

Abstract
Background:
CD6 is an important regulator of T cell function that interacts with the ligands CD166 and CD318. To further clarify the significance of CD6 in rheumatoid arthritis (RA), we examined the effects of targeting CD6 in the mouse collagen-induced arthritis (CIA) model, using CD6 knockout (KO) mice and CD6 humanized mice that express human CD6 in lieu of mouse CD6 on their T cells.
Methods:
We immunized wild type (Wt) and CD6 gene KO mice with a collagen emulsion to induce CIA. For treatment studies using humanized CD6 mice, mice were immunized similarly and UMCD6 (a mouse anti-human CD6 IgG) or control IgG was injected on days 7, 14, and 21. Joint tissues were evaluated for tissue damage, leukocyte infiltration, and local inflammatory cytokine production. Collagen-specific Th1, Th9 and Th17 responses and serum levels of collagen-specific IgG subclasses were also evaluated in CIA Wt and CD6 KO mice.
Results:
Absence of CD6 reduced 1) collagen-specific Th9 and Th17, but not Th1 responses, 2) many pro-inflammatory joint cytokines, 3) serum levels of collagen-reactive total IgG and IgG1, but not IgG2a and IgG3. Joint homogenate hemoglobin (Hb) content was significantly reduced in CIA CD6 KO compared to Wt mice (reduced angiogenesis). Moreover, treating CD6 humanized mice with mouse anti-human CD6 monoclonal antibody (mAb) was similarly effective in reducing joint inflammation in CIA.
Conclusions:
Taken together, these data suggest that interaction of CD6 with its ligands is important for perpetuation of CIA and other inflammatory arthritides that are T cell driven.
Keywords: CD6, CD318, CIA, rheumatoid arthritis
Introduction
CD6 is a 105–130kDa cell surface glycoprotein that is expressed by almost all mature T lymphocytes, many natural killer (NK) cells, and a small subset of B lymphocytes. Evidence for a role of CD6 in T cell activation includes effects of anti-CD6 monoclonal antibodies (mAbs) on proliferation of freshly isolated T cells and recall responses of T cell clones (1, 2), influence of CD6 on T cell receptor-associated signaling events (2–6), altered activation and differentiation of T cells from CD knockout (KO) mice (7, 8), and effects of CD6 on thymic selection of developing T cells (7, 9). Recent data also suggest that CD6 may be very important in T cell-driven autoimmune diseases both in humans and in animal models. Loci associated with CD6 affect susceptibility to multiple sclerosis (MS) (10), Behcet’s syndrome and severity of psoriasis (11). Notably, an anti-CD6 mAb is effective in the treatment of psoriasis (12).
Although CD6 was one of the earliest “CD antigens” to be described, it is only recently that attention has refocused on the potential for CD6 as a treatment target in immune-mediated diseases. CD166, also known as ALCAM (activated leukocyte cell adhesion molecule), was the first endogenous ligand of CD6 to be identified (13), and is expressed on the surface of numerous types of hematopoietic and non-hematopoietic cells including activated lymphocytes and antigen-presenting cells (13). Recently, CD318, a cell surface molecule expressed on various mesenchymal and epithelial cell types (but not on mature lymphoid or myeloid cells), was also shown to be a ligand of CD6 (14). CD166 and CD318 bind to different domains of CD6 and appear to have distinct functions in autoimmune conditions. For instance, CD166 KO mice developed worse experimental autoimmune encephalomyelitis (EAE) (15) while CD318 KO mice, similar to CD6 KO mice, were protected from EAE (14). Interestingly, CD6, CD166 and CD318 are all strongly expressed in the rheumatoid arthritis (RA) joint (14, 16–18).
In RA, both CD166 and CD318 are expressed by fibroblast-like synovial cells (FLS) and these ligands participate in T cell binding to FLS (14, 18, 19). FLS also shed some of their surface CD318 into RA synovial fluid, and we have shown that soluble CD318 (sCD318) is chemotactic for T cells at concentrations equivalent to the gradient between RA synovial fluid and serum (14). These findings imply a role for the CD6-CD6-ligand axis in the pathogenesis of RA. To test the potential effectiveness of CD6-targeted therapeutics in the treatment of inflammatory arthritis we used two mouse models of collagen-induced arthritis (CIA): genetically-altered mice that lack CD6 – and mice in which human CD6 has been substituted for mouse CD6 to allow in vivo testing of anti-human CD6 monoclonal antibodies.
Materials and Methods
Animals.
Wild type (Wt) and CD6 KO mice, and CD6 humanized mice (DBA-1J background) (8) were maintained under pathogen-free conditions in the animal facility of the Lerner Research Institute, Cleveland Clinic. All procedures involving mice were approved by the Institutional Animal Care and Use Committee of Cleveland Clinic, and all were done in accordance with the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals and institutional guidelines.
Induction of murine CIA.
We immunized age- and sex-matched Wt and CD6 KO mice with a collagen emulsion to induce CIA as previously described (20). Mice were immunized at the base of the tail with bovine Type II collagen (Chondrex, Redmond, WA) emulsified (2mg/mL) at 1: 1 ratio in a custom-made complete Freund’s Complete Adjuvant (CFA) containing 4mg/mL M. tuberculosis (BD Biosciences, San Jose, CA) in a total volume of 50µL.
Clinical assessment of CIA.
We used a scoring system identical to previously published CIA studies (21). Briefly, each joint was inspected and assessed for the severity of swelling using scores of 0 (normal appearance), 1 (mild), 2 (moderate), and 3 (severe), yielding a maximum score of 12 for each mouse. Swollen digits were noted but paws were only considered arthritic when the entire paw was inflamed for 2 consecutive days. The day of onset of arthritis was recorded for each mouse. During the chronic phase of the arthritis, paws and digits were inspected for distortion and manipulated to identify loss of flexion (ankylosis). Severity scores were obtained from the addition of inflammation, distortion, and ankylosis scores. Scores for each mouse were used to derive a mean arthritis severity score for the groups at each time point examined. Development of CIA in the CD6 gene KO and Wt control mice was monitored by visual scoring every other day starting at 21 days after immunization until day 28.
For treatment studies using humanized CD6 mice, we immunized similarly, and mice were randomly separated into two groups after immunization on day 7. One group was injected intraperitoneally (i.p.) with 0.4 mg of a mouse anti-human CD6 IgG (UMCD6) (22) on days 7, 14 and 21, and the other group received the same amount of purified mouse IgG (Jackson ImmunoResearch, West Grove, PA) as a control. Mice were then monitored and clinical scores recorded three times a week in a blinded fashion. At the end of experiments, mice were euthanized, the peripheral blood lymphocytes, splenocytes and draining lymph nodes were analyzed for percentages of CD4+,and CD6+ T cells. Splenocytes were used to carry out antigen-specific Th1, Th9, and Th17 recall assays, and paws/joints were analyzed using histological and histochemical assays.
Histology of mouse joint tissue.
Some of the ankles were embedded in OCT for sectioning, Briefly, serial synovial tissue (ST) sections (5–8µm) were cut frozen and stored at −80°C for future staining with hematoxylin and eosin (H&E) staining and immunofluorescence. Frozen slides were allowed to thaw and dry at room temperature and subsequently fixed in acetone for 10 minutes at 4°C. For immunohistochemistry, tissue slides were blocked with 5% donkey serum and 20% fetal bovine serum (FBS) in phosphate buffered saline (PBS) at 37°C for one hour. After blocking, FITC labeled anti-mouse CD3 antibody was added and incubated for 2 hours at room temperature. FITC tagged mouse IgG served as a control. DAPI was used at a 1:5000 dilution in PBS for 5 minutes. Slides where then mounted in Fluoromount-G and visualized under a fluorescence microscope.
Mouse joint homogenate preparation and multiplex assays.
Mice were sacrificed and joints and sera were collected. Front paws and hind joints were used in the study. Joints were removed directly below the hairline and snap frozen in liquid nitrogen. All joints were stored at −80°C before processing. Each joint was thawed on ice and quickly homogenized on ice in 1 to 2mL phosphate-buffered saline (PBS) containing a tablet of proteinase inhibitors (10-mL PBS/tablet; Boehringer Mannheim, Indianapolis, IN, USA). Homogenized tissues were centrifuged at 2,000 x g at 4°C for 10 minutes - then filtered, aliquoted and stored at −80°C until analysis by ELISA. Protein concentrations were determined by a BCA kit (thermo-fisher) and the concentrations of 32 different mouse cytokines/chemokines in the homogenates were measured by a multiplex assay (Eve Technology) and normalized against the total protein of each tissue lysate.
ELISA for murine CD318.
We used a direct ELISA assay to measure the levels of murine CDCP1 (CD318) in mouse tissue joint homogenates (R&D Systems).
Serum collagen-specific IgG measurement.
To measure Type II collagen-specific IgG levels in the serum, blood samples were collected from the tail vein of each mouse and analyzed by ELISA. In brief, sera were diluted (1:1000) then incubated in wells of a 96-well ELISA plate coated with 10μg/mL purified Type II collagen. After washing, HRP-conjugated rabbit anti-mouse IgG (1:4000; Southern Biotech, Birmingham, AL) was added into each well and the levels of Type II collagen-specific IgGs in the sera were determined by measuring OD450 after color development using a TMB substrate (Thermo Fisher Scientific, Waltham, MA). To determine the levels of different Type II collagen-specific IgG subclasses, similar ELISAs were performed with IgGs specific for mouse IgG1, IgG2a, IgG2b, and IgG3 (Sigma-Aldrich, St. Louis, MO) as the detecting antibodies. All samples were run in duplicate.
Type II collagen-specific Th1, Th9, and Th17 cell recall assays.
Spleens were collected and processed from Wt and CD6 KO mice with CIA - or from CD6-humanized mice treated with UMCD6 or control mouse IgG after CIA induction. 0.4×106 splenocytes were incubated with or without 10µg/mL Type II collagen or the same concentration of a non-relevant peptide (OVA 323–339) in RPMI 1640 complete medium with 10% (vol/vol) FBS in each well of a 96-well plate. After 72 hours, IFN-γ, IL-9 and IL-17A levels in the culture supernatants were measured by ELISA following standard protocols. In brief, ELISA plates were coated with 4μg/mL anti-mouse IFN-γ IgG (Biolegend, San Diego, CA) or anti-mouse IL-9 IgG (BD Biosciences, San Jose, CA) or anti-mouse IL-17A IgG (Biolegend, San Diego, CA) overnight and then blocked with 1mg/mL BSA. Next, culture supernatants were added to each well and incubated for 2 hours. After washing, 1μg/mL biotinylated anti-mouse IFN-γ or IL-9 or IL-17a IgG was added into each well and incubated for another 1 hour. The plates were then developed after incubation with streptavidin-HRP (1:4000; Biolegend, San Diego, CA) and TMB substrate (Thermo Fisher Scientific, Waltham, MA). The concentrations of IFN-γ, IL-9, and IL-17a in the supernatants were calculated by measuring OD450. All samples were run in duplicate.
Hemoglobin (Hb) measurement on hind joint homogenates.
For measurement of Hb concentration, a reflection of vascularity of the tissue, mouse joints were homogenized as previously described. Hb levels were measured by adding 25uL of homogenate mixed with 25uL of 3,3,5,5-tetramethylbenzidine reagent to 96-well plates. Samples were incubated for 5 minutes at room temperature and absorbance measured using a Microplate Manager enzyme-linked immunosorbent assay reader (BioTek Instruments) at 450nm. Hb concentrations were determined by comparison with a standard curve. Values were normalized by dividing the Hb concentration by the amount of total protein in the homogenate.
Statistical Analysis.
Experiments were repeated at least twice. To determine whether statistically significant differences existed between groups, clinical scores were analyzed by using ANOVA test - whereas other results were compared using the Student’s t-test (p-values<0.05 were considered statistically significant). Data is graphed as mean ± SEM unless otherwise indicated.
Results
CD6 KO mice exhibit reduced clinical CIA severity:
We immunized age- and sex-matched CD6 KO mice or Wt controls with collagen emulsion (Chondrex) to induce CIA. Beginning on day 21, CIA clinical scores were recorded by a blinded investigator until day 28, when the Wt mice developed severe arthritis and required euthanasia according to the IACUC-approved study protocol. CIA severity was scored in each joint (front and hind) from Wt and CD6 KO mice. A reduction in CIA severity was evident on days 23 and beyond (figure 1) as differences were significant at all-time points except early on at day 21. The lower right panel shows that day 28 Wt mice have a robust inflammatory response to collagen that is not evident in the CD6 KO mice. This finding indicates that CD6 deficiency partially protected mice from CIA.
Figure 1. Clinical scoring shows reduction in CIA severity in CD6 knockout (KO) mice compared to Wt mice.
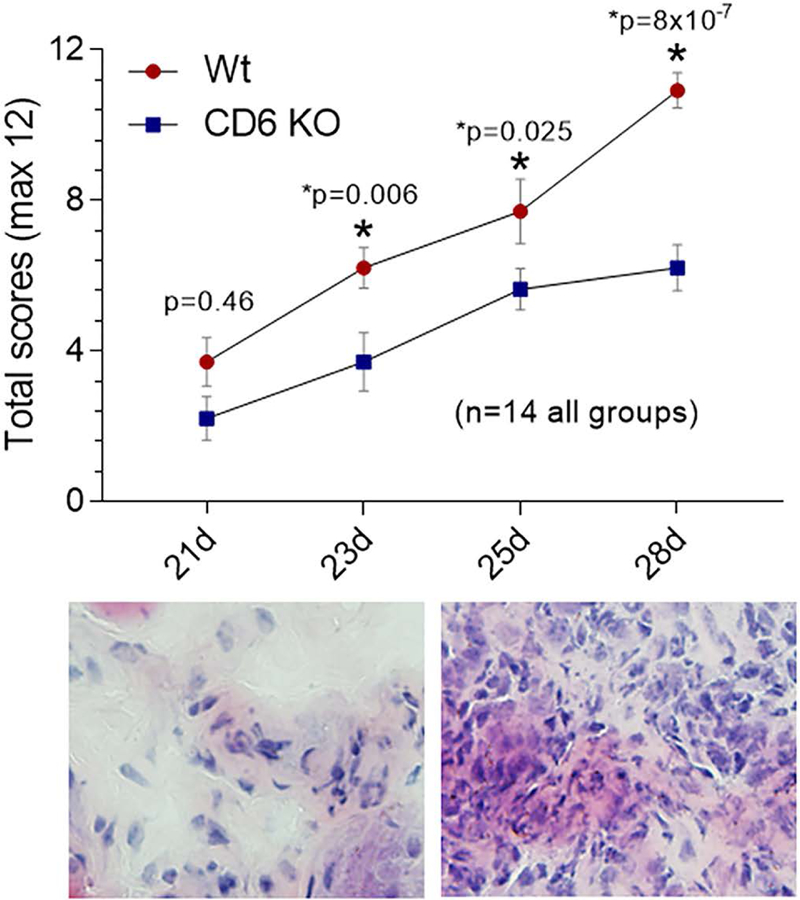
CIA severity was measured in each paw and ankle (front and hind joints). A total reduction in CIA severity was evident on days 23 and beyond. On day 28, Wt mice have a robust synovial inflammatory response (lower right panel) that is not evident in the CD6 KO mice (lower left panel, n=14 in each group). Data are presented as mean ± SEM (*p<0.05).
CD6 humanized mice administered with UMCD6 show attenuated clinical CIA severity.
To further assess CD6 as a potential target in the treatment of CIA, we induced arthritis in CD6 humanized DBA/1 mice and randomly divided these mice into two groups. The first group was injected i.p. with a mouse anti-human CD6 mAb (UMCD6) and a second group was injected similarly with purified mouse IgG on days 7, 14 and 21. To monitor CIA development, clinical scores were recorded in a blinded manner from day 21 to day 35. UMCD6 effectively mitigated the severity of joint swelling after day 21 (figure 2). Moreover, the lower left panel in figure 2 shows that mice administered IgG have demonstrable synovial lymphocyte staining (fluorescent green cells) on day 35 that is absent in the UMCD6-treated mice (lower right panel).
Figure 2. Clinical scoring shows a reduction of CIA severity in CD6-humanized mice administered UMCD6.

CIA severity was measured in each paw and ankle (front and hind joints) from CD6-humanized mice with CIA treated with either human IgG or UMCD6 antibody on days 7, 14 and 21. A total reduction of CIA severity was evident on days 28 and beyond. The lower left panel shows that mice administered IgG have demonstrable synovial CD3+ lymphocyte staining (fluorescent green cells) on day 35, that is not evident in the UMCD6 treated mice (lower right panel, n=14 in each group) data were mean ± SEM (*p<0.05).
CD6 humanized mice treated with UMCD6 exhibit reduced collagen-specific Th1/Th9/Th17 responses.
Collagen-specific Th1//Th9/Th17 recall assays using splenocytes from the UMCD6-treated CD6 humanized mice 35 days after CIA induction were used to evaluate pathogenic T cell responses. These experiments showed that levels of IFN-γ, IL-9 and IL-17a in the recall assays were significantly reduced in UMCD6-treated CIA mice compared to the IgG treated control CIA mice (figure 3). This demonstrates that UMCD6 profoundly impaired the pathogenic T cell responses in the CD6 humanized mice after CIA induction, which could be a mechanism underlying the reduced CIA clinical scores observed in the UMCD6-treated mice.
Figure 3. Reduced T cell cytokine production in response to collagen due to blocking or absence of CD6.
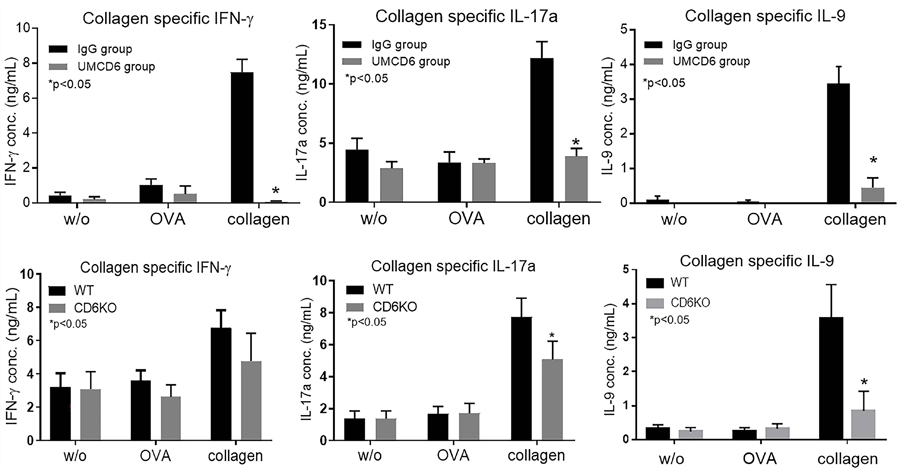
Th1//Th17/Th9 recall assays in UMCD6 treatment studies show that splenocytes from the IgG treated CIA group, but not the UMCD6 treated CIA mice, showed reactivity to collagen in vitro as measured by secretion of IFN-γ, IL-17 and IL-9. Controls include no antigen and a non-relevant antigen (OVA 323–339 peptide) (upper panel). We also performed collagen-specific recall assays using splenocytes collected from CD6 KO or Wt mice after CIA induction. These assays demonstrated significantly reduced collagen-specific Th17 and Th9 but not Th1 responses (lower panel). The ELISA assays have sensitivities in the picogram range for each of the cytokines tested. Data are mean ± SEM (*p<0.05).
CD6 KO mice exhibit reduced collagen-specific Th9 and Th17 responses following CIA induction:
We collected splenocytes from Wt and CD6 KO mice with CIA on day 28 to assess the collagen-specific Th1, Th9 and Th17 responses. The corresponding measurements of IFN-γ, IL-9 and IL-17a levels in the culture supernatants from recall assays indicated that the collagen-specific Th9 and Th17 responses of CD6 KO mice were significantly reduced compared to those of Wt mice. Although CD6 KO mice tended to also exhibit reduced collagen-specific Th1 responses, this difference was not statistically significant (figure 3).
CD6 KO mice develop reduced levels of collagen-specific total IgG and IgG1 but not IgG2a and IgG3.
Previous reports have implicated collagen-specific IgGs in the pathogenesis of CIA. Therefore, we used ELISAs to measure the levels of collagen-specific total IgG and collagen-specific IgG subclasses in sera collected on day 28. Notably, levels of collagen-specific IgGs were significantly reduced in sera from CD6 KO mice. In the IgG subclass analyses, we observed reduced levels of collagen-specific IgG1 in CD6 KO mice compared to Wt mice (figure 4), while the levels of IgG2a or IgG3 were not significantly affected (data not shown).
Figure 4. Total serum levels of IgG and IgG1 against collagen II are lower in CD6 KO CIA mice.
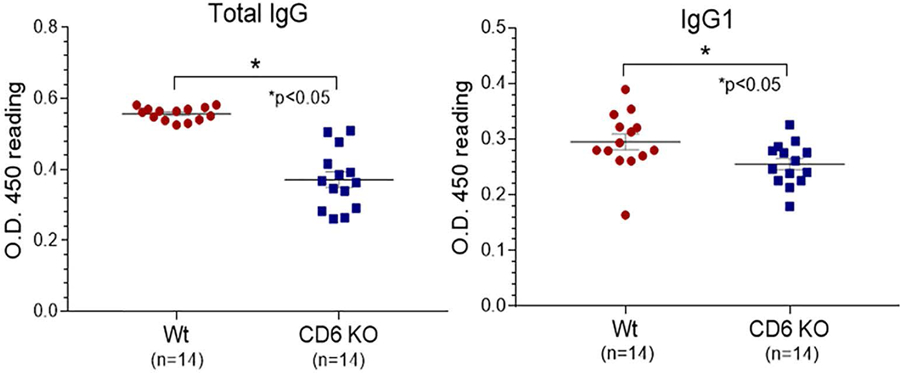
Total serum levels of IgG and IgG1 anti-collagen II were measured by ELISA in Wt and CD6 KO mice with CIA. Left panel: Total IgG anti-collagen II, Right panel: anti-collagen II IgG1. Data are mean ± SEM (*p<0.05).
CD6 KO mice show altered cytokine production profiles in the joints after CIA development.
Using a cytokine multiplex assay, the levels of 32 inflammatory cytokines from homogenates of collected paw and ankle joint tissues were measured and normalized against the concentrations of total proteins. The paws and hind joints from treated mice exhibited attenuated tissue damage and leukocyte infiltration consistent with the significantly reduced clinical scores in CD6 KO mice (figure 5 – upper and lower panels). In addition, the cytokine multiplex assays revealed that the levels of eotaxin, G-CSF, IFN-γ, IL-1β, IL-2, IL-5, IL-9, IL-12 (p70), IL-13, IL-17, IP-10, KC, MCP-1, M-CSF, MIP-1α, MIP-2, RANTES, TNF-α and VEGF were significantly lower in the CD6 KO group compared to the Wt group. In contrast, the levels of GM-CSF, IL-1α, IL-3, IL-4, IL-6, IL-7, IL-10, IL-12 (p40), IL-15, LIF, LIX, MIG and MIP-1β were comparable between the two groups of mice (figure 5).
Figure 5. Cytokine panels from joint homogenates of CIA CD6 KO and Wt mice.

CIA mouse joints from Wt and CD6 KO mice were homogenized and measured for protein concentrations. A cytokine multiplex assay was used to measure the levels of 32 cytokines in homogenates of each of the paw and ankle joint tissues. Cytokine levels were normalized to protein concentration for each joint tissue homogenate (pg cytokine/mg protein). Many pro-inflammatory cytokines were reduced in CD6 KO CIA mice compared to Wt CIA mice. The numbers listed below each cytokine are p-values for each cytokine measured. Numbers in red indicate p-values<0.05 (statistically significant).
UMCD6 modulates CD6 expression on CD4+ T cells without depleting this T cell population in treated mice.
On day 35 after CIA induction we obtained blood and draining lymph nodes from CD6-humanized mice treated with UMCD6 or mouse IgG and analyzed the percentages of CD4+ T cells and their CD6 expression levels by flow cytometry. The UMCD6-treated and mock-treated samples contained similar percentages of total CD4+ T cells. However, the percentage of CD4+/CD6+ T cells was significantly reduced in the UMCD6-treated mice, and this reduction was even more profound in the draining lymph nodes than in the peripheral blood (figure S1). A further analysis of CD4+ T cells from UMCD6-treated mice revealed that most T cells did not express detectable levels of CD6. Thus, UMCD6 treatment modulated the expression of CD6 on CD4+ T cells rather than depleting these T cells in CD6-humanized mice with CIA.
CD6 KO CIA mice have reduced Hb levels in their joint tissue homogenates.
Hb measurement from joint tissue homogenates revealed a significant reduction in Hb content in CIA CD6 KO mice compared to CIA Wt control mice - signifying attenuated angiogenic activity in the CD6 KO mice (figure S2). Reductions in Hb content was not observed in humanized CD6 CIA mice treated with UMCD6 compared to control mice (data not shown).
Discussion
CD6 is an important regulator of T cell function that mediates interactions of T cells with a wide variety of other cell types through engagement of its ligands CD166 and CD318. We previously examined CD318 expression in synovial tissue and sCD318 concentrations in synovial fluids (SFs) of RA patients and demonstrated a potential role for CD318 in recruitment and retention of T cells in the RA joints. To further clarify the significance of CD6 in RA, we examined the effects of targeting CD6 in the mouse CIA model using CD6 KO mice and CD6-humanized mice that express human CD6 in lieu of mouse CD6 on their T cells. Using these models, we analyzed the joints of the ankles and paws for tissue damage, leukocyte infiltration and local inflammatory cytokine production. We also compared collagen-specific Th1, Th9 and Th17 responses and serum levels of collagen-specific IgG subclasses and used cytokine multiplex assays to measure the levels of 32 cytokines in homogenates of joint tissues.
We found that CD6 KO mice had significantly lower clinical arthritis scores than Wt mice, suggesting that CD6 acts to tune the severity of joint inflammation in CIA. Absence of CD6 reduced 1) collagen-specific Th9 and Th17 responses, 2) many pro-inflammatory joint cytokines and chemokines, 3) serum levels of collagen-reactive total IgG and IgG1, but not IgG2a and IgG3, and 4) synovial angiogenesis in CD6 KO mice. Recall assays using collagen, or OVA 323–339 peptide as a non-relevant antigen, showed significant reductions in the production of IFN-γ, IL-17 and IL-9 in CIA mice when CD6 was targeted. These findings are in agreement with our previously published data that T cells from CD6 KO mice showed enhanced proliferation, but diminished Th1 and Th17 cell differentiation in response to anti-CD28 and anti-CD3 antibodies (8). Moreover, treating CD6-humanized mice with a mouse anti-human CD6 mAb was similarly effective in reducing joint inflammation in CIA. The reduction in inflammatory cytokines occurred without significant depletion of T cells and was even more profound in UMCD6 treated mice than in CD6 KO mice. Furthermore, we find that there were more synovial CD3+ lymphocytes in the tissue sections prepared from IgG-treated CIA mice compared to CIA mice receiving UMCD6 - likely due to an overall reduction in T cell recruitment to inflamed synovium in response to therapy.
The previously identified ligand of CD6 is ALCAM (CD166) - a member of the immunoglobulin gene superfamily which is widely expressed on cells of the immune system, including activated T and B lymphocytes, and on many types of mesenchymal, epithelial and endothelial cells in various organs and tissues. Indications that a second ligand of CD6 might exist arose initially from functional studies in which T cell adhesion to keratinocytes and synovial fibroblasts was found to be dependent on CD6 but not on CD166 (19). Moreover, these interactions involved a CD6 ligand whose expression was increased by IFN-γ, a cytokine that does not upregulate CD166 (17, 19). Subsequently, a mAb was generated that identified a 130 kDa molecule that also bound to a CD6-Ig fusion protein (18); we recently identified this CD6L as CD318 (14). CD318 is expressed by a variety of tissue cell types with which T cells interact in organ-targeted autoimmune diseases, but it is not expressed on mature cells of the immune system such as lymphocytes and myeloid cells (2, 18). CD166 and CD318 bind to distinct domains of CD6, and it appears possible that CD6 can bind both ligands concurrently (14).
Recent work suggests that the appearance of sCD318 in SF is a potential biomarker for RA and closely related conditions, and also highlights the importance of T cell interactions with FLS and with molecules shed from the FLS surface (14). UMCD6 treatment of CIA reduced lesional levels of sCD318, possibly through downregulation of expression of pro-inflammatory cytokines such as IFN-γ, which in turn upregulate CD318 expression. This effect on sCD318 levels was not observed in CD6 KO mice (unpublished). On the other hand, CD6 KO mice with CIA showed reduced synovial angiogenesis compared to Wt mice, but this effect was not seen in UMCD6-treated CD6-humanized mice. These differences suggest that, notwithstanding similar effects on T effector cell subset differentiation, genetic absence of CD6 versus use of anti-CD6 antibody impacts CIA by mechanisms that differ in subtle ways at the target organ - the synovium. Elucidation of the mechanistic basis for these differences will require additional experimental approaches, possibly including tissue-specific manipulation of the expression of CD6 ligands.
While no animal model is an exact replica of RA, the murine CIA model has proven to be very useful in studies of the pathogenesis of RA (20). CIA is typically induced in mice or rats by immunization with heterologous type II collagen, the principal type of collagen that is present in articular cartilage. In susceptible strains of rodents, an autoreactive response against type II collagen is induced, leading to synovitis and joint destruction over an interval of several weeks. As in human RA, multiple immune and inflammatory events and pathways are involved, including autoreactive Th1 and Th17 T cells, autoantibodies, pro-inflammatory cytokines and activated, tissue-destructive synovial cells (20). Moreover, synovial enrichment of Th9 cells shows a positive correlation with disease activity in RA, and synovial IL-9 prolongs the survival of neutrophils, increases their matrix metalloprotienase-9 production, and facilitates Th17 cell differentiation (23).
Since Hb levels have been correlated with joint inflammation and upregulated angiogenesis in RA, and in animal models of RA (24–26), we examined Hb levels as a measure of tissue vascularity in the joint tissue homogenates of the Wt and CD6 KO CIA mice. Our data shows significant reductions in joint Hb levels in the CD6 KO compared to Wt CIA mice – and this corresponds with reductions in several key angiogenic factors that we measured in our cytokine array, including vascular endothelial growth factor (VEGF).
In studies using CD6 KO mice, Orta-Mascaro and colleagues observed heightened autoimmunity and exacerbated disease in the CIA model (7). These experiments were performed in the C57BL/6 strain in which the severity of CIA is modest. In view of the apparent contradiction between our findings in EAE and these results in CIA, we performed experiments to assess the role of CD6 in CIA, using the DBA strain in which CIA is considerably more severe compared to C57BL/6 mice. We found that, as in EAE, absence of CD6 is indeed protective in CIA in the DBA strain, and that treatment with anti-CD6 has therapeutic effects.
CD6-humanized mice provide a useful pre-clinical in vivo model for demonstrating the effectiveness of anti-CD6 antibodies in the treatment of autoimmune conditions. UMCD6 significantly reduced inflammatory responses in the target organs of CIA, EAE and experimental autoimmune uveitis (EAU) in these mice (2, 27). Further, UMCD6 is an effective agent in suppressing differentiation of autoreactive lymphocytes into pathogenic, cytokine producing Th1 and Th17 cells. Similar alteration in the differentiation of Th1 and Th17 cells occurs in CD6 KO and CD318 KO mice (2).
Although there is some evidence that CD6 can augment signal transduction through the T cell receptor complex (3, 28), our recent studies in CD6-deficient mice indicate that CD6 attenuates the early stages of T cell activation and also enhances differentiation of Th1 and Th17 CD4 effector cells (8). These Th1 and Th17 CD4+ effectors then drive autoimmune disease(s), including RA. Precisely how CD6 signals upon engagement of its natural ligands is unknown, but the cytoplasmic region of CD6 can be phosphorylated by multiple kinases (29). Many details of how CD6 participates in T cell signaling remain unclear, including the positioning of CD6 with respect to the “immunological synapse” formed through contact of the T cell with antigen-presenting cells. Thus, the role of CD6 in fomenting organ-targeted autoimmunity may specifically reflect CD6 interaction with CD318, which is expressed on key tissue cells in these diseases including neurons and endothelial cells in the brain, synovial fibroblasts in the joint and keratinocytes in the skin (2, 18). In the 1980s, an IgM anti-CD6 was tested in clinical trials on MS (30) and allograft rejection (31), but the small size and uncontrolled design of those studies precluded determination of therapeutic efficacy. More recently, an anti-CD6 mAb termed itolizumab has been extensively studied in humans with psoriasis and was demonstrated to be of significant clinical benefit, leading to approval for treatment of psoriasis in India (12).
UMCD6 is a mAb that has very high affinity for CD6 (16) without depleting T cells in CD6-humanized mice. UMCD6 instead appears to strip CD6 from the T cell surface, likely through internalization. If anti-CD6 mAbs were similarly non-lymphocyte depleting in humans, host defenses against infection might be relatively well preserved compared to other immunosuppressive strategies, and could lead to a safety advantage for CD6-directed mAbs (2, 14). Our data herein, taken together with what is already known about the role of CD6 in other mouse models of autoimmune diseases, strongly suggests that CD6 is required for the development and perpetuation of inflammatory arthritis and other autoimmune disorders that are T cell driven, and that attenuation of such disorders is achievable through targeting of CD6.
Supplementary Material
Figure S1. Peripheral blood and lymph nodes from humanized CD6 CIA mice show a reduction in CD4+/CD6+ lymphocytes. CD4+ T cells from the draining lymph nodes (popliteal lymph nodes) and peripheral blood in UMCD6 or control IgG treated CIA mice were analyzed by flow cytometry after staining with a polyclonal anti-human CD6 mAb (R&D Systems) and a monoclonal anti-mouse CD4 mAb (Biolegend). The percentages of peripheral blood and lymph node T cells expressing CD4 did not show significant differences in CD6 humanized CIA mice when administered UMCD6 antibodies (upper and lower right panels). However, peripheral blood and lymph node T cells showed significant reductions in CD4+/CD6+ cells in CIA humanized mice, with profound reductions in lymph node CD4+ T cells expressing CD6 (upper and lower left panels). Thus, UMCD6 treatment did not reduce CD4+ percentages in the draining lymph nodes or peripheral blood, but it did reduce levels of CD6 on these T cells, potentially through endocytosis.
Figure S2. CD6 KO CIA mice have reduced Hb levels in their joint tissue homogenates. Hb measurement from joint tissue homogenates revealed a significant reduction in Hb content in CIA CD6 KO mice compared to CIA Wt mice - signifying attenuated angiogenic activity in these mice.
Acknowledgements
This work was supported by grants from the NIH (EY025373 to FL), Department of Defense (PR120641 to DAF), and from the Frederick G.L. Huetwell and William D. Robinson, M.D. Professorship in Rheumatology (DAF).
Footnotes
Conflicts of interest: none
References
- 1.Gangemi RM, Swack JA, Gaviria DM, Romain PL. Anti-T12, an anti-CD6 monoclonal antibody, can activate human T lymphocytes. J Immunol. 1989;143(8):2439–47. [PubMed] [Google Scholar]
- 2.Consuegra-Fernandez M, Lin F, Fox DA, Lozano F. Clinical and experimental evidence for targeting CD6 in immune-based disorders. Autoimmun Rev. 2018;17(5):493–503. [DOI] [PubMed] [Google Scholar]
- 3.Osorio LM, Ordonez C, Garcia CA, Jondal M, Chow SC. Evidence for protein tyrosine kinase involvement in CD6-induced T cell proliferation. Cell Immunol. 1995;166(1):44–52. [DOI] [PubMed] [Google Scholar]
- 4.Breuning J, Brown MH. T Cell Costimulation by CD6 Is Dependent on Bivalent Binding of a GADS/SLP-76 Complex. Mol Cell Biol. 2017;37(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hem CD, Ekornhol M, Granum S, Sundvold-Gjerstad V, Spurkland A. CD6 and Linker of Activated T Cells are Potential Interaction Partners for T Cell-Specific Adaptor Protein. Scand J Immunol. 2017;85(2):104–12. [DOI] [PubMed] [Google Scholar]
- 6.Roncagalli R, Hauri S, Fiore F, Liang Y, Chen Z, Sansoni A, et al. Quantitative proteomics analysis of signalosome dynamics in primary T cells identifies the surface receptor CD6 as a Lat adaptor-independent TCR signaling hub. Nat Immunol. 2014;15(4):384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orta-Mascaro M, Consuegra-Fernandez M, Carreras E, Roncagalli R, Carreras-Sureda A, Alvarez P, et al. CD6 modulates thymocyte selection and peripheral T cell homeostasis. J Exp Med. 2016;213(8):1387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Singer NG, Whitbred J, Bowen MA, Fox DA, Lin F. CD6 as a potential target for treating multiple sclerosis. Proc Natl Acad Sci U S A. 2017;114(10):2687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singer NG, Fox DA, Haqqi TM, Beretta L, Endres JS, Prohaska S, et al. CD6: expression during development, apoptosis and selection of human and mouse thymocytes. Int Immunol. 2002;14(6):585–97. [DOI] [PubMed] [Google Scholar]
- 10.De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41(7):776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Consuegra-Fernandez M, Julia M, Martinez-Florensa M, Aranda F, Catala C, Armiger-Borras N, et al. Genetic and experimental evidence for the involvement of the CD6 lymphocyte receptor in psoriasis. Cell Mol Immunol. 2018;15(10):898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krupashankar DS, Dogra S, Kura M, Saraswat A, Budamakuntla L, Sumathy TK, et al. Efficacy and safety of itolizumab, a novel anti-CD6 monoclonal antibody, in patients with moderate to severe chronic plaque psoriasis: results of a double-blind, randomized, placebo-controlled, phase-III study. J Am Acad Dermatol. 2014;71(3):484–92. [DOI] [PubMed] [Google Scholar]
- 13.Bowen MA, Patel DD, Li X, Modrell B, Malacko AR, Wang WC, et al. Cloning, mapping, and characterization of activated leukocyte-cell adhesion molecule (ALCAM), a CD6 ligand. J Exp Med. 1995;181(6):2213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enyindah-Asonye G, Li Y, Ruth JH, Spassov DS, Hebron KE, Zijlstra A, et al. CD318 is a ligand for CD6. Proc Natl Acad Sci U S A. 2017;114(33):E6912–E21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lecuyer MA, Saint-Laurent O, Bourbonniere L, Larouche S, Larochelle C, Michel L, et al. Dual role of ALCAM in neuroinflammation and blood-brain barrier homeostasis. Proc Natl Acad Sci U S A. 2017;114(4):E524–E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levesque MC, Heinly CS, Whichard LP, Patel DD. Cytokine-regulated expression of activated leukocyte cell adhesion molecule (CD166) on monocyte-lineage cells and in rheumatoid arthritis synovium. Arthritis Rheum. 1998;41(12):2221–9. [DOI] [PubMed] [Google Scholar]
- 17.Joo YS, Singer NG, Endres JL, Sarkar S, Kinne RW, Marks RM, et al. Evidence for the expression of a second CD6 ligand by synovial fibroblasts. Arthritis Rheum. 2000;43(2):329–35. [DOI] [PubMed] [Google Scholar]
- 18.Saifullah MK, Fox DA, Sarkar S, Abidi SM, Endres J, Piktel J, et al. Expression and characterization of a novel CD6 ligand in cells derived from joint and epithelial tissues. Journal of immunology. 2004;173(10):6125–33. [DOI] [PubMed] [Google Scholar]
- 19.Singer NG, Mitra R, Lialios F, Richardson BC, Marks RM, Pesando JM, et al. CD6 dependent interactions of T cells and keratinocytes: functional evidence for a second CD6 ligand on gamma-interferon activated keratinocytes. Immunol Lett. 1997;58(1):9–14. [DOI] [PubMed] [Google Scholar]
- 20.Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. 2007;2(5):1269–75. [DOI] [PubMed] [Google Scholar]
- 21.Ruth JH, Amin MA, Woods JM, He X, Samuel S, Yi N, et al. Accelerated development of arthritis in mice lacking endothelial selectins. Arthritis research and therapy. 2005;7(5):R959–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bott CM, Doshi JB, Morimoto C, Romain PL, Fox DA. Activation of human T cells through CD6: functional effects of a novel anti-CD6 monoclonal antibody and definition of four epitopes of the CD6 glycoprotein. Int Immunol. 1993;5(7):783–92. [DOI] [PubMed] [Google Scholar]
- 23.Chowdhury K, Kumar U, Das S, Chaudhuri J, Kumar P, Kanjilal M, et al. Synovial IL-9 facilitates neutrophil survival, function and differentiation of Th17 cells in rheumatoid arthritis. Arthritis Res Ther. 2018;20(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabquer BJ, Amin MA, Teegala N, Shaheen MK, Tsou P, Ruth JH, et al. Junctional adhesion molecule-C is a soluble mediator of angiogenesis. Journal of Immunology. 2010;185(3):1777–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isozaki T, Arbab AS, Haas CS, Amin MA, Arendt MD, Koch AE, et al. Evidence that CXCL16 is a potent mediator of angiogenesis and is involved in endothelial progenitor cell chemotaxis : studies in mice with K/BxN serum-induced arthritis. Arthritis Rheum. 2013;65(7):1736–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isozaki T, Amin MA, Arbab AS, Koch AE, Ha CM, Edhayan G, et al. Inhibitor of DNA binding 1 as a secreted angiogenic transcription factor in rheumatoid arthritis. Arthritis Res Ther. 2014;16(2):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Li Y, Qiu W, Bell BA, Dvorina N, Baldwin WM 3rd, et al. Targeting CD6 for the treatment of experimental autoimmune uveitis. J Autoimmun. 2018. [DOI] [PMC free article] [PubMed]
- 28.Ibanez A, Sarrias MR, Farnos M, Gimferrer I, Serra-Pages C, Vives J, et al. Mitogen-activated protein kinase pathway activation by the CD6 lymphocyte surface receptor. J Immunol. 2006;177(2):1152–9. [DOI] [PubMed] [Google Scholar]
- 29.Kobarg J, Whitney GS, Palmer D, Aruffo A, Bowen MA. Analysis of the tyrosine phosphorylation and calcium fluxing of human CD6 isoforms with different cytoplasmatic domains. Eur J Immunol. 1997;27(11):2971–80. [DOI] [PubMed] [Google Scholar]
- 30.Hafler DA, Fallis RJ, Dawson DM, Schlossman SF, Reinherz EL, Weiner HL. Immunologic responses of progressive multiple sclerosis patients treated with an anti-T-cell monoclonal antibody, anti-T12. Neurology. 1986;36(6):777–84. [DOI] [PubMed] [Google Scholar]
- 31.Carpenter CB, Milford EL, Reinherz EL, Schlossman SF, Tilney NL, Strom TB, et al. Monoclonal anti-T12 antibody as therapy for renal allograft rejection. Trans Assoc Am Physicians. 1983;96:84–92. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Peripheral blood and lymph nodes from humanized CD6 CIA mice show a reduction in CD4+/CD6+ lymphocytes. CD4+ T cells from the draining lymph nodes (popliteal lymph nodes) and peripheral blood in UMCD6 or control IgG treated CIA mice were analyzed by flow cytometry after staining with a polyclonal anti-human CD6 mAb (R&D Systems) and a monoclonal anti-mouse CD4 mAb (Biolegend). The percentages of peripheral blood and lymph node T cells expressing CD4 did not show significant differences in CD6 humanized CIA mice when administered UMCD6 antibodies (upper and lower right panels). However, peripheral blood and lymph node T cells showed significant reductions in CD4+/CD6+ cells in CIA humanized mice, with profound reductions in lymph node CD4+ T cells expressing CD6 (upper and lower left panels). Thus, UMCD6 treatment did not reduce CD4+ percentages in the draining lymph nodes or peripheral blood, but it did reduce levels of CD6 on these T cells, potentially through endocytosis.
Figure S2. CD6 KO CIA mice have reduced Hb levels in their joint tissue homogenates. Hb measurement from joint tissue homogenates revealed a significant reduction in Hb content in CIA CD6 KO mice compared to CIA Wt mice - signifying attenuated angiogenic activity in these mice.