

Abstract
Hepatocyte nuclear factor 4 alpha (HNF4α) is a master regulator of development and function of digestive tissues. The HNF4A gene uses two separate promoters P1 and P2, with P1 products predominant in adult liver whereas P2 products prevalent in fetal liver, pancreas, and liver/colon cancer. To date, the mechanisms for the regulation of HNF4A and the dynamic switch of P1 and P2-HNF4α during ontogenesis and carcinogenesis are still obscure. Our study validated the previously reported self-stimulation of P1-HNF4α but invalidated the reported synergism between HNF4α and HNF1α. HNF4A-AS1, a long-non-coding RNA, is localized between the P2 and P1 promoter of HNF4A. We identified critical roles of P1-HNF4α in regulating the expression of HNF4A-AS1 and its mouse ortholog Hnf4a-os. PAX6, a master regulator of pancreas development overexpressed in colon cancer, cooperated with HNF1α to induce P2-HNF4α but antagonized HNF4α in HNF4A-AS1 expression. Thus, PAX6 may be important in determining ontogenic and carcinogenic changes of P2-HNF4α and HNF4A-AS1 in the pancreas and intestine. We also interrogated transactivation activities on multiple gene targets by multiple known and novel HNF4α mutants identified in patients with maturity onset diabetes of the young1 (MODY1) and liver cancer. Particularly, HNF4α-D78A and HNF4α-G79S, two mutants found in liver cancer with mutations in DNA-binding domain, displayed highly gene-specific transactivation activities. Interestingly, HNF4α-Q277X, a MODY1 truncation mutant, antagonized the transactivation activities of HNF1α and FXR, key regulators of insulin secretion. Taken together, our study provides novel mechanistic insights regarding the transcriptional regulation and transactivation activity of HNF4α in digestive tissues.
Keywords: HNF4A, MODY1, LIVER CANCER, DIABETES, HNF1A, PAX6, D69A, Q268X
Hepatocyte nuclear factor 4 alpha (HNF4α) is a liver-enriched master regulator of liver development and liver function [Gonzalez, 2008; Walesky et al., 2013]. HNF4α also play key roles in regulating the development and function of pancreas and intestine [Garrison et al., 2006; Wang et al., 2000]. The HNF4A gene is composed of 12 exons and driven by two separated P1 and P2 promoters [Hwang-Verslues and Sladek, 2010]. Overall 9 transcriptional variants are generated via alternative splicing [Babeu and Boudreau, 2014; Lu, 2016]. Currently, HNF4α1–3 and HNF4α7–9 are experimentally verified P1 and P2 products. The activation function 1 (AF-1) domain at the N-terminus depicts the major difference between the P1 and P2 HNF4α. The P2-HNF4α lacks the AF-1 domain and therefore is weaker than P1-HNF4α regarding the transactivation activity [Babeu and Boudreau, 2014; Vuong et al., 2015]. Additionally, P1- and P2-HNF4α also differ in terms of tissue distribution. P1-HNF4α is predominantly expressed in adult liver and kidney, whereas P2-HNF4α is mainly prevalent in fetal liver and pancreas [Perilhou et al., 2008; Vuong et al., 2015]. Both products are detectable in adult colon and small intestine [Tanaka et al., 2006]. The dysregulation of P1- and P2-HNF4α is a hallmark in multiple carcinogenesis. Down-regulation of P1-HNF4α has been reported in hepatocellular carcinoma (HCC), colorectal carcinoma, and gastric carcinoma, during which P2 products are aberrantly elevated [Oshima et al., 2007; Tanaka et al., 2006]. P2-HNF4α has been shown to promote inflammation and carcinogenesis in colon [Chellappa et al., 2016; Vuong et al., 2015].
To date, the precise mechanism for maintaining the balance of P1- and P2-HNF4α expression in different tissues and tumors remains unknown. HNF4A-AS1 (NR_109949.1) is an antisense RNA transcribed by the complementary motif of HNF4A gene between the P1 and P2 promoter. Antisense RNA has a widespread occurrence within the human genome [Yelin et al., 2003], and many of which are known to inhibit gene expression by affecting DNA methylation, chromatin modification, isoform variation and RNA stability [Magistri et al., 2012; Pelechano and Steinmetz, 2013]. HNF4A-AS1 has been implicated as one of the biomarkers for the diagnosis of HCC and Crohn’s disease [Esposti et al., 2016; Haberman et al., 2018]. These findings strongly indicate the positive correlation between HNF4α and HNF4A-AS1, as loss of HNF4α is a hallmark of those diseases. In the present study, we found that P1-HNF4α strongly activated the proximal promoter of HNF4A-AS1, which indicated that HNF4A-AS1 may be a biomarker of the P1-HNF4α expression, and might be involved in regulating the expression and function of HNF4α isoforms.
The self-stimulation of HNF4α on its own promoter, and the reciprocal regulatory loop between HNF4α and HNF1α has been reported for more than two decades [Bailly et al., 1998; Hatzis and Talianidis, 2001; Spath and Weiss, 1997]. This positive feedback loop between HNF4α and HNF1α needs a break, such as self-inhibition of HNF1α [Ktistaki and Talianidis, 1997], to sustain constant intracellular levels of HNF4α and HNF1α in vivo. Surprisingly, a following study indicated that HNF4α was capable of not only self-activating its own promoter, but also inducing a highly synergistic self-activation in HeLa cells via interacting with HNF1α and HNF6 [Hatzis and Talianidis, 2001]. Such a strong self-activation by HNF4α is very unusual in biology. Thus, this study was aimed to interrogate the exact self-regulatory mechanisms of HNF4α using luciferase reporters that contain more complete regulatory genetic sequences of HNF4A in two cell lines, human embryonic kidney 293 (HEK293) cells and hepatocellular carcinoma HepG2 cells.
Many pathological mutations of HNF4α have been reported to compromise the transactivation activity of HNF4α. Maturity Onset Diabetes of the Young 1 (MODY1) is caused by the heterozygous loss-of-function mutations of HNF4α. However, most of these HNF4α mutants do not exert dominant-negative effects on the wild type (WT) HNF4α; it remains unclear how moderate decreases of HNF4α activity in patients with heterozygous HNF4α mutation cause MODY [Sato et al., 2017; Sladek et al., 1998; Wisely et al., 2002]. MODY1 currently is less commonly identified than MODY3, which accounts for the majority of MODY cases and is mainly caused by mutations of HNF1α [Forlani et al., 2010; Vaxillaire et al., 1997]. The pathologies of MODY1 and MODY3 are very similar. Based on the reported reciprocal regulatory loop of HNF4α and HNF1α, an HNF4α mutation may compromise its ability to transactivate HNF1α, resulting in the dysregulation of HNF1α and MODY. Additionally, it is noteworthy that HNF4α serves as a master regulator in liver via crosstalk with many other transcription factors [Lu, 2016]. A point mutation may disrupt one specific function of HNF4α protein (e.g. DNA binding, ligand binding, dimerization, etc), which may consequently compromise its transactivation activity for a subset of gene targets, or alter its crosstalk with other factors. Therefore, it is in a strong desire that the functional studies of HNF4α mutants should cover a broad range of gene-targets and their crosstalk with other key factors to fully depict the effect of a potential pathological mutation of HNF4α. In this study, we interrogated transactivation activities on multiple gene targets by multiple known and novel HNF4α mutants identified in patients with MODY1 and liver cancer. Our results illustrated highly gene-specific changes of transcriptional activities by two cancer mutants and markedly altered crosstalk with other transcription factors by a MODY1 mutant.
MATERIALS AND METHODS
VECTOR CONSTRUCTIONS
All reporter/expression vectors used in the study were generated by restriction digestion and ligation reactions or site-directed mutagenesis (SDM). The inserts with artificial restriction enzyme sites were obtained by PCR cloning or direct purchase from Integrated DNA Technologies (IDT) and GenScript. For vector construction, the luciferase reporter vectors were built on the pGL3-Basic/Promoter backbones, and the protein expression vectors were built on the pCDNA backbone. The detailed information regarding the engineered sequence was provided in supplemental table 1. The mammalian expression vectors for HNF1α (FR_HNF1A, #31104), P1-HNF4α (FR_HNF4A2, #31100), and P2-HNF4α (FR_HNF4A8, #31114) as well as luciferase reporter vector for the HNF4A P2 promoter (HNF4A_P2–2200, #31062) were purchased from Addgene. The expression vector for wildtype P1-HNF4α, pcDNA3-HNF4A2 was generated by PCR cloning of the HNF4A2 cDNA into the EcoRI/NotI site of pcDNA3 using the FR_HNF4A2 as the PCR template. All HNF4α mutations used in this study were generated by SDM with the Q5® Site-Directed Mutagenesis Kit (E0554S, New England BioLabs) using the pcDNA3-HNF4A2 as the template and verified by sequencing. The mammalian expression vectors for HNF1β (pBJ5 HNF1β, Dr.Gerald Crabtree, Stanford University), HNF3β (pHNF3β, Dr. David Waxman, Boston University), HNF6 (pCMV Flag HNF6, Dr. Frédéric Lemaigre, Université Catholique de Louvain), PAX6 (Dr. Ales Cvekl, Albert Einstein College of Medicine), and FXR (pCMX FXRα2, Dr. David Mangelsdorf, University of Texas Southwestern Medical Center) were generous gifts from other laboratories. The luciferase reporter p-1445-Luc for the promoter of human bile salt export pump (BSEP) was a gift from Dr. M. Ananthanarayanan (The Mount Sinai Medical Center).
TRANSIENT TRANSFECTION AND DUAL-LUCIFERASE ASSAY
Human embryonic kidney 293 (ATCC Cat# CRL-3216, RRID:CVCL_0063) and human hepatocellular carcinoma HepG2 (ATCC Cat# CRL-10741, RRID:CVCL_1098) were seeded in the 96-well-plate, After 24 h, cells in each well were transfected with 100 ng plasmids that included 28 ng firefly luciferase vectors, 2 ng renilla luciferase vectors (pRL-CMV), 10 ng eGFP expressing vectors (to monitor the transfection efficiency), and 60 ng other expression vectors (collection of co-transfecting factors), using Lipofectamine 2000 or 3000 (Invitrogen, 2000 for HEK293 cells and 3000 for HepG2 cells). Cells were harvested 24 h after transfection for dual-luciferase assay using Dual-Glo™ luciferase assay system (Promega) and GloMax Luminometer (Promega), following the manufacturer’s protocol. The ratios of firefly/renilla luciferases were calculated as the normalized reporter activities, with the control values set as 1.0.
REAL-TIME QUANTITATIVE PCR
Total RNAs from cells were isolated by RNA-STAT60 (Tel-Test) and quantified by Qubit RNA assay kit and Qubit 2.0 fluorometer (Life technology). The High-Capacity RNA-to-cDNA™ kit(Applied Biosystems®, life technologies) was used for cDNA synthesis. iQ™ SYBR® Green supermix (Bio-rad) was applied to quantify mRNAs using MyiQ2™ Two-Color Real-Time PCR System (Bio-rad). The amounts of mRNA were calculated using the comparative CT method, which determines the amount of target gene normalized to an introduced control.
WESTERN BLOT ASSAY
HEK293 cells in 6-well-plates were transfected with 500 ng HNF4α expression vectors and 500 ng pcDNA3-eGFP. Whole cell lysates were prepared 24 h after transfection. Proteins in cell lysates were resolved in sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Western blot quantification of HNF4α and EGFP was conducted with primary antibodies as follows: anti-HNF4α (Cell Signaling Technology Cat# 3113S, RRID:AB_2295208) and anti-GFP (Abcam Cat# ab290, RRID:AB_303395). Primary antibodies were revealed with HRP-conjugated secondary antibodies (Cell Signaling Technology Cat# 7076, RRID:AB_330924 & Cell Signaling Technology Cat# 7074, RRID:AB_2099233) and ECL Western Blotting Substrate (W1015, Promega). ChemiDocTM XRS+System (Bio-Rad) and software were used for band capturing.
STATISTICAL ANALYSIS
All values were expressed as mean ± S.E. The two-tailed Student’s test was used to determine the statistical difference between two groups, which was set as p < 0.05.
RESULTS
The 5’UTR coding motif is required for the self-stimulation of the proximal P1 promoter by P1-HNF4α.
We in the first place wanted to reproduce the self-stimulation of HNF4α. Our recent study discovered a critical role of the HNF4α 5’ UTR in inhibiting the protein expression of HNF4α [Guo and Lu, 2017]. In order to further investigate the mechanism of the self-stimulation of P1-HNF4A, we generated multiple luciferase reporter vectors carrying the P1-promoter (nt −985) and/or the coding sequence of 5’UTR (nt +89). For control purpose, we also included the empty vector (pGL3-Basic) and the 5’UTR only vector (pGL3T7–5’UTR-Luc). Surprisingly, in HEK293 cells, the P1-HNF4α enhanced the luciferase activity by 2.5 fold only in the presence of both the P1-promoter and the +89nt 5’UTR coding region (Fig.1A). The reporter vector that merely contains the P1 promoter (HNF4A-P1-Luc) or the 5’UTR was not activated by HNF4α (Fig.1A). Results of RT-PCR showed that P1-HNF4α did not alter the mRNA levels of the luciferase reporter (Fig.1B), indicating that the self-stimulation of HNF4α on its proximal P1 promoter may not be through the up-regulation of transcription. Similarly, HNF4α activated HNF4A-P1–5’UTR-Luc by two fold in HepG2 cells, but had no effect on HNF4A-P1-Luc (Fig.1C). In order to determine if the HNF4A-5’UTR is commonly required for the transcription-factor (TF)-modulated transactivation, we also tested HNF1α, a known HNF4A transactivator. Interestingly, in HEK293 cells, HNF1α activated HNF4A-P1 promoter by ~12 fold without requiring the 5’UTR coding motif (Fig.1D). Overall, these data confirmed the self-activation of HNF4α on its own proximal promoter, and indicated that the 5’UTR coding region (+89 nt) was indispensable for HNF4α-mediated activation of the P1 proximal promoter.
Fig. 1. Self-regulation of HNF4α P1 proximal promoter by HNF4α.
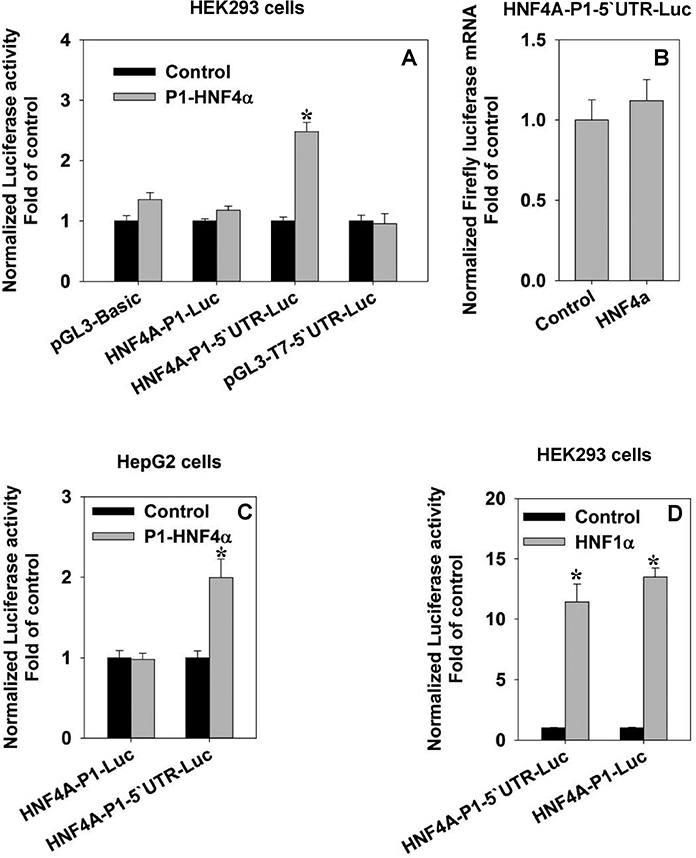
(A) Effects of HNF4α on the reporter activities of HNF4A-P1 promoter with/without 5’ UTR in HEK293 cells. N=4, mean ± SE. * p<0.05 vs control group; (B) Real-time PCR quantification of HNF4A-P1–5’UTR mRNA in the presence/absence of HNF4α. N=3, mean ± SE; (C) Effects of HNF4α on the reporter activities of HNF4A-P1 promoter with/without 5’ UTR in HepG2 cells. in. N=4, mean ± SE. * p<0.05 vs control group; (D) Effects of HNF1α on the reporter activities of HNF4A-P1 promoter with/without 5’ UTR in HEK293 cells. N=4, mean ± SE. * p<0.05 vs control group.
The proximal promoter of HNF4A-AS1 is strongly activated by P1-HNF4α but not P2-HNF4α
HNF4A-AS1 (NR_109949.1) encodes a 648-nt antisense non-coding-RNA that locates on the complementary strand of P2-HNF4A_intron 1–2. The overall length of HNF4A-AS1 coding sequence is ~17.9 kb that consists of four exons and three introns. The P1 proximal promoter is approximately 8 kb downstream (Fig.2A). The chromatin immunoprecipitation-sequencing (ChIP-seq) data from both human liver and HepG2 cells indicated that P1-HNF4α had strong bindings to the HNF4A-AS1 locus (Fig.2B). The same data also revealed the weak and strong bindings of HNF4α and HNF1α, respectively to the HNF4A-P1 proximal promoter, which was consistent with the reported studies that both factors activated the P1-HNF4A. Moreover, our RNA-sequencing data from primary human hepatocytes treated with the HNF4α inhibitor (BI6015) indicated that the transcripts of HNF4A-AS1 was reduced by inhibition of HNF4α, in accordance with the Na+-taurocholate cotransporting polypeptide (NTCP, also known as SLC10A1), an HNF4α-target gene (Fig.S1A). Last but not least, the HNF4A-AS1 is mainly transcribed in liver, kidney and intestine, which highly overlap with P1-HNF4α regarding the tissue distribution (Fig.S1B). Taken together, we hypothesized that HNF4α is capable of activating the transcription of HNF4A-AS1, which potentially regulates the P1- and P2-HNF4α expression. To test this hypothesis, we cloned the proximal promoter (Fig.2A, red arrow) of HNF4A-AS1 into the pGL3-Basic reporter, and found that 3 ng and 10 ng P1-HNF4α strongly activated the reporter vector HNF4A-AS1-Pro-Luc by 25 and 58 fold, respectively (Fig.3A). Conversely, P2-HNF4α had no activation (Fig.3A). To determine whether this HNF4A-AS1 promoter region could also regulate the transcription of P1-HNF4A (as a distal promoter), we cloned the reverse complementary sequence of the HNF4A-AS1 proximal promoter (Fig.2A, green arrow) into the 5’ upstream of the P1 proximal promoter in the HNF4A-P1–5’UTR-Luc. The newly generated chimerical reporter vector was named HNF4A-Dis-P1–5’UTR-Luc. Compared to the HNF4A-P1–5’UTR-Luc, insertion of the potential distal promoter at the upstream of the P1 promoter induced an additional 50% increase in the luciferase activity when stimulated by P1-HNF4α (Fig.3B). The same study was also performed in HepG2 cells, where introducing the distal promoter induced an additional 63% enhancement for the self-stimulation (Fig.3C). In conclusion, the proximal promoter of HNF4A-AS1 was strongly activated by P1-HNF4α but not P2-HNF4α. The reverse complementary motif of HNF4A-AS1 proximal promoter served as a distal promoter of P1-HNF4A that could augment the self-stimulation.
Fig. 2. Bioinformatics studies indicate the correlation between HNF4A-AS1 and HNF4α.
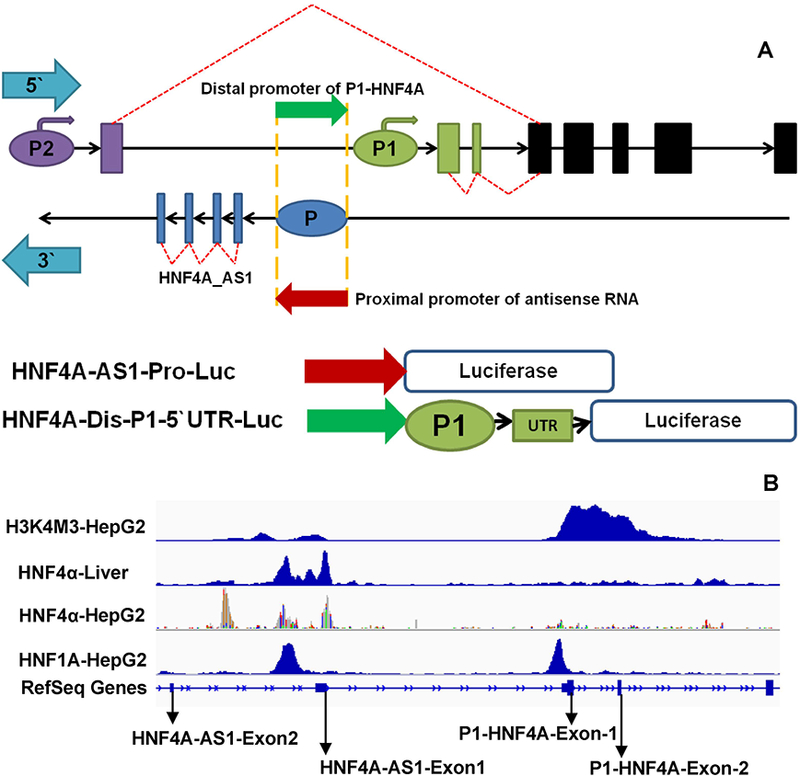
(A) A schematic view of HNF4A/HNF4A-AS1 genomic structure and the construction principle of reporter vectors with HNF4A-AS1. Purple ellipse: HNF4A-P2 promoter; Purple rectangle: P2-exon1; Cyan arrows: The indicator of 5’ and 3` orientation. Blue ellipse: The proximal promoter of HNF4A-AS1; Blue rectangle: HNF4A-AS1 exons; Red arrow: coding sequence of HNF4A-AS1 proximal promoter. Green arrow: The reverse complementary sequence of red arrow; (B) The ChIP-seq data showing the binding of HNF4α to the promoter of HNF4A-AS1 and P1-HNF4A in liver and HepG2 cells.
Fig. 3. Regulation of HNF4A-AS1 promoter by HNF4α.
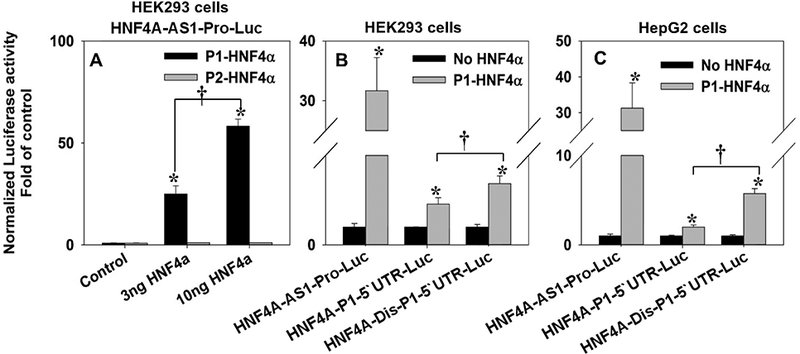
(A) Luciferase reporter activities of HNF4A-AS1-Pro-P1-Luc induced by 0, 3 and 10 ng P1 & P2 HNF4α in HEK293 cells. N=4, mean ± SE. * p<0.05 vs control group. † p<0.05 vs 3 ng P1-HNF4α; (B & C) Comparison of reporter activities of HNF4A-AS1-Pro-Luc, HNF4A-P1–5’UTR-Luc, and HNF4A-Dis-P1–5’UTR-Luc with/without HNF4α induction in HEK293 cells (B) and HepG2 cells (C). N=4, mean ± SE. * p<0.05 vs HNF4α-null group. † p<0.05 vs HNF4A-P1–5’UTR-Luc.
In addition, we found evidence that the ortholog of HNF4A-AS1 in mouse, Hnf4a-os (NR_027970.1), also had a strong correlation with Hnf4α. For instance, Hnf4α showed strong binding to Hnf4aos1-exon1 based on the ChIP-seq data from mouse liver (Fig.S2A). The RNA-seq data of mouse liver indicated that hepatic expression of Hnf4a_os started to rise after birth and reached the highest levels at adulthood. In adult mouse liver, Hnf4a_os had a moderate reduction in Hnf4α heterozygous mice and a remarkable down-regulation in Hnf4α knockout mice (Fig.S2B & 2C). All these changes of Hnf4a_os were consistent with the expression level of P1 Hnf4α. Moreover, we cloned the proximal promoter of Hnf4a_os into the reporter vector, and found that Hnf4α activated it by 4.2 fold (Fig.S2D). These findings suggest a conserved important role of HNF4A-AS1 in liver during evolution.
Paired box 6 (Pax6) activates the P2 promoter whereas antagonizes the P1-HNF4α-induced activation OF HNF4A-AS1
A previous study indicates that P1-HNF4α is expressed in human fetal pancreas [Harries et al., 2008], whereas P1-HNF4α and HNF4A-AS1 are barely detectable in adult pancreas. It remains unknown how the P2-HNF4α predominates in adult pancreas and colon cancer. Pax6 is a crucial transcriptional factor for the development and functional maintenance of pancreas [Hart et al., 2013]. We analyzed the ChIP-seq data of Pax6 in pancreas and found that Pax6 was capable of binding to HNF4A-P2 promoter (Fig.4A). Thus, we hypothesized that Pax6 is one of the key factors in pancreas that regulates the P2-HNF4A transcription. We determined the effect of Pax6 on HNF4A-P2-Luc in HEK293 and HepG2 cells. We also included HNF1α, which was known to activate the P2 promoter [Galan et al., 2011], as a positive control. HNF1α activated HNF4A-P2-Luc by 8 and 5 fold in HEK293 and HepG2 cells, respectively (Fig.4B). Pax6 activated the P2 promoter by 2 and 8.5 fold in HEK293 and HepG2 cells, respectively. The combination of Pax6 and HNF1α activated the P2 promoter by 13 and 21 fold, respectively (Fig.4B). We then further studied the effect of Pax6 on the activation of HNF4A-AS1 by P1-HNF4α. As expected, over-expression of Pax6 in HEK293 cells caused a 44% reduction of HNF4A-AS1 promoter activity activated by P1-HNF4α (Fig.4C). In summary, the Pax6 was capable of stimulating the P2 promoter and antagonizing the transactivation of HNF4A-AS1 induced by P1-HNF4α.
Fig. 4. Regulation of HNF4α P2 promoter and HNF4A-AS1 promoter by Pax6.
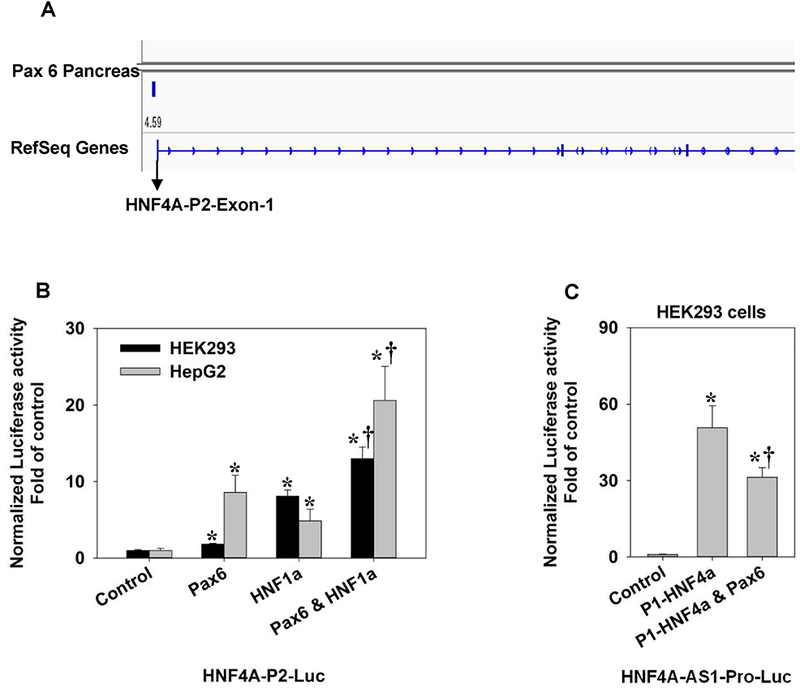
(A) The ChIP-seq data showing the binding of Pax6 to the HNF4A-P2 promoter in the pancreas; (B) Effects of Pax6 and HNF1α on the reporter activity of HNF4A-P2-Luc in HEK293 cells and HepG2 cells. N=4, mean ± SE. * p<0.05 vs control † p<0.05 vs Pax6 or HNF1α single treatment; (C) Effects of Pax6 and HNF4α on the reporter activity of HNF4A-AS1-Pro-Luc in HEK293 cells. N=4, mean ± SE. * p<0.05 vs control. † p<0.05 vs P1-HNF4α group.
HNF4α shows no synergistic effect with either HNF1α or HNF6 for the self-stimulation.
Previous studies indicated that HNF4α synergizes with HNF6 and HNF1α to activate its own promoter [Hatzis and Talianidis, 2001]. In order to validate the reported synergism, so as to systemically study the crosstalk of HNF4α with other TFs, we combined HNF4α with HNF1α, HNF1β, and HNF3β, which were all liver-enriched TFs, to determine the effects of their interactions on the HNF4A P1 promoter. In addition to the known induction by HNF1α and HNF4α, HNF1β also activated the P1 promoter by 1.5 fold (Fig.5A). HNF3β had no prominent effect. However, none of the combinations displayed a synergistic activation (Fig.5A). We repeated the combination of HNF4α and HNF1α on HNF4A-Dis-P1–5’UTR-Luc, and found no synergism either (Fig.5B). To test the synergism between HNF4α and HNF6, we introduced the reporter for the promoter of PDZ domain containing 1 (PDZK1), a known HNF4α-target gene [Zhang et al., 2014]. The transporter adaptor protein PDZK1 is required for proper membrane localization of certain key membrane transporters important for lipid metabolism [Kato et al., 2005; Kocher and Krieger, 2009]. The combination of HNF4α and HNF6 activated the PDZK1 promoter by 42 fold (Fig.5C), which indicated a synergism compared to the individual activation by HNF4α and HNF6 (12.8 fold and 2.2 fold, respectively). Nevertheless, co-expression of HNF4α and HNF6 only induced a 3.4-fold activation of HNF4A-P1–5’UTR-Luc (Fig.5C), which was less than the sum of the individual treatment (1.8 fold and 2.3 fold). Similar results were obtained with the HNF4A-Dis-P1–5’UTR-Luc: in HEK293 cells, the co-expression of HNF6 and HNF4α induced a 5-fold activation, and the individual treatments were 1.6 fold and 3.4 fold (Fig.5D). Likewise, in HepG2 cells, the combination induced an 8.7-fold activation, whereas the individual treatments were 4.3 fold and 5.7 fold, respectively (Fig.5D). Overall, we observed the synergism between HNF4α and HNF6/HNF1α on PDZK1 promoter (See Fig.8B for the synergistic activation of PDZK1 promoter by HNF4α and HNF1α). Even so, by the same combination we only observed an additive transactivation on the HNF4A P1 promoter by HNF4α and HNF6, and HNF4α tended to inhibit, rather than enhance the transactivation of the proximal P1 promoter by HNF1α (Fig.5A).
Fig. 5. Interactions of HNF4α with other liver-enriched transcription factors in the regulation of gene expression.
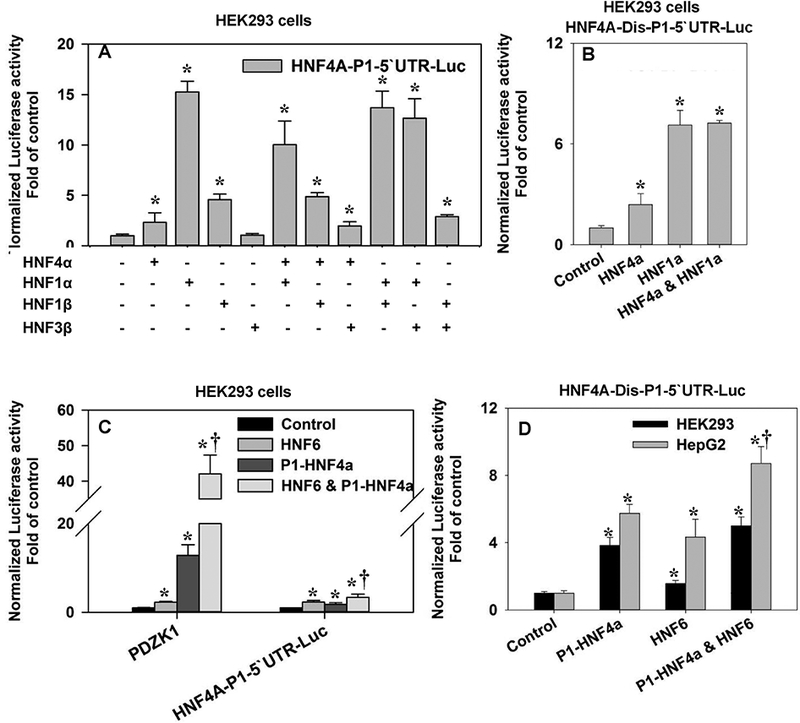
(A) Effects of HNF4α, HNF1α, HNF1β, and HNF3β on the reporter activities of HNF4A-P1–5’UTR-Luc in HEK293 cells. N=4, mean ± SE. * p<0.05 vs control; (B) Effects of HNF4α and HNF1α on the reporter activities of HNF4A-Dis-P1–5’UTR-Luc in HEK293 cells. N=4, mean ± SE. * p<0.05 vs control; (C & D) Effects of HNF4α and HNF6 on the reporter activities of PDZK1 and HNF4A-P1–5’UTR-Luc, in HEK293 cells (C) and HNF4A-Dis-P1–5’UTR-Luc in HEK293 and HepG2 cells (D). N=4, mean ± SE. * p<0.05 vs control. † p<0.05 vs HNF6 or P1-HNF4α single treatment.
Fig. 8. Interactions of HNF4α mutants with HNF1α and HNF6 in the regulation of PDZK1 promoter in HEK293 cells.
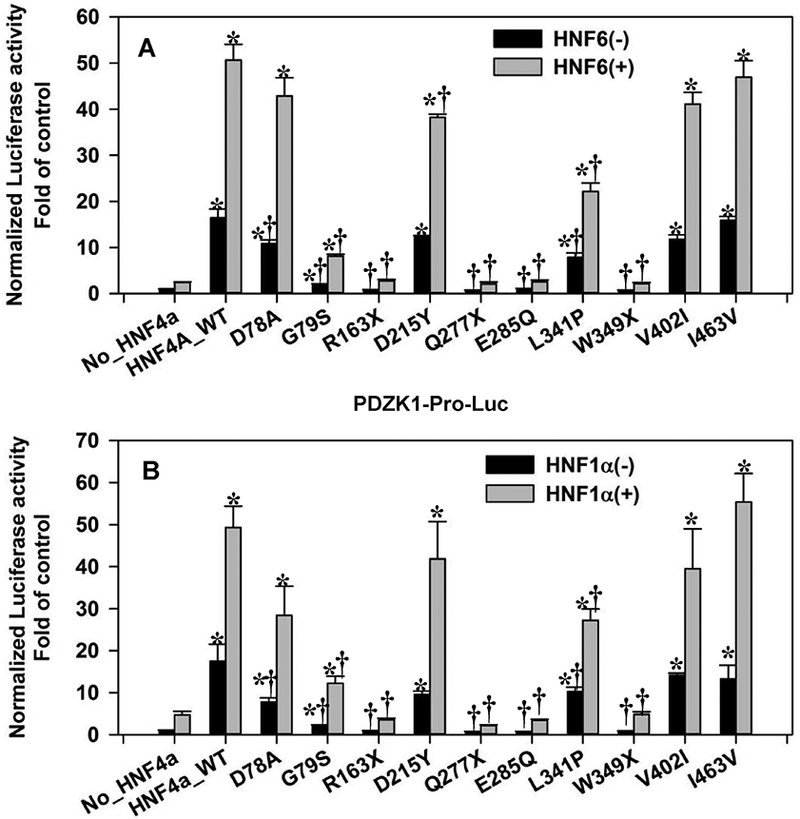
Reporter activities of PDZK1 promoter induced by WT/mutant P1-HNF4α in the presence/absence of HNF6 (A) or HNF1α (B). N=4, mean ± SE. * p<0.05 vs HNF4α-null group. † p<0.05 vs HNF4α_WT group.
A serial scan of HNF4A mutations revealed the gene-specific transactivation activity of multiple HNF4α mutants
To investigate how the pathological mutations of HNF4α affect their transactivation activities, we selected a set of P1-HNF4α mutations from MODY1 and liver cancer (Fig.6A), which include D78A (was D69A), G79S, R163X (was R154X), D215Y (was D206Y), Q277X (was Q268X), E285Q (was E276Q), L341P, W349X (was W340X), V402I (was V393I) and I463V (. 5A). D78A is a mutation originally found in HepG2 cells. This mutation alters the structure of the Zinc-finger in the DNA-binding domain (DBD), and has unchanged or enhanced transactivation activity on several promoters compared to WT-HNF4α [Lausen et al., 2000]. G79S (COSM4943131) thus far is only identified in cancers, including two cases of liver cancer, in the COSMIC human cancer mutations database. D215Y/D206Y is a MODY1 mutation that has been reported to occur near the binding pocket of the LXXLL motif, and has a subtle effect on the transactivation [Han et al., 2012]. R163X/R154X, Q277X/Q268X and W349X/W340X are all MODY1 non-sense mutations that produce a truncated HNF4α, which consequently results in the dysfunction of the HNF4α protein [Lausen et al., 2000; Raeder et al., 2006]. L341P/L332P is an uncharacterized MODY1 mutation [Pearson et al., 2005]. E285Q/E276Q and V402I/V393I are MODY1 mutations that have reduced transactivation activities [Hani et al., 1998; Lausen et al., 2000]. I463V/I454V has a mutation on the F domain, and whether this variant is responsible for MODY1 is still uncertain [Amara et al., 2015]. We created all mutant HNF4α expressing vectors by using the wild type HNF4α2, one of the major isoform of P1-HNF4α.
Fig. 6. Effects of HNF4α mutations on the transcriptional activities of HNF4α in HEK293 cells.
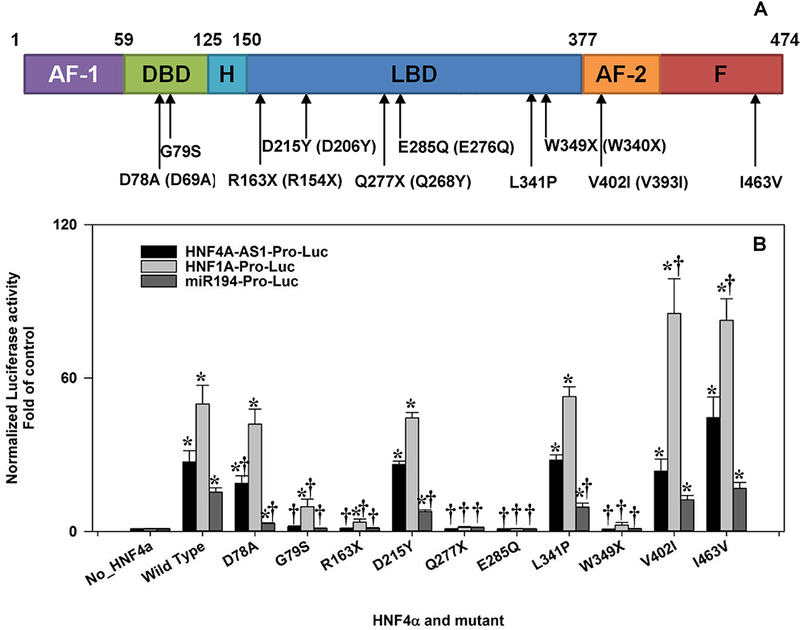
(A) A schematic view of HNF4α2 protein structure. AF-1 & AF-2: Activation Function domain 1 & 2; DBD: DNA Binding Domain; H: Hinge; LBD: Ligand Binding Domain; F: F domain. The mutants in parentheses indicated the prior known names of HNF4α mutations; (B) The reporter activities of HNF4A-AS1-Pro-Luc, HNF1A-Pro-Luc and miR-194-Pro-Luc induced by wild-type and mutant HNF4α2. N=4, mean ± SE. * p<0.05 vs control. † p<0.05 vs wildtype HNF4α (HNF4α_WT).
We investigated the effects of all these WT and mutant HNF4α on three HNF4α-target promoters which were HNF4A-AS1, HNF1A, and miR-194 [Lu et al., 2017]. The detailed changes of luciferase activities were presented in supplemental Table 2. Briefly, E285Q and the two nonsense mutations including Q277X and Q349X essentially lost the transactivation activity on all three promoters (Fig. 6B and supplemental Table 2). Noteworthy, R163X maintained a weak transactivation activity on the proximal promoter of HNF4A-AS1, even though it completely lost the activation of miR-194 and HNF1A promoters, similar to other truncated mutations (Fig. 6B). D215Y and L341P had the same performance with the wild type on the promoters of HNF4A-AS1 and HNF1A; however, both mutations had a moderately reduced transactivation on the miR-194 promoter (Fig. 6B and supplemental Table 2) In contrast, V402I and I463V either displayed no changes as the wild type, or a moderately enhanced activation on all three promoters (Fig. 6B and supplemental Table 2). The cancer mutation G79S only maintained a weak transactivation activity for HNF1A promoter, but lost the transactivation activity for the HNF4A-AS1 promoter and miR-194 promoter. Since no previous publications reported the functional study of G79S, we confirmed the protein expression of HNF4α-G79S in HEK293 cells, which showed a comparable expression with the wild type HNF4α (Fig. S4C). Interestingly, another cancer mutation that resides neighboring the G79S, namely D78A, displayed highly distinct transactivation activities on different gene targets. D78A had unchanged and slightly reduced activity on HNF1A and HNF4A-AS1 promoter, respectively, but remarkably (80%) decreased transactivation of the miR-194 promoter (Fig.6B and supplemental Table 2).
We further applied the combination of HNF4α mutants with HNF1α (or HNF6) on promoters of HNF4A-P1 (HNF4A-Dis-P1–5’UTR-Luc) and PDZK1. All detailed changes of values were summarized in supplemental Tables 3 & 4. Generally, the single treatment of HNF4α mutations on promoters of PDZK1 and HNF4A-P1 were similar to other three promoters that we have tested (Fig. 7 & Fig. 8A). Both D78A and G79S completely lost the transactivation activity for HNF4A-P1, but maintained a strong and moderate activation of PDZK1 promoter, respectively (Fig. 7 & Fig. 8A). For the combination of HNF1α and mutant HNF4α on HNF4A-P1, HNF1α again did not show any synergism with HNF4α, and none of the mutant HNF4α, except Q277X, compromised the transactivation of HNF4A-P1 by HNF1α (Fig. 7). For PDZK1 promoter, both combinations (HNF4α/HNF1α and HNF4α/HNF6) displayed a high synergism (Fig. 8A & 8B), and this synergism was diminished along with different mutations. For example, G79S and L341P had a decreased transactivation activity on PDZK1 promoter. Similarly, the corresponding combination of G79S and L341P with HNF1α or HNF6 also showed a reduced transactivation. R163X, W349X and E285Q completely lost the transactivation potential on PDZK1, and they showed no synergism when combined with HNF1α or HNF6 (Fig. 8A & 8B). Noteworthy, Q277X is the only mutation that showed an antagonizing effect on the HNF1α-induced activation of HNF4A-P1 and PDZK1 promoter. Although Q277X had no transactivation activity, the addition of Q277X HNF4α even further reduced the HNF1α-mediated activation of HNF4A-P1 and PDZK1 by 42 and 61% (Fig. 7 & Fig. 8B). In summary, our study reported the transactivation activities of multiple HNF4α mutations from MODY1 and liver cancer. Particularly, D78A and G79S displayed highly distinct gene-specific transactivation activities. Q277X appears to possess an antagonizing effect on HNF1α-mediated transactivation.
Fig. 7. Interactions of HNF4α mutants with HNF1α in the regulation of HNF4A-Dis-P1–5’UTR in HEK293 cells.
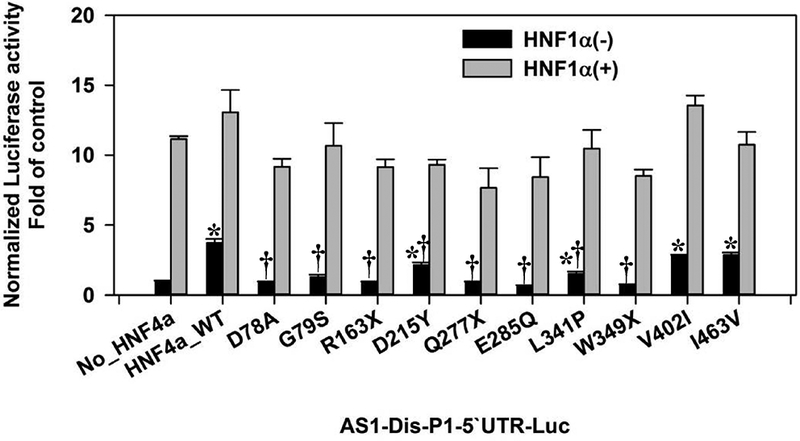
Reporter activities of HNF4A-Dis-P1–5’UTR-Luc induced by wildtype (WT)/mutant P1-HNF4α in the presence/absence of HNF1α. N=4, mean ± SE. * p<0.05 vs HNF4α-null group. † p<0.05 vs HNF4α_WT group.
DISCUSSION
In the current study, we validated the self-stimulation of HNF4α on its own promoter and 5’UTR coding motif. Further investigations uncovered the strong activation of HNF4A-AS1 promoter by the P1-HNF4α. We were unable to reproduce the previously reported synergistic activation of the HNF4A P1 promoter by HNF4α and HNF1α/HNF6. These data further prompted us to determine the crosstalk of HNF4α with other TFs on different gene targets, and our results elucidated highly gene-specific transactivation by multiple HNF4α mutations and a novel antagonism of HNF1α transactivation activity by HNF4α-Q277X, the first identified MODY1 mutant.
The present study discovers novel roles of the distal promoter and the 5’ UTR in the self-stimulation of HNF4α. HNF4α induces a moderate increase in the reporter activity of its own proximal promoter only when the 5’UTR coding motif is present. Previous results of ChIP-seq (Fig. 2B) and ChIP-qPCR [Hatzis and Talianidis, 2001] indicate a strong binding of HNF1α but a very weak binding of HNF4α to the P1 proximal promoter of HNF4α. Mutation analysis [Hatzis and Talianidis, 2001] indicates that the HNF1-binding site in sequence −97 to −119 in the P1 promoter is required for the transactivation by HNF1α, which may explain the independence of HNF1α transactivation on the HNF4α 5’ UTR. Previous EMSA results indicate a weak binding by HNF4α to bp −256 to −304 of P1 promoter [Hatzis and Talianidis, 2001]. Additionally, HNF4α can regulate gene expression through physical interaction with transcription factor SP1, which is independent of the direct binding of HNF4α to the DNA [Hwang-Verslues and Sladek, 2008]. Although there is no predicted HNF4-binding site in the 5’ UTR, there are multiple SP1 binding sites in the 5’ UTR and proximal promoter (Supplemental Table 1). Thus, HNF4α could either directly bind to the proximal promoter or interact with SP1 bound to the 5’ UTR or promoter regions. Surprisingly, the self-stimulation of HNF4α on its own proximal promoter does not change the mRNA level (Fig.1B). We recently discovered the presence of G-quadruplex within the 5’UTR of human HNF4A that induces a strong translational suppression [Guo and Lu, 2017]. In addition, our results indicate that the reporter activity of P1–5’UTR activated by the P1-HNF4α is comparable with the basal activity of the P1 promoter without the 5’UTR (Data not shown). Therefore, HNF4α may shift the transcriptional starting site (TSS) forward, which consequently shortens the 5’ UTR of HNF4A mRNA and thus attenuates the inhibitory effect by the 5’ UTR. Thus, HNF4α has no activation effect on the P1 proximal promoter when the 5’UTR is removed. Indeed, the TF-mediated TSS selection or shift is not rare [Goel et al., 2012; Richards et al., 2017], and we also find inconsistent reports on the TSS of P1-HNF4A in different databases. Overall, we propose that the HNF4α protein may induce the self-activation through enhancing the distal promoter activity and shifting the TSS to lessen the translational repression by its 5’ UTR.
The present data do not support the previously reported synergistic self-activation of HNF4α with other TFs. The previously reported synergism of HNF4α with HNF1α or HNF6 on HNF4A promoter is a bit surprising since it brings the positive feedback loop into a further cascade that would result in an aberrant high intracellular level of HNF4α. Our study did not observe the synergism between HNF4α with HNF1α/HNF6 on HNF4A-P1 promoter in two cell lines (Fig.5). This might be partly due to the difference of cell lines (HeLa cells were used in the previous study). Furthermore, our HNF4A-P1 reporter vector maintains the HNF4A promoter and 5’UTR motifs with a higher integrity (nt −985 to nt +89) compared to the prior construct (nt −500 to nt +63). The shortened promoter and the incomplete 5’UTR may have larger chances to generate false positive observations. Taken together, we propose that this synergism in self-stimulation of HNF4α reported in HeLa cells is unlikely of physiological relevance. We established this model by following additional considerations: HNF1α inhibits the transactivation of HNF1α promoter by HNF4α [Ktistaki and Talianidis, 1997], and our data showed that HNF4α tends to antagonize, rather than enhance, the activation of HNF4α P1 proximal promoter by HNF1α (Fig.5A). Overall, we propose a working model that during liver development, HNF4α, the upstream regulator of HNF1α, in the first place induces HNF1α and a moderate self-activation, and HNF1α in turn further transactivates HNF4α to achieve high expression levels of both HNF1α and HNF4α to promote hepatocellular differentiation and maturation. When hepatic expression of HNF1α arrives to a threshold, HNF1α and HNF4α antagonize each other on the transactivation of HNF1α and HNF4α proximal promoter to break the reciprocal transactivation between HNF1α and HNF4α, and thus hepatic expression of HNF1α and HNF4α will reach plateau. The additive transactivation of HNF4α by HNF4α and HNF6 (Fig.5D) will help ensure that hepatic expression of HNF4α can be independent of HNF1α.
To date, the dynamic change of P1 and P2 HNF4α during the embryonic development and oncogenesis is still puzzling. Both P1- and P2-HNF4α are detected in fetal liver and adult intestine. However, P2-HNF4α dramatically diminishes in adult liver, and re-expresses/over-expresses during the development of HCC and colon cancer [Babeu and Boudreau, 2014; Briancon et al., 2004; Lazarevich et al., 2010; Tanaka et al., 2006]. This dynamic switch has been explained partly by the finding that P1-HNF4α is capable of inhibiting P2-HNF4α [Briancon et al., 2004]; however, the underlining mechanism remains elusive. Additionally, how P2-HNF4α predominates over P1-HNF4α in adult pancreas is uncertain. The current study uncovers the strong correlation between P1-HNF4α and the corresponding antisense RNA, HNF4A-AS1: On one hand, the P1-HNF4α substantially activates the proximal promoter of HNF4A-AS1 (Fig.3A). On the other hand, the corresponding sense motif of the HNF4A-AS1 proximal promoter serves as a distal promoter that can further enhance the self-stimulation of P1-HNF4A (Fig.3B & 3C). Moreover, the special localization of HNF4A-AS1 that sits between the P2 and P1 promoters (Fig.2A) might enable the HNF4A-AS1 to selectively modulate the balance between P2- and P1-HNF4α. Interestingly, when writing this manuscript, it is reported that transient knockdown of HNF4A-AS1 using HNF4A-AS1 siRNAs in differentiated hepatoma cells does not affect the total mRNA levels of HNF4α, but moderately increases the expression of certain HNF4α-target genes [Chen et al., 2018]. The precise role of HNF4A-AS1 in regulating the balance of HNF4α isoforms and HNF4α function is still obscure and requires further investigation.
The present study provides the first evidence of interactions of PAX6 with HNF1α and HNF4α in regulating gene expression, in that PAX6 cooperates with HNF1α to promote P2-HNF4α expression but antagonizes HNF4α in HNF4A-AS1 expression. PAX6 is a master regulator of pancreas development [Hart et al., 2013]. In contrast, progressive overexpression of PAX6 contributes to colon carcinogenesis [Li et al., 2014] when P2-HNF4α expression is either maintained or increased [Chellappa et al., 2016; Vuong et al., 2015]. Thus, PAX6 may play important roles in determining the tissue distribution and carcinogenic changes of P2-HNF4α and HNF4A-AS1 in the pancreas and intestine.
The present study discovers highly gene-specific changes of transcriptional activities by multiple HNF4α mutations. Previous functional studies of some HNF4α mutations raise controversy. For instance, HNF4α-E285Q was originally reported to lose the ability of binding to HNF4 consensus sequence, and therefore completely lost the transactivation activity [Navas et al., 1999]. However, other studies reported the human HNF4α-E285Q was still transactive [Suaud et al., 1999]. This controversy was later explained by the species difference between human and rat HNF4α [Lausen et al., 2000]. Our results indicate that the HNF4α-E285Q does not maintain any transactivation activity for all promoters that we have tested. The other example is HNF4α-V402I, which has been reported to have a significantly reduced transactivation activity on HNF1α [Hani et al., 1998]. A later study of the HNF4α-I463V concluded that the I463V might not be responsible for MODY1 [Amara et al., 2015]. These authors also indicate that the I463V could have a similar effect with V402I. Our data are mostly in agreement with the conclusion that V402I and I463V may not be a direct cause of MODY1, as both mutations do not show a comprised transactivation activity. We have confirmed that all three nonsense mutations (R163X, Q277X and W349X) lost their transactivation activity, with the exception of a weak activation of HNF1A promoter by R163X. R163X maintains the AF-1 activation domain and DNA-binding activity. The Q277X cannot bind DNA or dimerize, and it has no dominant-negative effect on WT-HNF4α [Lausen et al., 2000; Sladek et al., 1998]. Our novel finding that the Q277X interferes with the transactivation by HNF1α may provide important mechanistic insight on the MODY1 pathogenesis induced by Q277X. Moreover, we found that Q277X strongly antagonized farnesoid X receptor (FXR) for the activation of the promoter of bile salt export pump (BSEP) (Fig.S3A). FXR can physically interact with HNF4α [Thomas et al., 2013], and FXR plays a key role in regulating insulin transcription and secretion in the pancreas [Renga et al., 2010]. The subcellular localization of HNF4α-Q277X is different from the WT-HNF4α [Ogata et al., 2012; Sladek et al., 1998]. Whether the Q277X mutant may alter the cellular localization of HNF1α and FXR to inhibit their transactivation requires further investigations. Overall, our study provides further evidence and mechanistic insights for the functional changes of multiple elusive HNF4α MODY1 mutations.
In addition to MODY1 mutants, we provide novel findings regarding the gene-specific effects of HNF4α-D78A and G79S, two DBD mutations found in liver cancer. Although G79S has a gene-specific pattern, it only maintains a weak transactivation activity on HNF1α and PDZK1 promoter. Heterozygous mutation of D78A is present in HepG2 cells, a widely used liver cancer cell line [Lausen et al., 2000]. D78A has been reported to bear an enhanced transactivation activity on specific promoters including the rat apolipoprotein AI promoter and the Xenopus HNF1α promoter [Lausen et al., 2000]. We found that the effects of D78A mutation on HNF4α-target genes are highly gene-specific, which varies from a complete loss of activation (on self-stimulation) to a moderately enhanced transactivation of mouse stearoyl-CoA desaturase-1 (Fig.S3B). The diminished transactivation of tumor suppressors (e.g. P1-HNF4α and miR-194) and increased transactivation of certain metabolic genes by HNF4α-D78A may help explain the unique feature of HepG2 cells being a cancer cell line with high metabolic activities. Currently, the mechanism of the highly gene-specific effects of D78A mutation remains unknown. A previous study using a D78E mutant of HNF4α demonstrates that D78 is required for the selective binding of HNF4α to certain “HNF4-specific” binding motif, but dispensable for its binding to and transactivation of genes that contain more canonical direct repeat-1 sites [Fang et al., 2012]. Thus, D78A may have selective loss of binding to certain “HNF4-specific” binding motif, and may also have decreased interaction with certain other transcription factors. In this regard, both the DBD and LBD of HNF4α are required for the physical interaction of HNF4α with SP1 [Kardassis et al., 2002]. Our previous study suggests that interaction of HNF4α with SP1 plays a key role in its transactivation of miR-194 [Lu et al., 2017]. Thus, the loss of self-activation activity and dramatically attenuated activation of miR-194 by the D78A mutant might be due to its loss/decrease of binding to “HNF4-specific” binding motif and/or the loss/decrease of interaction with SP1 or other transactivators. The DBD of nuclear receptors is important in DNA-binding, dimerization, and recruitment of co-activators [Helsen and Claessens, 2014]. Interestingly, a E157D DBD-mutant of peroxisome proliferator-activated receptor gamma (PPARγ) does not decrease the receptor’s affinity to classical PPAR response elements (PPRE), yet it severely reduces its target gene transcription; in contrast, this DBD mutation causes increased binding affinity and markedly higher activation of a subset of genes with cryptic PPRE than wildtype PPARγ [Campeau et al., 2012]. Thus, the D78A mutant will likely have increased affinity for certain DNA motif. Alternatively, the putative loss of binding of D78A to “HNF4-specific” binding motif might result in the relative enrichment of D78A to other sites, resulting in the increased transactivation of these genes by the D78A mutant. The inconsistency and discrepancy regarding the functional studies of certain HNF4α mutants have been noted in the past two decades. HNF4α crosstalks with diverse extracellular and intracellular signaling molecules to regulate a large battery of genes [Lu, 2016]. Thus, when performing functional studies of HNF4α mutations, a more extensive test of the HNF4α-target genes and interactions of the mutant HNF4α with other signaling pathways will be preferred to elucidate the precise changes in the transactivation activities of these HNF4α mutants.
In summary, the current study provides multiple novel insights regarding the regulatory mechanisms of the gene expression and transactivation activity of HNF4α. The 5’UTR-involved self-stimulation of HNF4α requires further investigation, which may be a potential novel target for up-regulating the gene expression of HNF4α. We have identified the antisense HNF4A-AS1 as a novel HNF4α-target gene. In addition to being a promising biomarker for diseases diagnosis, HNF4A-AS1 may play a role in regulating the P2 and P1 HNF4α expression/function in tissue distribution, ontogeny, and carcinogenesis, which warrant further investigation. Future studies on the interactions of PAX6 with HNF4α and HNF1α on the regulation of HNF4α and HNF4α/HNF1α-target genes will help understand the ontogeny and carcinogenesis of digestive tissues. Finally, the novel discoveries of the antagonism of HNF1α and FXR transactivation activities by the MODY1 mutant HNF4α-Q277X and the highly target-gene-specific effects of D78A and G79S, two DBD mutations of HNF4α provide important mechanistic insights on how HNF4α mutations lead to MODY1 and liver cancer.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Debashis Ghosh in Department of Pharmacology and Dr. Guirong Wang in Department of Surgery for discussion of experiments during this study.
Contract grant sponsor: National Institute of Health; Contract grant number: CA169877.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Amara A, Ben Charfeddine I, Ghedir H, Mamai O, Jemni-Yacoub S, Chaieb L, Saad A, Chadli-Chaieb M, Gribaa M. 2015. Frequency of HNF4A-P.I463V Variant in the Tunisian North-African Population and Its Relation with Diabetes Mellitus. Iran J Public Health 44:396–403. [PMC free article] [PubMed] [Google Scholar]
- Babeu JP, Boudreau F. 2014. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol 20:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly A, Spath G, Bender V, Weiss MC. 1998. Phenotypic effects of the forced expression of HNF4 and HNF1alpha are conditioned by properties of the recipient cell. J Cell Sci 111 ( Pt 16):2411–21. [DOI] [PubMed] [Google Scholar]
- Briancon N, Bailly A, Clotman F, Jacquemin P, Lemaigre FP, Weiss MC. 2004. Expression of the alpha7 isoform of hepatocyte nuclear factor (HNF) 4 is activated by HNF6/OC-2 and HNF1 and repressed by HNF4alpha1 in the liver. J Biol Chem 279:33398–408. [DOI] [PubMed] [Google Scholar]
- Campeau PM, Astapova O, Martins R, Bergeron J, Couture P, Hegele RA, Leff T, Gagne C. 2012. Clinical and molecular characterization of a severe form of partial lipodystrophy expanding the phenotype of PPARgamma deficiency. J Lipid Res 53:1968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappa K, Deol P, Evans JR, Vuong LM, Chen G, Briancon N, Bolotin E, Lytle C, Nair MG, Sladek FM. 2016. Opposing roles of nuclear receptor HNF4alpha isoforms in colitis and colitis-associated colon cancer. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Bao Y, Piekos SC, Zhu K, Zhang L, Zhong XB. 2018. A Transcriptional Regulatory Network Containing Nuclear Receptors and Long Noncoding RNAs Controls Basal and Drug-Induced Expression of Cytochrome P450s in HepaRG Cells. Mol Pharmacol 94:749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposti DD, Hernandez-Vargas H, Voegele C, Fernandez-Jimenez N, Forey N, Bancel B, Le Calvez-Kelm F, McKay J, Merle P, Herceg Z. 2016. Identification of novel long non-coding RNAs deregulated in hepatocellular carcinoma using RNA-sequencing. Oncotarget 7:31862–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B, Mane-Padros D, Bolotin E, Jiang T, Sladek FM. 2012. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res 40:5343–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlani G, Zucchini S, Di Rocco A, Di Luzio R, Scipione M, Marasco E, Romeo G, Marchesini G, Mantovani V. 2010. Double heterozygous mutations involving both HNF1A/MODY3 and HNF4A/MODY1 genes: a case report. Diabetes Care 33:2336–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan M, Garcia-Herrero CM, Azriel S, Gargallo M, Duran M, Gorgojo JJ, Andia VM, Navas MA. 2011. Differential effects of HNF-1alpha mutations associated with familial young-onset diabetes on target gene regulation. Mol Med 17:256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison WD, Battle MA, Yang C, Kaestner KH, Sladek FM, Duncan SA. 2006. Hepatocyte nuclear factor 4alpha is essential for embryonic development of the mouse colon. Gastroenterology 130:1207–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel S, Krishnamurthy S, Hampsey M. 2012. Mechanism of start site selection by RNA polymerase II: interplay between TFIIB and Ssl2/XPB helicase subunit of TFIIH. J Biol Chem 287:557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez FJ. 2008. Regulation of hepatocyte nuclear factor 4 alpha-mediated transcription. Drug Metab Pharmacokinet 23:2–7. [DOI] [PubMed] [Google Scholar]
- Guo S, Lu H. 2017. Conjunction of potential G-quadruplex and adjacent cis-elements in the 5’ UTR of hepatocyte nuclear factor 4-alpha strongly inhibit protein expression. Sci Rep 7:17444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman Y, BenShoshan M, Di Segni A, Dexheimer PJ, Braun T, Weiss B, Walters TD, Baldassano RN, Noe JD, Markowitz J, Rosh J, Heyman MB, Griffiths AM, Crandall WV, Mack DR, Baker SS, Kellermayer R, Patel A, Otley A, Steiner SJ, Gulati AS, Guthery SL, LeLeiko N, Moulton D, Kirschner BS, Snapper S, Avivi C, Barshack I, Oliva-Hemker M, Cohen SA, Keljo DJ, Ziring D, Anikster Y, Aronow B, Hyams JS, Kugathasan S, Denson LA. 2018. Long ncRNA Landscape in the Ileum of Treatment-Naive Early-Onset Crohn Disease. Inflamm Bowel Dis 24:346–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han EH, Rha GB, Chi YI. 2012. MED25 is a mediator component of HNF4alpha-driven transcription leading to insulin secretion in pancreatic beta-cells. PLoS One 7:e44007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hani EH, Suaud L, Boutin P, Chevre JC, Durand E, Philippi A, Demenais F, Vionnet N, Furuta H, Velho G, Bell GI, Laine B, Froguel P. 1998. A missense mutation in hepatocyte nuclear factor-4 alpha, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. J Clin Invest 101:521–6.9449683 [Google Scholar]
- Harries LW, Locke JM, Shields B, Hanley NA, Hanley KP, Steele A, Njolstad PR, Ellard S, Hattersley AT. 2008. The diabetic phenotype in HNF4A mutation carriers is moderated by the expression of HNF4A isoforms from the P1 promoter during fetal development. Diabetes 57:1745–52. [DOI] [PubMed] [Google Scholar]
- Hart AW, Mella S, Mendrychowski J, van Heyningen V, Kleinjan DA. 2013. The developmental regulator Pax6 is essential for maintenance of islet cell function in the adult mouse pancreas. PLoS One 8:e54173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis P, Talianidis I. 2001. Regulatory mechanisms controlling human hepatocyte nuclear factor 4alpha gene expression. Mol Cell Biol 21:7320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsen C, Claessens F. 2014. Looking at nuclear receptors from a new angle. Mol Cell Endocrinol 382:97–106. [DOI] [PubMed] [Google Scholar]
- Hwang-Verslues WW, Sladek FM. 2008. Nuclear receptor hepatocyte nuclear factor 4alpha1 competes with oncoprotein c-Myc for control of the p21/WAF1 promoter. Mol Endocrinol 22:78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang-Verslues WW, Sladek FM. 2010. HNF4alpha--role in drug metabolism and potential drug target? Curr Opin Pharmacol 10:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardassis D, Falvey E, Tsantili P, Hadzopoulou-Cladaras M, Zannis V. 2002. Direct physical interactions between HNF-4 and Sp1 mediate synergistic transactivation of the apolipoprotein CIII promoter. Biochemistry 41:1217–28. [DOI] [PubMed] [Google Scholar]
- Kato Y, Sai Y, Yoshida K, Watanabe C, Hirata T, Tsuji A. 2005. PDZK1 directly regulates the function of organic cation/carnitine transporter OCTN2. Mol Pharmacol 67:734–43. [DOI] [PubMed] [Google Scholar]
- Kocher O, Krieger M. 2009. Role of the adaptor protein PDZK1 in controlling the HDL receptor SR-BI. Curr Opin Lipidol 20:236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistaki E, Talianidis I. 1997. Modulation of hepatic gene expression by hepatocyte nuclear factor 1. Science 277:109–12. [DOI] [PubMed] [Google Scholar]
- Lausen J, Thomas H, Lemm I, Bulman M, Borgschulze M, Lingott A, Hattersley AT, Ryffel GU. 2000. Naturally occurring mutations in the human HNF4alpha gene impair the function of the transcription factor to a varying degree. Nucleic Acids Res 28:430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevich NL, Shavochkina DA, Fleishman DI, Kustova IF, Morozova OV, Chuchuev ES, Patyutko YI. 2010. Deregulation of hepatocyte nuclear factor 4 (HNF4)as a marker of epithelial tumors progression. Exp Oncol 32:167–71. [PubMed] [Google Scholar]
- Li Y, Liu Y, Xie P, Li F, Li G. 2014. PAX6, a novel target of microRNA-7, promotes cellular proliferation and invasion in human colorectal cancer cells. Dig Dis Sci 59:598–606. [DOI] [PubMed] [Google Scholar]
- Lu H 2016. Crosstalk of HNF4alpha with extracellular and intracellular signaling pathways in the regulation of hepatic metabolism of drugs and lipids. Acta Pharm Sin B 6:393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Lei X, Liu J, Klaassen CD. 2017. Regulation of hepatic microRNA expression by hepatocyte nuclear factor 4 alpha. World J Hepatol 9:191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistri M, Faghihi MA, St Laurent G 3rd, Wahlestedt C 2012. Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet 28:389–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas MA, Munoz-Elias EJ, Kim J, Shih D, Stoffel M. 1999. Functional characterization of the MODY1 gene mutations HNF4(R127W), HNF4(V255M), and HNF4(E276Q). Diabetes 48:1459–65. [DOI] [PubMed] [Google Scholar]
- Ogata M, Awaji T, Iwasaki N, Fujimaki R, Takizawa M, Maruyama K, Bell GI, Iwamoto Y, Uchigata Y. 2012. Localization of hepatocyte nuclear factor-4alpha in the nucleolus and nucleus is regulated by its C-terminus. J Diabetes Investig 3:449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima T, Kawasaki T, Ohashi R, Hasegawa G, Jiang S, Umezu H, Aoyagi Y, Iwanari H, Tanaka T, Hamakubo T, Kodama T, Naito M. 2007. Downregulated P1 promoter-driven hepatocyte nuclear factor-4alpha expression in human colorectal carcinoma is a new prognostic factor against liver metastasis. Pathol Int 57:82–90. [DOI] [PubMed] [Google Scholar]
- Pearson ER, Pruhova S, Tack CJ, Johansen A, Castleden HA, Lumb PJ, Wierzbicki AS, Clark PM, Lebl J, Pedersen O, Ellard S, Hansen T, Hattersley AT. 2005. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 48:878–85. [DOI] [PubMed] [Google Scholar]
- Pelechano V, Steinmetz LM. 2013. Gene regulation by antisense transcription. Nat Rev Genet 14:880–93. [DOI] [PubMed] [Google Scholar]
- Perilhou A, Tourrel-Cuzin C, Zhang P, Kharroubi I, Wang H, Fauveau V, Scott DK, Wollheim CB, Vasseur-Cognet M. 2008. The MODY1 gene for hepatocyte nuclear factor 4alpha and a feedback loop control COUP-TFII expression in pancreatic beta cells. Mol Cell Biol 28:4588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeder H, Bjorkhaug L, Johansson S, Mangseth K, Sagen JV, Hunting A, Folling I, Johansen O, Bjorgaas M, Paus PN, Sovik O, Molven A, Njolstad PR. 2006. A hepatocyte nuclear factor-4 alpha gene (HNF4A) P2 promoter haplotype linked with late-onset diabetes: studies of HNF4A variants in the Norwegian MODY registry. Diabetes 55:1899–903. [DOI] [PubMed] [Google Scholar]
- Renga B, Mencarelli A, Vavassori P, Brancaleone V, Fiorucci S. 2010. The bile acid sensor FXR regulates insulin transcription and secretion. Biochim Biophys Acta 1802:363–72. [DOI] [PubMed] [Google Scholar]
- Richards AL, Watza D, Findley A, Alazizi A, Wen X, Pai AA, Pique-Regi R, Luca F. 2017. Environmental perturbations lead to extensive directional shifts in RNA processing. PLoS Genet 13:e1006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Tsuyama T, Sato C, Karim MF, Yoshizawa T, Inoue M, Yamagata K. 2017. Hypoxia reduces HNF4alpha/MODY1 protein expression in pancreatic beta-cells by activating AMP-activated protein kinase. J Biol Chem 292:8716–8728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek FM, Dallas-Yang Q, Nepomuceno L. 1998. MODY1 mutation Q268X in hepatocyte nuclear factor 4alpha allows for dimerization in solution but causes abnormal subcellular localization. Diabetes 47:985–90. [DOI] [PubMed] [Google Scholar]
- Spath GF, Weiss MC. 1997. Hepatocyte nuclear factor 4 expression overcomes repression of the hepatic phenotype in dedifferentiated hepatoma cells. Mol Cell Biol 17:1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suaud L, Hemimou Y, Formstecher P, Laine B. 1999. Functional study of the E276Q mutant hepatocyte nuclear factor-4alpha found in type 1 maturity-onset diabetes of the young: impaired synergy with chicken ovalbumin upstream promoter transcription factor II on the hepatocyte nuclear factor-1 promoter. Diabetes 48:1162–7. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Jiang S, Hotta H, Takano K, Iwanari H, Sumi K, Daigo K, Ohashi R, Sugai M, Ikegame C, Umezu H, Hirayama Y, Midorikawa Y, Hippo Y, Watanabe A, Uchiyama Y, Hasegawa G, Reid P, Aburatani H, Hamakubo T, Sakai J, Naito M, Kodama T. 2006. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha in the pathogenesis of human cancer. J Pathol 208:662–72. [DOI] [PubMed] [Google Scholar]
- Thomas AM, Hart SN, Li G, Lu H, Fang Y, Fang J, Zhong XB, Guo GL. 2013. Hepatocyte nuclear factor 4 alpha and farnesoid X receptor co-regulates gene transcription in mouse livers on a genome-wide scale. Pharm Res 30:2188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaxillaire M, Rouard M, Yamagata K, Oda N, Kaisaki PJ, Boriraj VV, Chevre JC, Boccio V, Cox RD, Lathrop GM, Dussoix P, Philippe J, Timsit J, Charpentier G, Velho G, Bell GI, Froguel P. 1997. Identification of nine novel mutations in the hepatocyte nuclear factor 1 alpha gene associated with maturity-onset diabetes of the young (MODY3). Hum Mol Genet 6:583–6. [DOI] [PubMed] [Google Scholar]
- Vuong LM, Chellappa K, Dhahbi JM, Deans JR, Fang B, Bolotin E, Titova NV, Hoverter NP, Spindler SR, Waterman ML, Sladek FM. 2015. Differential Effects of Hepatocyte Nuclear Factor 4alpha Isoforms on Tumor Growth and T-Cell Factor 4/AP-1 Interactions in Human Colorectal Cancer Cells. Mol Cell Biol 35:3471–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walesky C, Edwards G, Borude P, Gunewardena S, O’Neil M, Yoo B, Apte U. 2013. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology 57:2480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB. 2000. Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic beta -cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J Biol Chem 275:35953–9. [DOI] [PubMed] [Google Scholar]
- Wisely GB, Miller AB, Davis RG, Thornquest AD, Johnson R, Spitzer T, Sefler A Shearer B, Moore JT, Willson TM, Williams SP. 2002. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure 10:1225–34. [DOI] [PubMed] [Google Scholar]
- Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, Nemzer S, Pinner E, Walach S, Bernstein J, Savitsky K, Rotman G. 2003. Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol 21:379–86. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Lei X, Lu H. 2014. Alterations of Epigenetic Signatures in Hepatocyte Nuclear Factor 4alpha Deficient Mouse Liver Determined by Improved ChIP-qPCR and (h)MeDIP-qPCR Assays. PLoS ONE 9:e84925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.