

Abstract
The development of HIV-1 dual inhibitors is a highly innovative approach aimed at reducing drug toxic side effects as well as therapeutic costs. HIV-1 integrase (IN) and reverse transcriptase-associated ribonuclease H (RNase H) are both selective targets for HIV-1 chemotherapy, and the identification of dual IN/RNase H inhibitors is an attractive strategy for new drug development. We newly synthesized pyrrolyl derivatives that exhibited good potency against IN and a moderate inhibition of the RNase H function of RT, confirming the possibility of developing dual HIV-1 IN/RNase H inhibitors and obtaining new information for the further development of more effective dual HIV-1 inhibitors.
Graphcal Abstract
INTRODUCTION
The development of dual-action drugs is a promising approach to ameliorate drug–drug interactions, reduce toxic side effects, and suppress viral resistance selection.1–4 Among dual-action drugs, dual inhibitors are single compounds that are able to inhibit two enzyme activities. Several reports have shown that dual inhibitors may have a role in the treatment of different diseases such as Alzheimer,5 Parkinson,6 inflammation,7 and cancer.1,8,9 This approach had been attempted also in the virological arena, aiming to inhibit rhinovirus replication.10 Recently, tropolones,11–13 madurahydroxylactone,14 and 2-hydroxyisoquinolin-1,3(2H,4H)-diones15,16 have been reported to act as dual inhibitors against HIV-1, targeting viral integrase (IN) and reverse transcriptase (RT) ribonuclease H (RNase H) activities.
HIV-1 IN is the viral enzyme responsible for the integration of the proviral dsDNA into the cell host chromosome through two coordinated enzyme functions, both accomplished by the same active site.17 In the first reaction, termed 3′-end processing (3′-P), IN removes the two terminal nucleotides (GT) from each 3′-end of the dsDNA.17 In the second reaction, termed strand-transfer (ST), IN catalyzes a nucleophilic attack by the free 3′-OH of the viral processed DNA to the target chromosomal DNA, resulting in covalent joining of the two molecules. Several classes of integrase inhibitors have been identified;18 among these the diketoacids (DKAs) showed greatest promise, and the first DKA bioisoster, raltegravir (1), has been approved in 2007 for HIV-1 therapy.19
HIV-1 IN belongs to the functionally diverse superfamily of DDE(D) nucleotidyltransferases, whose other notable members include RNaseH and MuA, Tn5, and Mos1 transposases.17 The active sites of these enzymes typically contain three essential carboxylates that coordinate a pair of divalent metal cations (usually Mg2+). Thus, chelating inhibitors can be active across several classes of viral metal-dependent enzymes and chelation has been successfully used in drug design, also of dual inhibitors.20 In particular, DKAs have been reported to chelate the divalent cations in the IN active site,17,18 and notably, DKAs originally developed against HIV-1 IN have been also reported to inhibit the HIV-1 RNase H.21,22 The HIV-1 RT-associated RNase H function hydrolyzes the RNA strand of the replicative intermediate RNA:DNA hybrid and, hence, is essential for viral replication.23 Even though several compounds have been recently identified to inhibit this RT activity,24–30 up to today no RNase H inhibitor has been approved for HIV therapy. Therefore, this viral function is a very attractive target for drug development.
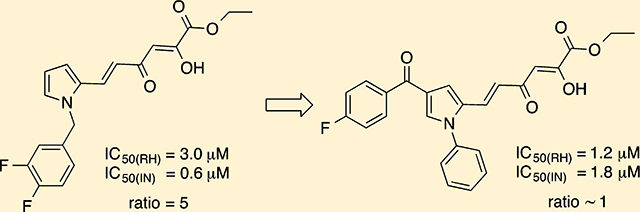
The first DKA IN inhibitor later described also as RNase H inhibitor was the 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid (BTDBA, 2) discovered by Merck.21 Recently we reported that 6-[1-(4-fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester (RDS 1643, 3) inhibited the HIV-1 RT-associated RNase H function in biochemical assays with an IC50 value of 8 μM, the HIV-1 IN ST (the IC50 value was 98 μM) and blocked the HIV-1 replication in cell based assays with an EC50 value of <0.2 μM (Chart 1).22 Starting from these observations, more recently we designed a small library of 3 analogues with the aim of obtaining dual IN/RNase H HIV-1 inhibitors and found compounds active at micromolar concentration against RNase H and low nanomolar IC50 values against IN in recombinant assays.31 The best dual inhibition was found for compounds showing a diketo ester group and fluorine atoms as substituents on the benzyl portion, as exemplified by compound 4 (Chart 1, IC50,IN = 0.6 μM, IC50,RH = 3.0 μM, EC50 = 2 μM). Furthermore, we extended these results to a quinolonyl diketo acid series, in which less marked dual activity was found, having in the best case IC50,IN = 26.2 μM, IC50,RH = 2.4 μM, and EC50 = 3.6 μM (compound 5).32
Chart 1.

Selective Inhibitor of IN Enzyme (1), First Described Dual IN/RNase H Inhibitor (2), and Recently Discovered Dual IN/RNase H Inhibitors 3–5
Herein we present the design, synthesis, and biological evaluation of new compounds related to 3 to better define the structure–activity relationship (SAR) within the most interesting pyrrolyl diketo acid series. All the newly designed pyrrolyl DKAs, both ester 6 and acid 7 derivatives, are shown in Chart 2. Basically, starting from 3, we fixed the pyrrole ring and the DKA chain while wider tranformations included one or more of the following modifications: (i) introduction of aromatic substituent in position 4 of the pyrrole ring; (ii) shift of the diketo hexenoic chain from 2 to 3 position of the pyrrole moiety; (iii) shortening of the diketo hexenoic branch into a diketo butanoic group; (iv) introduction of alkyl or aryl group replacing the fluorobenzyl moiety; (v) replacement of carboxylic function with a triazole ring; (vi) introduction of alkyl group within the DKA branch; (vii) replacement of keto group of DKA moiety with NH2 function. Notably, among the compounds described in this paper, 7k has been the first DKA derivative reported by Merck as selective ST IN inhibitor.33 We decided to include this compound in this study to define its properties as dual inhibitor of IN and RNase H.
Chart 2.

Newly Designed Pyrrolyl DKA Derivatives 6a–l and 7a–m
RESULTS AND DISCUSSION
Chemistry.
The synthesis of derivatives 6a–l and 7a–m is outlined in Schemes 1–4. Derivatives 6a–d and 7a–d were synthesized according to the pathway described in Scheme 1. The acetyl or propionyl pyrrole intermediates 9a–c were obtained by two different procedures: (1) the alkylation with 4-fluorobenzyl bromide in alkaline medium (K2CO3) of 1-(4-phenyl-1H-pyrrol-3-yl)ethanone34 or derivatives 8, achieved via toluene-4-sulfonylmethyl isocyanide (TosMIC) reaction from (E)-1-phenylpent-1-en-3-one;35 (2) termal transposition of the acetyl chain of 2-acetyl-1-[(4-fluorophenyl)methyl]pyrrole36 from 2- to 3-position of the pyrrole ring in the presence of CF3COOH. Derivatives 9a–c were condensed in turn with diethyl oxalate in the presence of sodium ethoxide to provide the diketobutanoic ethyl esters 6a–c. Compound 6a was used as substrate to provide (i) 6d, obtained by reacting the enol 6a with ammonium acetate in acid medium (CH3COOH) following a known procedure reported for DKA derivarives,37 and (ii) 7a, achieved by hydrolysis of ester 6a in the presence of 6 N NaOH. The last conditions have also been used to obtain 7b–c starting from 6b–c, respectively. A sligthly different condition has been used to obtain 7d, as previously described.37
Scheme 1.

Synthetic Route to Pyrrolyl DKAs 6a–d and 7a–da
aReagents and conditions: (i) TosMIC, NaH, Et2O/DMSO, room temp, 1 h; (ii) trifluoroacetic acid, 80 °C, 24 h; (iii) 4-F-benzyl bromide, K2CO3, DMF, 100 °C, 24 h; (iv) diethyl oxalate, C2H5ONa, THF, room temp, 2 h; (v) CH3COONH4, benzene, glacial acetic acid, reflux, 20 h; (vi) 1 N NaOH, THF/CH3OH, room temp, 1 h.
Scheme 4.

Synthetic Route to Pyrrolyl DKAs 6k,l and 7k,la
aReagents and conditions: (i) NIS, acetone, −78 °C, 96 h; (ii) phenylboronic acid, Cs2CO3, P(t-But)3, Pd2(dba)3, dioxane, 80 °C, 24 h; (iii) diethyl oxalate, C2H5ONa, THF, room temp, 2 h; (iv) 1-trityl-1H-[1,2,4]triazole-3-carboxylic acid ethyl ester,36 n-BuLi, THF, from −78 to 0 °C, 3.5 h; (v) 3 M HCl sol, 1,4-dioxane, 60 °C, 30 min; (vi) 1 N NaOH, THF/CH3OH, room temp, 1 h.
Derivatives 6e–h and 7e–h were obtained according to Scheme 2. The pyrroles 10 and 11 were used as starting materials to obtain the key intermediates 12e–h. Compound 10 was obtained by alkylation of commercially available 4-iodo-2-formylpyrrole in alkaline medium (K2CO3) with 4-F-benzyl bromide.
Scheme 2.

Synthetic Route to Pyrrolyl DKAs 6e–h and 7e–ha
aReagents and conditions: (i) alkylating agent, K2CO3, DMF, 100 °C, 24 h; (ii) phenylboronic acid, Cs2CO3, P(t-But)3, Pd2(dba)3, dioxane, 80 °C, 24 h; (iii) (1) DMF, 1,2-dichloroethane dry, oxalyl chloride, 0 °C, 15 min, room temp, 15 min; (2) 4-F-benzoyl chloride, AlCl3, room temp, 4 h; (iv) phenylboronic acid, copper(II) acetate anhydrous, pyridine/NMP 1:1, microwave at 60 W, 120 °C, 6 min; (v) acetone, 4 N NaOH, room temp, 24 h; (vi) diethyl oxalate, C2H5ONa, THF, room temp, 2 h; (vii) 1 N NaOH, THF/CH3OH, room temp, 1 h.
Derivative 11 was obtained starting from pyrrole that underwent a two-step one-pot reaction comprising (i) formylation in the presence of oxalyl chloride and DMF and (ii) Friedel–Crafts reaction with 4-F-benzoyl chloride. Thus, compound 11 was arylated to 12f or alkylated on nitrogen atom of pyrrole ring with the appropriate alkyl halide furnishing 12e and 12g, while intermediate 10 underwent a Suzuki coupling in position 4 to obtain 12h. The formylpyrroles 12e–h were condensed with acetone in alkaline medium (4 N NaOH) to obtain α,β-insaturated ketones 13e–h. The last compounds were in turn condensed with diethyl oxalate in the presence of sodium ethoxide to provide the diketobutanoic ethyl esters 6e–h that were finally hydrolyzed by reaction with 1 N NaOH to give the corresponding carboxylic acids 7e–h.
The synthetic pathway to obtain derivatives 6i,j and 7i,j is outlined in Scheme 3. The enones 17i,j were obtained starting from 15 and 16, respectively. Pyrrole 15 has been achieved starting from pyrrole-2-carboxaldehyde that was alkylated in alkaline medium (K2CO3) to obtain 14. The termal transposition of the formyl chain from position 2 to 3 of the pyrrole ring in the presence of trifluoroacetic acid led to derivative 15. In a parallel pathway (3E,5E)-6-phenylhexa-3,5-dien-2-one38 underwent ring closure by reacting with TosMIC, giving the pyrrole derivatives 16. Interestingly, the attack of the TosMIC reagent was specific on the 5,6 double bond of the starting dienone. Afterward, derivative 15 was condensed with acetone, affording 17i; conversely, compound 16 was converted into 17j by reaction with 4-F-benzyl bromide in alkaline medium (K2CO3). Finally, intermediates 17i,j were converted into esters 6i,j by condensation with diethyl oxalate and then converted to acids 7i,j by alkaline hydrolysis.
Scheme 3.

Synthetic Route to Pyrrolyl DKAs 6i,j and 7i,ja
aReagents and conditions: (i) 4-F-benzyl bromide, K2CO3, DMF, 100 °C, 24 h; (ii) trifluoroacetic acid, 80 °C, 24 h; (iii) acetone, 4 N NaOH, room temp, 24 h; (iv) Et2O/DMSO, NaH, TosMIC, room temp, 1 h; (v) 4-F-benzyl bromide, K2CO3, DMF, 100 °C, 24 h; (vi) diethyl oxalate, C2H5ONa, THF, room temp, 2 h; (vii) 1 N NaOH, THF/CH3OH, room temp, 1 h.
Derivatives 6k,l and 7k,l were obtained according to the pathway described in Scheme 4. The iodination of 2-acetyl-1-[(4-fluorophenyl)methyl]pyrrole in the presence of N-iodosuccinimide (NIS) afforded derivative 18, which underwent a Suzuki coupling reaction to give 19. Intermediates 19 and 2-acetyl-1-[(4-fluorophenyl)methyl]pyrrole were converted into the diketobutanoic acid derivatives 7k,l through the ethyl esters 6k,l, according to the condensation/hydrolysis procedure described above.
Finally, 2-acetyl-1-[(4-fluorophenyl)methyl]pyrrole was subjected to a condensation reaction in the presence of 1-trityl-1H-[1,2,4]triazole-3-carboxylic acid ethyl ester39 to afford the triazole-protected derivative 20, which was deprotected by hydrolysis in the presence of 3 M HCl to obtain the triazole derivative 7m.
Evaluation of Biological Activities.
All newly synthesized compounds 6a–l and 7a–m were tested in vitro in enzyme assays against both recombinant RNase H and IN. The IC50 values obtained for each compound in the inhibition of both the IN ST reaction and HIV-1 RT-associated RNase H function were plotted against each other in correlation plots (Figure 1). In these plots, single dots correspond to single compounds. The compounds are distributed around two perpendicular axes crossing the IN IC50 axis (X axis) at 1 μM and the RNase H IC50 axis (Y axis) at 10 μM (bolded crosshair in the center of each graph, Figure 1).
Figure 1.

Scatter plot for the inhibition of RNase H and IN enzymes. (A) Compounds are categorized according to their acidic or ester function. (B) Compounds are categorized according to the nature of their R substitution. (C) Compounds are categorized according to the nature of their R1 substitution. (D) Compounds are categorized according to the nature of their R2 substitution. Compounds with one IC50 value missing such as 6d have been left out of the plot, and compounds with IC50 values above 111 μM have been arbitrary positioned at the 100 μM value.
These two axes splice the graph into four quarters corresponding to RNase H/IN dual inhibitors (lower left quarter), RNase H-selective inhibitors (lower right quarter), IN selective inhibitors (upper left quarter), and inhibitors of lower potency (upper right quarter). As seen in Figure 1, these graphs do not show any particular correlation between RNase H and IN inhibition.
The newly synthesized pyrrolyl derivatives 6a–l and 7a–m exhibited good potency of inhibition when tested on the HIV-1 IN ST (Table 1). In general, as reported previously,31,32 the acid derivatives 7a–m were endowed with the highest potency showing IC50 values in the range of 26–0.019 μM. In our assay Merck compound 7k was confirmed as potent IN ST inhibitor showing IC50 = 0.057 μM (literature data IC50 = 0.08 μM).33 As seen in Figure 1A, 80% of the acid compounds (red dots) are distributed in the left half of the graph, suggesting the acid function is critical for IN inhibition but not critical for RNase H inhibition. Among them, seven compounds (7a,c,d,e,h,k,l) were proven to have submicromolar activity (IC50 value were in the range of 66–19 nM), while two derivatives (7b,i) can be considered less active to almost inactive (IC50 values were 111 and 26 μM, respectively). The most active compound was the 4-phenylpyrrole derivative 7l with an IC50 value of 19 nM, 3 times less active than 1 in inhibiting of the ST reaction of IN. Interestingly, even though not all the synthesized analogues were tested on the HIV-1 3′-P activity, results showed that the newly synthesized acids were selective inhibitors of the ST step, with the IC50 values on the 3′-P step 2–3 orders of magnitude higher with respect to the ones obtained on ST, thus confirming that the DKAs are selective ST inhibitors (data not shown).
Table 1.
Cytotoxicity, Enzymatic, and Antiviral Activities of Compounds 6a–l and 7a–m
| activity in enzyme assay, IC50
a |
antiviral activity |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R | R1 | R2 | X | RHb | STc | EC50 d | CC50 e | SIf |
| 6a | Ph | H | OH | Et | 10 | 0.42 | 0.56 | 11 | 19.6 |
| 6b | Ph | Me | OH | Et | |||||
| 6c | H | H | OH | Et | >100 | 1.6 | 0.90 | >50 | >55 |
| 6d | Ph | H | NH2 | Et | 72 | NT | 19 | >50 | |
| 6e | 4-FBzg | Bnh | Et | >100 | 4.3 | 19 | >50 | ||
| 6f | 4-FBzg | Ph | Et | 1.8 | 1.2 | 20 | >50 | ||
| 6g | 4-FBzg | Me | Et | 28 | >333 | 48 | >50 | ||
| 6h | Ph | 4-FBnh | Et | 13.4 | 2.5 | ||||
| 6i | H | Et | 55 | 90 | 50 | >50 | |||
| 6j | Ph | Et | 3.0 | >21 | 4.3 | 26.9 | 6.3 | ||
| 6k | H | COOEt | 21 | 0.51 | 1.2 | 33 | 27 | ||
| 6l | Ph | COOEt | 6.0 | 0.79 | 0.70 | 3.9 | 6 | ||
| 7a | Ph | H | OH | H | 7.5 | 0.022 | 0.66 | >50 | >75 |
| 7b | Ph | CH3 | OH | H | 64 | >111 | >50 | >50 | |
| 7c | H | H | OH | H | 41 | 0.024 | 0.58 | >50 | >86 |
| 7d | Ph | H | NH2 | H | 2.0 | 0.043 | 0.63 | >50 | >79 |
| 7e | 4-FBzg | Bnh | H | 7.5 | 0.063 | >50 | |||
| 7f | 4-FBzg | Ph | H | 100 | 0.59 | >50 | |||
| 7g | 4-FBzg | Me | H | 20 | |||||
| 7h | Ph | 4-FBnh | H | 23 | 0.066 | ||||
| 7i | H | H | 69 | 26 | >50 | >50 | |||
| 7j | Ph | H | 7.0 | 0.73 | 17.2 | >50 | >2.9 | ||
| 7ki | H | COOH | 54 | 0.057 | 1.0 | 28 | 28 | ||
| 7l | Ph | COOH | 14 | 0.019 | 0.7 | >50 | >72 | ||
| 7m | H | Trj | 26 | 0.11 | 20.4 | >50 | |||
| 1 | 0.007 | 0.016 | >250 | >15000 | |||||
| 2 | 3.2 | 1.9 | >50 | ||||||
| 3 | 8 | 98 | <0.2 | >50 | >250 | ||||
| 4 | 3 | 0.60 | 2 | >50 | >25 | ||||
| 5 | 26.2 | 2.4 | 3.6 | >50 | >13.8 | ||||
Inhibitory concentration 50% (μM) determined from dose–response curves.
Experiments performed against HIV-1 RT-associated RNase H activity.
Experiments performed against HIV-1 IN ST activity.
Effective concentration 50% (μM).
Cytotoxic concentration 50% (μM).
SI = CC50/EC50.
Bz, benzoyl.
Bn, benzyl.
See also ref 33.
Tr, triazolyl.
The newly synthesized pyrrolyl DKA derivatives 6a–l and 7a–m can be divided into two classes: the diketobutanoic (6a–d,k,l and 7a–d,k,l) and the diketohexenoic (6e–j and 7e–j) derivatives. In the diketobutanoic structures of derivatives 7a–d, 7k,l, two main differences involving the substitution of the pyrrole ring emerged: the diketobutanoic chain can be linked on the pyrrole ring into two different positions (2- or 3-position), along with a phenyl ring, which can be substituted in position 4 of the pyrrole ring. The two variables did not influence the IN inhibitory activity. Only the phenyl substitution at position 4 of the pyrrole ring (R substituent) seems to favor slightly IN vs RNase H inhibition with a majority of compounds bearing this substitution distributed in the two left quarters of the correlation graphs (green dots, Figure 1B). In fact, from a comparison of the inhibition data of diketobutanoic/diketohexenoic 7a/7l and 7c/7k, which are characterized respectively by the presence of the phenyl ring and by its absence, a correspondence in the orders of magnitude of their IC50 values was observed. Moreover, 7a and 7c linked the diketobutanoic chain in position 3 of the pyrrole ring, while 7k and 7l linked it in 2-position of the same ring. All these compounds were characterized by similar potencies (IC50 values of 22, 24, 57, and 19 nM, respectively). The 4-fluorobenzoyl substitution at position 4 of the pyrrole ring (R substituent) does not seem to favor the inhibition of either enzymes (orange dots, Figure 1B).
The other three modifications involved exclusively the diketobutanoic chain: (1) the substitution of the enolic OH with a NH2 (7d), (2) the introduction of a methyl group in 3-position (7b), and (3) the substitution of the carboxylic acid function with its bioisoster triazole ring (7m). The NH2 (7d) and triazole (7m) derivatives were 2-fold less active with respect to their OH (7a) and COOH (7h) counterparts, respectively (IC50 values were 43, 110, 22, and 57 nM, respectively). On the contrary, the 3-methylbutenoate derivative (7b) completely lost its ability to inhibit IN enzyme (IC50 values of >111 μM). It appears that IN seems to tolerate a wide variety of substitutions at the R1 position, including the absence of substituent, with a majority of compounds with such substitutions distributed in the two left quarters of the correlation graphs (Figure 1C). No preferential inhibition pattern can be observed for compounds with substitutions at the R2 position on the correlation plot presented in Figure 1 D.
When the two series of diketobutanoic and the diketohexenoic esters and acids were tested on the HIV-1 RT-associated RNase H function in biochemical assays, the most active derivatives were the diketohexenoic ester 6f and the diketobutanoic acid 7d, with IC50 value of 1.8, and 2 μM, respectively. Interestingly, both compounds were more active then the references 3 and 4. Noteworthy, the structure of 6f is related to the reference 3, since both are pyrrolyl diketohexenoic derivatives, but 6f bring a phenyl ring on nitrogen replacing the 4-F-benzyl group of 3 and a 4-F-benzoyl moiety linked in 4 position of the pyrrole ring. In the matter of compound 7d, it is a 3-pyrrolyl diketobutanoic acid derivative characterized by the presence of an amine function that replaces the enol OH in position 3 of the diketobutanoic chain. Since 7d is a carboxylic acid, its stronger inhibition toward IN than RNase H function was expected; conversely, the ester 6f is the best dual inhibitor IN/RNase H of this series. From a first analysis of the results we can state that (i) as known, the acid function confers a better inhibitory activity on IN, (ii) the ester function is amenable for inhibition of RNase H function of RT, confirming the results recently reported by the means of docking studies and mutagenesis experiments,40 and (iii) contemporaneously, the ester function is necessary for a dual inhibition IN/RNase H.
Among the ester derivatives (6a–l), the diketohexenoic compounds (6e–j, IC50 values in the range of 1.8–55 μM) were able to inhibit the HIV-1 RNase H function with a slightly higher potency then the diketobutanoic counterpart (6a–d,k,l, IC50 values in the range of 6–72 μM). Differently, when the acid derivatives (7a–m) were tested, this difference was not observed.
In both the diketohexenoic and the diketobutanoic ester and acid derivatives, the presence of a phenyl substituent in position 4 of the pyrrole ring influenced the inhibitory activity. In fact, the 4-phenyl substituted diketobutanoic ester and acid 6a/7a (IC50 values of 10 μM, 7.5 μM, respectively) and diketohexenoic 6j/7j (IC50 values of 3.0 μM, 7.0 μM, respectively) were consistently more active than the unsubstituted counterparts 6c/7c and 6i/7i (IC50 values of >100, 41, 55, and 69 μM, respectively).
In general, when the DKA chain was shifted from 2- to 3-position, a 2- to 4-fold increase in potency of inhibition was observed (compare 6h, 7h, and 7l with 6j, 7j, and 7a: IC50 values of 13.4, 23, 14, 3, 7, and 7.5 μM, respectively), with the sole exception of 6a and 6l, which showed comparable potency (IC50 values of 10 and 6 μM, respectively).
The substitution of the enolic OH on the diketobutanoic chain (6a/7a) with a NH2 group (6d/7d) in the ester series led to a 7-fold decrease of the potency of RNase H inhibition (6d, IC50 values of 72 μM) with respect to the unmodified counterpart (6a, IC50 value of 10 μM), while within the acid series, the NH2 derivative improved its potency of inhibition (7d, IC50 value of 2 μM; 7a, IC50 value of 7.5 μM). The introduction of a methyl group in position 3 of the diketobutanoic chain (7b) reduced the inhibition (7b and 7a IC50 values of 64 and 7.5 μM, respectively) and likewise the substitution of the carboxylic acid function with its bioisoster triazole (7a and 7m, IC50 values of 7.5 μM and 26 μM, respectively).
Among all the newly synthesized derivatives, the 2-pyrrolyl diketohexenoic ester 6f, athough not very potent, emerged as dual inhibitor showing similar IC50 values against both IN enzyme and RNase H function of RT (1.2 and 1.8 μM, respectively). This compound retains the ester function that is demonstrated to be necessary for the dual inhibiton31 and is characterized by a phenyl ring on nitrogen replacing the 4-F-benzyl group of 3 and by a 4-F-benzoyl moiety linked in 4 position of the pyrrole ring.
Cell-Based Assays.
Among the newly synthesized pyrrolyl DKA derivatives 6a–l and 7a–m, seven derivatives (6a,c,l and 7a,c,d,l) were characterized by a good anti-HIV activity showing a EC50 values in the submicromolar concentration (EC50 in the range of 0.56–0.9 μM) and a good selectivity index (SI). Three compounds, the 6k,j and 7k derivatives, showed an anti-HIV activity in the range of the micromolar concentration (EC50 in the range of 1–4.3 μM), while 10 compounds were less active or inactive (6d–i and 7b,e,f,i,m, EC50 in the range of 17.2 to >50 μM). In general, all the compounds had a low citotoxicity index (CC50 > 50 μM). Compound 7c, characterized by the diketobutanoic chain in 3-position of the pyrrole ring, showed the best antiviral efficacy on HIV-1 infected cell (EC50 = 0.58 μM) and a low cytotoxicity (CC50 > 50 μM, SI > 86). The best dual inhibitor derivative 6f that showed activity at micromolar concentration in biochemical assays was 20 times less potent in cell-based assays.
CONCLUSION
The development of HIV-1 dual inhibitors is a highly innovative approach aimed at reducing drug toxic side effects and therapeutic costs. Since HIV-1 IN and RNase H are both selective targets for HIV-1 chemotherapy, the identification of dual IN/RNase H inhibitors is an attractive strategy for new drug development. The newly synthesized pyrrolyl derivatives 6a–l and 7a–m exhibit good potency against IN and a moderate inhibition of the RNase H function of RT. In general, by comparison of the inhibition data among the ester and the acid derivatives, a different behavior was observed. As expected, the acid derivatives showed a higher potency of IN inhibition with respect to the corresponding esters, while the latter compounds have been often found more potent than the corresponding acids in inhibiting the RNase H function of RT enzyme. Notably, compound 6f, although not very potent on HIV-infected cell, showed a good correlation between HIV-1 IN and RNase H inhibition. It is characterized by a diketoester function, a phenyl ring on nitrogen, and a 4-F-benzoyl moiety linked in 4 position of the pyrrole ring. We can state that although the acid function confers a better inhibitory activity on IN, an ester function is amenable for inhibition of RNase H function of RT, and as a consequence, the ester function is necessary for a dual inhibition IN/RNase H. These basic chemical features should be considered for development of more potent dual inhibitors. Overally, the data reported in this work confirm the possibility of developing dual HIV-1 IN/RNase H inhibitors and give new information for the further development of effective dual HIV-1 inhibitors.
EXPERIMENTAL SECTION
Chemistry. General.
Melting points were determined on a Bobby Stuart Scientific SMP1 melting point apparatus and are uncorrected. Compound purities were always >95% determined by high pressure liquid chromatography (HPLC). HPLC analyses were carried out with a Shimadzu LC-10AD VP CTO-10AC VP. Column used was generally Discovery Bio Wide Pore C18 (10 cm × 4.6 mm, 3 μm). Infrared (IR) spectra were recorded on a PerkinElmer Spectrum-One spectrophotometer. 1H NMR spectra were recorded at 400 MHz on a Bruker AC 400 Ultrashield 10 spectrophotometer (400 MHz). Dimethyl sulfoxide-d6 99.9% (code 44,139–2) and deuterochloroform 98.8% (code 41,675–4) of isotopic purity (Aldrich) were used. Column chromatographies were performed on silica gel (Merck; 70–230 mesh) column or aluminum oxide (Sigma-Aldrich; 150 mesh) column. All compounds were routinely checked on TLC by using aluminum-baked silica gel plates (Fluka DC-Alufolien Kieselgel 60 F254) or aluminum oxide (Fluka DC-Alufolien). Developed plates were visualized by UV light. Solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of rotary evaporator (Büchi) operating at a reduced pressure (∼20 Torr). Organic solutions were dried over anhydrous sodium sulfate (Merck). All reactions were carried out under nitrogen. All solvents were freshly distilled under nitrogen and stored over molecular sieves for at least 3 h prior to use.
Microwave Irradiation Experiments.
Microwave reactions were conduced using a CEM Discover system unit (CEM. Corp., Matthews, NC). The machine consists of a continuous focused microwave-power delivery system with operator selectable power output from 0 to 300 W. The temperature of the contents of the vessel was monitored using a calibrated infrared temperature control mounted under the reaction vessel. All experiments were performed using a stirring option whereby the contents of the vessel are stirred by means of a rotating magnetic plate located below the floor of the microwave cavity and a Teflon-coated magnetic stir bar in the vessel.
General Procedure A (GP-A): Synthesis of Pyrrole Nucleus.
A solution of α,β-unsaturated ketone (5.42 mmol) and toluene-4-sulfonylmethyl isocyanide (1.16 g, 5.96 mmol, 1.1 equiv) dissolved in a mixture of anhydrous dimethyl sulfoxide/ethyl ether (14:30 mL) was added dropwise into a well-stirred suspension of sodium hydride (60% in paraffine oil; 0.48 g, 11.93 mmol, 2.2 equiv) in dry ethyl ether (30 mL) under argon atmosphere. After the addition the mixture was stirred at room temperature for 1 h. The reaction was treated with water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum. The crude product was purified by chromatography on aluminum oxide (chloroform as eluent) to afford the pure product. Yield (%), melting point (°C), recrystallization solvent, IR, and 1H NMR are reported for each compound.
General Procedure B (GP-B): Alkylation of the Pyrrole Nitrogen.
A mixture of the appropriate pyrrole (1.1 mmol), alkylating agent (3.3 mmol), and anhydrous K2CO3 (210 mg, 1.5 mmol) in dry DMF (10 mL) was stirred at 100 °C for 2 h. Then the mixture was cooled, treated with water (40 mL), and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum. The crude product was purified by chromatography on silica gel to afford the pure product. Chromatography eluent, yield (%), melting point (°C), recrystallization solvent, IR, and 1H NMR are reported for each compound.
General Procedure C (GP-C): Condensation of Pyrrole Carboxaldehyde with Acetone.
The proper pyrrole carboxaldehyde (0.075 mol) was dissolved in 250 mL of acetone. To this mixture was added 4 N NaOH (110 mL), and the mixture was stirred at room temperature for 24 h. After this period water (300 mL) and ethyl acetate (250 mL) were added. The organic layer was separated, washed with water (2 × 100 mL), dried over sodium sulfate, filtered, and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel to obtain pure products. Chromatography eluent, yield (%), melting point (°C), recrystallization solvent, IR, and 1H NMR are reported for each compound.
General Procedure (GP-D): Acetyl Transposition.
A mixture of opportune α acetyl-substituted pyrrole (1.23 mmol) in trifluoroacetic acid (5 mL) was heated at 80 °C for 20 h. After this period the reaction was quenched with water (30 mL) and extracted with ethyl acetate (2 × 50 mL). The organic layers were collected, dried over sodium sulfate, filtered, and evaporated under vacuum. The crude product was purified by chromatography on silica gel (chloroform as eluent) to afford pure product as a brown oil. Yield (%), melting point (°C), recrystallization solvent, IR, and 1H NMR are reported for each compound.
General Procedure E (GP-E): Suzuki Reaction.
Pd2(dba)3 (0.1 g, 1.7 mmol) was added into a well stirred mixture of appropriate 4-iodopyrrole (1.7 mmol), phenylboronic acid (0.85 g, 7.0 mmol), Cs2CO3 (0.665 g, 2.0 mmol), and P(t-But)3 in dioxane (20 mL). The mixture was stirred at 80 °C for 24 h under argon atmosphere. Then the mixture was cooled to room temperature, filtered, and washed with dioxane. The organic layer was evaporated under vacuum. The raw material was extracted with water (50 mL) and ethyl acetate (50 mL). The organic phase was separated, dried over sodium sulfate, filtered, and evaporated under vacuum. The raw material was purified by silica gel chromatography. Chromatography eluent, yield (%), melting point (°C), recrystallization solvent, IR, and 1H NMR are reported for each compound.
General Procedure F (GP-F): Synthesis of Diketo Esters.
Freshly prepared sodium ethoxide (390 mg, 5.5 mmol) was added into a well-stirred mixture of the appropriate acetyl derivative (2.7 mmol) and diethyl oxalate (790 mg, 5.4 mmol) in anhydrous THF (2.7 mL) under nitrogen atmosphere. The mixture was stirred at room temperature for 2 h and then was poured into n-hexane (50 mL). The collected precipitate was vigorously stirred for 30 min in 1 N HCl (50 mL). The yellow solid that formed was filtered, washed with water, and dried under IR lamp to afford the pure diketo esters. Yield (%), melting point (°C), IR, and 1H NMR are reported for each compound.
General Procedure G (GP-G): Synthesis of Diketo Acids.
A mixture of 1 N NaOH (6.5 mL) and the appropriate ester (1.3 mmol) in 1:1 THF/methanol (12 mL) was stirred at room temperature for 40 min and then poured onto crushed ice. The aqueous layer was treated with 1 N HCl until pH 3 (pH 7 for 1d) was obtained, and the solid that formed was collected by filtration, then washed with water and dried under warming lamp to afford pure acids. Yield (%), melting point (°C), IR, and 1H NMR are reported for each compound.
1-(4-Phenyl-1H-pyrrol-3-yl)propan-1-one (8).41
Compound 8 was prepared from (E)-1-phenylpent-1-en-3-one35 by means of GP-A. 79% as a yellow solid; 169–170 °C; toluene. Anal. (C13H13NO) C, H, N.
1-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)ethanone (9a).
Compound 9a was prepared from 1-(4-phenyl-1H-pyrrol-3-yl)ethanone32 by means of GP-B, using 4-fluorobenzyl bromide as alkylatig agent. Chloroform; 100% as brown oil; IR ν 1705 (C=O ketone) cm−1; 1H NMR (DMSO d6) δ 2.26 (s, 3H, CH3), 5.04 (s, 2H, CH2), 6.63 (s, 1H, pyrrole α-proton), 7.06 (m, 2H, benzyl H), 7.19 (m, 2H, benzyl H), 7.25–7.41 (m, 6H, benzene H and pyrrole α-proton). Anal. (C19H16FNO) C, H, N, F.
1-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)propan-1-one (9b).
Compound 9b was prepared from 8 by means of GP-B, using 4-fluorobenzyl bromide as alkylating agent. Chloroform/acetate 50:1; 66% as brown oil; IR ν 1656 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 1.04 (t, 3H, J = 8 Hz, CH2CH3), 2.75 (q, 2H, J = 8 Hz, CH2CH3), 5.18 (s, 2H, CH2), 7.07 (d, 1H, J = 2.2 Hz, pyrrole C5-H), 7.2–7.3 (m, 3H, benzene H), 7.32 (t, 2H, benzyl H), 7.4 (m, 2H, benzene H), 7.47 (m, 2H, benzyl H), 7.87 (d, 1H, J = 2 Hz, pyrrole C2-H). Anal. (C20H18FNO) C, H, N, F.
1-(1-(4-Fluorobenzyl)-1H-pyrrol-3-yl)ethanone (9c).
Compound 9c was prepared from 1-(1-(4-fluorobenzyl)-1H-pyrrol-2-yl)ethanone36 by means of GP-D. 50% as brown oil; IR ν 1655 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.37 (s, 3H, CH3), 5.03 (s, 2H, CH2), 6.60–6.63 (m, 2H, pyrrole C4-H and C5-H), 7.03 (t, 2H,benzene H), 7.12 (m, 2H, benzene H), 7.28 (t, 1H, J = 2.0 Hz, pyrrole C2-H,). Anal. (C13H14FNO) C, H, N, F.
1-(4-Fluorobenzyl)-4-iodo-1H-pyrrole-2-carboxaldehyde (10).
Compound 10 was prepared from commercially available 4-iodopyrrole-2-carboxaldehyde by means of GP-B, using 4-fluorobenzyl bromide as alkylating agent. Chloroform/n-hexane 4:1; 39% as brown oil; IR ν 1651 (C=O) cm−1. 1H NMR (CDCl3) δ 5.47 (s, 2H), 6.9–7.0 (m, 4H, pyrrole α-proton, pyrrole β-proton, and benzene H), 7.02 (m, 2H, benzene H), 9.48 (s, 1H, CHO). Anal. (C12H9FINO) C, H, N, F, I.
4-(4-Fluorobenzoyl)-1H-pyrrole-2-carboxaldehyde (11).
To a well stirred solution of DMF (3.9 mL, 50 mmol) in 1,2-dichloroethane (100 mL) refrigerated in an ice bath was added dropwise a solution of oxalyl chloride (6.35 g, 50 mmol) in 1,2 dichloroethane (100 mL) in a period of 15 min. After addition, the suspension was stirred at room temperature for 15 min. After this time the reaction mixture was refrigerated in ice bath and treated with a solution of pyrrole (49.9 mmol) in 1,2-dichloroethane (100 mL). The mixture was stirred for 15 min at room temperature and then treated with AlCl3 (14.6 g, 109 mmol) and 4-fluorobenzoyl chloride (50 mmol). The reaction was maintained at room temperature for 4 h. After this period the reaction was quenched with ice and water and extracted with ethyl acetate, dried over sodium sulfate, filtered, and evaporated under vacuum. The crude product was purified with chromatography on aluminum oxide (1:1 ethyl acetate–chloroform as eluent) to afford 5 as yellow solid. 85%; 110–111 °C; benzene/cyclohexane; IR ν 2900 (enol), 1660 (C=O ketone) 1640 (C=O) cm−1. 1H NMR (CDCl3) δ 7.1–7.4 (m, 3H, benzene H and pyrrole β-proton), 7.45 (s, 1H, pyrrole α-proton), 7.8–7.9 (m, 2H, benzene H), 9.60 (s, 1H, CHO), 12 (sb, 1H, NH). Anal. (C12H8FNO2) C, H, N, F.
1-Benzyl-4-(4-fluorobenzoyl)-1H-pyrrole-2-carbaldehyde (12e).
Compound 12e was prepared from 11 by means of GP-B, using benzyl bromide as alkylatig agent. Acetate/n-hexane 1:2; 34% as brown oil; IR ν 1672 (C=O aldehyde), 1638 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 5.62 (s, 2H, CH2), 7.1–7.2 (m, 4H, benzyl H and pyrrole β-proton), 7.3–7.4 (m, 4H, benzyl H and benzoyl H), 7.58 (s, 1H, pyrrole α-proton), 7.8–7.9 (m, 2H, benzoyl H), 9.62 (s, 1H, CHO). Anal. (C19H14FNO2) C, H, N, F.
4-(4-Fluorobenzoyl)-1-phenyl-1H-pyrrole-2-carbaldehyde (12f).
Compound 11, phenylboronic acid, pyridine, and copper(II) acetate anhydrous were dissolved in NMP (2.4 mL) in a microwave reactor tube and left to react at 60 W, 120 °C for 6 min. After this period the reaction was quenched with water and extracted with ethyl acetate (5 × 20 mL), washed with water (5 × 20 mL), dried over sodium sulfate, filtered, and evaporated under vacuum. The crude product was purified with chromatography on silica gel (1:1 ethyl acetate–hexane as eluent) to afford 12f as brown solid (63% yield). 130–131 °C. Benzene/cyclohexane; IR ν 1680 (C=O aldehyde), 1632 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 7.21 (t, 2H, benzoyl H), 7.4–7.5 (m, 2H, benzene H), 7.5–7.6 (m, 4H, benzene H and pyrrole β-proton), 7.67 (d, 1H, J = 2 Hz, pyrrole α-proton), 7.9–8.0 (m, 2H, benzoyl H), 9.68 (s, 1H, CHO). Anal. (C18H12FNO2) C, H, N, F.
1-Methyl-4-(4-fluorobenzoyl)-1H-pyrrole-2-carboxaldehyde (12g).
Compound 12g was prepared from 11 by means of GP-B, using iodomethane as alkylatig agent. Chloroform; 90% as gray solid; 115–116 °C; benzene/cyclohexane; IR ν 1660 (C=O aldehyde), 1640 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 4.03 (s, 3H, N–CH3), 7.2 (m, 2H, benzoyl H), 7.4 (d, 1H, pyrrole β-proton), 7.5 (s, 1H, pyrrole α-proton), 7.8–7.9 (m, 2H, benzoyl H), 9.60 (s, 1H, CHO). Anal. (C13H10FNO2) C, H, N, F.
1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrole-2-carbaldehyde (12h).
Compound 12h was prepared from 10 by means of GP-E. 1:1 ethyl acetate–hexane as eluent; 95% as brown oil; IR ν 1642 (C=O aldehyde) cm−1. 1H NMR (CDCl3) δ 5.54 (s, 2H, CH2), 6.82 (d, 2H, J = 7.0 Hz, benzene H), 6.91 (t, 1H, J = 7.0 Hz, benzene H), 6.99 (t, 2H, benzyl H), 7.16–7.24 (m, 4H, pyrrole β-proton, pyrrole α-proton, benzyl H), 7.48 (d, 2H, J = 7 Hz, benzene H), 9.58 (s, 1H, CHO). Anal. (C18H14FNO) C, H, N, F.
4-(1-Benzyl-4-(4-fluorobenzoyl)-1H-pyrrol-2-yl)but-3-en-2-one (13e).
Compound 13e was prepared from 12e by means of GP-C. 71% as yellow solid; 94–95 °C; benzene/cyclohexane; IR ν 1675 (C=O ketone), 1637 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.26 (s, 3H, CH3), 5.29 (s, 2H, CH2), 6.60 (d, 1H, J = 16 Hz, butenone C3-H), 7.1–7.2 (m, 5H, butenone C4-H, benzyl H and pyrrole β-proton), 7.3–7.4 (m, 4H, benzyl H and benzoyl H), 7.46 (d, 1H, J = 2 Hz, pyrrole α-proton), 7.90 (m, 2H, benzoyl H). Anal. (C22H18FNO2) C, H, N, F.
4-(1-Phenyl-4-(4-fluorobenzoyl)-1H-pyrrol-2-yl)but-3-en-2-one (13f).
Compound 13f was prepared from 12f by means of GP-C. 30% as yellow solid; 145–146 °C; benzene/cyclohexane; IR ν 1680 (C=O ketone), 1634 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.24 (s, 3H, CH3), 6.56 (d, 1H, J = 16 Hz, butenone C3-H), 7.19 (t, 2H, benzoyl H), 7.24 (d, 1H, J = 16 Hz, butenone C4-H), 7.35 (d, 1H, J = 2 Hz, pyrrole β-proton), 7.38 (d, 1H, J = 2 Hz, pyrrole α-proton), 7.5–7.6 (m, 5H, benzene H), 7.94 (m, 2H, benzoyl H). Anal. (C21H16FNO2) C, H, N, F.
4-(1-Methyl-4-(4-fluorobenzoyl)-1H-pyrrol-2-yl)but-3-en-2-one (13g).
Compound 13g was prepared from 12g by means of GP-C. 70% as yellow solid; 117–118 °C; benzene/cyclohexane; IR ν 1660 (C=O ketone), 1640 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.32 (s, 3H, CH3), 3.78 (s, 3H, N-CH3), 6.62 (d, 1H, J = 16 Hz, butenone C3-H), 7.1–7.2 (m, 3H, benzene H and pyrrole β-proton), 7.35 (d, 1H, J = 3.7 Hz, pyrrole α-proton), 7.43 (d, 1H, J = 16 Hz, butenone C4-H), 7.8–7.9 (m, 2H, benzene H). Anal. (C16H14FNO2) C, H, N, F.
4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-2-yl)but-3-en-2-one (13h).
Compound 13h was prepared from 12h by means of GP-C. 15% as yellow solid; 160–161 °C; benzene/cyclohexane; IR ν 1604 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.28 (s, 3H, CH3), 5.24 (s, 2H, CH2), 6.57 (d, 1H, J = 16 Hz, butenone C3-H), 7.0–7.1 (m, 5H, pyrrole β-proton and benzyl H), 7.18 (d, 1H, J = 2 Hz, pyrrole α-proton,), 7.24 (t, 1H, J = 7 Hz, benzene H), 7.3–7.4 (m, 3H, butenone C4-H and benzene H), 7.5–7.6 (m, 2H, benzene H). Anal. (C21H18FNO) C, H, N, F.
1-(4-Fluorobenzyl)-1H-pyrrole-2-carboxaldehyde (14).
Compound 14 was prepared from commercially available pyrrole-2-carboxaldehyde by means of GP-B, using 4-fluorobenzyl bromide as alkylating agent. Chloroform; 80% as brown oil; IR ν 1640 (C=O) cm−1. 1H NMR (CDCl3) δ 5.54 (s, 2H, CH2), 6.30 (t, 1H, J = 4 Hz, pyrrole C4-H), 6.9–7.0 (m, 4H, benzene H), 7.15 (d, 1H, J = 4 Hz, pyrrole C3-H), 7.17 (d, 1H, J = 4 Hz, pyrrole C5-H), 9.57 (s, 1H, CHO). Anal. (C12H10FNO) C, H, N, F.
1-(4-Fluorobenzyl)-1H-pyrrole-3-carboxaldehyde (10).
Compound 10 was prepared from 9 by means of GP-D. 55% as brown oil; IR ν 1640 (C=O aldehyde) cm−1. 1H NMR (CDCl3) δ 5.08 (s, 2H, CH2), 6.6 (s, 1H, pyrrole C4-H), 6.7 (s, 1H, pyrrole C-2H), 7.0–7.2 (m, 4H, benzene H), 7.31 (s, 1H, pyrrole C2-H), 9.75 (s, 1H, CHO). Anal. (C12H10FNO) C, H, N, F.
4-(4-Phenyl-1H-pyrrol-3-yl)but-3-en-2-one (16).42
Compound 16 was prepared from (3E,5E)-6-phenylhexa-3,5-dien-2-one38 by means of GP-A. 56% as brown solid; toluene; IR ν 1640 (C=O ketone) cm−1. Anal. (C14H13NO) C, H, N.
4-(1-(4-Fluorobenzyl)-1H-pyrrol-3-yl)but-3-en-2-one (17i).
Compound 17i was prepared from 15 by means of GP-C. 70% as brown oil; IR ν 1655 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.34 (s, 3H, CH3), 5.07 (s, 2H, CH2), 6.43–6.48 (m, 2H, pyrrole C4-H and butenone C3-H), 6.71 (s, 1H, pyrrole C2-H), 7.0 (s, 1H, pyrrole C5-H), 7.06–7.12 (m, 2H, benzene H), 7.15–7.18 (m, 2H, benzene H), 7.49 (d, 1H, butenone C4-H, J = 16 Hz). Anal. (C15H14FNO) C, H, N, F.
4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)but-3-en-2-one (17j).
Compound 17j was prepared from 16 by means of GP-B, using 4-fluorobenzyl bromide as alkylating agent. Chloroform; 63% as brown oil; IR ν 1640 (C=O) cm−1. 1H NMR (CDCl3) δ 2.44 (s, 3H, CH3), 5.07 (s, 2H, CH2), 6.82 (s, 1H, J = 16.3 Hz, butenone C3-H), 6.90 (s, 1H,), 6.99–7.54 (m, 11H, pyrrole C2-H, pyrrole C5-H, benzene H and benzyl H), 7.80 (s, 1H, J = 16.3 Hz, butenone C4-H). Anal. (C21H18FNO) C, H, N, F.
1-(1-(4-Fluorobenzyl)-4-iodo-1H-pyrrol-2-yl)ethanone (18).
A mixture of 1-(1-(4-fluorobenzyl)-1H-pyrrol-2-yl)ethanone36 (4 g, 18.4 mmol) in dry acetone (100 mL) was cooled at −78 °C. NIS (4.98 g, 22.1 mmol) was added. The mixture was stirred, and the temperature was slowly increased to 25 °C in a period of 96 h. After this period the mixture was evaporated, and ethyl acetate (50 mL) and NaHCO3(aq) (50 mL) were added. The organic phase was separated, dried over sodium sulfate, filtered, and evaporated under vacuum. The raw material was purified with a column chromatography on silica gel (1:10 ethyl acetate–hexane as eluent) to afford 18 as white solid with a yield of 40%. 81–82 °C; n-hexane; IR ν 1640 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.40 (s, 3H, CH3), 5.50 (s, 2H, CH2), 6.92 (d, 1H, J = 2.0 Hz, pyrrole C3-H), 7.00 (t, 2H, benzene H), 7.07 (d, 1H, J = 2.0 Hz, pyrrole C5-H), 7.1–7.2 (m, 2H, benzene H). Anal. (C13H11FINO) C, H, N, F, I.
1-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-2-yl)ethanone (19).
Compound 19 was prepared from 18 by means of GP-E. 1:7 ethyl acetate–hexane as eluent. 43% as colorless oil; IR ν 1650 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 2.48 (s, 3H, CH3), 5.58 (s, 2H, CH2), 6.97–7.02 (t, 2H, benzyl H), 7.14–7.27 (m, 5H, benzene H, pyrrole C3-H and pyrrole C5-H), 7.35–7.39 (t, 2H, benzyl H), 7.51 (d, 2H,benzene H). Anal. (C18H21FNO) C, H, N, F.
1-(1-(4-Fluorobenzyl)-1H-pyrrol-2-yl)-3-hydroxy-3-(1-trityl-1H-1,2,4-triazol-3-yl)prop-2-en-1-one (20).
A solution of 1-[1-(4-fluorobenzyl)-1H-pyrrol-2-yl]ethanone36 (1 g, 4.6 mmol) in anhydrous THF (5 mL) was thermostated at −32 °C. LHMDS (9.2 mL) was added, and the mixture was stirred at the some temperature for 2 h. A solution of 1-trityl-1H-[1,2,4]triazole-3-carboxylic acid ethyl ester39 (2 g, 5.3 mmol) in anhydrous THF (18 mL) was added dropwise to the solution thermostated at −32 °C. After the addition, the reaction mixture was stirred for 1.5 h at room temperature. The reaction was poured into 1 N HCl (100 mL) and extracted with ethyl acetate. The organic phase was separated, washed with water, dried over sodium sulfate, filtered, and evaporated under vacuum obtaining 2.6 g of crude product as light yellow solid. The raw material was purified by recrystallization from benzene/cyclohexane, obtaining 1.84 g of pure 20. 54%; 110–112 °C; benzene/cyclohexane; IR ν 2954 (OH enol), 1626 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 5.62 (s, 2H, CH2), 6.3 (t, 1H, pyrrole C4-H), 6.9–7.0 (m, 4H, butenoate C3-H, pyrrole C3-H, benzyl H), 7.1–7.2 (m, 9H, benzyl H, pyrrole C5–H and benzene H), 7.3–7.4 (m, 9H, benzene H), 8.01 (s, 1H, triazole H), 15 (br s, 1H, OH enol). Anal. (C35H27FN4O2) C, H, N, F.
Ethyl 4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-2-hydroxy-4-oxobut-2-enoate (6a).
Compound 6a was prepared from 9a by means of GP-F. 88%; 111–112 °C; benzene; IR ν 2900 (OH enol), 1720 (C=O ester), 1620 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 1.29 (t, 3H, CH3CH2), 4.25 (q, 2H, CH2CH3), 5.06 (s, 2H, CH2 benzyl), 6.48 (s, 1H, butenoate C3-H), 6.67 (d, 1H, J = 1.5 Hz, pyrrole C5-H), 7.02–7.46 (m, 10H, pyrrole C2-H, benzene H and benzyl H), 15 (br s, 1H, OH enol). Anal. (C23H20FNO4) C, H, N, F.
Ethyl 4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-2-hydroxy-3-methyl-4-oxobut-2-enoate (6b).
Compound 6b was prepared from 9b by means of GP-F. 6b was extracted with ethyl acetate. The organic phase was separated, washed with brine, dried over sodium sulfate, filtered, and evaporated under vacuum obtaining a crude product that was purified with column chromatography on silica gel (ethyl acetate/n-hexane 1:2). 30% as yellow oil; IR ν 1730 (C=O ester), 1650 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 1.14 (d, 3H, J = 7 Hz, CH3), 1.34 (t, 3H, J = 7.5 Hz, CH3CH2), 4.33 (q, 2H, J = 7.5 Hz, CH2CH3), 5.25 (s, 2H, CH2), 7.14 (d, 1H, J = 1.9 Hz, pyrrole C2-H), 7.22–7.51 (m, 9H, benzene H and benzyl H), 8.08 (s, 1H, J = 1.9 Hz, pyrrole C5-H), 14 (br s, 1H, enol). Anal. (C24H22FNO4) C, H, N, F.
Ethyl 4-(1-(4-Fluorobenzyl)-1H-pyrrol-3-yl)-2-hydroxy-4-oxobut-2-enoate (6c).
Compound 6c was prepared from 9c by means of GP-F. 93% as yellow solid; 63–65 °C; ligroin; IR ν 3500–2500 (OH enol), 1726 (C=O ester), 1633 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 1.28 (t, 3H, J = 7 Hz, CH3CH2), 4.27 (q, 2H, J = 7 Hz, CH2CH3), 5.17 (s, 2H, CH2), 6.62 (t, 1H, pyrrole C4-H), 6.73 (s, 1H, butenoate C3-H), 7.02 (t, 1H, pyrrole C5-H), 7.18 (t, 2H, benzene H), 7.36 (dd, 2H, benzene H), 8.07 (s, 1H, pyrrole C2-H), 15 (br s, 1H, enol). Anal. (C17H18FNO4) C, H, N, F.
Ethyl 2-Amino-4-(1-(4-fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-4-oxobut-2-enoate (6d).
To a stirred mixture of 6a (1 g, 2.5 mmol) and ammonium acetate (0.22 g, 2.9 mmol) in benzene (30 mL) was added acetic acid glacial (0.2 mL, 3.9 mmol). The mixture was stirred at reflux for 20 h with a Dean–Stark apparatus. After this period the mixture was cooled to room temp and washed with a saturated solution of NaHCO3 (50 mL). The organic layer was separated, dried over sodium sulfate, filtered, and evaporated under vacuum. The raw material was purified with chromatography on aluminum oxide (chloroform as eluent) to afford 6d as yellow solid with a yield of 50%. 135–136 °C. benzene; IR ν 3500 (NH2), 1720 (C=O ester), 1620 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 1.27 (t, 3H, CH3CH2), 4.25 (q, 2H, CH3CH2), 5.08 (s, 2H, CH2), 6.13 (s, 1H, butenoate C3-H), 6.67 (d, 1H, J = 1.5 Hz, pyrrole C5-H), 7.0–7.6 (m, 10H, benzene H, benzyl H, and pyrrole C2-H), 9 (br s, 2H, NH2). Anal. (C23H21FN2O2) C, H, N, F.
Ethyl 6-(1-Benzyl-4-(4-fluorobenzoyl)-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoate (6e).
Compound 6e was prepared from 13e by means of GP-F. 62% as yellow solid; 154–155 °C; benzene/cyclohexane; IR ν 1730 (C=O ester), 1636 (C=O ketone) cm−1. 1H NMR (acetone-d6) δ 1.34 (t, 3H, CH3CH2), 4.32 (q, 2H, CH2CH3), 5.60 (s, 2H, CH2), 6.86 (d, 1H, J = 15.6 Hz, hexanoate C5-H), 7.2–7.5 (m, 9H, J = 1.6 Hz, benzene H, pyrrole β-proton, benzoyl H, and hexanoate C3-H), 7.72 (d, 1H, J = 15.6 Hz, hexanoate C6-H), 7.90 (d, 1H, J = 1.6 Hz, pyrrole α-proton), 7.9–8.0 (m, 2H, benzoyl H), 14 (bs, 1H, enol). Anal. (C26H22FNO5) C, H, N, F.
Ethyl 6-(4-(4-Fluorobenzoyl)-1-phenyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoate (6f).
Compound 6f was prepared from 13f by means of GP-F. 80% as yellow solid; 129–130 °C; benzene/cyclohexane; IR ν 3500 (OH enol), 1720 (C=O ester), 159 (C=O ketone) cm−1. 1H NMR (CD3OD) δ 1.36 (t, 3H, J = 7.5 Hz, CH3CH2), 4.31 (q, 2H, J = 7.5 Hz, CH2CH3), 7.26–7.48 (m, 3H, benzoyl H and hexanoate C5-H), 7.39 (d, 1H, J = 15.6 Hz, hexanoate C6-H), 7.43–7.48 (m, 3H, benzene H and hexanoate C3-H), 7.58–7.62 (m, 4H, benzene H and pyrrole β-proton), 7.72 (s, 1H, pyrrole α-proton), 7.97–8.01 (m, 2H, benzoyl H), 14 (bs, 1H, OH enol). Anal. (C25H20FNO5) C, H, N, F.
Ethyl 6-(4-(4-Fluorobenzoyl)-1-methyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoate (6g).
Compound 6g was prepared from 13g by means of GP-F. 83% as yellow solid; 166–167 °C; benzene/cyclohexane; IR ν 1 2900 (OH enol), 1720 (C=O ester), 1650 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 1.39 (t, 3H, J = 7.5 Hz, CH3CH2), 3.81 (s, 3H, N-CH3), 4.39 (q, 2H, J = 7.5 Hz, CH2CH3), 6.45 (s, 1H, hexanoate C3-H), 6.50 (d, 1H, J = 15.4 Hz, hexanoate C5-H), 7.12–7.20 (m, 3H, benzene H and pyrrole β-proton), 7.36–39 (m, 1H, pyrrole α-proton), 7.63 (d, 1H, J = 15.4 Hz, hexanoate C6-H), 7.83–7.91 (m, 2H, benzene H), 15 (bs, 1H, enol). Anal. (C20H18FNO5) C, H, N, F.
Ethyl 6-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoate (6h).
Compound 6h was prepared from 13h by means of GP-F. 92% as red solid; 169–170 °C; benzene; IR ν 2900 (OH enol), 1723 (C=O ester), 1602 (C=O ketone) cm−1. 1H NMR (acetone-d6) δ 1.35 (t, 3H, J = 7.5 Hz, CH3CH2), 4.32 (q, 2H, J = 7.5 Hz, CH2CH3), 5.53 (s, 2H, CH2), 6.46 (s, 1H, hexanoate C3-H), 6.74 (d, 1H, J = 16 Hz, hexanoate C5-H), 7.13–7.24 (m, 3H, benzyl H and benzene H), 7.30–39 (m, 4H, benzyl H, hexanoate C6-H, and pyrrole β-proton), 7.43 (s, 1H, pyrrole α-proton), 7.65 (d, 2H, benzene H), 7.7–7.8 (m, 2H, benzene H), 15 (bs, 1H, enol). Anal. (C25H22FNO4) C, H, N, F.
Ester 6-(1-(4-Fluorobenzyl)-1H-pyrrol-3-yl)-2-hydroxy-4-oxohexa-2,5-dienoate (6i).
Compound 6i was prepared from 17i by means of GP-F. 41% as yellow solid; 88–90 °C; ligroin; IR ν 3400 (OH enol), 1725 (C=O ester), 1625 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 1.44 (t, 3H, J = 7 Hz, CH3CH2), 4.41 (q, 2H, J = 7 Hz, CH2CH3), 5.08 (s, 2H, CH2), 6.39 (s, 1H, J = 16 Hz, hexanoate C5-H), 6.5–6.6 (m, 2H, pyrrole C4-H and hexanoate C3-H), 6.7 (t, 1H, pyrrole C5-H), 7.0 (t, 1H, pyrrole C2-H), 7.08–7.20 (m, 4H, benzene H), 7.75 (s, 1H, J = 16 Hz, hexanoate C6-H), 14 (br s, 1H, enol). Anal. (C19H18FNO4) C, H, N, F.
Ethyl 6-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-2-hydroxy-4-oxohexa-2,5-dienoate (6j).
Compound 6j was prepared from 17j by means of GP-F. 56% as yellow solid; 121–122 °C; cyclohexane; IR ν 2900 (OH enol), 1720 (C=O ester), 1620 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 1.43 (t, 3H, CH3CH2), 4.40 (q, 2H, CH2CH3), 5.09 (s, 2H, CH2), 6.73 (s, 1H, hexanoate C3-H), 6.87 (d, 1H, J = 16.6 Hz, hexanoate C5-H), 7.01 (s, 1H, pyrrole C5-H), 7.08–7.54 (m, 10H, pyrrole C2-H, benzene H and benzyl H), 7.74 (d, 1H, hexanoate C6-H), 15 (bs, 1H, enol). Anal. (C25H21FNO4) C, H, N, F.
Ethyl 4-(1-(4-Fluorobenzyl)-1H-pyrrol-2-yl)-2-hydroxy-4-oxobut-2-enoate (6k).
Compound 6k was prepared from 1-(1-(4-fluorobenzyl)-1H-pyrrol-2-yl)ethanone36 by means of GP-F. 41% as yellow solid; 88–89 °C; ligroin; IR ν 2900 (OH enol), 1720 (C=O ester), 1620 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 1.26 (t, 3H, J = 7.5 Hz, CH3CH2), 4.25 (q, 2H, J = 7.5 Hz, CH2CH3), 5.59 (s, 2H, CH2), 6.32 (dd, 1H, J = 2.5 Hz, J = 3.5 Hz, pyrrole C4-H), 6.84 (s, 1H, butenoate C3-H), 7.0–7.2 (m, 4H, benzene H), 7.43 (d, 1H, J = 3.5 Hz, pyrrole C3-H), 7.53 (s, 1H, pyrrole C5-H), 14 (br s, 1H, enol). Anal. (C17H16FNO4) C, H, N, F.
Ethyl 4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxobut-2-enoate (6l).
Compound 6l was prepared from 19 by means of GP-F. 95% as yellow solid; 102–103 °C; benzene/cyclohexane; IR ν 3400 (OH enol), 1720 (C=O ester), 1620 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 1.34 (t, 3H, J = 7.5 Hz, CH3CH2), 4.33 (q, 2H, J = 7.5 Hz, CH2CH3), 5.68 (s, 2H, CH2), 7.06 (s, 1H, butenoate C3-H), 7.17–7.31 (m, 5H, benzene H and benzyl H), 7.40–7.44 (m, 2H, benzene H), 7.75 (d, 2H, benzene H), 8.01 (s, 1H, pyrrole C5-H), 8.13 (s, 1H, pyrrole C3-H), 14 (bs, 1H, enol). Anal. (C22H20FNO4) C, H, N, F.
4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-2-hydroxy-4-oxobut-2-enoic Acid (7a).
Compound 7a was prepared from 6a by means of GP-G. 98% as brown solid; 109–110 °C; toluene; IR ν 3500–2000 (OH acid and enol), 1740 (C=O acid), 1620 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 5.06 (s, 2H, CH2), 6.48 (s, 1H, butenoate C3-H), 6.67 (d, 1H, J = 1.5 Hz, pyrrole C5-H), 7.06–7.10 (t, 2H, benzyl H), 7.18–7.25 (m, 2H, benzyl H), 7.30–7.39 (m, 5H, benzene H), 7.49 (s, 1H, pyrrole C2-H), 14 (br s, 2H, OH acid and enol). Anal. (C21H16FNO4) C, H, N, F.
4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-2-hydroxy-3-methyl-4-oxobut-2-enoic Acid (7b).
Compound 7b was prepared from 6b by means of GP-G. 38% as white solid; 233–234 °C; benzene; IR ν 3500–2500 (OH acid and enol), 1700 (C=O acid), 1640 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 1.14 (d, 3H, CH3, J = 7 Hz), 5.13 (s, 2H, CH2), 7.1–7.2 (m, 3H, benzyl H and pyrrole C5-H), 7.34 (t, 2H, benzyl H), 7.47 (s, 1H, pyrrole C2-H), 7.51 (m, 3H, benzene H), 7.9–8.0 (m, 2H, benzene H), 14 (br s, 2H, enol and acid). Anal. (C22H18FNO4) C, H, N, F.
4-(1-(4-Fluorobenzyl)-1H-pyrrol-3-yl)-2-hydroxy-4-oxobut-2-enoic Acid (7c).
Compound 7c was prepared from 6c by means of GP-G. 57% as yellow solid; 146–147 °C; toluene; IR ν 3500–2500 (OH acid and enol), 1727 (C=O acid), 1621 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.16 (s, 2H, CH2), 6.60 (dd, 1H, J1 = 1.5 Hz, J2 = 3.0 Hz, pyrrole C4-H), 6.72 (s, 1H, butenoate C3-H), 7.00 (dd, 1H, J1 = 1.5 Hz, J2 = 3 Hz, pyrrole C5-H), 7.18 (t, 2H, benzene H), 7.36 (dd, 2H, benzene H), 8.04 (s, 1H, pyrrole C2-H), 14 (br s, 1H, enol), 15 (br s, 1H, COOH). Anal. (C15H12FNO4) C, H, N, F.
6-(1-Benzyl-4-(4-fluorobenzoyl)-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoic Acid (7e).
Compound 7e was prepared from 6e by means of GP-G. 87% as orange solid; 165–166 °C; benzene; IR ν 3500–2500 (OH acid and enol), 1727 (C=O acid), 1630 (C=O ketone) cm−1. 1H NMR (CD3OD) δ 5.22 (s, 2H, CH2), 6.69 (s, 1H, J = 15.2 Hz, hexanoate C5-H), 7.15–7.44 (m, 9H, hexanoate C3-H, benzyl H, benzoyl H, and pyrrole β-proton), 7.65 (s, 1H, J = 15.2 Hz, hexanoate C6-H), 7.79 (s, 1H, pyrrole α-proton), 7.92–7.97 (m, 2H, benzoyl H). Anal. (C24H18FNO5) C, H, N, F.
6-(4-(4-Fluorobenzoyl)-1-phenyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoic Acid (7f).
Compound 7f was prepared from 6f by means of GP-G. 85% as yellow solid; 183–184 °C; benzene; IR ν 3500–2500 (OH acid and enol), 1724 (C=O acid), 1596 (C=O ketone) cm−1. 1H NMR (CD3OD) δ 6.69 (s, 1H, J = 16 Hz, hexanoate C5-H), 7.26 (t, 2H, benzoyl H), 7.41 (d, 1H, hexanoate C6-H), 7.47–7.49 (m, 2H, pyrrole β-proton and hexanoate C3-H), 7.73 (s, 1H, pyrrole α-proton), 7.98–8.01 (m, 2H, benzoyl H). Anal. (C23H16FNO5) C, H, N, F.
6-(4-(4-Fluorobenzoyl)-1-methyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoic Acid (7g).
Compound 7g was prepared from 6g by means of GP-G. 95% as orange solid; 162–163 °C; ethanol; IR ν 3500–2500 (OH acid and enol), 1700 (C=O acid), 1600 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 3.32 (s, 3H, N-CH3), 6.52 (s, 1H, hexanoate C3-H), 6.93 (d, 1H, hexanoate C5-H), 7.32–7.86 (m, 7H, benzene H, pyrrole β-proton, pyrrole α-proton, and hexanoate C6-H), 14 (br s, 2H, OH enol and acid). Anal. (C18H14FNO5) C, H, N, F.
6-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxohexa-2,5-dienoic Acid (7h).
Compound 7h was prepared from 6h by means of GP-G. 73% as red solid; decompose; benzene; IR ν 3500–2500 (OH acid and enol), 1707 (C=O acid), 1571 (C=O ketone) cm−1. 1H NMR (acetone-d6) δ 5.51 (s, 2H, CH2), 6.49 (s, 1H, hexanoate C3-H), 6.7, (d, 1H, hexanoate C5-H), 7.07–7.42 (m, 7H, benzyl H, pyrrole β-proton, pyrrole α-proton, and hexanoate C6-H), 7.6–7.8 (m, 5H, benzene H), 14 (br s, 2H, enol and acid). Anal. (C23H18FNO4) C, H, N, F.
6-(1-(4-Fluorobenzyl)-1H-pyrrol-3-yl)-2-hydroxy-4-oxohexa-2,5-dienoic Acid (7i).
Compound 7i was prepared from 6i by means of GP-G. 92% as yellow solid; >300 °C; DMF/H2O; IR ν 3500–2500 (OH acid and enol), 1720 (C=O acid), 1630 (C=O ketone) cm−1. 1H NMR (DMF-d7) δ 5.34 (s, 2H, benzyl), 6.39 (s, 1H, hexanoate C3-H), 6.54 (d, 1H, hexanoate C5-H), 6.7 (bs, 1H, pyrrole C4-H), 7.38–7.67 (m, 7H, pyrrole C2-H, pyrrole C5-H, benzene H, and hexanoate C6-H), 14 (br s, 2H, OH enol and acid). Anal. (C17H16FNO4) C, H, N, F.
6-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-2-hydroxy-4-oxohexa-2,5-dienoic Acid (7j).
Compound 7j was prepared from 6j by means of GP-G. 75% as yellow solid; 128–129 °C; benzene; IR ν 3500–2500 (OH acid and enol), 1700 (C=O acid), 1600 (C=O ketone) cm−1. 1H NMR (CDCl3) δ 5.20 (s, 2H, CH2), 6.75 (bs, 1H, hexanoate C3-H), 6.80 (d, 1H, hexanoate C5-H), 7.1–7.6 (m, 12H, pyrrole C2-H, pyrrole C5-H, benzene H, benzyl H, and hexanoate C6-H), 14 (br s, 2H, enol and acid). Anal. (C23H17FNO4) C, H, N, F.
4-(1-(4-Fluorobenzyl)-1H-pyrrol-2-yl)-2-hydroxy-4-oxobut-2-enoic Acid (7k).
Compound 7k was prepared from 6k by means of GP-G 80% as yellow solid; 156–157 °C; benzene; IR ν 3500–2500 (OH acid and enol), 1700 (C=O acid), 1620 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 5.59 (s, 2H, CH2), 6.30 (dd, 1H, J = 2.5 Hz, J = 3.5 Hz, pyrrole C4-H), 6.81 (s, 1H, butenoate C3-H), 7.0–7.09–7.16 (m, 4H, benzene H), 7.39 (d, 1H, J = 3.5 Hz, pyrrole C3-H), 7.51 (s, 1H, pyrrole C5-H) 14 (br s, 2H, OH enol and acid). Anal. (C15H12FNO4) C, H, N, F.
4-(1-(4-Fluorobenzyl)-4-phenyl-1H-pyrrol-2-yl)-2-hydroxy-4-oxobut-2-enoic Acid (7l).
Compound 7l was prepared from 6l by means of GP-G. 82% as yellow solid; 195–196 °C; toluene; IR ν 3500–2000 (OH acid and enol), 1740 (C=O acid), 1620 (C=O ketone) cm−1. 1H NMR (DMSO d6) δ 5.68 (s, 2H, CH2 benzyl), 7.05 (s, 1H, butenoate C3-H), 7.1–7.3 (m, 5H, benzyl H and benzene H), 7.41 (t, 2H, benzene H), 7.76 (d, 2H, benzene H), 8.01 (s, 1H, pyrrole C3-H), 8.13 (s, 1H, pyrrole C5-H) 14 (br s, 1H, OH enol), 15 (bs, 2H, OH acid). Anal. (C20H16FNO4) C, H, N, F.
(E/Z)-2-Amino-4-(1-(4-fluorobenzyl)-4-phenyl-1H-pyrrol-3-yl)-4-oxobut-2-enoic Acid (7d).
Compound 6d (340 mg, 0.9 mmol) was dissolved in anhydrous THF (4.5 mL) under argon atmosphere and cooled to 0 °C. To this was added dropwise a solution of 0.5 N KOH (2 mL, 1.0 mmol), and the mixture was allowed to stir at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue partitioned between water and ethyl acetate. The aqueous layer was cooled on ice and acidified with 1 N HCl. After chilling at 4 °C for 2 h, the resulting precipitate was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and concentrated under vacuum to yield the acid 7d (250 mg). 76% yellow solid; 86–88 °C; toluene; IR ν 3000–2500 (OH acid and enol), 1700 (C=O acid) cm−1. 1H NMR (CDCl3) δ 4.95 (m, 2H, CH2 E/Z form), 6.70 (s, 1H, butenoate C3-H), 6.8–7.43 (m, 11H, pyrrole C2-H, pyrrole C5-H, benzene H, and benzyl H), 14 (br s, 2H, acid and enol). Anal. (C21H17FN2O3) C, H, N, F.
1-(1-(4-Fluorobenzyl)-1H-pyrrol-2-yl)-3-hydroxy-3-(1H-1,2,4-triazol-3-yl)prop-2-en-1-one (7m).
20 (1.84 g, 1.8 mmol) was suspended in 12 mL of dioxane and treated with 4.4 mL of 1 N HCl. The reaction mixture was stirred at 70 °C for 4 h. After cooling, the mixture was poured into 4.4 mL of 1.5 N NaOH. The formed precipitate was filtered and portioned between ethyl acetate and 1 N NaOH. The water phase was separated, and acidification until pH 4 was obtained was done with concentrated HCl. The formed solid was filtered, washed with water, and recrystallized from absolute ethanol, obtaining 430 mg of pure 7m. 64%; 188–189 °C; ethanol; IR ν 3200–2400 (NH, OH enol, and acid), 1712 (C=O ketone) cm−1. 1H NMR (DMSO-d6) δ 4.50 (s, 1H, butanoate C3-H), 5.47 and 5.62 (s, 2H, CH2 keto and enol form), 6.21 and 6.29 (t, 1H, pyrrole C4-H), 6.91 (s, 1H, butenoate C3-H), 7.06–7.16 (m, 6H, benzene H keto and enol form), 7.22 and 7.29 (m, 1H, pyrrole C3-H), 7.36 and 7.45 (s, 1H, pyrrole C2-H keto and enol form), 8.59 (bs, 1H, NH), 14 (br s, 1H, OH enol and acid). Anal. (C16H13FN4O2) C, H, N, F.
Biological Methods.
RT Expression and Purification.
The recombinant HIV-1 RT protein, whose coding gene was subcloned in the p6HRT_prot plasmid, was expressed in E. coli strain M15.43,44 The bacteria cells were grown up to an OD600 of 0.8 and induced with 1.7 mM IPTG for 5 h. HIV-1 RT purification was carried out as described. Briefly, cell pellets were resuspended in lyses buffer (20 mM Hepes, pH 7.5, 0.5 M NaCl, 5 mM β-mercaptoethanol, 5 mM imidazole, 0.4 mg/mL lysozime), incubated on ice for 20 min, sonicated, and centrifuged at 30 000g for 1 h. The supernatant was applied to a His-binding resin column and washed thoroughly with wash buffer (20 mM Hepes, pH 7.5, 0.3 M NaCl, 5 mM β-mercaptoethanol, 60 mM imidazole, 10% glycerol). The RT protein was eluted with elute buffer. The enzyme-containing fractions were pooled, dialyzed, and aliquots were stored at −80 °C.
HIV-1 RT RNase H Inhibition.
The RT-associated RNase H function was measured in a polymerase-independent cleavage assay, in which the poly(dC)-[3H]poly(rG) hybrid was used as reaction substrate as previously described.43
HIV-1 IN Inhibition.
HIV-1 IN gel-based assays were carried out as previously published.45
HIV-1 Replication Inhibition.
The antiviral activity of compounds was determined in a cell-based assay according to the procedure described previously46 and modified as follows. HeLa-CD4-LTR-β-gal cells were maintained in DMEM with 10% serum and 0.5 mg/mL G418. The day prior to experimentation, 96-well plates were prepared to contain 10 000 cells per well in 100 μL of Dulbecco’s modified Eagle medium (DMEM) complemented with 10% serum. On day 1, each drug was serial diluted directly on cells following a 3-fold dilution over 6 points, and each well was then filled to 200 μL with either fresh medium or concentrated viral supernatant (HIV-1(IIIB), Advanced Biotechnologies Inc.). The highest compound concentration tested was 50 μM. On day 2, cells were washed three times with PBS before adding 200 μL of a solution containing 50 mM Tris-HCl, pH 7.5, 100 mM β-mercaptoethanol, 0.05% Triton X100, and 5 mM 4-methylumbelliferyl-β-d-galactopyranoside (4-MUG, Sigma). On day 3, sealed plates were read in a SpectraMax GEMINI-XS (Molecular Devices) with λex = 360 nm and λem = 460 nm.
Cellular Toxicity.
Similar to the antiviral assays, plates were prepared with 10 000 HeLa-CD4-LTR-β-gal cells per well and a serial dilution of compounds in 100 μL. After 24 h of culture, 100 μL of ATPlite reagent (PerkinElmer) was added to each well. After 5 min at room temperature, the plates’ luminescence was quantified using an EnVision multilabel reader (PerkinElmer) according to the manufacturer’s instructions.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Italian MIUR for financial support, ISS 40H4, PRIN 2010–2011 (Grant 2010W2KM5L_002). R.D.S. and R.C. thank the FP7 CHAARM project for support. This work was also supported by the NIH Intramural Research Program, Center for Cancer Research, National Cancer Institute.
ABBREVIATIONS USED
- IN
integrase
- RT
reverse transcriptase
- RNase H
ribonuclease H
- 3′-P
3′-processing
- ST
strand transfer
- DKA
diketo acid
- BTDBA
4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid
- SI
selectivity index
- TosMIC
toluene-4-sulfonylmethyl isocyanide
- NIS
N-iodosuccinimide
- GP
general procedure
- DMEM
Dulbecco’s modified Eagle medium
Footnotes
ASSOCIATED CONTENT
Supporting Information
Elemental analysis results of compounds 6a–l and 7a–m. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Patyar S; Prakash A; Medhi B Dual inhibition: a novel promising pharmacological approach for different disease conditions. J. Pharm. Pharmacol. 2011, 63, 459–471. [DOI] [PubMed] [Google Scholar]
- (2).Mellors JW; Dutschman GE; Im GJ; Tramontano E; Winkler SR; Cheng Y-C In vitro selection and molecular characterization of human immunodeficiency virus-1 resistant to non-nucleoside inhibitors of reverse transcriptase. Mol. Pharmacol. 1992, 41, 446–451. [PubMed] [Google Scholar]
- (3).Mellors J; Im GJ; Tramontano E; Winkler SR; Medina DJ; Dutschman GE; Bazmi HZ; Piras G; Gonzales CJ; Cheng Y-C A single conservative amino acid substitution in the reverse transcriptase of human immunodeficiency virus type 1 confers resistance to (+)-(5S)-4,5,6,7-tetrahydro-5-methyl-6-(3-methyl-2butenyl)imidazo[4,5,1,jk][1,4]benzodiazepin-2(1H)-thione (TIBO R82150). Mol. Pharmacol. 1993, 43, 11–16. [PubMed] [Google Scholar]
- (4).Mehellou Y; De Clercq E Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [DOI] [PubMed] [Google Scholar]
- (5).Brühlmann C; Ooms F; Carrupt PA; Testa B; Catto M; Leonetti F; Altomare C; Carotti A Coumarins derivatives as dual inhibitors of acetylcholinesterase and monoamine oxidase. J. Med. Chem. 2001, 44, 3195–3198. [DOI] [PubMed] [Google Scholar]
- (6).Pretorius J; Malan SF; Castagnoli N Jr.; Bergh JJ; Petzer JP Dual inhibition of monoamine oxidase B and antagonism of the adenosine A(2A) receptor by (E,E)-8-(4-phenylbutadien-1-yl)caffeine analogues. Bioorg. Med. Chem. 2008, 16, 8676–8684. [DOI] [PubMed] [Google Scholar]
- (7).Altavilla D; Squadrito F; Bitto A; Polito F; Burnett BP; Di Stefano V; Minutoli L Flavocoxid, a dual inhibitor of cyclooxygenase and 5-lipoxygenase, blunts pro-inflammatory phenotype activation in endotoxin-stimulated macrophages. Br. J. Pharmacol. 2009, 157, 1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Park S; Chapuis N; Bardet V; Tamburini J; Gallay N; Willems L; Knight ZA; Shokat KM; Azar N; Viguié F; Ifrah N; Dreyfus F; Mayeux P; Lacombe C; Bouscary D. PI-103, a dual inhibitor of class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 2008, 22, 1698–1706. [DOI] [PubMed] [Google Scholar]
- (9).Li T; Wang J; Wang X; Yang N; Chen SM; Tong LJ; Yang CH; Meng LH; Ding J WJD008, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the proliferation of transformed cells with oncogenic PI3K mutant. J. Pharmacol. Exp. Ther. 2010, 334, 830–838. [DOI] [PubMed] [Google Scholar]
- (10).Wang QM; Johnson RB; Jungheim LN; Cohen JD; Villarreal EC Dual inhibition of human rhinovirus 2A and 3C proteases by homophthalimides. Antimicrob. Agents Chemother. 1998, 42, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Didierjean J; Isel C; Querre F; Mouscadet JF; Aubertin AM; Valnot JY; Piettre SR; Marquet R Inhibition of human immunodeficiency virus type 1 reverse trascriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob. Agents Chemother. 2005, 49, 4884–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Budihas S; Gorshkova I; Gaidamakov S; Wamiru A; Bona M; Parniak K; Crouch R; McMahon J; Beutler J; Le Grice S Selective inhibition of HIV-1 reverse trascriptase-associated ribonuclase H activity by hydroxylated tropolones. Nucleic Acids Res. 2005, 33, 1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Semenova EA; Johnson AA; Marchand C; Davis DA; Yarchoan R; Pommier Y Preferential inhibition of the magnesium dependent strand transfer reaction of HIV-1 integrase by α-hydroxytropolones. Mol. Pharmacol. 2006, 69, 1454–1460. [DOI] [PubMed] [Google Scholar]
- (14).Marchand C; Beutler JA; Wamiru A; Budihas S; Möllmann U; Heinisch L; Mellors JW; Le Grice SF; Pommier Y. Madurahydroxylactone derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and RNase H. Antimicrob. Agents Chemother. 2008, 52, 361–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Billamboz M; Bailly F; Barreca ML; De Luca L; Mouscadet JF; Calmels C; Andreola ML; Witvrouw M; Christ F; Debyser Z; Cotelle P Design, synthesis, and biological evaluation of a series of 2-hydroxyquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and reverse trascriptase RNase H domain. J. Med. Chem. 2008, 51, 7717–7730. [DOI] [PubMed] [Google Scholar]
- (16).Billamboz M; Bailly F; Lion C; Calmels C; Andreola ML; Witvrouw M; Christ F; Debyser Z; De Luca L; Chimirri A; Cotelle P 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: influence of the alkylation of position 4. Eur. J. Med. Chem. 2011, 46, 535–546. [DOI] [PubMed] [Google Scholar]
- (17).Cherepanov P; Maertens GN; Hare S Structural insights into the retroviral DNA integration apparatus. Curr. Opin. Struct. Biol. 2011, 21, 249–256. [DOI] [PubMed] [Google Scholar]
- (18).Beare KD; Coster MJ; Rutledge PJ Diketoacid inhibitors of HIV-1 integrase: from L-708,906 to raltegravir and beyond. Curr. Med. Chem. 2012, 19, 1177–11792. [DOI] [PubMed] [Google Scholar]
- (19).Cocohoba J; Dong BJ Raltegravir: the first HIV integrase inhibitor. Clin. Ther. 2008, 30, 1747–1765. [DOI] [PubMed] [Google Scholar]
- (20) (a).Kirschberg T; Parrish J Metal chelators as antiviral agents. Curr. Opin. Drug Discovery Dev 2007, 10, 460–472. [PubMed] [Google Scholar]; (b) Rogolino D; Carcelli M; Sechi M; Neamati N. Viral enzymes containing magnesium: metal binding as a successful strategy in drug design. Coord. Chem. Rev 2012, 256, 3063–3086. [Google Scholar]; (c) Rouffet M; Cohen SM Emerging trends in metalloprotein inhibition. Dalton Trans 2011, 40, 3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shaw-Reid CA; Munshi V; Graham P; Wolfe A; Witmer M; Danzeisen R; Olsen DB; Carroll SS; Embrey M; Wai JS; Miller MD; Cole JL; Hazuda DJ Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 2003, 278, 2777–2780. [DOI] [PubMed] [Google Scholar]
- (22).Tramontano E; Esposito F; Badas R; Di Santo R; Costi R; La Colla P 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2, 4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 2005, 65, 117–124. [DOI] [PubMed] [Google Scholar]
- (23).Esposito F; Corona A; Tramontano E HIV-1 reverse transcriptase still remains a new drug target: structure, function, classical inhibitors and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012, 2012, 586401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tramontano E HIV-1 RNase H: Recent progress in an exciting, yet little explored, drug target. Mini-Rev. Med. Chem. 2006, 6, 727–737. [DOI] [PubMed] [Google Scholar]
- (25).Tramontano E; Di Santo R HIV-1 RT-associated RNase H function inhibitors: recent advances in drug development. Curr. Med. Chem. 2010, 17, 2837–2853. [DOI] [PubMed] [Google Scholar]
- (26).Kharlamova T; Esposito F; Zinzula L; Floris G; Cheng Y-C; E. Dutschman G; Tramontano E. Inhibition of HIV-1 ribonuclease H activity by novel frangula-emodine derivatives. Med. Chem 2009, 5, 398–410. [DOI] [PubMed] [Google Scholar]
- (27).Tramontano E; Kharlamova T; Zinzula L; Esposito F Effects of new quinizarin derivatives on both HCV NS5B RNA polymerase and HIV-1 reverse transcriptase associated ribonuclease H activities. J. Chemother 2011, 23, 273–276. [DOI] [PubMed] [Google Scholar]
- (28).Esposito F; Corona A; Zinzula L; Kharlamova T; Tramontano E New anthraquinone derivatives as inhibitors of the HIV-1 reverse transcriptase-associated ribonuclease H function. Chemotherapy 2012, 58, 299–307 DOI: 10.1159/000343101. [DOI] [PubMed] [Google Scholar]
- (29).Suchaud V; Bailly F; Lion C; Tramontano E; Esposito F; Corona A; Christ F; Debyser Z; Cotelle P Development of a series of 3-hydroxyquinolin-2(1H)-ones as selective inhibitors of HIV-1 reverse transcriptase associated RNase H activity. Bioorg. Med. Chem. Lett. 2012, 22, 3988–3992. [DOI] [PubMed] [Google Scholar]
- (30).Distinto S; Esposito F; Kirchmair J; Cardia MC; Gaspari M; Maccioni E; Alcaro S; Markt P; Wolber G; Zinzula L; Tramontano E Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur. J. Med. Chem. 2012, 50, 1–14. [DOI] [PubMed] [Google Scholar]
- (31).Costi R; Métifiot M; Esposito F; Cuzzucoli Crucitti G; Pescatori L; Messore A; Scipione L; Tortorella S; Zinzula L; Novellino E; Pommier Y; Tramontano E; Marchand C; Di Santo R. 6-(1-Benzyl-1H-pyrrol-2-yl)-2,4-dioxo-5-hexenoic acids as dual inhibitors of recombinant HIV-1 integrase and ribonuclease H, synthesized by a parallel synthesis approach. J. Med. Chem. 2013, 56, 8588–8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Costi R; Métifiot M; Suhman C; Cuzzucoli Crucitti G; Maddali K; Pescatori L; Messore A; Madia VN; Pupo G; Scipione L; Tortorella S; Di Leva FS; Cosconati S; Marinelli L; Novellino E; Le Grice SFJ; Corona A; Pommier Y; Marchand C; Di Santo R. Basic quinolinonyl diketo acid derivatives as inhibitors of HIV integrase and their activity against RNase H function of reverse transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hazuda DJ; Felock P; Witmer M; Wolfe A; Stillmock K; Grobler JA; Espeseth A; Gabryelski L; Schleif W; Blau C; Miller MD Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [DOI] [PubMed] [Google Scholar]
- (34).Van Leusen AM; Siderius H; Hoogenboom BE; Van Leusen D A new simple synthesis of the pyrrole ring system from Michael acceptors and tosylmethylisocianides. Tetrahedron Lett. 1972, 52, 5337–5340. [Google Scholar]
- (35).Anguillera A; Alcantara AR; Marinas MJ; Sinistera V Ba(OH)2 as the catalyst in organic reaction. Part XIV. Mechanism of Claisen–Schmidt condensation in solid liquid conditions. Can. J. Chem 1987, 65, 1165–1171. [Google Scholar]
- (36).Sechi M; Bacchi A; Carcelli M; Sechi M; Bacchi A; Carcelli M; Compari C; Duce E; Fisicaro E; Rogolino D; Gates P; Derudas M; Al-Mawsawi LQ; Neamati N From ligand to complexes. Inhibition of human immunodeficiency virus tipe 1 by β-diketo acid metal complexes. J. Med. Chem. 2006, 49, 4248–4260. [DOI] [PubMed] [Google Scholar]
- (37).Drysdale MJ; Hind SL; Jansen M; Reinhard JF Jr. Synthesis and SAR of 4-aryl-2-hydroxy-4-oxobut-2-enoic acids and esters and 2-amino-4-aryl-4-oxobut-2-enoic acids and esters: potent inhibitors of kynurenine-3-hydroxylase as potential neuroprotective agents. J. Med. Chem. 2000, 43, 123–127. [DOI] [PubMed] [Google Scholar]
- (38).Nongkhlaw RL; Nongrum R; Myrboh B Synthesis of substituted hexa-3,5-dienoic acid methyl esters from conjugated dienones. J. Chem. Soc., Perkin Trans 1 2001, 1300–1303. [Google Scholar]
- (39).Fujishita T; Yoshinaga T; Sato A PCT Int. Appl. WO-00/39086, 2000. [Google Scholar]
- (40).Corona A; Di Leva FS; Thierry S; Pescatori L; Cuzzucoli Crucitti G; Subra F; Delelis O; Esposito F; Rigogliuso G; Costi R; Cosconati S; Novellino E; Di Santo R; Tramontano E Identification of highly conserved residues involved in inhibition of HIV-1 RNase H function by diketo acid derivatives. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Alberola A; Jose MA; Gonzalez A; Pedrosa R; Vicente M Differential reactivity of β-amino enones and 3-(dimethylamino)-acrylaldehyde towards α-amino derivatives. J. Chem. Soc., Perkin Trans. 1 1990, 10, 2681–2685. [Google Scholar]
- (42).Di Santo R; Costi R; Artico M; Ragno R; Greco G; Novellino E; Marchand C; Pommier Y Design, synthesis and biological evaluation of heteroaryl diketohexenoic and diketobutanoic acids as HIV-1 integrase inhibitors endowed with antiretroviral activity. Farmaco 2005, 60, 409–417. [DOI] [PubMed] [Google Scholar]
- (43).Tramontano E; Cheng Y-C HIV-1 reverse transcriptase inhibition by a dipyridodiazepinone derivative: BI-RG-587. Biochem. Pharmacol. 1992, 43, 1371–1376. [DOI] [PubMed] [Google Scholar]
- (44).Esposito F; Kharlamova T; Distinto S; Zinzula L; Cheng Y-C; Dutschman G; Floris G; Markt P; Corona A; Tramontano E Alizarine derivatives as new dual inhibitors of the HIV-1 reverse transcriptase(RT)-associated DNA polymerase and ribonuclease H (RNase H) activities effective also on the RNase H activity of non-nucleoside resistant RTs. FEBS J 2011, 278, 1444–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Métifiot M; Maddali K; Johnson BC; Hare S; Smith SJ; Zhao XZ; Marchand C; Burke TR Jr.; Hughes SH; Cherepanov P; Pommier Y Activities, crystal structures, and molecular dynamics of dihydro-1H-isoindole derivatives, inhibitors of HIV-1 integrase. ACS Chem. Biol. 2013, 8, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Chung S; Wendeler M; Rausch JW; Beilhartz G; Gotte M; O’Keefe BR; Bermingham A; Beutler JA; Liu S; Zhuang X; Le Grice SF Structure–activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 2010, 54, 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.