

SUMMARY
Tumor evolution from a single cell into a malignant, heterogeneous tissue remains poorly understood. Here, we profile single-cell transcriptomes of genetically engineered mouse lung tumors at seven stages, from pre-neoplastic hyperplasia to adenocarcinoma. The diversity of transcriptional states increases over time and is reproducible across tumors and mice. Cancer cells progressively adopt alternate lineage identities, computationally predicted to be mediated through a common transitional, high-plasticity cell state (HPCS). Accordingly, HPCS cells prospectively isolated from mouse tumors and human patient-derived xenografts display high capacity for differentiation and proliferation. The HPCS program is associated with poor survival across human cancers and demonstrates chemoresistance in mice. Our study reveals a central principle underpinning intra-tumoral heterogeneity and motivates therapeutic targeting of the HPCS.
Keywords: Tumor heterogeneity, plasticity, tumor evolution, cell state transition, lung cancer, single-cell transcriptomics
Graphical abstract
INTRODUCTION
Tumors are cellular societies in which the phenotype, or state, of each cancer cell is influenced by multiple cell-intrinsic and cell-extrinsic factors. The diversity of cancer cell states within tumors poses a challenge for effective cancer therapies (Lawson et al., 2018). The nature and sequence of the genetic events that define some common cancers have been characterized in detail over the past three decades (Fearon and Vogelstein, 1990; Hutter and Zenklusen, 2018), as have the expression profiles of bulk mouse and human tumors in late stages of tumor progression (Ambrogio et al., 2016; Campbell et al., 2016; Feldser et al., 2010; Winslow et al., 2011). However, our increasingly fine understanding of genetic events occurring during tumorigenesis is not yet matched by a similar understanding of the progression of cancer cell states at the molecular and functional levels, especially for early microscopic neoplasias that cannot be readily detected in patients. In particular, we do not know the diversity of these states at different points along tumorigenesis, how reproducibly they would arise in a defined genetic context, how the states of different cells in the same tumor relate to, support, or compete with each other, and what role they may play in driving tumor progression or response to therapy.
Genetically engineered mouse models (GEMM) of human cancer and single cell RNA-Seq (scRNA-Seq) can together help address this gap. ScRNA-Seq is a powerful tool for characterizing the molecular identity of individual cells in tissues, including in solid tumors (Lambrechts et al., 2018; Patel et al., 2014; Tirosh et al., 2016a; Tirosh et al., 2016b; Zilionis et al., 2019). However, it has typically been applied to advanced tumors in humans analyzed at a single point in time, thus limiting one’s ability to infer temporal changes over processes that take years in patients. In particular, the spectra of cell states that exist in advanced human tumors may represent transitions that occurred over short or far longer time scales (Neftel et al., 2019). This limitation can be addressed by studying cancer GEMMs, which allow spatiotemporal control over tumor development in the context of mammalian physiology.
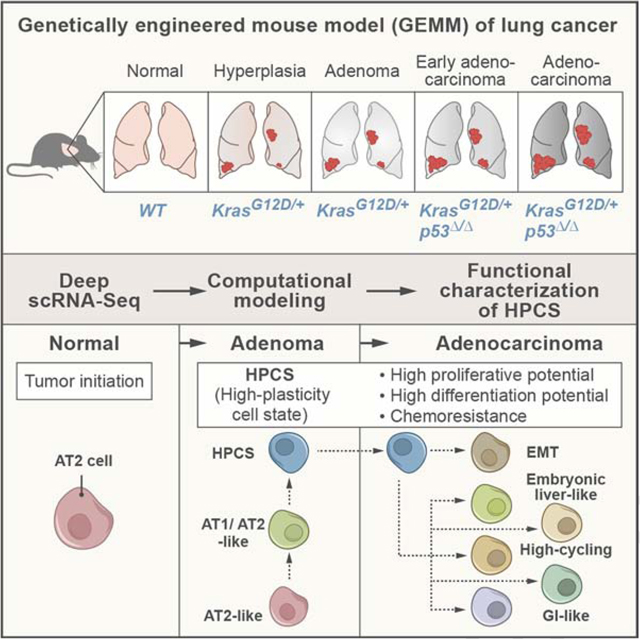
Emerging evidence indicates that LUAD predominantly arises from a subset of alveolar type 2 (AT2) cells (Desai et al., 2014; Nabhan et al., 2018; Sutherland et al., 2014; Treutlein et al., 2014; Zacharias et al., 2018). In GEMMs of lung adenocarcinoma (LUAD), viral expression of Cre recombinase in AT2 cells leads to somatic activation of oncogenic KRAS-G12D with or without deletion of the p53 tumor suppressor (referred to here as “K” and “KP” models, respectively) (Jackson et al., 2005; Jackson et al., 2001; Sutherland et al., 2014). K tumors rarely progress beyond adenomas, whereas the KP tumors evolve over a span of >12 weeks into advanced LUADs. These models accurately mimic human lung adenoma and adenocarcinoma progression at the molecular and histopathological levels (Jackson et al., 2005; Jackson et al., 2001; Winslow et al., 2011), as well as in their response to chemotherapy (Oliver et al., 2010), making them well-suited for studying tumor evolution, heterogeneity and treatment responses.
RESULTS
LUAD progression is characterized by a dramatic and reproducible increase in phenotypic diversity
To initiate lung tumors, we delivered adenoviral vectors encoding Cre recombinase under the control of an AT2 cell-specific surfactant protein-C promoter [AdSPC-Cre; (Sutherland et al., 2014)] into the lungs of Rosa26LSL-tdTomato/+ (“T”), KT, or KPT mice. We isolated live tdTomato+/CD45−/CD11b−/TER119−/CD31− cells (Tammela et al., 2017) at defined time points and performed full-length scRNA-Seq using a modified SMART-Seq2 protocol (Figure 1A–C; STAR Methods). To characterize malignant cell diversity along tumorigenesis, we collected 3,891 high quality, single cell full-length transcriptomes from 39 mice at eight distinct stages of LUAD evolution, defined by genetic perturbation and time point, starting with normal AT2 cells and ending with fully formed LUADs (Figure 1A–C; Figure S1A–C; STAR Methods).
Figure 1. Increased transcriptional heterogeneity in mouse lung adenocarcinoma (LUAD) evolution is reproducible across individual tumors and mice, but cannot fully be explained by gene copy number variation (CNV).

(A) Experimental pipeline. (B) Tumor evolution in a LUAD GEMM. Top: genetic constructs of three mouse models profiled by scRNA-Seq at different time points. Middle and bottom: schematic (middle) and hematoxylin & eosin staining of tissue sections (bottom) at different phases of tumor progression. AT1: normal alveolar type 1 (AT1) cells; AT2: normal alveolar type 2 (AT2) cells, AAH: atypical adenomatous hyperplasia. Scale bar: 100 μm. (C) PHATE map embedding (STAR Methods) of scRNA-Seq profiles (dots) collected from the models and time points in (B) (labels, top). Colored dots: Cells collected from the indicated sample; grey dots: all other cells. (D) Increased diversity of cell clusters with progression. Left: The fraction of cells (y axis) in each cluster (x axis) that are derived from each sample type (genotype and time point; colored as in (C)). Middle and Right: matched t-stochastic neighbor embedding (tSNE, left plot, STAR Methods) and PHATE map embedding (right plot, as in (C)) colored by either sample type (middle pair) or cluster number (STAR Methods) (right pair). (E) Reduced transcriptional homogeneity within time point with progression. Transcriptional heterogeneity is inversely proportional to the Normalized Mutual Information (NMI, y axis) between cells within in each sample type (genotype/time point combination, x axis), for either whole lung samples or microdissected single tumors. Box plots: upper, median, and lower quartile of 1,000 bootstrap samples, of 50 cells each, from the indicated time point; whiskers: 1.5 interquartile range. * p < 0.05, ** p < 0.01, *** p < 0.001 (STAR Methods). (F) Fraction of cells (y axis) in sample (x axis) that are members of each cluster (color code, as in D, right). The number of clusters observed in each individually plucked tumor is indicated at the top of the bars. (G) CNVs (red: amplifications, blue: deletions) across the chromosomes (columns) inferred from the scRNA-Seq of each cell (rows) from 12 KP tumors at the 30-week time point (STAR Methods). Color: the cluster membership of each cell. (H) Congruence between CNV profiles inferred from scDNA-Seq and scRNA-Seq. CNVs shown as in (G) for single cells (rows) of one individually microdissected KPT tumor at 30 weeks profiled by scDNA-Seq (top-left) or scRNA-Seq (bottom-left). Left color bar: Predominant clonotypes identified from scDNA-Seq (top-left) and assigned to scRNA-Seq cells (bottom-left). Far left color bar in scRNA-Seq panels: cell cluster membership as in (G). (I) A single clonotype matches multiple transcriptional states. PHATE map as in Figure 1D, colored by clonotype. See also related Figure S1.
The single cell expression profiles spanned 12 clusters with distinct expression patterns discovered by unsupervised clustering (Shekhar et al., 2016) (Figure S1D; STAR Methods), showing increasing cellular phenotypic heterogeneity with tumor progression (Figure 1C, D). The growing diversity was reflected in that cells from later time points (late adenoma and LUAD) were members of a larger number of clusters (Figure 1C, D; Figure S1C, E) and showed a more diverse expression pattern (Figure 1E; Figure S1F; STAR Methods). Cells from p53 mutant KPT tumors were the most heterogeneous, consistent with the established role of p53 in restricting cancer progression and safeguarding lineage commitment (Kastenhuber and Lowe, 2017).
The increased cell state heterogeneity during tumor progression was remarkably reproducible from tumor to tumor within and across mice, and was in line with each tumor’s histopathological progression (Figure 1B). Late-stage adenocarcinomas contained the unique “late onset” subpopulations (clusters 10, 11, and 12; Figure 1D; Figure S1E) in addition to all cellular states detected at the earlier steps of tumor initiation, including the very earliest cell states found in normal AT2 cells and in early neoplasias. Furthermore, most of the cancer cell phenotypes were present in each of the individually micro-dissected KPT tumors at 30 weeks (Figure 1F; Figure S1G). Notably, cluster 5 and 9 cells were present in every tumor analyzed, both across and within mice and individual tumors. Thus, in this genetically defined animal model, tumors undergo a relatively ordered and reproducible diversification of transcriptional states.
Diversity in gene copy number variation is not a sufficient determinant of phenotypic heterogeneity in LUAD
We next tested whether genetic heterogeneity underlies the phenotypic diversity in advanced adenocarcinomas (KPT 30 weeks), which had the largest number of cell states (Figure 1E, F; Figure S1E, G). Previous studies have demonstrated that the mutational landscape of K and KP tumors is dominated by chromosomal copy number alterations and that the tumors do not develop recurrent point mutations (Chung et al., 2017; McFadden et al., 2016; Westcott et al., 2015). We therefore inferred chromosomal copy number variations (CNVs) from each cell’s scRNA-Seq profile (Figure 1G; Figure S1H–J), using a method we previously demonstrated and validated in multiple human tumors (STAR Methods) (Jerby-Arnon et al., 2018; Patel et al., 2014; Puram et al., 2017; Tirosh et al., 2016a; Tirosh et al., 2016b; Venteicher et al., 2017). KPT cells harbored more CNVs when compared to KT tumors at corresponding time points (Figure S1H, I), consistent with previously published results and the established role of p53 in maintaining genome integrity (Chung et al., 2017; Kastenhuber and Lowe, 2017; McFadden et al., 2016; Westcott et al., 2015). In a subset of the KPT tumors at 30 weeks we estimated DNA copy number by whole genome sequencing (WGS) of individual tumor cells (scDNA-Seq) (Figure 1H, I; Figure S1K, L; n = 3), which was highly concordant with the scRNA-Seq-based inference. There was considerable inter- and intratumoral heterogeneity in the single cell CNV patterns, which increased with tumor progression (Figure S1H, I). Prominent shared CNVs across mice and tumors implicated common clonal founders (“trunks”) for each tumor (Figure 1G, H; Figure S1J, K).
We classified the CNV patterns into subtypes based on scDNA-Seq data (Figure 1H; Figure S1K), and assigned each cell analyzed by scRNA-Seq into these clonotypes (Figure S1L). Surprisingly, cell subtypes defined by CNV patterns did not directly align with the transcriptional classes (Figure 1H, I; Figure S1K–M). Specifically, cells harboring highly similar CNV patterns were members of multiple transcriptionally distinct clusters (Figure 1I; Figure S1L, M) and cells with different CNVs belonging to different clonotypes were members of the same transcriptional cluster (Figure 1I; Figure S1L, M). These results suggest that substantial phenotypic heterogeneity in the KP tumors is reproducibly acquired and not simply a result of gene CNV.
Loss of alveolar identity and acquisition of features associated with lung progenitors, embryonic endoderm, and epithelial-to-mesenchymal transition during LUAD progression
The 12 transcriptional clusters were associated with distinct expression signatures (Table S1) that corresponded to known mouse cell identity programs, with more divergent states emerging in advanced tumors, suggesting a reversal of the lung developmental trajectory (Figure 2A; Figure S2A). We characterized each cellular subset with a signature of differentially expressed genes (Table S1; STAR Methods), which we compared to a published Mouse Cell Atlas scRNA-Seq dataset (Han et al., 2018) (Figure 2A). Cells in the early-emerging clusters 1 and 2 expressed features of normal AT2 cells and were present in most tumors throughout LUAD progression (Figure 2A; Figure 1D). Distinct subpopulations that emerged first in adenomas (clusters 3 and 4) lost some AT2 transcriptional identity, but retained features of the lung epithelial lineage (Figure 2A). Most populations that emerged in adenocarcinomas (clusters 6–10 and 12, Figure 1D) had features of intestinal and/or gastric or embryonic liver epithelium – all endodermal tissues derived from the embryonic primordial gut (Cao et al., 2019; Nowotschin et al., 2019) (Figure 2A). This suggests that LUAD evolution is characterized by a loss of fidelity of the lung lineage and emergence of alternative related fates. Indeed, features of embryonic lineages more primitive than the primordial gut emerged in multiple subsets of lung tumor cells during tumor progression (Cao et al., 2019; Nowotschin et al., 2019) (Figure 2B). These changes were associated with the previously described loss of expression of the lung lineage-defining transcription factor Nkx2–1 as well as loss of the AT2 markers Sftpc and Lyz2, correlating with induction of developmental master regulators Hnf4a (primordial gut) and Hmga2 (primordial gut, developing lung) (Snyder et al., 2013; Winslow et al., 2011) (Figure 2C; Figure S2A).
Figure 2. Loss of lung lineage fidelity in LUAD progression and emergence of a highly mixed identity program.

(A, B) Signature score (color bar, STAR Methods) of either adult [(Han et al., 2018; Zhang et al., 2019); (A), z-score)] or embryonic [(Cao et al., 2019; Nowotschin et al., 2019); (B), z-score)] mouse cell signatures in the cells of each cluster (columns). In (B), signatures (rows) are ordered from most differentiated (top) to most primitive (bottom) cells. (C) PHATE maps (as in Figure 1D), with cells (dots) colored by expression (Log2(TPX+1), color bar) of Nkx2–1, Hnf4a, and Hmga2. (D, E) Five key gene programs highlight alternative cell type programs, two key transition states and an EMT-like state. PHATE map (as in Figure 1D), with cells (dots) colored either by the activity of each program (D, NMF loading, color bar, see Figure S2C for additional programs, STAR Methods) or by the expression level (E, Log2(TPX+1), color bar) of a selected marker from the corresponding program. (F) Immunofluorescence for Lysozyme (AT2-like program), Claudin-2 (hepatocyte-like program), and Claudin-4 (highly mixed program). Pink numbered arrowheads indicate cell states or transitions in (D-F): 1 - AT2-like (lysozyme) to Embryonic liver-like (Claudin-2) transition; 2 – Embryonic liver-like (Claudin-2) to Highly mixed (Claudin-4) transition; 3 - Highly mixed program (Claudin-4). Scale bar: 20 μm. (G) Cells from cluster 5 show significantly elevated activity of the Highly mixed NMF program (t-test, p < 1×10 −16). (H) Cell scores for Highly mixed program (y axis) and a cluster 5 signature (x axis). Pearson R2 = 0.78. Lighter dot color indicates higher cell density. (I) PHATE map embedding as in Figure 1D, with cells (dots) colored by score of the highly mixed program (left) or by cluster 5 membership (blue, middle). Right: Proportion of cells (y axis) from cluster 5 (blue) in each sample (mouse or tumor; x axis), ordered by tumor progression. See also related Figure S2 and Table S2.
Interestingly, one late-emerging subpopulation (cluster 11, Figure 1D) bore no resemblance to epithelial cells, adopting a mouse embryonic fibroblast-like state and an expression program consistent with epithelial-to-mesenchymal transition (EMT) (Dongre and Weinberg, 2019) (Figure 2A). Only late-stage adenocarcinomas contained a subpopulation that had fully undergone EMT, indicating that LUAD tumors remain largely epithelial until late stages. Finally, our analysis confirmed heterogeneous expression of previously published markers of LUAD cell subpopulations (Guinot et al., 2016; Tammela et al., 2017; Zheng et al., 2013) (Figure S2B).
A highly mixed program emerges during LUAD evolution
As our results pointed to a highly dynamic acquisition of cell states across the tumor evolution continuum, we next explored continuous changes in transcriptional programs and cell-state transitions using non-negative matrix factorization (NMF) (Kotliar et al., 2019; Lee and Seung, 1999; Puram et al., 2017) (STAR Methods). We uncovered 11 transcriptional programs, five of which particularly highlighted gradual phenotypic changes during tumor progression (Figure 2D, E; Figure S2C; Table S2). Three of the five programs were consistent with the emergence of the different cell identity programs we uncovered above: a program associated with AT2 cell features present at the onset of LUAD development, an Embryonic liver-like program, and an EMT program emerging at a later stage (Figure 2D).
In addition, we uncovered two previously unknown cell programs, an early program associated with a mix of AT1 and AT2 cell features (“Mixed AT1/AT2” state) and another program that did not match a consistent, defined cell identity program (“Highly mixed” state; Figure 2D; Table S2). The Mixed AT1/AT2 program was characterized by co-expression of AT1 markers, such as Hopx and Pdpn, together with AT2 markers Sftpc and Lyz2 (Figure 2D; Figure S2A; Table S2). This AT1/AT2-like program may mimic common alveolar progenitors in development or bi-potent alveolar progenitor cells in mature lungs (Desai et al., 2014; Nabhan et al., 2018; Treutlein et al., 2014; Zacharias et al., 2018). Conversely, the Highly mixed program displayed features of drastically different cell types, ranging from trophoblast stem cells to chondroblasts and kidney tubular epithelium (Table S2), suggesting that cells in this state are capable of exploring a broad phenotypic space. Interestingly, a subset of cells expressing the Highly mixed program also expressed a portion of the late-emerging EMT program (Figure 2D).
We performed immunostaining for highly specific markers for these programs (Figure 2E, F; Figure S2D), including one marking the Highly mixed program (claudin-4, encoded by Cldn4, e.g. Figure 2E, F “3”). Interestingly, we detected cells that co-expressed markers of distinct programs, suggesting that these cells may be in the process of transitioning from one state to another. For example, some cells co-expressed lysozyme (encoded by Lyz1 and Lyz2) and claudin-2 (encoded by Cldn2, e.g. Figure 2E, F “1”) and may thus be in transition between the AT2-like state and the Embryonic liver-like state. Other cells (e.g., Figure 2E, F “2”) expressed both claudin-2 and claudin-4, suggesting that they are in transition between the Embryonic liver-like and the Highly mixed state.
Relating the clusters and programs, we found that of the 12 clusters, cluster 5 was strongly enriched for the Highly mixed program (Figure 2G, H; Figure S2E). Notably, cluster 5 cells were present in both early adenomas and fully formed LUADs across all mice and tumors (Figure 2I; Figure S2F) and distinctly expressed Slc4a11, a gene associated with poor overall survival in grade 3/4 serous ovarian cancers (Qin et al., 2017) (Figure S2H–J).
An optimal transport model predicts that the Highly mixed program marks a high-plasticity cell state (HPCS) forming a key transition point between other states
Based on the timing of cluster 5’s emergence, its expression of the Highly mixed program, and its particular persistence across tumors, we hypothesized that cells in cluster 5 may form a key transition point and give rise to the heterogeneity observed in advanced tumors. To explore this hypothesis, we modeled the likelihood of transitions between cell states as a temporal coupling between cells along a time course using our Waddington-Optimal Transport (Waddington-OT) algorithm (Schiebinger et al., 2019) (STAR Methods). Where some clusters were transcriptional “sinks”, having low probabilities of giving rise to other states (in particular clusters 3 and 11), others (clusters 2, 4, 5, 6 and 9) had both higher potential to give rise to other cellular states and a substantial number of incoming trajectories, suggesting they may be important transition points in tumor evolution. Cluster 5 had the most abundant and robust connections with other cellular states across the time course (Figure 3A; Figure S3A). This was evident even when compared to other clusters of a similar “age distribution” such as cluster 2, 3 or 4 (Figure S3A). Given this prediction and that cluster 5 contained cells with a highly mixed cellular identity, we designated this cell state a high-plasticity cell state (HPCS).
Figure 3. Identification of a highly plastic cell state with a distinct chromatin accessibility profile.

(A) Probability of cell state transitions as predicted by an optimal transport model. Two cell clusters (nodes, proportional to ‘pagerank’ score – proportion of time spent at node on a random walk) A and B are connected by a directed edge from A to B, if the cells in cluster A at time point t (color code, as in Figure 1B,C) are predicted by the optimal transport model to be ancestors of cells in cluster B at the next time point in that model. Edge thickness is proportional to the probability of the transition predicted by the model (low probability edges < 0.1, are excluded for graphical clarity). Right: Sub-graphs showing only edges between clusters for selected time couplings (labels, top) are on the right. Line width is proportional to the probability of transition ranges from <0.01 for the thinnest line to 0.65 for the thickest line. Dot size is proportional to the pagerank importance of each node, i.e. the amount of “time” spent in a random walk on the graph in any given node. (B) tSNE of cell profiles from primary tumor cells sorted as TIGIT+ (top) and TIGIT− (bottom) sampled to the same cell numbers, colored by membership in cluster 5 (blue). Cells sorted from n = 12 mice. (C) Distribution of cluster 5/HPCS signature score (y axis) in TIGIT+ and TIGIT− cells (p = 3.08 × 10−25; Mann-Whitney U test). (D) UMAP embedding of scATAC-Seq profiles from 164 TIGIT+ (blue) and 3,787 TIGIT− (grey) cells from dissociated primary tumors of n = 5 mice (E) UMAP as in (D) but with cells colored by cluster 5/HPCS gene activity signature scores. (F) Distribution of cluster 5/HPCS gene activity signature score (y axis) in scATAC-Seq profiles of TIGIT+ and TIGIT− cells from n = 5 mice (p = 1.8×10−6, Wilcoxon rank-sum test). (G) Activity scores (color bar) of chromatin state modules (rows, from LaFave et al.) in TIGIT+ and TIGIT− sorted cells (columns) from n = 5 mice. See also related Figure S3 and Table S3.
The LUAD cell subset comprising the HPCS can be prospectively isolated based on TIGIT expression
To functionally interrogate cluster 5 cells comprising the HPCS state, we queried our data for surface markers whose expression is enriched in this subset (Figure S2G). Surprisingly, the Tigit (T cell immunoreceptor with IgG and ITIM domains) gene was a marker of the HPCS subset (Figure S3B top). TIGIT is a co-inhibitory immunoreceptor typically expressed in lymphocytes, and has been studied in the context of autoimmunity, viral immunity, and cancer (Manieri et al., 2017).
We validated the association between Tigit expression and the HPCS (cluster 5 cells) by prospective isolation of TIGIT+ and TIGIT− KP LUAD cells from primary autochthonous tumors at 20 weeks post-initiation, followed by droplet based scRNA-Seq of 26,739 cells. This analysis indicated a strong association of TIGIT+ cells with the HPCS signature (Figure 3B, C). Quantitative PCR (qPCR) indicated robust enrichment of Tigit and the most specific cluster 5/HPCS marker, Slc4a11, in the TIGIT+ KP LUAD cell fraction (Figure S3D). We also confirmed by qPCR for Epcam that the isolated cells were of epithelial (tumor) origin, rather than immune cells (Figure S3D).
The HPCS has a distinct chromatin accessibility profile
We hypothesized that the HPCS may represent a distinct program reflected in a unique chromatin state. To test this hypothesis, we profiled cluster 5 cells by performing single-cell assay for transposase-accessible chromatin sequencing (scATAC-Seq) on TIGIT+ and TIGIT− cells, along with bulk ATAC-Seq of matching populations. As expected, TIGIT+ tumor cells had increased accessibility at genes defining the cluster 5 signature (Figure 3D–F; Figure S3D; Table S3). We further scored the chromatin accessibility signatures identified in the accompanying article (LaFave et al., 2020) and found that TIGIT+ cells had a higher module accessibility score for modules characterized by low Nkx2.1 accessibility (module 1), late stage of progression (module 9), and high Runx2 (module 2) (Figure 3G; Figure S3E). Consistently, we found that Nkx2–1 expression was lower in HPCS cells (Figure 2C), the Runx2 locus was more accessible in TIGIT+ cells (by bulk ATAC-Seq, Figure S3F), and Runx2 expression was higher in HPCS cells (Figure S3G). Notably, LaFave et al. identified RUNX2 as a driver of the metastatic phenotype in the primary tumors (LaFave et al., 2020) and CD109 signaling activity through the Jak/Stat pathway has been shown to contribute to this phenotype (Chuang et al., 2017). Consistently, we found that CD109 marks cluster 11 (EMT, Figure S2G). Thus our findings suggest that the HPCS likely serves as a precursor to the EMT state that acquires metastatic capacity in the primary tumor (Chuang et al., 2017).
TIGIT+ KP LUAD cells are highly plastic in vitro and in vivo
Besides giving rise to EMT (cluster 11), our Waddington-OT model predicted that the HPCS cells are capable of giving rise to multiple other cell states (clusters) (Figure 4A). To functionally evaluate the phenotypic plasticity of cluster 5 cells, we evaluated the diversity of isolated primary TIGIT+ LUAD cells in 3D tumor sphere cultures by scRNA-Seq (Figure 4B). As comparators, we profiled cells from tumor sphere cultures of (i) all TIGIT− cells; and (ii) the CD109+ EMT cell state (cluster 11), which was predicted to be fixed (Figure 4A; Figure S3B bottom). Overall, tumor spheres arising from the TIGIT+ population had the greatest diversity of cell states, followed by the TIGIT− cells (a population depleted of HPCS cells) and finally the CD109+ EMT-like cells (cluster 11) (Figure 4C, D; STAR Methods), consistent with the Waddington-OT model (Figure 4A).
Figure 4. Prospectively isolated HPCS cells display high differentiation potential in vitro and in vivo.

(A) Prediction of plastic and static cell states by the optimal transport model. Graph as in Figure 3A, but showing all transitions (aggregate across all time points) to and from cluster 5 (left) or 11 (right) cells. (B) Experimental design. TIGIT+ HPCS/cluster 5 cells (blue), CD109+(cluster 11) cells (gold), and all non-HPCS TIGIT− cells (grey) were sorted from 17–22 week old LUAD tumors, and grown as tumor spheres for 11 days, followed by scRNA-Seq. (C) tSNE of scRNA-Seq profiles of cells from tumor spheres arising from TIGIT+ (blue), CD109+ (gold) and TIGIT− (grey) KP or KPT LUAD cells at 11 days after cell plating (n = 7 mice). (D) Transcriptional homogeneity. Normalized Mutual Information (NMI, y axis) between each of the three populations. Box plots: upper, median, lower quartile of 1,000 bootstrap samples, of 50 cells each, from the indicated time point; whiskers: 1.5 interquartile range. * p < 0.05, *** p < 0.001 (STAR Methods). (E) Experimental design. TIGIT+ HPCS/cluster 5 cells (blue) and all non-HPCS TIGIT− cells (grey) were sorted from 18–21 week LUAD tumors, and orthotopically transplanted to lungs of NSG mice. (F) Normalized Mutual Information (NMI, y axis) within TIGIT+ and TIGIT− populations. Box plots: upper, median, lower quartile of 1,000 bootstrap samples, of 100 cells each, from the indicated time point; whiskers: 1.5 interquartile range. * p < 0.05 (n = 2 biological replicates, STAR Methods) (n = 6 mice). (G) NMI (y axis) between each population. Box plots: upper, median, lower quartile of 1,000 bootstrap samples, of 50 cells each, from the indicated time point; whiskers: 1.5 interquartile range. * p < 0.05. (H) Relative proportion of cells from TIGIT+ and TIGIT− transplanted primary tumor cells in each cluster (n = 5 TIGIT− vs 3 TIGIT+ allotransplant mice).
To investigate the differentiation potential of HPCS cells in vivo, we isolated primary TIGIT+ and TIGIT− LUAD tumor cells by FACS from mice harboring autochthonous KP tumors and transplanted the subsets intratracheally into the lungs of immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. We assessed the diversity of the cells both pre-transplantation and in the resulting tumors by droplet-based scRNA-Seq (Figure 4E). As expected, TIGIT+ HPCS cells were more homogenous pre-transplantation when compared to the TIGIT− cells (Figure 4F). Yet, transplanted tumors derived from TIGIT+ HPCS-enriched cells had higher diversity than those derived from TIGIT− cells (Figure 4G–I; STAR Methods). Collectively, our findings indicate that cluster 5 represents a high-plasticity cell state with robust potential for cell state transitions in vitro and in vivo.
LUAD cells enriched for the HPCS show high proliferative potential and marked chemoresistance
We found that isolated HPCS (TIGIT+) cells were more efficient at forming tumor spheres than TIGIT− cells in 3D cultures (Figure 5A, B). To examine the tumor-propagating potential of the HPCS cells in vivo, we isolated TIGIT+ and TIGIT− cells from autochthonous KP LUAD tumors (STAR Methods) and transplanted them orthotopically into the lungs of immunodeficient NSG recipient mice (Figure 5C). The HPCS cells grew faster and propagated a greater number of tumors than the TIGIT− cells (Figure 5D, E).
Figure 5. LUAD cells in the HPCS show high growth potential in vitro and in vivo, and are chemoresistant in vivo.

(A) Experimental design. TIGIT+ HPCS/cluster 5 cells (blue) and all non-HPSC TIGIT− cells (grey) were sorted from 17–22 week LUAD tumors, and grown as tumor spheres for 11 days, as in Figure 4B. (B) Number of tumor spheres per 500 cells plated (y axis) arising in individual tumor spheres (dots) from TIGIT+ vs. TIGIT− KPT LUAD cells after 11 days in 3D culture (x axis). Data plotted as mean ± S.D. Two independent biological replicates are shown. ** p < 0.01; *** p < 0.001 (unpaired t-test). (C) Experimental design. TIGIT+ HPCS/cluster 5 cells (blue) and all non-HPCS TIGIT− LUAD cells (grey) expressing firefly luciferase were sorted from 18–21-week tumors and orthotopically allotransplanted into immunodeficient NSG mice. Bioluminescence imaging and tumor harvest were performed at 39 days post-transplantation. (D) Average radiance (y axis) in allotransplanted tumors derived from TIGIT+ and TIGIT− sorted cells. Data plotted as mean ± S.D. * p < 0.05 (t-test; n = 4 TIGIT+ vs 11 TIGIT− allotransplants). (E) Number of surface tumors per 10,000 transplanted cells (y axis) for TIGIT+ or TIGIT− cells in lungs of recipient mice. Data plotted as mean ± S.D. *** p < 0.001 (t-test). (F) Experimental design. Mice with 20-week LUAD tumors were subjected to treatment with vehicle or cisplatin (7 mg/kg); tumors were harvested after 72 hours. (G) tSNE of scRNA-Seq profiles from 20-week KPT LUAD tumors, collected 72 hours after administration of vehicle or cisplatin, colored by predicted membership (STAR Methods) in cluster 5 (blue) or 8 (green). Two independent mice were used per condition. (H) Relative enrichment (y axis, Pearson’s residual: , STAR Methods) of cells in different clusters (x axis), after cisplatin treatment in KPT LUAD tumors in vivo. See also related Figure S4.
We next examined the relative ability of HPCS cells to resist chemotherapy by scRNA-Seq of advanced KPT LUAD tumors 72 h after a single dose of cisplatin, a component of first-line chemotherapies for advanced-stage LUAD patients (Gandhi et al., 2018; Schiller et al., 2002) and a well-characterized chemotherapy agent in the KP LUAD model (Oliver et al., 2010). Annotating the post-treatment cells with the previously identified cell cluster labels from the tumor progression time course (Figure 1D; Figure S4A), we observed a significant compositional difference between cells treated with cisplatin vs. vehicle control (Figure 5F–H; Figure S4B, p < 1×10−20 for association between cluster 5 and cisplatin treatment, Fisher’s exact test). Notably, out of all 12 clusters found in advanced KP LUAD tumors, cells in the HPCS (cluster 5) exhibited the lowest cell cycle score (Figure S4C). This may in part explain why the HPCS cells are resistant to chemotherapy, which targets proliferating cells.
Our results suggest that the HPCS is associated with particularly aggressive features, including robust potential for differentiation and proliferation as well as drug resistance. Such aggressive features are frequently associated with cancer stem-like cells (CSCs) (Batlle and Clevers, 2017; Kreso and Dick, 2014). To interrogate whether the HPCS correlates with known stem cell types, we performed a comparison of the HPCS signature with 1,197 previously published cancer and normal tissue stem cell signatures. We found weak, but significant correlations between only eight of these signatures and the HPCS, suggesting that the HPCS is largely distinct from known stem cell identities (Figure S4D; Table S4; STAR Methods).
Cancer cells in a similar high plasticity cell state are present in human LUAD tumors and associate with poor survival
Finally, we explored the relevance of the HPCS in human LUAD tumors, finding important correspondence to our observations in the mouse model. First, immunostaining of human LUAD tissues for markers of the different programs revealed cells representing the transitions observed in the mouse model (Figure S5A; Figure 2F). Additionally, an analysis of 9,543 scRNA-Seq profiles of malignant cells from 20 human LUAD tumors across three published datasets (Lambrechts et al., 2018; Laughney et al., 2020; Zilionis et al., 2019) showed that cells with the Highly mixed/HPCS program assignment were present in each of these tumors (Figure 6A; Figure S5B–G).
Figure 6. HPCS-like program is expressed in human tumors and associates with poor survival.

(A) The high-plasticity program is expressed in individual malignant cells from human LUAD tumors. Left: PHATE map of the mouse LUAD cells (as in Figure 2D), colored by the program score. Right: For each of three scRNA-Seq studies of cancer cells from human LUAD tumors, shown are the violin plot (left) of the distribution of the Highly mixed/HPCS program scores (y axis) in the cancer cells of each tumor (x axis), and a tSNE of the profiles, with cells (dots) colored by their program scores. (B) Hazard ratio (HR, x axis, mean HR and 95%-confidence interval) for each NMF program (y axis) in LUAD patients in the TCGA as predicted by a Cox proportional hazards model independently fit to each NMF activity term as a continuous variable (n = 403; STAR Methods). (C) Hazard ratio (HR, x axis, mean HR and 95%-confidence interval) for each cluster (y axis) in LUAD patients in the TCGA as predicted by a Cox proportional hazards model independently fit to each cluster activity term as a continuous variable (n = 403; STAR Methods). (D) Hazard ratio (HR, x axis, mean HR and 95%-confidence interval) for each NMF program (y axis) across all tumors with tumor purity information in TCGA (n = 5723) as predicted by a Cox proportional hazards model independently fit to each NMF activity term as a continuous variable (STAR Methods). (E) Hazard ratio (HR, x axis, mean HR and 95%-confidence interval) for each cluster (y axis) in all cancer patients in the TCGA as predicted by a Cox proportional hazards model independently fit to each cluster activity term as a continuous variable (n = 5723; STAR Methods). See also related Figure S5 and Table S5.
Importantly, in an analysis of The Cancer Genome Atlas (TCGA) bulk RNA-Seq data (The Cancer Genome Atlas Research, 2014), LUAD tumors that express the Highly mixed, EMT, High-cycling, and GI-epithelium programs were associated with worse survival, whereas the AT2-like, Embryonic liver-like, and Mixed AT1/AT-like states were associated with a more favorable prognosis (Figure 6B; Table S5; p = 2.4×10−4, 4.2×10−3, 3.6×10−2, 2.4×10−2, 5.6×10−3 respectively, Cox proportional hazards model; p = 4×10−4 in the full model including all NMF programs). A cluster-based analysis of the same TCGA LUAD data also demonstrated worse survival for cluster 5/HPCS (Figure 6C; Table S5; p = 2.35×10−2, Cox proportional hazards model). Notably, the significance of association of the Highly mixed program did not require KRAS or TP53 mutations (Figure S5H; Table S5). Accordingly, high CLDN4 expression, a marker of the Highly mixed state, predicted poor outcomes in human LUAD (Figure S5I) (Gyorffy et al., 2013). The Highly mixed state and cluster 5/HPCS signatures also predicted poor outcomes in a pan-cancer analysis across the pooled TCGA collection (Figure 6D, E; Table S5; p < 2×10−16 for a model including all NMFs, Cox proportional hazards model), suggesting that features of the HPCS may generally define aggressive cancers. As in mouse lung adenomas and LUAD tumors, some cells expressing the HPCS program were present in each of the 15 primary human LUAD tumors and in five metastases examined by scRNA-Seq (Lambrechts et al., 2018; Laughney et al., 2020; Zilionis et al., 2019) (Figure 7A). Notably, SLC4A11 was a marker of the cell state in both mouse and human LUAD tissues (Figure S6A, B).
Figure 7. Integrin α2Hi LUAD HPCS cells isolated from human patient-derived xenografts harbor high growth potential and plasticity.

(A) Cells in the HPCS are present across all profiled tumors. Fraction of cells that were mappable (y axis) from each tumor (x axis) that express the HPCS-like program. (B) Histograms showing the distribution of the fraction of pan-cytokeratin positive cells in human LUAD tissues staining for: Claudin-4 (left), Integrin α2 (middle), and both together (right). Vertical dotted lines represent the point at which at least 10% of a tumor stained strongly positive. (C) Experimental design. Integrin α2Hi and integrin α2Lo LUAD cells were isolated from three distinct human patient-derived xenograft (PDX) models, followed by 3D tumor sphere culture for 22 days. (D) Pan-cytokeratin and integrin α2 immunofluorescence in one of the PDX models. Scale bar: 50 μm. (E) Fold change in growth (y axis) of tumor spheres derived from integrin α2Hi and integrin α2Lo cells. Data plotted as mean ± S.D. * p = 0.0216 (t test of the log2 transform of the shown fold change; n = 3 biological replicates) (F) NMI (y axis) between each population. Box plots: upper, median, lower quartile of 1,000 bootstrap samples, of 50 cells each, from the indicated time point; whiskers: 1.5 interquartile range. * p < 0.05 (STAR Methods). See also related Figure S6.
We next evaluated whether HPCS-like cells in human LUAD tumors contained cell surface markers compatible with flow cytometry. We did not detect TIGIT mRNA or protein in human LUAD (data not shown), suggesting that some features of the HPCS signature are species-specific. Instead, we identified alternative putative cell surface markers based on the expression profiles of human LUAD cells (from three published datasets) that showed the highest overlap with the mouse HPCS signature (Figure 7A; Figure S6C; STAR Methods). In particular, ITGA2, encoding integrin α2 (CD49B), a subunit of the integrin α2β1 collagen receptor (Hynes and Naba, 2012; Tuckwell et al., 1995), was expressed at high levels in both human and mouse LUAD HPCS cells (Figure S6D; Table S1; Table S3).
We next surveyed integrin α2 expression in 135 human LUAD patient tissues and identified heterogeneity in integrin α2 signal, with 39.3% of patients (53 of 135) with tumor samples containing at least 10% integrin α2Hi tumor cells (defined as the top 15% of integrin α2 expressing cells). Notably, 40.7% of patients (55 of 135) had tumor samples with at least 10% claudin-4Hi tumor cells (defined as the top 15% of claudin-4 staining), and 19.3% (26 of 135) had tumor samples with at least 10% of tumor cells staining both claudin-4Hi and integrin α2Hi (Figure 7B; Figure S6E, F). These results suggest that the Highly mixed cell state and the HPCS is present in a significant fraction of LUAD patients.
Finally, we tested whether the integrin α2Hi human LUAD cells functionally recapitulate features of the mouse LUAD HPCS, including high plasticity and the capacity to proliferate. We found that integrin α2Hi tumor cells prospectively isolated from three independent PDX models formed significantly more tumor spheres compared to integrin α2Lo cells (Figure 7C–E; Figure S6E; STAR Methods). We also performed droplet-based scRNA-Seq on tumor spheres and observed that the integrin α2Hi human LUAD cells gave rise to spheres with higher transcriptional diversity than the integrin α2Lo bulk of the tumor (Figure 7F). Taken together, these results suggest that a HPCS-like state also exists in human LUAD and may have significant implications as a driver and biomarker of tumor progression and drug resistance in the clinic.
DISCUSSION
Here, we used scRNA-Seq to study cell state changes during tumor evolution in a mouse model of LUAD mimicking the oncogenic transformation processes observed in human disease (Jackson et al., 2005; Jackson et al., 2001), where mutations in oncogenes, such as KRAS, are thought to occur early, followed by inactivation of the p53 pathway (Campbell et al., 2016; The Cancer Genome Atlas Research, 2014). Transcriptional heterogeneity grew dramatically during tumor progression, but the process was stereotypic and reproducible across individual tumors within a mouse and between mice. Further, some states were shared between the K and KP genotypes. Thus, phenotypic diversity, as captured by transcriptional states, is reproducible in this cancer model, suggesting the existence of deterministic programs governing the emergence and maintenance of heterogeneity.
One straightforward hypothesis was that this cell state variation is a direct outcome of underlying genetic variation, consistent with a model of tumor progression where every step is governed by the acquisition of a novel driver mutation (Fearon and Vogelstein, 1990). However, the CNV patterns and transcriptional states of individual cells were not directly aligned in the KP tumors, suggesting that additional factors besides genetic drivers, such as tumor microenvironment and epigenetic changes (LaFave et al., 2020), influence cell states during tumor evolution.
In contrast to embryogenesis, where new states emerge and preceding states are lost (Cao et al., 2019; Nowotschin et al., 2019), we found that during tumor progression new states are acquired and preceding states are maintained even in advanced tumors. Our results suggest that disruption of normal developmental programs is a major organizing principle in the acquisition of new states: we first observed alternative lung epithelial programs, followed by several alternative programs mimicking the primordial gut, and finally the emergence of cells with a mesenchymal state, indicating a complete EMT (Figure S6G). Whereas each of these cell states emerged at a different characteristic time, all persisted in tumors once they arose, such that more advanced tumors were composed of a growing assortment of cells with an increasing diversity of states.
Our analysis highlighted one particular cell state, which was not similar to any defined or previously reported program, as the hub of cell state transitions in the tumor. This high-plasticity cell state (HPCS) was enriched in cluster 5, the only cluster whose cells were present in a significant fraction in all mouse adenomas and LUAD tumors analyzed, as well as in scRNA-Seq profiles of human LUAD tumors (Lambrechts et al., 2018; Laughney et al., 2020; Zilionis et al., 2019).
Interestingly, we found that the HPCS develops not only in advanced KrasG12D mutant, p53 deficient KPT adenocarcinomas, but also in early stage KrasG12D mutant, p53 proficient KT adenomas. Thus, it is the cell states downstream of the HPCS rather than the HPCS itself that depend on p53 status or, more broadly, the stage of tumor progression: The HPCS can give rise to more diversity and more aggressive cell states, such as EMT, in advanced p53 mutant adenocarcinomas when compared to p53 wild-type adenomas (Figure S6G). These findings cast p53 as a guardian of lineage fidelity, whose deletion enables cancer cells to sample a broader range of phenotypic space. However, growth signals that naturally drive tissue regeneration – that become co-opted by oncogene activation upon transformation – may suffice to give rise to at least some plasticity even in p53 proficient cells, as suggested by our results and recent work on wound healing and tumorigenesis in the skin (Ge et al., 2017).
Our findings are surprising, as they do not support an intuitive model whereby lineage erosion occurs gradually from a “leading edge” of progressively more de-differentiated cells. Rather, LUAD heterogeneity appears to arise from a highly plastic cell state that emerges rapidly in tumorigenesis and persists in advanced tumors. Furthermore, we found that isolated HPCS cells can functionally give rise to the entire diversity of observed cell states in the tumor in vivo, spanning a range of defined cancer cell states in established tumors. For instance, a subset of the HPCS-expressing cells partially activated the EMT program, suggesting that the HPCS may be a prerequisite to EMT. Skin and mammary tumor models (Pastushenko et al., 2018) and human head and neck cancers (Puram et al., 2017) were recently shown to contain a “pre-EMT” state, which may in fact represent a HPCS in these cancer types.
Cell plasticity has been postulated to contribute to failure of chemo-, targeted- and immunotherapies (Arozarena and Wellbrock, 2019; Gupta et al., 2019; Horn et al., 2020). A particularly fascinating example is the conversion of lung and prostate adenocarcinomas to a neuroendocrine lineage, which is occasionally observed as a response to highly effective targeted therapies. This lineage conversion causes the tumors to lose dependence on oncogene activity and become resistant to oncoprotein-targeted therapies (Beltran et al., 2019; Quintanal-Villalonga et al., 2020). Given that HPCS cells were enriched shortly following platinum-based chemotherapies, it is possible that the acquisition of the neuroendocrine lineage during extreme therapeutic pressure occurs through a HPCS. Our results implicate the HPCS as a cell state that is strongly associated with LUAD treatment resistance, motivating its therapeutic targeting.
The HPCS shares functional features of both normal tissue stem cells and CSCs, including robust growth and differentiation potential (Batlle and Clevers, 2017; Kreso and Dick, 2014). However, the HPCS gene expression signature was largely distinct from published normal and cancer stem cell signatures. CSCs have classically been identified and studied using candidate markers derived from normal stem cells. In contrast, we discovered the HPCS using an unsupervised profiling approach and computational modeling, which led us to uncover unexpected markers for this cell state that have previously not been implicated in CSCs (e.g. TIGIT, integrin α2 and Slc4a11). These results suggest that the HPCS represents a truly distinct cell state with importance in human LUAD and, possibly, human cancers more broadly. Indeed, the HPCS predicted poor survival not just in LUAD, but even in an analysis pooling all cancers represented in TCGA, suggesting that features that define the HPCS are particularly malignant or that similar HPCSs may exist across the spectrum of human cancers. Thus, the HPCS signature may enable identification of similar plastic states in other cancer types and biological contexts.
In conclusion, we have shown that increased transcriptional heterogeneity coupled with lineage infidelity and plasticity are hallmarks of tumor progression in a mouse model of LUAD, and that these features are present in human tumors. Whereas increased plasticity is highly reproducible and greater in tumors where p53 is inactivated, the phenotypic variation itself is largely independent of specific genetic alterations. In addition to programs reflecting lung and other epithelial cell states, a high-plasticity cell state appears at the nexus of these developmental cell state transitions, and is associated with resistance to chemotherapy, high growth potential, and poor survival in patients. Our work casts the HPCS as a key driver of tumor progression and intra-tumoral heterogeneity, underscoring the importance of targeting plastic cell states in cancer therapy.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tuomas Tammela (tammelat@mskcc.org).
Materials availability
This study did not generate new unique reagents.
Data availability
Processed scRNAseq data is available for download or interactive exploration at the Broad Single Cell Portal at the following URLs:
https://singlecell.broadinstitute.org/single_cell/study/SCP971 https://singlecell.broadinstitute.org/single_cell/study/SCP972/ https://singlecell.broadinstitute.org/single_cell/study/SCP973/
Raw data for SmartSeq2 scRNA-Seq, 10x scRNA-Seq, CNV, scDNA-Seq, and Bulk ATAC available from GEO: GSE152607
Code availability
Relevant code and instruction, which may be used to reproduce the principle results presented here is provided on GitHub: https://github.com/matanhofree/lungTumorEvolution.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Briefly, to generate Wnt-conditioned media, the L-WRN mouse fibroblast cell line (ATCC, catalog #CRL-3276) was grown to confluency on a tissue culture dish using D-MEM (Gibco, catalog #10313039) supplemented with 1% penicillin/streptomycin and 1% glutamine (see Key Resources Table) at 37 C. The media was then aspirated and replaced with fresh supplemented D-MEM. Media was then collected and refreshed after every 3rd day for two harvests. Harvested media was filtered through a 0.45μM filter, aliquoted, and kept frozen at −80 C for future use.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat Anti-Mouse CD16 / CD32 Monoclonal Antibody | BD Biosciences | Clone 2.4G2, Cat# 553142, RRID:AB_394657 |
| Human TruStain FcX | Biolegend | Cat# 422301, RRID:AB_2847850 |
| Rat Anti-Mouse CD45 Monoclonal Antibody, APC | BD Biosciences | Clone 30-F11, Cat# 559864, RRID:AB_398672 |
| Rat Anti-Mouse CD31 Monoclonal Antibody, APC | BD Biosciences | Clone MEC 13.3, Cat# 561814, RRID:AB_10893351 |
| Rat Anti-Mouse CD11b Monoclonal Antibody, APC | BD Biosciences | Clone M1/70, Cat# 561690, RRID:AB_10897015 |
| Rat Anti-Mouse TER-119 Monoclonal Antibody, APC | BD Biosciences | Clone TER-119, Cat# 561033, RRID:AB_10584336 |
| Rat Anti-Mouse CD45, FITC | Invitrogen | Clone 30-F11, Cat# 11-0451-82, RRID:AB_465050 |
| Rat Anti-Mouse CD31, FITC | Invitrogen | Clone 390, Cat# 11-0311-82, RRID:AB_465012 |
| Rat Anti-Mouse CD11b, FITC | Invitrogen | Clone M1/70, Cat# 11-0112-82, RRID:AB_464935 |
| Hamster Anti-Mouse CD11c, FITC | Biolegend | Clone N418, Cat# 117305, RRID:AB_313774 |
| Rat Anti-Mouse F4/80, FITC | Invitrogen | Clone BM8, Cat# 11-4801-82, RRID:AB_2637191 |
| Rat Anti-Mouse TER-119/Erythroid Cells, FITC | Biolegend | Clone TER-119, Cat# 116206, RRID:AB_313707 |
| Mouse Anti-Mouse CD109, AF647 | Santa Cruz Biotechnology | Clone C-9, Cat# sc-271085 AF647, RRID:AB_2847851 |
| Rat Anti-Mouse CD45, APC | Invitrogen | Clone 30-F11, Cat# 17-0451-82, RRID:AB_469392 |
| Rat Anti-Mouse CD31, APC | Invitrogen | Clone 390, Cat# 17-0311-82, RRID:AB_657735 |
| Rat Anti-Mouse CD11b, APC | Invitrogen | Clone M1/70, Cat# 17-0112-82, RRID:AB_469343 |
| Hamster Anti-Mouse CD11c, APC | Biolegend | Clone N418, Cat# 117310, RRID:AB_313779 |
| Rat Anti-Mouse F4/80, APC | Invitrogen | Clone BM8, Cat# 17-4801-82, RRID:AB_2784648 |
| Rat Anti-Mouse TER-119, APC | Invitrogen | Clone TER-119, Cat# 17-5921-82, RRID:AB_469473 |
| Rat Anti-Mouse CD326 (EpCAM), PE | Invitrogen | Clone G8.8, Cat# 12-5791-82, RRID:AB_953615 |
| Mouse Anti-Mouse TIGIT, BV421 | Biolegend | Clone 1G9, Cat# 142111, RRID:AB_2687311 |
| Mouse Anti-Human CD45, FITC | Invitrogen | Clone HI30, Cat# 11-0459-42, RRID:AB_10852703 |
| Mouse Anti-Human CD31, FITC | Biolegend | Clone WM59, Cat# 303104, RRID:AB_314330 |
| Mouse Anti-Human CD11b, FITC | Biolegend | Clone ICRF44, Cat# 301330, RRID:AB_2561703 |
| Mouse Anti-Human CD11c, FITC | Biolegend | Clone 3.9, Cat# 301604, RRID:AB_314174 |
| Mouse Anti-Mouse H-2Kd, FITC | Biolegend | Clone SF1-1.1, Cat# 116606, RRID:AB_313741 |
| Mouse Anti-Human CD326 (EpCAM), PE/Cy7 | Biolegend | Clone 9C4, Cat# 324222, RRID:AB_2561506 |
| Mouse Anti-Human CD49b (Integrin α2), APC | Biolegend | Clone P1E6-C5, Cat# 359309, RRID:AB_2564198 |
| Mouse Anti-Claudin 2 | Invitrogen | Clone 12H12, Cat# 32-5600, RRID:AB_2533085 |
| Rabbit Anti-Claudin 4 | Invitrogen | Clone ZMD.306, Cat# 36-4800, RRID:AB_2533262 |
| Rabbit Anti-Lysozyme | DAKO | Clone EC3.2.1.17, Cat# A0099, RRID:AB_2341230 |
| Rabbit Anti-Prosurfactant Protein C | Millipore | Cat# AB3786, RRID:AB_91588 |
| Rabbit Anti-Integrin α2 | Abcam | Clone EPR17338, Cat# 181548, RRID:AB_2847852 |
| Rabbit Anti-RFP | Rockland | Cat# 600-401-379, RRID:AB_2209751 |
| Bacterial and Virus Strains | ||
| Ad5mSPC | Viral Vector Core, University of Iowa | N/A |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Sigma-Aldrich | D9542-1MG |
| YOPRO-1 | Invitrogen | Y3603 |
| Advanced DMEM/F12 | Gibco | 12634028 |
| DMEM | Gibco | 10313039 |
| B27 Supplement | Gibco | 17504044 |
| FGF-7 (KGF) | PeproTech | 100-19 |
| FGF-10 | PeproTech | 100-26-50ug |
| Noggin | PeproTech | 120-10C-50ug |
| EGF | PeproTech | AF-100-15-100ug |
| N-Acetylcysteine | Sigma-Aldrich | A9165-5G |
| Nicotinamide | Sigma-Aldrich | N0636-100G |
| SB431542 | SelleckChem | S1067 |
| CHIR99021 | Sigma-Aldrich | SML1046-5MG |
| HEPES | Gibco | 15630080 |
| Penicillin/Streptomycin | Gibco | 15140163 |
| L/glutamine | Gibco | 35050061 |
| Y-27632 | SelleckChem | S1049 |
| D-Luciferin | Perkin Elmer | 122799 |
| S-MEM | Gibco | 11380037 |
| Dispase II | Gibco | 17105-041 |
| Collagenase Type IV | Thermo Fisher Scientific | 17104019 |
| DNase I | Sigma-Aldritch | 69182-3 |
| Gentamicin | Gibco | 15750-060 |
| ACK | Thermo Fisher Scientific; Lonza | A1049201; 10-548E |
| TCL Buffer | Qiagen | 1031576 |
| Maxima Reverse Transcriptase | Thermo Fisher Scientific | EP0752 |
| Digitonin | Promega | G9441 |
| Illumina Tagment DNA Enzyme | Illumina | 15027865 |
| BamBanker Cell Freezing Medium | Lymphotec | 302-14681 |
| Matrigel | Corning | CB-40230C |
| Penicillin-Streptomycin | Gibco | 15140163 |
| Corning Dispase | Corning | 354235 |
| TrypLE | Gibco | 12604013 |
| Cisplatin | EMD-Millipore | 232120 |
| Shandon Zinc Formal-Fixx | Thermo Scientific | 6764255 |
| Vectashield with DAPI | Vector Labs | H-1200 |
| ImmPACT DAB Peroxidase (HRP) Substrate | Vector Labs | SK-4105 |
| Critical Commercial Assays | ||
| RNAscope® 2.5 HD Detection Reagents-RED | ACDBio | 322360 |
| DapB ISH Probe | ACDBio | 310043 |
| Ppib Mouse ISH Probe | ACDBio | 313911 |
| Slc4a11 Mouse ISH Probe | ACDBio | 559521 |
| Tigit Mouse ISH Probe | ACDBio | 319751 |
| PPIB Human ISH Probe | ACDBio | 313901 |
| SLC4A11 Human ISH Probe | ACDBio | 583931 |
| Lung Dissociation Kit | Miltenyi Biotech | 130-095-927 |
| Tumor Dissociation Kit | Miltenyi Biotech | 130-095-929 |
| Agencourt RNAClean XP beads | Beckman Coulter | A63881 |
| KAPA HiFi HotStart ReadyMix | KAPA Biosystems | KK2601 |
| Agencourt AMPure XP beads | Beckman Coulter | A63881 |
| Nextera XT Library Prep kit | Illumina | FC-131-1096 |
| GenomePlex Single Cell Whole Genome Amplification Kit | Sigma | 254-457-8 |
| Qiagen MinElute PCR Purification Kit | Qiagen | 28004 |
| Chromium Single Cell ATAC Library Kit v1 chemistry | 10x Genomics | PN-1000083 |
| Qiagen RNeasy Plus Mini kit | Qiagen | 74136 |
| Qiagen RNeasy Plus Micro kit | Qiagen | 74034 |
| SuperScript VILO cDNA synthesis kit | Invitrogen | 11754050 |
| PrimeScript RT Reagent kit | Takara | RR037B |
| Powerup SYBR mix | Applied Biosystems | A25778 |
| ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit | Vector Labs | MP-7401-50 |
| Mouse-on-Mouse ImmPRESS HRP (Peroxidase) Polymer Kit | Vector Labs | MP-2400 |
| Deposited Data | ||
| Single cell RNAseq (SmatrtSeq2,10X, DNA, ATAC) & Bulk ATAC | This paper | GEO: GSE152607 |
| Human single cell lung adeno tumors | Zilionis et al. | GEO: GSE127465 |
| Human single cell lung adeno tumors | Lambrechts et al. | E-MTAB-6653, E-MTAB-6653 |
| Human single cell lung adeno tumors | Laughney et al. | GEO: GSE123903 |
| TCGA LUAD | MC3 | |
| TCGA PanCan | LinkedOmics | http://www.linkedomics.org/ |
| Experimental Models: Cell Lines | ||
| Mouse L-WRN cell line | ATCC | CRL-3276 |
| Experimental Models: Organisms/Strains | ||
| Mouse/B6.129: KrasLSL-G12D | Jackson et al., 2001; The Jackson Laboratory | 008179 |
| Mouse/B6.129: Trp53flox/flox | Marino et al., 2000; The Jackson Laboratory | 008462 |
| Mouse/B6.129: Rosa26LSL-tdTomato | Madisen et al., 2010; The Jackson Laboratory | 007905 |
| Mouse/B6.129: Rosa26LSL-Luciferase | Safran et al., 2003; The Jackson Laboratory | 005125 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (aka NSG mice) | Ishikawa et al., 2005, The Jackson Laboratory | 005557 |
| Oligonucleotides | ||
| GusB qPCR F - CCGACCTCTCGAACAACCG | Roche Universal Probe Library | N/A |
| GusB qPCR R - GCTTCCCGTTCATACCACACC | Roche Universal Probe Library | N/A |
| Tigit qPCR F - TGCCTTCCTCGCTACAGG | Roche Universal Probe Library | N/A |
| Tigit qPCR R - TGCAGAGATGTTCCTCTTTGTATC | Roche Universal Probe Library | N/A |
| Slc4a11 qPCR F - CGAGGATCCAGAACAGACCT | Roche Universal Probe Library | N/A |
| Slc4a11 qPCR R - GAGATGTTTGTGCAAAGAAGGA | Roche Universal Probe Library | N/A |
| Epcam qPCR F - TGTCATTTGCTCCAAACTGG | Roche Universal Probe Library | N/A |
| Epcam qPCR R - GTTCTGGATCGCCCCTTC | Roche Universal Probe Library | N/A |
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| Code generated as part of this study | This paper | https://github.com/matanhofree/lungTumorEvolution |
| Matlab (The Mathworks) | https://www.mathworks.com/ | R2019a |
| R | https://cran.r-project.org/ | v3.6.1 |
| RSEM | https://github.com/deweylab/RSEM | v1.3.0 |
| Cellranger (10X Genomics) | https://support.10xgenomics.com/ | v3.1.0 |
| Cellranger ATAC (10X Genomics) | https://support.10xgenomics.com/ | v1.2.0 |
| CNVkit | https://github.com/etal/cnvkit/ | v0.9.6 |
| fastp | https://github.com/OpenGene/ | v0.20.0 |
| ATAC-seq-pipeline | https://github.com/ENCODE-DCC/atac-seq-pipeline/ | v1.5.4 |
| Other | ||
| N/A | ||
Mouse 3-dimensional tumor sphere cultures
Primary 3D tumor sphere cultures were generated from tumors isolated from 30–34 week-old mice bearing 17–22 week old LUAD tumors. The entire primary culture was used in the downstream experiments as described in the manuscript.
Mouse cultures were plated on Matrigel as previously described (Tammela et al., 2017). Briefly, 350–1000 KP primary mouse LUAD cells were mixed in 50% Matrigel (Corning, catalog #CB-40230C) and 50% Advanced DMEM/F12 (Gibco, catalog #12634028) and plated on 10–12 μl of Matrigel on an 8 chambered coverglass (Thermofisher, catalog #155379). The solution was allowed to solidify at 37° C and then Advanced DMEM/F12 supplemented with Gentamicin, Penicillin-Streptomycin (Gibco, catalog #15140163), 10 mM HEPES (Gibco, catalog #15630080), and 2% heat-inactivated fetal bovine serum was added to fully cover the Matrigel plug. Cultures were grown in standard tissue culture conditions at 37° C. Media was refreshed every 1–3 days.
Human 3-dimensional tumor sphere cultures
Primary 3D tumor sphere cultures were generated from patient-derived xenografts (PDXs) implanted into NSG mice as described below.
Human tumor sphere cultures were plated using tissue culture treated plates with inserts (Plates: Falcon, catalog #353504; Inserts: Falcon, catalog #353095). Briefly, up to 10,000 primary patient-derived xenograft LUAD cells were mixed in 50% Matrigel and 50% Advanced DMEM/F12 and plated on the insert. Human organoid media with appropriate supplements (Table S6) was added to the well before the addition of the insert. Cultures were grown in standard tissue culture conditions at 37° C. Media was refreshed every 2–3 days.
Mice
We used C57BL/6 x Sv129 mixed background mice from the following previously published strains: KrasLSL-G12D (Jackson et al., 2001), Trp53flox/flox (Marino et al., 2000), Rosa26LSL-tdTomato (Madisen et al., 2010), and Rosa26LSL-Luciferase (Safran et al., 2003). In addition, we used NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (aka NSG mice) (Ishikawa et al., 2005) (The Jackson Laboratory, catalog #005557) in our allotransplant and patient-derived xenograft studies. Tumors were induced in K or KP mice with 0.5–2.5 × 108 PFU of AdSPC-Cre (Sutherland et al., 2011) or 2 × 108 PFU of AdCMV-FlpO (Iowa). Mice in all experiments were monitored by the investigators and veterinary staff at the Department of Comparative Medicine at Massachusetts Institute of Technology (MIT), MA or by the staff at the Research Animal Resource Center at Memorial Sloan Kettering Cancer Center (MSKCC), NY with food and water provided ad libitum. Mice were treated in accordance to all relevant institutional and national guidelines and regulations. Animal studies were approved by the Committee for Animal Care at MIT, MA (institutional animal welfare assurance no. A-3125–01) or the Institutional Animal Care and Use Committee at MSKCC, NY (protocol #17–11-008). For all mouse experiments, sex did not appear to significantly influence the resulting tumor transcriptome analysis. A complete list of mice along with age, sex, and age of tumor used in experiments is available (Table S6).
Human samples.
Histologic human LUAD samples from MSKCC were obtained under MSKCC IRB #06–107 and IRB #12–245. MSK-IMPACT profiling (Samstein et al., 2019) was previously performed and the cBioPortal (Cerami et al., 2012; Gao et al., 2013) was used to identify LUAD patient samples with KRAS and TP53 mutations. Human samples assembled in tissue microarrays in this study from Vanderbilt University Medical Center and the Tennessee Valley Health Care Systems were collected with informed consent from subjects enrolled on Institutional Review Board-approved protocol 000616 that complies with all relevant ethical regulations at Vanderbilt University Medical Center and the Tennessee Valley Health Care Systems, Nashville Campus, TN.
Primary tumors for generation of PDX models were obtained with informed consent from patients under protocols approved by the MSKCC Institutional Review Board as above as well as MSKCC IRB #14–091. MSK-LX984 was derived from a 70-year-old man with lung adenocarcinoma harboring somatic TP53R283P, CDKN2AA36Pfs*17, DNMT3BX558_splice, MAXH81Pfs*5, and PALB2E1120* mutations as well as an amplification in MAPK1 and deletions in MAP2K4, FLCN, and NCOR1. MSK-LX1012 was derived from a 52-year-old woman with lung adenocarcinoma harboring somatic mutations KRASG12A, EGFRL858R, and PIK3CAH1047R along with amplifications in CDK4, MDM2, and TERT. MSK-LX1182 was derived from a 68-year-old woman with a lung adenocarcinoma harboring somatic mutations NF1Q2721*fs6, TP53S241F, RASA1S247Lfs*6, and KEAP1E343* as well as an amplification of MET. A complete list of annotated tissue sections and PDXs used is available (Table S6).
METHOD DETAILS
Isolating cells from lung adenocarcinomas
Mice with LUAD tumors were euthanized at 2, 12, 20, or 30 weeks following tumor induction. We chose these time points because they reflect key stages in LUAD progression: atypical adenomatous hyperplasia (AAH) (KT and KPT at 2 weeks), adenoma (KT at 12 and 30 weeks), adenoma-to-LUAD transition (KPT at 12 weeks) and LUAD (KPT at 20 and 30 weeks). We micro-dissected large KPT tumors individually at 20 and 30 weeks, whereas all other samples were harvested by dissociating entire lungs containing mixtures of neoplasias in various stages of tumor progression. Following euthanasia, mice were perfused with S-MEM (Gibco, catalog #11380037) through the right ventricle of the heart. Dissected lungs or microdissected tumors were dissociated either with protease and DNAse solution in the Lung Dissociation Kit (Miltenyi Biotech, catalog #130–095-927) followed by mechanical dissociation using gentleMACS “C” columns (Miltenyi Biotech, catalog #130–093-237) according to the manufacturer’s instructions (Tammela et al., 2017), or by a mixture of Dispase II (Gibco, catalog #17105–041, final concentration 0.6 U/ml), Collagenase Type IV (Thermo Fisher Scientific, catalog #17104019; final concentration 0.083 U/ml), and DNase I (Sigma-Aldrich, catalog #69182–3; final concentration 10 U/ml) in S-MEM solution containing Gentamicin (Gibco, catalog #15750–060, final concentration 20 μg/ml) at 37°C for 30 min (Table S6). The dissociated cells were filtered using a 100 μm strainer and spun at 300 g for 5 min at room temperature. The supernatant was removed by aspiration and red blood cell lysis was performed using ACK (Thermo Fisher Scientific, catalog #A1049201). Cells were then washed with media and pelleted at 300 g for 5 min at 4°C. The supernatant was removed, and the pellet resuspended in Fluorescence-Activated Cell Sorting (FACS) buffer media (200 mM EDTA with 250 μl heat-inactivated FBS in PBS) before being passed through a 40 μm strainer and counted for use in FACS below.
Dissociation of patient-derived xenografts
Primary tumors collected for generation of patient-derived xenografts (PDX) models were obtained with informed consent from patients under protocols approved by the MSKCC Institutional Review Board. Subcutaneous flank tumors were generated as described previously (Daniel et al., 2009).
PDX tumors were dissected off the flank of immunocompromised NSG mice (Jackson Laboratory, catalog #005557). Tumor samples were minced using fresh razor blades in a sterile dish. Tumors were then transferred to a gentleMACS C tube (Miltenyi Biotech, catalog #130–093-237) with 7 ml of RPMI and TDK enzymes (Miltenyi Biotech, catalog #130–095-929). The tube was then placed inverted on a gentleMACS dissociator (Miltenyi Biotech, catalog #130–096-427) with a heater attached. A pre-selected program (37C_h_TDK_3) was used for dissociation. After dissociation (~1hr), the dissociated tumor cells were transferred to a 50 ml tube with a 70 μm MACS SmartStrainer (Miltenyi Biotech, catalog # 130–098-462) and washed with 20–25 ml of FACS buffer. The sample was then centrifuged at 300 g for 5 min and the supernatant discarded. The cell pellet was resuspended in up to 5 ml of ACK Lysing Buffer (Lonza, catalog #10–548E) and kept at room temperature for 2 min. 20–25 ml of FACS buffer was added and another spin at 300 g for 5 min was performed. The supernatant was then discarded and cells resuspended in PBS.
Fluorescence-activated cell sorting (FACS)
Cells were prepared as above and Fc block was added on ice for 5 min prior to being stained with the appropriate antibody panel (Table S6). Cells were stained for 20 min before washing twice with FACS buffer media. Five-min, 300 g spins at 4°C were performed in between washes to pellet the cells. YO-PRO-1 (Invitrogen, catalog #Y3603) or DAPI (final concentration 1 μg/ml) was added to each sample to identify dead cells and FACS was performed at either the Swanson Biotechnology Center Flow Cytometry Core Facility at the Koch Institute for Integrative Cancer Research or the Flow Cytometry Core Facility at Sloan Kettering Institute/MSKCC, using a BD FACS Aria Sorter. Cells for single cell experiments were sorted using the ‘single cell purity’ mode; cells for culture and allotransplant were sorted using ‘yield’ mode. Cancer cells in the LUAD progression study were sorted as (CD45/CD31/CD11b/TER119)−/tdTomato+/DAPI− live cells. To isolate TIGIT+ cancer cells, dissociated tumor cells were stained and sorted for live (CD45/CD31/CD11b/CD11c/F4/80/TER119)−/EPCAM+/YO-PRO1−/TIGIT+ cells. TIGIT− cells were sorted as live (CD45/CD31/CD11b/CD11c/(F4/80)/TER119)−/EPCAM+/YO-PRO1−/TIGIT− cells. CD109+ cells were sorted from tumors generated in KPT mice and gated as (CD45/CD31/CD11b/CD11c/(F4/80)/TER119)−/tdTomato+/YO-PRO-1−/CD109+ live cells. We confirmed that the isolated TIGIT+ cells belonged to cluster 5/HPCS by qPCR (described below) for cluster 5 markers (Tigit, Epcam, and Slc4a11). Gusb was used as a housekeeping control. qPCR primer sequences are available (Table S6 and Key Resources Table).
Integrin α2Hi and integrin α2Lo cells were isolated from patient-derived xenografts grown in NSG mice by flow cytometry. Tumors were dissociated as above and sorted as live (anti-human CD45, CD31, CD11b, CD11c) −/(anti-mouse CD45/TER119/H-2Kd/CD31)−/(anti-human EPCAM)+/DAPI−/Integrin α2Hi) cells. Integrin α2Hi cells were defined as the top 15% of the integrin α2-expressing cells; integrin α2Lo cells represented the rest of the tumor.
Plate-based scRNA-Seq
Cells were dissociated as above, stained with DAPI and live cells were sorted as described above into 96 well plates containing 5 μl of TCL Buffer (Qiagen, catalog #1031576) with 1% beta-mercaptoethanol. Plates were processed by a modified SMART-Seq2 protocol (Picelli et al., 2013), with the following modifications: RNA from single cells was first purified with Agencourt RNAClean XP beads (Beckman Coulter, catalog #A63881) using Bravo Automated Liquid Handling Platform prior to oligo-dT primed reverse transcription with Maxima reverse transcriptase (Thermo Fischer, catalog #EP0752) and locked TSO oligonucleotide (Exiqon, custom made), which was followed by a 21 cycle PCR amplification using KAPA HiFi HotStart ReadyMix (KAPA Biosystems, catalog #KK2601). The WTA product was purified using Agencourt AMPure XP beads (Beckman-Coulter, catalog #A63881) and a Bravo Automated Liquid Handling Platform. Libraries were tagmented using the Nextera XT Library Prep kit (Illumina, catalog #FC-131-1096) with custom barcode adapters (Table S6). Libraries from 384 cells with unique barcodes were combined and sequenced using a NextSeq 500 sequencer (Illumina, catalog #FC-404-2005) at the Broad Genomics Platform.
Droplet-based scRNA-Seq
Mice with LUAD tumors were prepared and stained as above. Live cells were collected and processed directly by droplet based scRNA-Seq using the 10X genomics Chromium Single Cell 3’ Library & Gel bead Kit V2 according to manufacturer’s protocol. An input of 6,000 cells was added to each 10x channel with a median recovery of 3,266 cells. Libraries were sequenced on an Illumina Nextseq (Illumina, catalog #FC-20024907) or HiSeqX (132 bp reads) at the Broad Genomics Platform.
Single-cell DNA sequencing
Single tumor cells were isolated by microaspiration after tumor dissociation, and genomic DNA was amplified with the GenomePlex Single Cell Whole Genome Amplification Kit (Sigma, catalog #254–457-8). Amplified DNA was purified, barcoded and pooled, and sequenced on an Illumina HiSeq2000 at the MIT Bio-Micro Center.
Bulk ATAC-Seq
Bulk assay for transposase-accessible chromatin sequencing (ATAC-Seq) via Omni-ATAC was performed as described previously (Corces et al., 2017) with slight modifications: Briefly, ~10,000 cells were resuspended in 1 ml of cold ATAC resuspension buffer (RSB; 10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2 in water). Cells were centrifuged at 500 g for 5 min in a pre-chilled (4 °C) fixed-angle centrifuge. After centrifugation, the supernatant was carefully aspirated not to perturb the cell pellet. Cell pellets were then resuspended in 35 μl of ATAC-lysis buffer (ATAC-RSB containing 0.1% NP40, 0.1% Tween-20, and 0.01% digitonin (Promega, catalog #G9441)) by pipetting up and down. This cell lysis reaction was incubated on ice for 3 min. After lysis, 1 ml of ATAC-wash buffer (ATAC-RSB containing 0.1% Tween-20 (without NP40 or digitonin)) was added, and the tubes were inverted to mix. Nuclei were then centrifuged for 10 min at 500 g in a pre-chilled (4 °C) fixed-angle centrifuge. Supernatant was removed and nuclei were resuspended in 10 μl of transposition mix (25 μl 2× TD buffer, 2.5 μl transposase (Illumina, catalog #15027865), 16.5 μl PBS, 0.5 μl 1% digitonin, 0.5 μl 10% Tween-20, and 5 μl water) by pipetting up and down six times. Transposition reactions were incubated at 37 °C for 30 min in a thermomixer with shaking at 1,000 rpm. Reactions were cleaned up with Qiagen MinElute PCR Purification Kit (Qiagen, catalog #28004). ATAC-Seq libraries were amplified with 10 PCR cycles and sequenced on NextSeq 550 (paired-end 35 bp).
Single-cell ATAC-Seq
Samples for single-cell ATAC-sequencing were isolated from primary tumors by flow cytometry as above and then frozen in Bambanker Cell Freezing Medium (Lymphotec, catalog #302-14681) for at least 24 h. Cells were then thawed and processed as per manufacturer’s guidelines (Chromium Single Cell ATAC Reagent Kit v1 chemistry, catalog #1000083).
Quantitative PCR (qPCR)
RNA was isolated from whole tumors or sorted cell populations using either the Qiagen RNeasy Plus Mini kit (catalog #74136) or Micro kit (catalog #74034) as appropriate per manufacturer’s instructions. cDNA was synthesized using either the SuperScript VILO cDNA synthesis kit (Invitrogen, catalog #11754050) or the PrimeScript RT Reagent kit (Takara, catalog #RR037B). qPCR was performed in quadruplicate with 1–2 μl of cDNA (diluted 1:10 if necessary) using the Powerup SYBR mix (Applied Biosystems, catalog #A25778) and run on the QuantStudio 7 Flex Real-Time PCR System. The ΔΔCT method was used to compare markers of interest and expression was normalized to Gusb. All oligonucleotides used in this study are listed in Table S6.
Isolation of mouse LUAD tumor spheres
TIGIT+, CD109+, and TIGIT−/CD109− cells were isolated from 17–19 week LUAD tumors using FACS as above and plated at a density of <1000 cells per well on an 8-chamber coverglass (Thermofisher, catalog #155379 with Matrigel as above. Tumor spheres were grown for 11 days before counting and dissociation for scRNA-Seq.
Isolation of LUAD PDX tumor spheres
Integrin α2Hi and Integrin α2Lo cells were isolated from three PDXs (MSK-LX984, MSK-LX1012, MSK-LX1182) using FACS as above and plated on tissue culture-treated plates with inserts (Plates: Falcon, catalog #353504; Inserts: Falcon, catalog #353095) at a density of up to 10,000 cells per well. Tumor spheres were grown for 22 days before quantification.
Dissociation of tumor spheres
For dissociation of the organoids for single cell sequencing in a 24 well plate, media was replaced with 200 μl dissociation mix (50 μl Corning Dispase, catalog #354235; 150 μl Advanced DMEM/F12 supplemented media as above) per well and the plate incubated at 37°C for 30 min. 1 ml of cold PBS was added to each well and the media transferred to a 15 ml tube PBS was added to the tube to increase the volume to 10 ml, followed by a 300 g 5min spin at 4°C. The supernatant was gently aspirated, with the goal of leaving about 300–500 μl of supernatant. 500 μl of TrypLE (Gibco, catalog #12604013) was added and the tube incubated at 37°C for 5 min. Serum containing Advanced DMEM/F12 was then added and the contents transferred to a sterile filter top tube. The cells were pelleted by a 300 g 5min spin at 4°C and the supernatant carefully removed.
Generation of orthotopic mouse LUAD allotransplants
TIGIT+ and TIGIT− cells containing an active Rosa26LSL-Luciferase allele were sorted from 18–21-week old LUAD tumors using FACS as above in yield mode and orthotopically allotransplanted into three (receiving TIGIT+ cells) and five (receiving TIGIT− cells) 35-week old immunodeficient NSG mice at 28,000 transplanted cells per mouse. After 8 weeks, tumors were harvested. scRNA-Seq was performed both pre-transplantation (using ‘purity’ mode for enrichment) with the remaining cells harvested (using ‘yield’ mode) for transplantation. Tumor cells were harvested 8 weeks post-transplantation (using ‘purity’ mode).
Chemotherapy
The response of the KP model to cisplatin chemotherapy has been carefully characterized in a previous study (Oliver et al., 2010): the tumors undergo a nadir in proliferation and the peak of a second wave of apoptosis at 72 h following a single dose of cisplatin. Mice with 20 week old LUAD tumors were treated with freshly prepared cisplatin (EMD-Millipore, catalog #232120) in PBS at 7 mg/kg body weight intraperitoneally as previously described (Oliver et al., 2010). Tumors were extracted at 72 h following cisplatin or vehicle administration and isolated for scRNA-Seq.
In vivo bioluminescence
TIGIT+ and TIGIT− cells containing an active Rosa26LSL-Luciferase allele were sorted from 18–21-week old LUAD tumors and orthotopically allotransplanted into fourteen 8-week old immunodeficient NSG mice at 50,000 transplanted cells per mouse. After 39 days, mice were administered 100 mg kg D-Luciferin (Perkin Elmer, catalog #122799) via intraperitoneal (IP) injection. Ten min after injection, mice were imaged on an IVIS Spectrum imaging system (Perkin Elmer, catalog #124262) with 1-min exposure and medium binning. Average radiance was recorded in ROIs surrounding the chest cavity of each mouse.
Immunohistochemistry
Tissues were fixed in either Shandon Zinc Formal-Fixx (Thermo Scientific, catalog #6764255), 10% neutral buffered formalin, or 4% PFA for 24–48 h at 4° C and embedded in paraffin. Manual immunohistochemistry was performed using Vector Labs reagents (ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit, catalog #MP-7401–50; Mouse-on-Mouse ImmPRESS HRP (Peroxidase) Polymer Kit, catalog #MP-2400; ImmPACT DAB Peroxidase (HRP) Substrate, catalog # SK-4105) as per manufacturer protocols. Antibodies and dilutions used are available in Table S6.
Multiplexed IF
Automated immunofluorescence (IF) staining was performed at the Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using a Discovery XT processor (Ventana Medical Systems). The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 min with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 min (Ventana Medical Systems). Multiplexed immunofluorescence stainings were performed as previously described (Yarilin et al., 2015). Staining was performed in the following order: Anti-Claudin-4 (Invitrogen, catalog #36–4800, 5 μg/ml), anti-Claudin-2 (Invitrogen, catalog #32–5600, 5 μg/ml), anti-Lysozyme (DAKO, catalog #A0099, 2 μg/ml). After staining slides were counterstained with DAPI (Sigma Aldrich, catalog #D9542, 5 μg/ml) for 10 min and coverslipped with Mowiol mounting reagent. Secondary antibodies used for visualization were AF488 (Claudin-4), AF594 (Claudin-2), and AF546 (Lysozyme). Slides were scanned to acquire fluorescence signal.
Single-molecule mRNA in situ hybridization
Single-molecule mRNA in situ hybridization was performed on formalin-fixed paraffin-embedded tissues using the manual Advanced Cell Diagnostics RNAscope 2.5 HD Detection Kit (catalog # 322360) per the manufacturer’s instructions. Antigen retrieval times were 20 min for mouse and human tissues. Protease digestion times were 15 min for mouse LUAD tumor tissues and 30 min for human LUAD tumor tissues. Probes are listed in Table S6.
COMPUTATIONAL ANALYSIS
scRNA-Seq processing and quality filtering
For plate-based scRNA-Seq by SMART-Seq2, reads were aligned against Gencode GRCm38.p5 (M15) mouse reference using STAR (v2.5.4b), and transcript abundance was quantified using RSEM (v.1.3.0). For each cell bam, Picard-Tools CollectRnaSeqMetrics was run on each genome aligned bam and summary statistics were collected (Table S7). Cell were excluded from further analysis based on the following criteria: (1) Fewer than 1000 genes; (2) Fewer than 500,000 reads aligned. Additionally, for each plate we exclude cells deviating by >2 times the interquartile range (IQR) above/bellow the upper/lower quartile for: (1) number of genes-expressed; (2) total read counts (3) or mean expression of housekeeping panel (Tirosh et al., 2016a). Similarly, we exclude cells per plate deviating by >2×IQR above the top quartile for proportion of mitochondrial reads, proportion of intergenic reads, or total count of ribosomal RNA reads, and by >2×IQR below the bottom quartile for proportion aligned reads and expression of tdTomato marker transcript. Next, gene level read count summaries were sampled (with replacement) to a uniform depth of 500k reads per cell. In order to further account for differences in amplification efficiency and sequencing depth, read counts were transformed to log2(100k+1) normalized abundance, which was used for all downstream analysis unless indicated otherwise.
For droplet-based scRNA-Seq, Cellranger v3.1 was used to align reads to the mm10 mouse reference sequence, and its output processed using the dropletUtils R package for excluding chimeric reads, and identification and exclusion of empty cell droplets (Griffiths et al., 2018; Lun et al., 2019). We excluded any chimeric read that had less than 80% assignment to cell barcode. Cell barcodes were tested for emptiness against a background generated based on barcodes with 1000 to 10 UMIs, with cutoffs determined dynamically based on channel specific characteristics. We further estimate the saturation of UMIs and genes in individual cells by subsampling reads without replacement in each cell barcode, in incremental fractions of 2%, with 20 repeats. A saturation function of the form was fit based on the number of UMIs observed (Table S7). We excluded cell barcodes based on any one of following criteria: (1) Fewer than 500 genes; (2) Fewer than 5,000 reads; (3) Fewer than 1,000 transcript UMIs, (4) Less than 30% reads mapping; (5) Per cell estimated sequencing saturation less than 0.3; (6) Non-empty droplet FDR > 0.1; (7) Expression of tdTomato > 8 TP10k. In addition, a subset of 10x channels reaching high UMI sequencing saturation (Table S7), were filtered to retain only UMIs captured by 2 or more reads.
Dimensionality reduction and clustering
We clustered the plate-based scRNA-seq profiles across all time points using a non-negative matrix factorization (NMF) and a graph clustering-based approach, as follows. First, we identified transcriptionally over-dispersed genes within each experimental batch by examining the difference of the coefficient of variation (CV) with the median CV for other genes with a similar mean expression (Satija et al., 2015). A robust set of ~2,000 genes is retained based on an elbow-based criterion, applied to the median of over-dispersed difference statistic based on 200 samples of 75% of cells. Next, subsampling 80% of genes and samples, we used NMF to reduce the dimension of the full dataset to between 20 and 40 dimensions (Lee and Seung, 1999). The loading matrices (i.e., activations) of these NMF components were used to calculate a cosine similarity k-nearest-neighbors (k-NN) graph (k=21). This graph was clustered using stability optimizing graph clustering (Delvenne et al., 2010; Shekhar et al., 2016). A final clustering of 14 subsets was determined based on an elbow-criteria of mean cluster silhouette. Two clusters of 44 and 35 cells were excluded from further analysis as either suspected doublets and or recombination in off-target cells (club cells).
Visualization of single cell profiles
We generated tSNE plots from NMF loading matrices, with a perplexity value of 30 and the Barnes-Hut approximation method (Van Der Maaten, 2014).
We generated PHATE maps (Moon et al., 2019) using normalized single cell expression profiles of the same top over-dispersed genes as used for clustering (above), and the following input parameters: k=21 nearest neighbors, square root potential heat diffusion kernel (pot_method=‘sqrt’), 4,000 feature landmarks for metric multi-dimensional scaling (n_landmarks=4000, 30 input principle components (npca=30), distance=‘cov’).
Differentially expressed genes
Differentially expressed genes (DEG) were identified using a Poisson-Tweedie model on unscaled transcript counts normalized to uniform counts by sampling reads. Genes were identified as differentially expressed in a particular set of cells if they met all of the following criteria: (1) Benjamini-Hochberg FDR < 0.1; (2) Minimum expression in at least 10% of cells; and (3) Area under a receiver operating curve (AUROC) > 0.60, (4) log fold change vs. cells in all other subsets > 1.5, and (5) log-fold change vs. cells in any other subset is highest within the set.
Pearson residuals of contingency tables
The Pearson residual is a measure of relative enrichment for cells in a contingency table. It is calculated here as: , where the expected value is calculated as the product of row and column marginal probabilities by the total count.
Estimating heterogeneity within a timepoint
Heterogeneity of single cell profiles within a timepoint was quantified by examining the average pairwise Normalized Mutual Information (NMI) between the profiles within each time point. Using 100 differentially expressed genes per each of 12 subtype clusters and top 100 NMF genes per each of 11 NMF programs (above; Differentially expressed genes, and below; Novel expression programs by NMF) (total of 2,374 genes), we discretized expression per gene into 10 bins. In order to account for differences in the number of cells across samples, we subsampled 100 cells from each time-point (or mouse) 100 times and calculated the median NMI across each within-timepoint sampled pair. NMI was calculated between each pair of cells x and y by first calculating the mutual information and then normalizing it by the entropy of each cell: . To estimate a p-value for the difference in NMI value between two groups we compare the number of sub-samples from in A group vs all those in another B group and report P = (∑i ∑j |Ai < Bj | + 1)/(|A| ∙ |B| + 1).
Single-cell DNA copy number quantification
Sequencing reads were aligned to reference GRCm38.p mouse genome reference using BWA (0.7.17). Duplicate reads were marked with SAMBLASTER (v0.1.24). CNVkit (v0.9.6) was used to quantify read abundance in genomic window of 200 kb, and normalized for GC content and mappability, excluding outlier bins. Segmentation was performed using a three state HMM for amplified and deleted regions (Talevich et al., 2016).
Copy number inference from scRNA-Seq profiles
Single cell copy number was estimated following our previously published method (Tirosh et al., 2016a). Briefly, we square root log transformed TP100k expression values to stabilize variance (Anscombe transform), and capped per-cell, and per-gene expression to the 99th percentile to reduce the effect of outliers (i.e., for each cell, genes expressed above the 99th percentile are set to the 99th percentile, next for each gene, cells expressed above 99th percentile are set to the 99th percentile). Next, genes were assigned to each of 20 expression bins by mean expression in a reference normal, here assigned to be all cells from the “T” only timepoint. For each chromosome, all genes were ordered by the location of the Transcription Start Site (TSS), and the mean expression value in a sliding window of 25 million bases was calculated (with a step size of one million bases), corresponding to ~100 genes windows. For each cell and window, we compared the mean expression to a null distribution of gene samples drawn to match the normal mean expression, i.e., for a window of k genes, we drew k genes from matching expression bins in the normal reference sets (as in single-cell gene set enrichment below). The raw mean expression per cell and window was normalized by subtracting the mean of the resampling-based null distribution. Additionally, an empirical p value was calculated by comparing to the null distribution and used to filter for likely spurious CNV events.
Matching RNA inferred CNV to DNA CNVs
For three 20-week KP tumors for which we had both single cell DNA-Seq and single cell RNA-Seq, we matched between DNA-based single cell CNVs and RNA-inferred single cell CNVs, by relating each single cell RNA-based inferred CNV profile to the most similar DNA-based single cell CNV profile by the L1-norm distance.
Single cell gene set enrichment
We performed single cell gene-set enrichment as previously described (Chihara et al., 2018; Tirosh et al., 2016a). Briefly, genes were split into 20 bins by mean expression across all cells, where the 20 bins were defined based on the distribution of all genes’ expression. Gene expression was centered, scaled, and transformed using the logistic function to the [0,1] range. Given a gene set signature of k genes, the mean of the normalized expression for the set was calculated in each cell as a raw signature score. This score was then compared against a null distribution of 1,000 randomly selected signatures, each consisting of k genes, drawn such that the mean expression of each of the k genes matches the same global mean expression bin of a gene in the original signature. The final per cell activity score is calculated as the per cell raw score centered by the mean score of the signatures from the null distribution. This final score is subsequently normalized to have mean of zero and standard deviation of one (z-score). We calculate an empirical p value of association with the clustering to 12 subtypes (of Figure 1D) by comparing an ANOVA F-statistic for the true raw score, with the distribution of the F-statistic of the randomly selected signatures. The tested gene sets, and their sources are listed in Table S8.
Novel expression programs by NMF
To identify robust transcriptional programs, we adapted a consensus NMF procedure (Kotliar et al., 2019). We used as input 1,346 NMF expression weight components identified across 50 subsampled repeats used for clustering, as described above (see section on Dimensionality reduction and clustering). We excluded outlier components by sorting components by their cosine distance to the 20th nearest neighbor and excluding components with unusually high distance by an elbow-based criterion. Next, we constructed a symmetric k-nearest neighbors (k-NN) graph (k=30), and identified clusters of highly similar components in this graph, using stability optimizing graph clustering, with an exponentially varied scale parameter (0.1 to 10, resulting in 42 to 3 clusters). The components in each cluster were median-averaged into a single component, resulting in a short list of “consensus NMF” components. These were used as the initialization component matrix for a second round of NMF of all cells and highly variable genes (as described in Dimensionality reduction and clustering). We selected a solution with 11 NMF components based on an elbow criterion of reconstruction error of the input data matrix.
To characterize the novel transcriptional programs identified with this procedure, we used the top 100 genes in each of the 11 components, ranked by the following weighting scheme: For the ith gene and jth component we define the scaled weight as follows: . This weighting scheme prioritizes for high weight (highly expressed) and unique genes in each component. We tested for enrichment of the top 100 genes in each program in a compendium of gene sets listed in Table S8, with the hypergeometric test. P values were adjusted by a Benjamini-Hochberg false discovery rate procedure.
Optimal transport
To estimate robust transport maps of single cell profiles we adapted the Waddington-Optimal Transport (Waddington-OT) approach that we previously reported (Schiebinger et al., 2019). Briefly, Waddington-OT estimates, for a set of cells C at a given time point, its “descendant distribution” at a later time point as the mass distribution over all cells at that later time point. This is estimated by transporting C according to a temporal coupling between cells learned by the model. Similarly, the cell set C’s “ancestor distribution” at an earlier time is the mass distribution over all cells at that earlier time point, obtained by “rewinding” time according to the temporal coupling. In our case, after learning the model over the cells in our data, we used it to examine the connection between cell clusters across consecutive time points, by defining the sets C by membership in the 12 clusters in the respective time point (Figure 3A; Figure S3A).
We calculated transport maps between cells in each pair of consecutive time points, except that we merged KT and KPT 2-week samples due to low numbers of healthy cells in the KPT sample (Figure 1G), such that we had the following transitions: T→{KT2,KPT2}, {KT2,KPT2}→KT12, KT12→KT30, {KT2,KPT2}→KPT12, KPT12→KPT20, and KPT20→KPT30. For each pair of time points we use the cosine similarity of NMF loading matrices for each cell (as described in section Novel expression programs by NMF), as the input distance measure for inferring a transport map from each cell in the starting time point to a distribution of cells in the subsequent time point, with parameters lambda1 = 1, lambda2 = 25, and a uniform growth rate. We performed the OT inference procedure 20 times using random seeds and the mean across runs was used as the OT map estimate for each pair of time points.
Bulk ATAC-Seq
Analysis was performed using the ENCODE ATAC-Seq pipeline (v1.5.4, https://github.com/ENCODE-DCC/atac-seq-pipeline), with default parameters, for initial quality control analysis. The pipeline was run once for each condition, inputting FASTQ files from the mouse replicates (n = 4). A final peak list was generated by processing the resulting BAM files generated by the ENCODE ATAC-Seq pipeline with Samtools (v1.8; http://www.htslib.org/) to: (1) filter the BAM files to contain only the main chromosomes, (2) subsample each BAM file to the minimum number of reads observed across all replicates and conditions, and (3) merge BAM files from each replicate for each condition. MACS2 (v2.2.6) (Zhang et al., 2008) was used to call peaks, bedtools (v2.26.0; https://bedtools.readthedocs.io/en/latest/) to filter blacklisted regions (as defined by the ENCODE project) and merge the peak files from the experimental conditions, and the featureCounts function from the Subread package (v2.0.0; http://subread.sourceforge.net/) to generate a matrix of peak counts from the merged peak list and filtered BAM files. DESeq2 (v1.26.0) (Love et al., 2014)was used to call differentially accessible peaks in R (v3.6), with ~Mouse + Tigit_status as the design variable. Peaks were considered differentially accessible if they had an FDR adjusted p value less than 0.1. HOMER (v4.11) (Heinz et al., 2010) was used to annotate peaks. The UCSC Genome Browser (Kent et al., 2002) was used to visualize peaks.
scATAC-Seq data processing
We used the Cell Ranger ATAC (v1.2) pipeline (10x Genomics) to generate single-cell accessibility counts. First, we used cellranger-atac mkfastq to generate demultiplexed FASTQ files from the raw sequencing reads. We then aligned these reads to the mouse mm10 genome and quantified chromatin accessibility counts using cellranger-atac count. This pipeline performs barcode error correction, PCR duplicate marking, peak calling and cell calling, and produces both a filtered peak cell barcode matrix, and a fragment file containing all fragments assigned to single cells.
scATAC-Seq quality control
Starting with the filtered peak cell barcode matrix, we further filtered out low quality cells using five per-cell quality control metrics: the total number of fragments overlapping peaks, the percent of fragments mapping to peaks, the percent of fragments overlapping blacklisted regions as defined by the ENCODE project, the ratio of mononucleosomal to nucleosome-free fragments, and the transcriptional start site (TSS) enrichment score as defined by the ENCODE project (https://www.encodeproject.org/data-standards/terms/). We retained cells with between 1000 and 50000 fragments overlapping peaks, with at least 20% of the fragments mapping to peaks, with fewer than 5% of fragments mapping to blacklisted regions, with the ratio of mononucleosomal to nucleosome-free fragments less than five, and with TSS enrichment score greater than two.
scATAC-Seq dimensionality reduction
We analyzed the cells passing quality control using the R packages Signac (v0.2.1) (https://github.com/timoast/signac) and Seurat (v3.1.2) (https://github.com/satijalab/seurat) (Butler et al., 2019). We performed term frequency inverse document frequency (TF-IDF) normalization on the peak cell barcode matrix using RunTFIDF, which normalizes across both cells and peaks, to control for differences in cell sequencing depth and to increase values for peaks that occur more rarely across cells. We chose features (peaks) for dimensionality reduction and clustering using FindTopFeatures, which ranks peaks based on the total number of fragments in a peak across all cells. We retained the top 90% of peaks. We next performed a singular value decomposition to reduce dimensionality of the data, with the function RunSVD, and retained the left and right singular vectors associated with the 30 largest singular values. We performed graph-based Louvain clustering using FindNeighbors and FindClusters, with k = 20 for the k-nearest neighbor algorithm and the resolution parameter set to 0.8. We visualized gene activity and clustering results on Uniform manifold Approximation and Projection (UMAP) using RunUMAP. The UMAP was calculated from the first 30 singular vectors of the dimensionally reduced data with the following settings: n.neighbors=30, min.dist=0.3, and spread=1.
Chromatin accessibility data was used to estimate a gene’s activity by assuming that gene expression is correlated with promoter accessibility. For each gene, we extracted its gene coordinates from the mouse genome using EnsDb.Mmusculus.v79, and then extended the resulting coordinates 2 kb upstream so that they covered both the gene body and promoter. The activity of each gene was estimated by counting how many fragments within each cell mapped to this extended region. To examine the activity of entire gene modules or signatures within single cells, we scored signature activity levels using AddModuleScore. This function calculates the average activity levels of the genes in a signature and then subtracts off the average activity levels of control gene sets (Tirosh et al., 2016a). The genes in the control sets are randomly chosen with the constraint that they have similar activity levels to the genes in the signature. This approach controls for technical differences in cell quality and library complexity across single cells that contribute to a signature’s activity level.
scATAC-Seq data integration
To integrate the TIGIT+ and TIGIT− scATAC-Seq datasets, we restricted analysis to peak regions that overlapped across both datasets using MergeWithRegions and performed the same dimensionality reduction and clustering analysis described above. To integrate the data while correcting for technical batch effects, we use Seurat v3 integration, which identifies correspondences between cells in the two datasets and applies a correction matrix to the peak cell barcode matrix (Stuart et al., 2019). We identified the corresponding cell subsets using FindIntegrationAnchors, where the dimensionality of both datasets was first reduced using canonical correlation analysis and the first 30 canonical correlation vectors were retained. We then calculated and applied a correction to the peak barcode matrix using IntegrateData, with the weight.reduction parameter set to use the dimensional reduction space calculated above. Finally, we took this corrected peak cell barcode matrix and applied the same dimensional reduction, clustering, and UMAP visualization described above.
Comparison to human scRNA-Seq data
Processed scRNA-Seq profiles from human LUAD tumors were downloaded from GSE127465, E-MTAB-6149, and E-MTAB-6653 (Lambrechts et al., 2018; Laughney et al., 2020; Zilionis et al., 2019). Analysis was limited to lung adenocarcinoma samples and we examined only cells annotated by the authors as cancer cells.
Cross-cohort activity of NMF gene programs
Activity of NMF programs defined in the mouse time course study (“source dataset”) was estimated in additional secondary datasets from mouse or human (“target dataset”). For human, 1-to-1 gene orthologs were mapped between mouse and human using an ortholog table downloaded from Ensemble BioMart (v.96, downloaded June 11, 2019), retaining only 1:1 orthologs. For both human and mouse, the analysis was limited to 100 differentially expressed genes per each of 12 subtype clusters (Figure 1D) and top 200 NMF genes per each of 11 NMF programs (total of 2,374 genes). The distribution of each gene was matched between the source and target cohort based on a matching of the empirical cumulative distribution functions (eCDF) of the gene in the target dataset to the eCDF of the gene in the source dataset, while ignoring zero values – that is, for a given gene the cell expressed at the nth percentile in the target cohort is assigned the expression of the nth percentile cell in the source. We excluded from analysis genes expressed in less than 1% of the cells in the target dataset, as well as genes showing a large deviation in mean expression between the two cohorts after normalization (defined as genes deviating from the predicted expression at an alpha < 0.0005, using a Gaussian process regression of the source mean expression to the target mean expression). The remaining genes were used to estimate the activity matrix (H) in the target cohort, using a nonnegative least-squares (NNLS) fit of the source NMF gene program (W) matrix on the transformed and normalized expression values of the target dataset. NNLS fit was performed using the Block Principal Pivoting method for solving the equation: minH>0‖X − WH‖F, where X is the input matrix for the target dataset, and W was a matrix of NMF gene programs (gene by k) learned from the source dataset (Kim and Park, 2008).
Cross-cohort cluster assignments
To transfer cluster assignments, we use a similar procedure to that for estimating NMF activities (section Cross-cohort activity of NMF gene programs). The procedure above was applied to each of 50 NMF activity matrices (H) for the target dataset generated by subsampling the source dataset, resulting in a matrix of 1,346 activity features in the target dataset. Next, a multiclass gradient boosting tree classifier was trained to on the activity feature matrices to predict cluster type (using the XGBoost package v. 0.82.0.1 in R v3.5.3). This classifier was used to predict cluster assignments in the target dataset on the set of NMF activity features.
Comparison of HPCS to stem cell signatures
We quantified enrichment between our HPCS cluster or the highly mixed state/HPCS and known signatures for normal and cancer stem cells using the GeneOverlap R package (v1.22.0) (Shen and Sinai, 2019), which is based on the hypergeometric distribution. To build a set of stemness signatures, we collected 1197 gene sets from the Molecular Signatures Database (MSigDB, v6.2) (Liberzon et al., 2015; Liberzon et al., 2011; Subramanian et al., 2005) and CellMarker (downloaded on 2019/10/22) (Zhang et al., 2019), mapped them to mouse genes using the orthology mapping from Mouse Genome Informatics (http://www.informatics.jax.org/), and filtered the signatures to retain only those with “_stem_” in their name and at least four genes in the gene set; our final set of stemness signatures contained 1,197 gene sets. We defined our HPCS gene set by the set of 406 differentially expressed genes marking cluster 5, and our highly mixed state/ HPCS gene set as the 103 genes defining this NMF program. We calculated enrichment using newGeneOverlap and testGeneOverlap, with a genomic background of 25,656 – the number of genes in our RNA expression data expressed in 10 or more cells. P values were adjusted for multiple comparisons using p.adjust in R, with the ‘fdr’ correction method. All analyses were carried out in R (v3.6). Gene sets that showed significate enrichment (Padj ≤ 0.01), were manually curated to validate that they are truly enriched in normal or cancer stem cells and that the signature did not represent an experimental perturbation that may have confounded the conclusion. Gene sets from the following studies were identified: (Bystrykh et al., 2005; Gal et al., 2006; Gattinoni et al., 2011; Ramirez et al., 2012; Villanueva et al., 2011); in addition a gene ontology set “GO_POSITIVE_REGULATION_OF_STEM_CELL_PROLIFERATION” was identified. Curated gene sets are plotted based on Padj and Jaccard Index in Figure S4D. The Jaccard index was calculated by the number of intersecting genes between the two gene sets divided by the union of the two gene sets. We only found significant correlations between the HPCS and eight of these signatures, including several hematopoietic stem cell signatures, an adult stem cell signature, as well as an embryonic stem cell signature (Bystrykh et al., 2005; Gal et al., 2006; Gattinoni et al., 2011; Ramirez et al., 2012; Villanueva et al., 2011), with the largest overlap including only 14 (8.24%) of the 170 genes in the signature (Figure S4D; Table S4).
Human clinical data analyses
Processed RNA-seq expression data was downloaded from https://gdc.cancer.gov/about-data/publications/pancanatlas. Clinical annotations were downloaded from http://www.linkedomics.org/data_download/. All survival outcomes data was transformed to months. We excluded patients older than 85 at time of diagnosis, or having reported post-surgery residual disease (LUAD analysis only), the latter because this appeared to be a strong confounder of outcome with few observations. When calculating 5-year survival we capped the survival period at 60 months and right censored patients with longer survival. Survival analysis was performed using a Cox proportional hazards model including terms for age, tumor purity, and stratified for stage (early – stage I or stage II, vs. advanced – stage III or stage IV) for LUAD and stratified by cancer type for PANCAN. Kaplan-Meier plots were drawn by dividing the NMF activities or cluster gene signatures into 3 equal sized bins. NMF activities or cluster signature activities (calculated as described above in Cross-cohort activity of NMF gene programs and Single cell gene set enrichment), are used as continuous predictors in a cox proportional-hazards model. Reported p values are for a likelihood-ratio test comparing the full model to one including only the baseline parameters (age, tumors purity and stage or cancer type). Genetic mutation information was downloaded from cBioportal on Feb 24th 2020. When testing for association with outcome in the context of genetic state, samples were considered mutated if these were annotated for any non-silent mutation or copy number amplification/deletion.
Computational tools
Software used for analysis of data during this project included, GraphPad Prism (version >= 8) MATLAB (version >= 9.2.0.556344-R2017a), R (version >= 3.4), and Python (versions >= 2.7 and >= 3.6).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in the figure legends and main text above. Analysis was performed using Matlab, R and Python. For most small-scale experiments, significance was determined to control the family-wise type I error rate with α < 0.05 (Bonferroni procedure). When appropriate, multiple hypothesis testing correction was employed using the Benjamini-Hochberg procedure at a false discovery rate (FDR) < 0.1.
Supplementary Material
Supplementary Table S1. [Cluster gene signatures, and their enrichment in publicly available gene sets], Related to Figure 2
Supplementary Table S2. [NMF gene signatures and their enrichment in publicly available gene sets], Related to Figure 2
Supplementary Table S3. [Differentially accessible chromatin peaks in TIGIT− and TIGIT+ populations], Related to Figure 3 and Figure S3
Supplementary Table S4. [Enrichment of the stem signatures in HPCS], Related to Figure S4 and STAR Methods: Comparison of HPCS to stem cell signatures
Supplementary Table S5. [Enrichment of the HPCS gene signature in human data], Related to Figure 6
Supplementary Table S6. [Reagents and Samples used in experiments], Related to the experimental STAR Methods
Supplementary Table S7. [Summary quality statistics], Related to STAR Methods: scRNA-Seq processing and quality filtering
Supplementary Table S8. [Gene sets tested for enrichment], Related to STAR Methods: Single cell gene set enrichment
SIGNIFICANCE.
Cellular states capable of promoting tumor progression and resisting therapies invariably exist in heterogeneous tumors. Thus, understanding how intra-tumoral heterogeneity is generated and maintained during tumor evolution is of tremendous importance for the development of effective cancer therapies. We discovered that the emergence and maintenance of cellular heterogeneity is driven by a high-plasticity cell state (HPCS) that is common to mouse and human lung tumors. Furthermore, we find that the HPCS harbors high tumorigenic capacity, is drug resistant and associates with poor patient prognosis. We expect the HPCS program to prove useful in identifying highly plastic cell states in other cancers and biological contexts. Targeting the HPCS may enable therapeutic strategies to suppress tumor heterogeneity and treatment resistance.
Lung cancer progression is accompanied by a stereotypic expansion of heterogeneity
Cell state heterogeneity arises largely independently of genetic variation
State transitions occur via a HPCS harboring high differentiation & growth capacity
The HPCS is drug resistant and portends poor patient survival across all cancers
Cellular states capable of promoting tumor progression and resisting therapies exist in heterogeneous tumors. Marjanovic et al. discover that a high-plasticity cell state common to mouse and human lung tumors drives cellular heterogeneity, and is highly tumorigenic and drug resistant, as well as associates with poor patient prognosis.
ACKNOWLEDGEMENTS
We thank K. Daniels, R. Gardner, M. Griffin, M. Jennings and G. Paradis for FACS support; K. Manova, M. Turkekul, and D. Yarilin for histology and microscopy support; S. Fujisawa and A. Santella for help with quantitative analysis of immunostained tissue sections; H. Mummey and X. Zhuang for help with experiments; A. Berns for development of the Ad5mSPC-Cre virus; and L. Gaffney and A. Hupalowska for help in preparing the figures. This work was supported by the Transcend Program and Janssen Pharmaceuticals, the Howard Hughes Medical Institute, and, in part, by the NIH/NCI Cancer Center Support Grants P30-CA08748 (MSKCC) and P30-CA14051 (Koch Institute). T.T. is supported by American Cancer Society, Rita Allen, Josie Robertson Scholar, and V Foundation Scholarships and the American Association for Cancer Research Next Generation Transformative Research Award; the American Lung Association; the Stanley and Fiona Druckenmiller Center for Lung Cancer Research; and NCI-CA187317. T.J. is supported by NCI-PO1CA42063. A.R. is supported by the Klarman Cell Observatory. J.E.C is supported by the MSK T32 Investigational Cancer Therapeutics Training Program Grant (NIH MSK ICTTP T32-CA009207). P.P.M. is supported by NCI-CA196405. L.M. is supported by The Alan and Sandra Gerry Foundation. We acknowledge the use of the Integrated Genomics Operation Core, funded by CCSG P30-CA08748, Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology at MSKCC; the Flow Cytometry and Histology Core Facilities at the Swanson Biotechnology Center at the Koch Institute; and the MIT Bio-Micro Center. A.R., T.J. and A.A. are Howard Hughes Medical Institute Investigators; T.J. is a David H. Koch Professor of Biology, and a Daniel K. Ludwig Scholar.
DECLARATION OF INTERESTS
T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, and a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. Dr. Jacks’s laboratory currently also receives funding from Calico, but this funding did not support the research described in this manuscript. A.R. is a co-founder and equity holder in Celsius Therapeutics and a SAB member for Thermo Fisher, Asimov, Neogene Therapeutics, and Syros Pharmaceuticals, and an equity holder of Immunitas Therapeutics. C.R. serves on the SAB of Bridge Medicines and Harpoon Therapeutics, and has consulted regarding oncology drug development with AbbVie, Amgen, Ascentage, Bicycle, Celgene, Daiichi Sankyo, Genentech, Ipsen, Loxo, Pharmamar, and Vavotek. None of the affiliations listed above represent a conflict of interest with the design or execution of this study or interpretation of data presented in this manuscript. Other authors have nothing to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ambrogio C, Gomez-Lopez G, Falcone M, Vidal A, Nadal E, Crosetto N, Blasco RB, Fernandez-Marcos PJ, Sanchez-Cespedes M, Ren X, et al. (2016). Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med 22, 270–277. [DOI] [PubMed] [Google Scholar]
- Arozarena I, and Wellbrock C. (2019). Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer 19, 377–391. [DOI] [PubMed] [Google Scholar]
- Batlle E, and Clevers H. (2017). Cancer stem cells revisited. Nat Med 23, 1124–1134. [DOI] [PubMed] [Google Scholar]
- Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, Kelly K, Lin D, Dicker A, Arnold J, et al. (2019). The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res 25, 6916–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrykh L, Weersing E, Dontje B, Sutton S, Pletcher MT, Wiltshire T, Su AI, Vellenga E, Wang J, Manly KF, et al. (2005). Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat Genet 37, 225–232. [DOI] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. (2016). Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, et al. (2018). Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CH, Greenside PG, Rogers ZN, Brady JJ, Yang D, Ma RK, Caswell DR, Chiou SH, Winters AF, Gruner BM, et al. (2017). Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat Med 23, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Daemen A, Cheng JH, Long JE, Cooper JE, Wang BE, Tran C, Singh M, Gnad F, Modrusan Z, et al. (2017). Kras mutant genetically engineered mouse models of human cancers are genomically heterogeneous. Proc Natl Acad Sci U S A 114, E10947-E10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD, et al. (2009). A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res 69, 3364–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvenne JC, Yaliraki SN, and Barahona M. (2010). Stability of graph communities across time scales. Proc Natl Acad Sci U S A 107, 12755–12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai TJ, Brownfield DG, and Krasnow MA (2014). Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, and Weinberg RA (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20, 69–84. [DOI] [PubMed] [Google Scholar]
- Fearon ER, and Vogelstein B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, Sanchez-Rivera FJ, Resnick R, Bronson R, Hemann MT, et al. (2010). Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468, 572–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, Nagler A, Tavor S, Ein-Dor L, Lapidot T, et al. (2006). Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia 20, 2147–2154. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. (2018). Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 378, 2078–2092. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. (2011). A human memory T cell subset with stem cell-like properties. Nat Med 17, 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Gomez NC, Adam RC, Nikolova M, Yang H, Verma A, Lu CP, Polak L, Yuan S, Elemento O, et al. (2017). Stem Cell Lineage Infidelity Drives Wound Repair and Cancer. Cell 169, 636–650 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths JA, Richard AC, Bach K, Lun ATL, and Marioni JC (2018). Detection and removal of barcode swapping in single-cell RNA-seq data. Nat Commun 9, 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinot A, Oeztuerk-Winder F, and Ventura JJ (2016). miR-17–92/p38alpha Dysregulation Enhances Wnt Signaling and Selects Lgr6+ Cancer Stem-like Cells during Lung Adenocarcinoma Progression. Cancer Res 76, 4012–4022. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Pastushenko I, Skibinski A, Blanpain C, and Kuperwasser C. (2019). Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 24, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, and Lanczky A. (2013). Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8, e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107 e1017. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn LA, Fousek K, and Palena C. (2020). Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 6, 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter C, and Zenklusen JC (2018). The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 173, 283–285. [DOI] [PubMed] [Google Scholar]
- Hynes RO, and Naba A. (2012). Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol 4, a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, and Harada M. (2005). Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 106, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, and Jacks T. (2005). The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 65, 10280–10288. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development 15, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, and Lowe SW (2017). Putting p53 in Context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D. (2002). The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, and Park H. (2008). Toward Faster Nonnegative Matrix Factorization: A New Algorithm and Comparisons. Proceedings of the 2008 Eighth IEEE International Conference on Data Mining, 353–362. [Google Scholar]
- Kotliar D, Veres A, Nagy MA, Tabrizi S, Hodis E, Melton DA, and Sabeti PC (2019). Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreso A, and Dick JE (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291. [DOI] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwe H, Pircher A, Van den Eynde K, et al. (2018). Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24, 1277–1289. [DOI] [PubMed] [Google Scholar]
- Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallee VP, Xie Y, Masilionis I, Carr AJ, Kottapalli S, et al. (2020). Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 26, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, and Werb Z. (2018). Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol 20, 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DD, and Seung HS (1999). Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, participants in the 1st Human Cell Atlas, J., and Marioni JC (2019). EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol 20, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manieri NA, Chiang EY, and Grogan JL (2017). TIGIT: A Key Inhibitor of the Cancer Immunity Cycle. Trends Immunol 38, 20–28. [DOI] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A. (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, et al. (2016). Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A 113, E6409-E6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, van den Elzen A, Hirn MJ, Coifman RR, et al. (2019). Visualizing Structure and Transitions for Biological Data Exploration. bioRxiv, 120378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, and Desai TJ (2018). Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Setty M, Kuo YY, Liu V, Garg V, Sharma R, Simon CS, Saiz N, Gardner R, Boutet SC, et al. (2019). The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature 569, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Mercer KL, Sayles LC, Burke JR, Mendus D, Lovejoy KS, Cheng MH, Subramanian A, Mu D, Powers S, et al. (2010). Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer. Genes Dev 24, 837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, et al. (2018). Identification of the tumour transition states occurring during EMT. Nature 556, 463–468. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, and Sandberg R. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624 e1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Li T, and Liu Y. (2017). High SLC4A11 expression is an independent predictor for poor overall survival in grade 3/4 serous ovarian cancer. PLoS One 12, e0187385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanal-Villalonga A, Chan JM, Yu HA, Pe’er D, Sawyers CL, Triparna S, and Rudin CM (2020). Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez K, Chandler KJ, Spaulding C, Zandi S, Sigvardsson M, Graves BJ, and Kee BL (2012). Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity 36, 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M, Kim WY, Kung AL, Horner JW, DePinho RA, and Kaelin WG Jr. (2003). Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Mol Imaging 2, 297–302. [DOI] [PubMed] [Google Scholar]
- Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebinger G, Shu J, Tabaka M, Cleary B, Subramanian V, Solomon A, Gould J, Liu S, Lin S, Berube P, et al. (2019). Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 176, 928–943 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH, and Eastern Cooperative Oncology G. (2002). Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346, 92–98. [DOI] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323 e1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, and Sinai M. (2019). Test and visualize gene overlaps. R package version 1.22.0. [Google Scholar]
- Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG, Crowley D, Whittaker CA, Meyerson M, Kimura S, et al. (2013). Nkx2–1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell 50, 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R. (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, and Berns A. (2011). Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764. [DOI] [PubMed] [Google Scholar]
- Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, and Berns A. (2014). Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci U S A 111, 4952–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12, e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammela T, Sanchez-Rivera FJ, Cetinbas NM, Wu K, Joshi NS, Helenius K, Park Y, Azimi R, Kerper NR, Wesselhoeft RA, et al. (2017). A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 545, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research, N. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016a). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016b). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, and Quake SR (2014). Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckwell D, Calderwood DA, Green LJ, and Humphries MJ (1995). Integrin alpha 2 I-domain is a binding site for collagens. J Cell Sci 108 ( Pt 4), 1629–1637. [DOI] [PubMed] [Google Scholar]
- Van Der Maaten L. (2014). Accelerating t-SNE using tree-based algorithms. The Journal of Machine Learning Research 15, 3221–3245. [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A, Hoshida Y, Battiston C, Tovar V, Sia D, Alsinet C, Cornella H, Liberzon A, Kobayashi M, Kumada H, et al. (2011). Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology 140, 1501–1512 e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, et al. (2015). The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 517, 489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, et al. (2011). Suppression of lung adenocarcinoma progression by Nkx2–1. Nature 473, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarilin D, Xu K, Turkekul M, Fan N, Romin Y, Fijisawa S, Barlas A, and Manova-Todorova K. (2015). Machine-based method for multiplex in situ molecular characterization of tissues by immunofluorescence detection. Sci Rep 5, 9534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, and Morrisey EE (2018). Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M, et al. (2019). CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 47, D721-D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, de la Cruz CC, Sayles LC, Alleyne-Chin C, Vaka D, Knaak TD, Bigos M, Xu Y, Hoang CD, Shrager JB, et al. (2013). A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell 24, 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. [Cluster gene signatures, and their enrichment in publicly available gene sets], Related to Figure 2
Supplementary Table S2. [NMF gene signatures and their enrichment in publicly available gene sets], Related to Figure 2
Supplementary Table S3. [Differentially accessible chromatin peaks in TIGIT− and TIGIT+ populations], Related to Figure 3 and Figure S3
Supplementary Table S4. [Enrichment of the stem signatures in HPCS], Related to Figure S4 and STAR Methods: Comparison of HPCS to stem cell signatures
Supplementary Table S5. [Enrichment of the HPCS gene signature in human data], Related to Figure 6
Supplementary Table S6. [Reagents and Samples used in experiments], Related to the experimental STAR Methods
Supplementary Table S7. [Summary quality statistics], Related to STAR Methods: scRNA-Seq processing and quality filtering
Supplementary Table S8. [Gene sets tested for enrichment], Related to STAR Methods: Single cell gene set enrichment
Data Availability Statement
Processed scRNAseq data is available for download or interactive exploration at the Broad Single Cell Portal at the following URLs:
https://singlecell.broadinstitute.org/single_cell/study/SCP971 https://singlecell.broadinstitute.org/single_cell/study/SCP972/ https://singlecell.broadinstitute.org/single_cell/study/SCP973/
Raw data for SmartSeq2 scRNA-Seq, 10x scRNA-Seq, CNV, scDNA-Seq, and Bulk ATAC available from GEO: GSE152607