 6 | 3 | 1 | 88.19 | 0 |
| 2 | 336.38 | 3.02 | –3.85 | 60.33 | 7 | 3 | 1 | 88.19 | 0 |
| 3 | 415.28 | 3.78 | –4.76 | 60.33 | 7 | 3 | 1 | 88.19 | 0 |
| 4 | 386.44 | 4.17 | –4.99 | 60.33 | 7 | 3 | 1 | 88.19 | 0 |
| 5 | 373.24 | 3.91 | –4.72 | 54.12 | 6 | 2 | 2 | 90.33 | 0 |
Abbreviations: %ABS: percentage of oral absorption; cLogP: calculated octanol/water partition coefficient; cLogS: solubility parameter; HBA: number of hydrogen bond acceptors; HBD: number of hydrogen bond donors; Lipinski’s violation: 0 is good and 4 is bad; MW: molecular weight; NORTB: number of rotatable bonds; TPSA: topological polar surface area.
The cLogP and TPSA values of the compounds were plotted to predict human intestinal absorption (HIA) and blood brain barrier (BBB) access (Figure 2). The egg-shaped plot is divided into 3 parts including a grey region (no HIA or BBB access), a white area (HIA) and a yolk (BBB access). MLT, 5-MT and all derivatives are present in the yolk part of the plot, indicating that the molecules probably permeated the BBB. This boiled-egg model also predicted whether MLT, 5-MT and derivatives were substrates of P-glycoprotein (PGP). Red dots (PGP–) represent compounds that are not substrates of the PGP CNS efflux transporter, while, blue dots (PGP+) represent compounds that are substrates of PGP and predicted to pass through the CNS. As shown in Figure 2, MLT, 5-MT and all derivative compounds are represented by red dots and not PGP substrates. Therefore, all derivatives passed the pharmacokinetic requirements for drug-like compound behaviour, suggesting that these compounds had good oral bioavailability and could be considered as CNS drugs.
Figure 2.
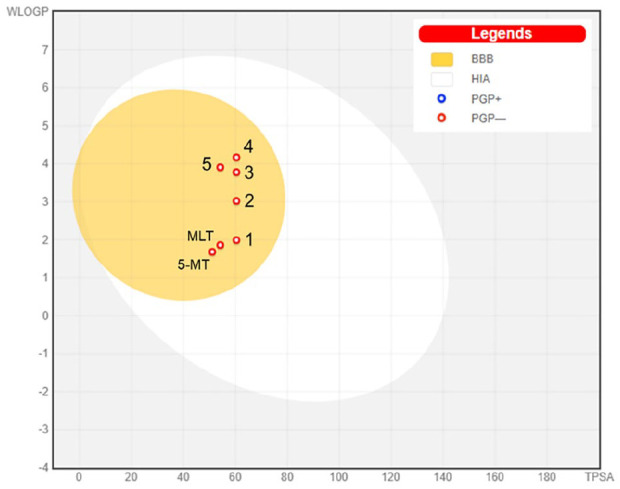
The boiled-egg plot of MLT, 5-MT and compounds 1-5.
Molecular docking
It has been reported that CYP1A2 principally hydroxylates MLT at the 6-position of the indole ring in in vitro and in vivo studies and in clinical trials.7,25-27 Therefore, this hepatic enzyme was selected for metabolism prediction by computer modelling. Moreover, 5-MT which is a metabolite of tryptophan is also known as a substrate of this enzyme.7 All derivatives were investigated for their coordination with the metabolising enzyme CYP1A2 to determine the effect of the lipophilic substituents on metabolism. The coordination protein-ligand binding energies and the distances between the SOM and the Fe(II) ion of heme are given in Table 2, while the representative binding modes of each coordinated complex are illustrated in Figure 3. The molecular docking results showed that MLT could undergo metabolism at the 6-position of the indole ring to form a 6-hydroxylated metabolite with binding energy of –9.2 kcal/mol. The NH of MLT formed hydrogen bonding interaction with a backbone oxygen of Asn312. The indole part made a face to face π-π interaction with Phe226 while acetamide moiety established hydrophobic interactions with sidechains of Phe125 and Ile117. Moreover, hydrophobic interactions between methoxyl group linking to indole ring and the non-polar residues Ile386 Leu497 were also observed. The coordinated distance between the SOM and the Fe(II) ion of heme was 8.60 Å. This accords with the study of Kesharwani et al.19 which showed that the plot of the experimental distance against the frames from the molecular dynamic simulations showed that this distance could fluctuate between 6 and 8 Å. The orientation of MLT showed that 5-methoxyl group faced to the heme in the binding pocket and the indole ring interacted with Phe residues. In addition, the distance between the SOM and the Fe(II) ion of heme could be lower to 4.15 Å when different crystal structure (PDB code: 1FAG) was used.26,28
Table 2.
Binding energy and distance between SOM of MLT derivatives and Fe atom of heme of the representative pose.
| Compounds | ΔG (kcal/mol) | Distance between SOM (C6) and Fe (II) atom (Å) |
|---|---|---|
| MLT | –9.2 | 8.60 |
| 5-MT | –8.2 | 8.60 |
| 1 | –8.5 | 7.32 |
| 2 | –11.4 | 9.24 |
| 3 | –7.4 | 10.82 |
| 4 | –8.6 | 9.34 |
| 5 | –11.2 | 5.14 |
| Indomethacin | –7.6 | 11.49 |
Figure 3.

Selected pose to represent the CYP1A2-ligand binding. Some surrounding amino acids near to the ligand are shown in wireframe while the ligand and the heme are represented by stick model.
The modelling showed that the acetylated derivative (1) would also be metabolised at the 6-position of the indole ring with a docking score –8.5 and a coordinated distance between the SOM and the Fe(II) ion of the distance of 7.31 Å. Compound 1 adopted different binding orientation to that of MLT, therefore, the hydrogen bonding interaction with Asn312 was not observed. The indole ring formed a face to face π-π interaction with Phe226. Moreover, the extended acetyl group in the indole part formed a non-polar interaction with the alkyl sidechain of Thr118. Similar to those detected MLT, hydrophobic interactions between methoxyl group linking to indole part and the non-polar residues Ile386 Leu497 were conserved. As mentioned earlier, 5-MT is a substrate of CYP1A2 and the modelling indicates that it could be metabolised at the same position as MLT with a docking score of –8.2 and a coordinated distance of 8.60 Å. This compound exhibited similar interaction profiles to those of MLT except that the hydrophobic interaction between ligand and Phe125 could not be detected. The substitution of a 4-bromobenzoyl group at the amine side chain of 5-MT (derivative 5) resulted in the completely different bound orientation from those of MLT related compounds. In this case, the hydrophobic interaction between the benzoyl group (instead of indole ring) and Phe226 were detected. Moreover, the oxygen of the methoxyl group formed hydrogen bonding interaction with the hydroxyl moiety of Thr124. However, the metabolic site of the compound was still the 6-position of the indole ring with a docking score of –11.2 and a coordinated distance of 5.14 Å. Therefore, compounds 1 and 5 are proposed to be metabolised in the same way as their parent compound, and therefore the half-life of these derivatives would not be different from MLT.
In contrast, the modelling indicated that the lipophilic substituted derivatives (2, 3 and 4) would not undergo hydroxylation at the 6-position of the indole ring. The addition of lipophilic groups hindered the Fe(II) atom’s access to the site of metabolism thus preventing hydroxylation. Major π-π interaction between indole ring of compound 2 and Phe226 was still preserved. The additional benzoyl group pointed towards heme and it was surrounded by non-polar residues Ala317, Ile386 and Leu497. Although the indole ring of compound 3 adopted somewhat different orientation from that of compound 2, the main π-π interaction with Phe226 was still conserved. The methoxyl moiety could possibly interact with Phe256 and Phe260 through hydrophobic interaction. Moreover, the 4-bromobenzoyl moiety oriented to the heme and this was similar to the case of compound 2 and the similar situation was also observed for compound 4. Hence, compounds 2, 3 and 4 would not be metabolised by CYP1A2 and their half-life would be prolonged compared to their parent compound. This phenomenon also occurs with indomethacin, a well-known anti-inflammatory drug, in which the chlorinated aromatic group of indomethacin hinders access to the site of metabolism. Indomethacin displayed bound conformation similar to that of compound 3 in which the halogenated benzoyl group located close to heme molecule. The indole ring also formed interacted with Phe226 through π-π interaction. The methoxyl moiety made hydrophobic interactions with the non-polar residues Ile117 and Phe226. The study of Nakajima et al.29 reported that indomethacin is mainly metabolised by CYP2C9 and only slightly metabolised by CYP1A2 and O-demethylation is the major metabolic pathway to form O-desmethylindomethacin. The biological half-life of indomethacin is about 5 to 10 hours.30 The possible metabolic pathway of compounds 2, 3 and 4 require further study.
Hydrolysis of MLT derivatives
All compounds were investigated for their ability to release MLT using 80% human plasma. Each compound was incubated with 80% human plasma in PBS (pH 7.4). An aliquot of 400 µL from each compound mixture was withdrawn after 0.5, 1, 2, 3 and 4 hours and the MLT releasing rate was determined by HPLC. Under the chromatographic conditions used, a melatonin peak was clearly detected at the retention time of 3.8 minutes. For the blank plasma, no endogenous compounds were seen at the MLT retention time. After 4 hours of incubation, the percentage of MLT released from the derivatives was less than 2.5% for all compounds (Figure 4). Compounds 1 and 3 released the most MLT, achieving release rates of 2.33 ± 0.55 and 2.13 ± 0.13%, respectively after 4-hour incubation (Figure 5). Compounds 2 and 4 released MLT at rates of 1.96 ± 0.16 and 1.63% ± 0.56%, respectively after 4-hour incubation. Therefore, it seemed that the N-amide derivatives did not behave like prodrugs of MLT.
Figure 4.
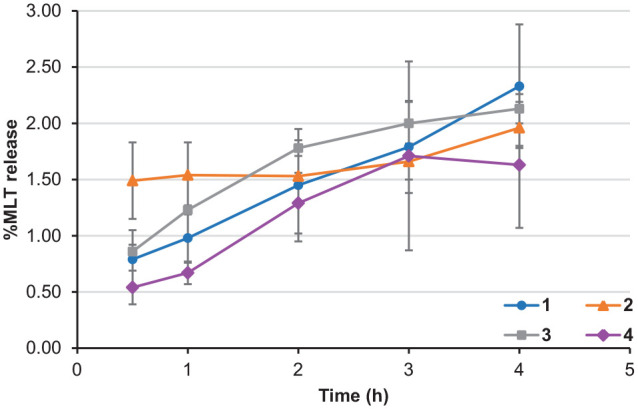
Hydrolysis profile of MLT derivatives after each interval of time.
Figure 5.

Selected chromatograms of blank plasma, MLT and compound 3 after incubation for 4 hours.
Conclusion
Lipophilic substituted MLT derivatives were designed to overcome the pharmacokinetic limitations of MLT. The N-amide derivatives (1-4) were relatively stable and released MLT at a rate of less than 2.33% after 4 hours in human plasma. It seemed that these derivatives preferred to behave like drugs rather than prodrugs of MLT. Pharmacokinetic analysis revealed that all derivatives (1-5) met the requirements of Lipinski’s and Veber’s rules of drug-likeness. Moreover, all compounds were predicted to cross the BBB and were not substrates of the PGP CNS efflux transporter. The addition of lipophilic groups (derivatives 2, 3 and 4) obstructed hydroxylation of MLT at the 6-position of the indole ring via CYP1A2. Therefore, compounds 2, 3 and 4 are potential orally bioavailable CNS MLT drugs.
Acknowledgments
The authors thank Dr. Glenn Borlace, Faculty of Pharmaceutical Sciences, Khon Kaen University for English language assistance.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Research and Academic Services from Khon Kaen University (RP63002), Graduate Research Fund Academic Year 2019 from National Research Council of Thailand (NRCT), The Office of the Higher Education Commission (Total Synthesis of Melatonin Project) and The Thailand Research Fund (DBG6080006), Thailand.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: PP (Ploenthip Puthongking) study conceptualization, methodology, formal analysis, investigation, resources, writing—original draft preparation, writing—review and editing, visualization, supervision, project administration and funding acquisition; PP (Panyada Panyatip) formal analysis, investigation, resources, writing—original draft preparation and writing—review and editing; NN methodology, formal analysis, investigation, resources, writing—original draft preparation and visualization.
ORCID iDs: Panyada Panyatip  https://orcid.org/0000-0003-4675-7132
https://orcid.org/0000-0003-4675-7132
Ploenthip Puthongking
https://orcid.org/0000-0002-5069-2071
References
- 1. Ye T, Yin X, Yu L, et al. Metabolic analysis of the melatonin biosynthesis pathway using chemical labeling coupled with liquid chromatography-mass spectrometry. J Pineal Res. 2019;66:e12531. [DOI] [PubMed] [Google Scholar]
- 2. Tan DX, Manchester LC, Terron MP, Flores LJ, Tamura H, Reiter RJ. Melatonin as a naturally occurring co-substrate of quinone reductase-2, the putative MT3 melatonin membrane receptor: hypothesis and significance. J Pineal Res. 2007;43:317-320. [DOI] [PubMed] [Google Scholar]
- 3. Paradies G, Petrosillo G, Paradies V, Reiter RJ, Ruggiero FM. Melatonin, cardiolipin and mitochondrial bioenergetics in health and disease. J Pineal Res. 2010;48:297-310. [DOI] [PubMed] [Google Scholar]
- 4. Tan DX, Manchester LC, Qin L, Reiter RJ. Melatonin: a mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int J Mol Sci. 2016;17:2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang JZ, Wang ZF. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol Sin. 2006;27:41-49. [DOI] [PubMed] [Google Scholar]
- 6. Cardinali DP. Melatonin: clinical perspectives in neurodegeneration. Front Endocrinol. 2019;10:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma X, Idle JR, Krausz KW, Gonzalez FJ. Metabolism of melatonin by human cytochromes p450. Drug Metab Dispos. 2005;33:489-494. [DOI] [PubMed] [Google Scholar]
- 8. Zetner D, Andersen LPH, Rosenberg J. Pharmacokinetics of alternative administration routes of melatonin: a systematic review. Drug Res. 2016;66:169-173. [DOI] [PubMed] [Google Scholar]
- 9. Farzaei MH, Hajialyani M, Naseri R. Melatonin. In: Nabavi SM, Silva AS, eds. Nonvitamin and Nonmineral Nutritional Supplements. Academic Press; 2019: 99-105. [Google Scholar]
- 10. Panyatip P, Puthongking P, Tadtong S. Neuroprotective and neuritogenic activities of melatonin and N-acetyl substituent derivative. IJPS. 2015;11:14-19. [Google Scholar]
- 11. Panyatip P, Johns NP, Priprem A, Nakagawa K, Puthongking P. Effect of N-Amide substitution on antioxidative activities of melatonin derivatives. Sci Pharm. 2020;88:3. [Google Scholar]
- 12. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao YH, Abraham MH, Le J, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res. 2002;19:1446-1457. [DOI] [PubMed] [Google Scholar]
- 14. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31: 455-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sansen S, Yano JK, Reynald RL, et al. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J Biol Chem. 2007;282:14348-14355. [DOI] [PubMed] [Google Scholar]
- 16. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 03, Revision C.02, Gaussian, Inc; 2004. [Google Scholar]
- 17. Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J Comput Aided Mol Des. 1996;10:293-304. [DOI] [PubMed] [Google Scholar]
- 18. Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron. 1980; 36:3219-3228. [Google Scholar]
- 19. Kesharwani SS, Nandekar PP, Pragyan P, Rathod V, Sangamwar AT. Characterization of differences in substrate specificity among CYP1A1, CYP1A2 and CYP1B1: an integrated approach employing molecular docking and molecular dynamics simulations. J Mol Recognit. 2016;29:370-390. [DOI] [PubMed] [Google Scholar]
- 20. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605-1612. [DOI] [PubMed] [Google Scholar]
- 21. Thoại PV, Nam NH. Design and synthesis of sustain-acting melatonin prodrugs. J Chem. 2013;2013:1-6. [Google Scholar]
- 22. Jenjirattithigarn N, Phunikom K, Kongpan T, Kanjanawart S, Johns JR, Tassaneeyakul W. Determination of melatonin in plasma by liquid-liquid extraction and high performance liquid chromatography coupled with fluorescence detection and its application for melatonin pharmacokinetic study in humans. Thai J Pharmacol. 2014;36:5-18. [Google Scholar]
- 23. Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337-341. [DOI] [PubMed] [Google Scholar]
- 24. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615-2623. [DOI] [PubMed] [Google Scholar]
- 25. Facciolá G, Hidestrand M, von Bahr C, Tybring G. Cytochrome P 450 isoforms involved in melatonin metabolism in human liver microsomes. Eur J Clin Pharmacol. 2001;56:881-888. [DOI] [PubMed] [Google Scholar]
- 26. Skene DJ, Papagiannidou E, Hashemi E, et al. Contribution of CYP1A2 in the hepatic metabolism of melatonin: studies with isolated microsomal preparations and liver slices. J Pineal Res. 2001;31:333-342. [DOI] [PubMed] [Google Scholar]
- 27. Härtter S, Ursing C, Morita S, et al. Orally given melatonin may serve as a probe drug for cytochrome P450 1A2 activity in vivo: a pilot study. Clin Pharmacol Ther. 2001;70:10-16. [DOI] [PubMed] [Google Scholar]
- 28. Lewis DF, Lake BG, George SG, et al. P.Molecular modelling of CYP1 family enzymes CYP1A1, CYP1A2, CYP1A6 and CYP1B1 based on sequence homology with CYP102. Toxicology. 1999;139:53-79. [DOI] [PubMed] [Google Scholar]
- 29. Nakajima M, Inoue T, Shimada N, Tokudome S, Yamamoto T, Kuroiwa Y. Cytochrome P450 2C9 catalyzes IndomethacinO-Demethylation in human liver microsomes. Drug Metab Dispos. 1998;26:261-266. [PubMed] [Google Scholar]
- 30. Helleberg L. Clinical pharmacokinetics of indomethacin. Clin Pharmacokinet. 1981;6:245-258. [DOI] [PubMed] [Google Scholar]