

Abstract
The incidence of hepatocellular carcinoma (HCC) has been increasing for decades. This disease has now risen to become the sixth most common malignancy overall, while ranking as the third most frequent cause of cancer mortality. While several surgical interventions and loco-regional treatment options are available, up to 80% of patients present with advanced disease not amenable to standard therapies. Indeed, traditional cytotoxic chemotherapeutic agents are notoriously ineffective and essentially play no role in the management of affected patients. This has led to an enormous need for more effective systemic therapeutic options. In recent years, immunotherapy has emerged as a potentially viable and exciting new alternative for the treatment of HCC. Although the current immunotherapeutic options remain imperfect, various strategies can be employed to further improve their efficacy. New findings have revealed epigenetic modulation can be effective as a new approach for improving HCC immunotherapy. Studying the gut microbiome (gut-liver axis) can also be an interesting subject in this regard. Here, we explore the latest insights into the role of immunotherapy treatmenting HCC, both mono and in combination with other agents. We also focus on the impact of epigenetic drugs and the microbiome in the overall effectiveness of HCC immunotherapy.
Keywords: Hepatocellular carcinoma (HCC), Immunotherapy, Gut microbiota, Epigenetic drugs, Combination therapy
1. Introduction
Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer. As the sixth most common cancer throughout the world, and the third leading cause of cancer-related mortality, it represents a remarkable healthcare burden.1 Globally, HCC leads to over 800,000 deaths annually.2 Many common liver diseases, such as viral hepatitis B and C, non-alcoholic fatty liver disease, cirrhosis due to alcohol and hereditary haemochromatosis, have all been associated with an increased risk of HCC occurrence.3 Unfortunately, most patients present with clinical symptoms which usually occur only with advanced disease. Since most patients also suffer from underlying cirrhosis, the vast majority are very poor candidates for definitive interventions, such as surgical resection, transplantation, or even loco-regional therapies.4 Traditional systemic cytotoxic chemotherapeutic agents are ineffective and play essentially no role in the management of this disease.5 In recent years, drugs inhibiting protein kinases have offered a systemic but more targeted assault on HCC. Such drugs include sorafenib and regorafenib, which represent first-and second-line therapy, respectively. A third agent, lenvatinib, presents a broader inhibition of vascular endothelial growth factor (VEGF) receptors, and is also only offered as a salvage therapy. Regrettably, these agents are only indicated in the treatment of advanced disease not amenable to any other intervention and have a collective impact on patient survival that is measured only in weeks or months at best.6–9
In this setting of such limited therapeutic options for the vast majority of patients with HCC, immunotherapy is emerging as a hopeful new alternative.7,10,11 New evidence suggests a promising potential for significant benefits of cancer immunotherapies which utilize immune checkpoint inhibitors for programmed death 1 (PD-1) and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4). Combined immune checkpoint inhibitor-based methods have shown encouraging outcomes, while other categories of immunotherapies, such as adoptive cell transfer, cancer vaccines, and oncolytic virotherapy have also evolved to the point of evaluation in both preclinical and clinical settings.10
Because overall response rates to current immunotherapeutic regimens for HCC remain relatively low, several recent studies have been undertaken with a goal to uncover the mechanisms or factors that might improve their efficacy. Recent publications have shown that epigenetic drugs can impact the therapeutic effectiveness of immunotherapy against HCC in both mice and humans.12 Another approach can be studying the gut microbiome. Although the role of the gut microbiome in modulating immunotherapy efficacy has been widely considered in many types of cancers, its role in the immunotherapy of HCC is not yet clear.13–17 To the best of our knowledge, there is only one study in this regard.18
In this review, we provide an overview of both the ongoing advancements in the HCC immunotherapy, and the impacts of epigenetic drugs and gut microbiome on immunotherapy.
2. Current approaches of immunotherapy
2.1. Early results with checkpoint inhibitors: phase I/II
PD-1 is a cell surface protein belonging to the CD28 family and is expressed on numerous immune cell types.19,20 Specifically, PD-1 can be up-regulated after T cell activation. The binding of PD-1 and programmed death-ligand 1 or 2 (PD-L1, PD-L2) on target cells has the effect of suppressing effector T cell reactions and therefore causes peripheral tolerance, which facilitates tumor perpetuation. Intervening to block the binding of PD-1 from its receptor can thus avoid this inhibitory signal and thereby enable the adaptive targeted response against tumor cells. Indeed, recent clinical studies have shown that checkpoint blockade immunotherapy, targeting PD-1, can be safe in treating patients with advanced HCC.21,22 The CheckMate 040 study was an open-label, non-comparative, phase 1/2 dose escalation and expansion trial in patients with advanced HCC, assessing the PD-1 checkpoint inhibitor nivolumab. The trial results indicated an acceptable safety profile and tolerability in this new immunotherapeutic approach.21 In addition, the results of the KEYNOTE-224 trial (a non-randomized, open-label phase 2 study) found that a similar drug, pembrolizumab, was well tolerated in patients with advanced HCC who were previously treated with sorafenib.22 In that trial, 62% of those treated patients had either an objective response or stable disease.
2.1.1. Efficacy of checkpoint inhibitors: phase III
More recently, at least two multi-center, randomized trials have been carried out to assess the more important phase III endpoints of overall response and survival to therapeutic intervention. The KEYNOTE-240, phase III, placebo-controlled trial of pembrolizumab was an expedited follow-up to the KEYNOTE-224. In this trial, patients treated with pembrolizumab had a reduced risk of death of 22%. Although the co-primary endpoints of progression-free survival and overall survival were both encouraging, they unfortunately did not meet the specified statistical significance required.23
The CheckMate-459 study was a phase III randomized controlled trial that compared nivolumab versus sorafenib as first-line therapies in patients with advanced-stage HCC. Although there was a clear trend toward an improvement in overall survival, statistical significance was again not reached for this pre-specified primary endpoint.24 Together, the objective response rate of PD-1/PD-L1 blockade as a single therapy for HCC appears to be in the range of only 20 to 30%. Although potentially relevant in this cohort of patients with advanced HCC, it remains relatively low compared to outcomes seen with this therapy among other solid tumors. This is possibly due to the heterogeneity of HCC, or perhaps other parameters yet to be discerned.25 In another trial (NCT03434379), the investigators showed that atezolizumab in combination with bevacizumab resulted in better overall and progression-free survival outcomes than sorafenib in patients with unresectable HCC.26 Currently, there are several additional ongoing phase III trials investigating PD-1 (nivolumab, pembrolizumab, camrelizumab and sintilimab), PD-L1 (durvalumab and atezolizumab), CTLA-4 (tremelimumab) or VEGF (bevacizumab) blockade as mono- or combination therapy for patients with HCC (Trial identifiers: NCT03794440, NCT03298451, NCT03764293, NCT02702401, NCT02576509, NCT03847428, NCT03755739, NCT03062358, NCT03713593).
2.1.2. Checkpoint inhibitors combination therapy
Although these recent clinical trials of checkpoint inhibitor efficacy per se have not been entirely encouraging, there is evidence nevertheless, that this strategy may still play a role in concert with other interventions.27 Indeed, PD-1/PD-L1 blockade can be combined with VEGF inhibition and loco-regional treatments or surgical resection, or one of the checkpoint inhibitors.28 Additional targets can include T-cell immunoglobulin, mucin-domain containing-3 (Tim-3), lymphocyte activation gene 3, and transforming growth factor-β in combination with PD-1/PD-L1 or CTLA-4 blockade.29–31 For example, phase I basket trials are currently underway to assess the dual effects to target PD1 plus lymphocyte activation gene 3 as well as Tim-3 plus PD-L1 in HCC cases.10 Unfortunately, combination therapies using checkpoint inhibitors have also been recognized to potentially increase undesirable side effects, such as immune-mediated hepatitis.32
2.1.3. Novel combination therapy
Novel PD-1/PD-L1 blockade-based combination treatments have also been studied. Poliovirus receptor-related-1 (PVRL-1, also known as nectin-1 and CD111) is up-regulated by HCC cells. This interacts with inhibitory molecules on CD8+ memory T-cells, which, in turn, suppresses the anti-tumor immune response. Inhibitors of PVRL-1, anti-PD1, and anti-T cell immuno-receptor with Ig and immuno-receptor tyrosine-based inhibitory motif were used to treat HCC in Trp53KO/C-MycOE mice and mice with tumors grown from Hepa1–6 cells. An increased ratio of cytotoxic to regulatory T-cells, reduced tumor growth and prolonged survival were observed.33 In another study, sunitinib malate, a small molecule inhibitor, was found to increase PD-1 and PD-L1 levels in the tumor microenvironment (TME). Sunitinib malate in combination with anti-PD-1 therapy significantly reduced tumor burden greater than monotherapy in female C57BL/6 mice with tumors grown from Hepa1–6 cells.34 In yet another study, a Listeria-based HCC vaccine, in combination with PD-1 blockade, caused a synergistic anti-tumor effect by the modification of tumor-associated macrophages in the TME. This combination therapy eliminated T-cell inhibitory signals to provide a novel, feasible approach to treating HCC in a Hepa1–6/multiple peptide fusing genes tumor-bearing mouse model.35
2.2. Other categories of immunotherapy
Adoptive cell transfers can lead to an improvement in HCC treatment outcomes via the passive administration of autologous lymphocytes after ex vivo cultivation.36 The broad cell subsets that have been utilized in such studies include natural killer (NK) cells, cytokine-induced killer (CIK) cells, tumor-infiltrating lymphocytes (TIL), mucosal-associated invariant T-cells, chimeric antigen receptor T-cells (CAR-T cells), and T-cell receptor-engineered T-cells (TCR-T cells). Among them, TCR-T cells, CIKs, and CAR-T cells are the major strategies. TCR-T cells are produced by cloning tumor antigen-specific TCR into T cells. CIKs are generated by ex vivo expansion of peripheral blood mononuclear cells in the presence of anti-CD3, IL-2, IL-1α, and interferon (IFN)-γ. CARs have two essential domains; one recognizes tumor antigens, and another transmits activation and proliferation signals into cells. After leukapheresis and expansion, these cells are transfused into host to target and eliminate tumor cells by different mechanisms (Fig. 1).37–39 Such approaches, individually or in combination, can provide new insights into methods to achieve improved clinical outcomes in HCC therapy.9
Fig. 1. Diagram of HCC immunotherapy using different techniques of adoptive cell transfer.
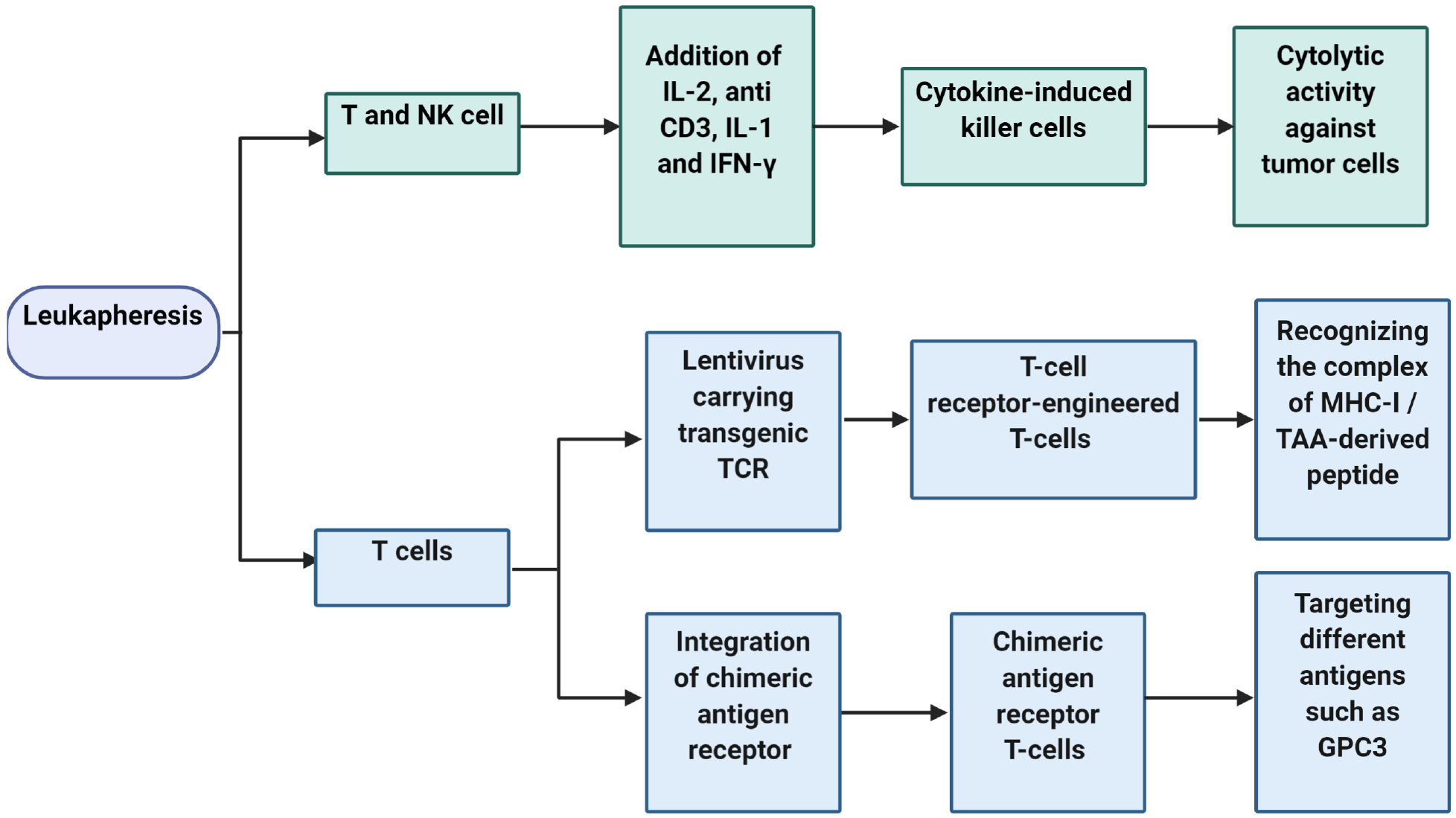
After leukapheresis, T-cell receptor-engineered T-cells/chimeric antigen receptor T-cells, and cytokine-induced killer cells is produced from T cells and NK cells, respectively. Following expansion, these cells are transfused into host to target and eliminate tumor cells by different mechanisms. Abbreviations: NK, natural killer; IFN, interferon; TCR, T-cell receptor; MHC-I, major histocompatibility complex-I; GPC3, glypican 3; TAA, tumor-associated antigen.
The selection of appropriate tumor-associated antigens (TAAs) and neo-antigens is essential for the development of new immuno-therapeutic candidate therapies. Neo-antigens are encouraging targets in cancer treatment, but their use for the immunotherapy of HCC has proven to be challenging, time-consuming, and costly with a success rate that is, thus far, quite low.40,41 Although to date, a limited number of relevant HCC TAAs have been identified; two of that have shown promise include glypican 3 (GPC3) and galectin-1 (Gal-1). In the following sections, we mainly focus on adoptive cell transfer, antibody mediated inhibition, peptide vaccine, and small molecule inhibition of these TAAs. Other therapies, such as DNA vaccines, immunotoxins, and genetic therapies have been reviewed elsewhere.42,43
2.2.1. GPC3
2.2.1.1. GPC3-targeted adoptive cell transfer
GPC3 belongs to the glypican-related integral membrane proteoglycan family that is crucial to the regulation of cell division and development.44 GPC3 expression is not seen in normal liver tissue; however, it appears to be reactivated in approximately 75% of human HCC. This makes GPC3 an appropriate target antigen for immunotherapy in this disease.45,46 A therapeutic method using CAR-T cells has been evaluated in a xenograft model of mice in which human Huh7 HCC cells were inoculated subcutaneously.47 Using this animal model, the third-generation CAR-T cells, GPC3–28BBZ, could efficiently eradicate GPC3-positive Huh7 cells, but not those that were GPC3-negative.47
A clinical phase I trial has been carried out in a small number of patients with advanced HCC in which GPC3-positive tumors were either refractory to therapy or had relapsed after treatment. Anti-GPC3 CAR-T cells were administered with or without lymphodepletion therapy. Those treated without lymphodepletion all showed disease progression. For the sub-group treated with lymphodepletion, there was some evidence of efficacy, with a partial response and others with disease stability.48
In another study, GPC3-transduced dendritic cells were co-cultured with autologous CIKs. These cells were found to have a GPC3-specific marked immune response and potent antitumor activity to GPC3-expressing HCC cells, both in vitro and in vivo.49 Currently, there are at least four phase I/II trials, ongoing or with recruitment completed, utilizing CAR-T cells targeting GPC3 (Trial identifiers: NCT03198546, NCT03130712, NCT02715362, and NCT02723942).32
To study possible mechanisms of action, mice with either intraperitoneal or orthotopic hepatic xenograft tumors were given injections of Hep3B or HepG2 cells, respectively. The results indicated that CAR (hYP7)-T cells decrease GPC3-positive HCC cell populations possibly through two different mechanisms. One mechanism appears to be an effect on cancer signaling by decreasing Wnt and Yes-associated protein (YAP), an important effector molecule in the Hippo pathway. The other mechanism appears to be a result of T cell signaling by the activation of polyfunctional T cells (e.g. CD8+ cytotoxic T cells), which eradicate tumor cells by inducing perforin/granzyme apoptosis pathway (Fig. 2).50
Fig. 2. Proposed mechanism of action of GPC3-targeted CAR (hYP7)-T cells for HCC therapy in mice with xenograft and orthotopic liver tumors.
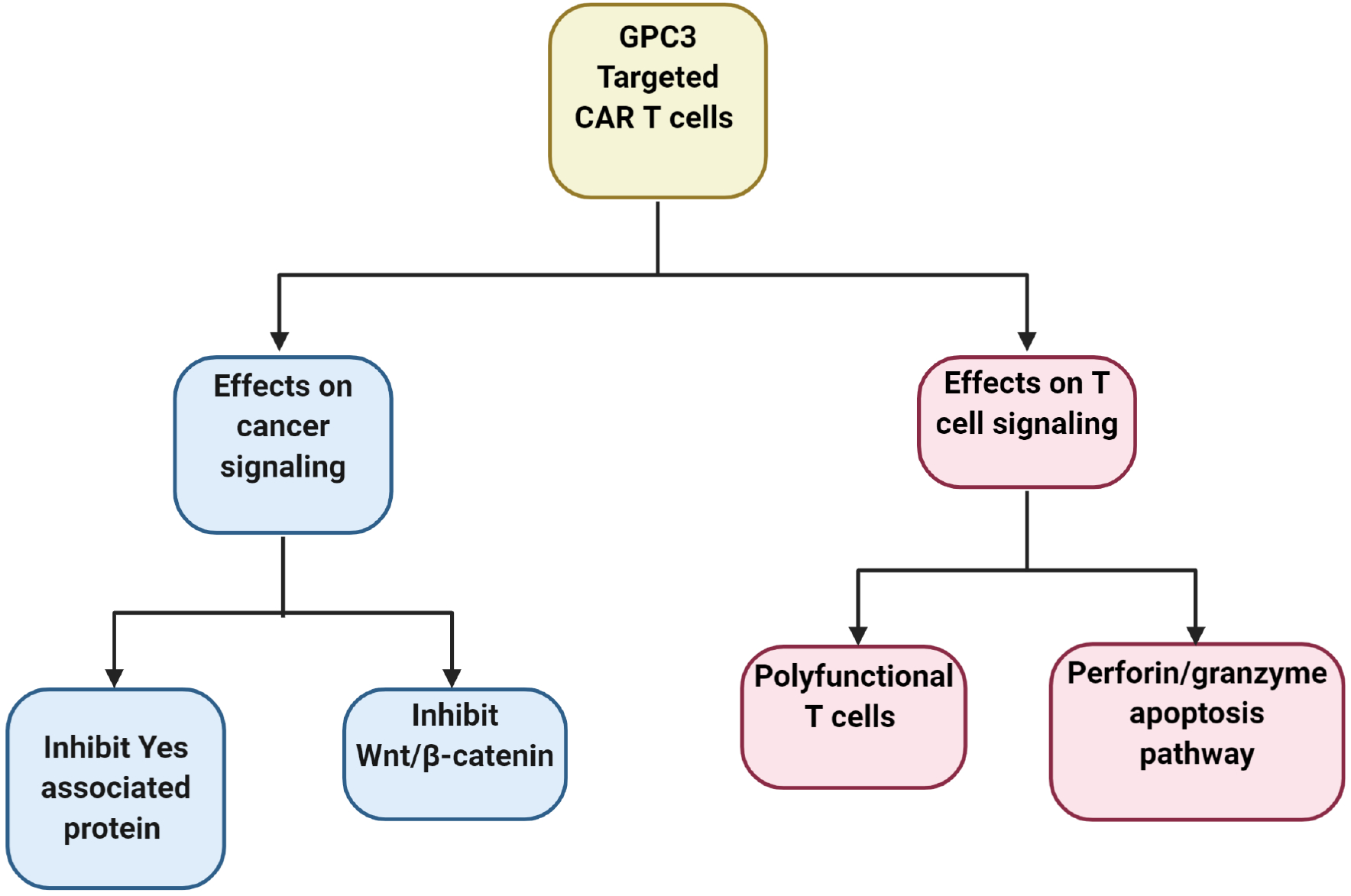
CAR (hYP7)-T cells decrease GPC3-positive HCC cell populations possibly via polyfunctional CD8+ cytotoxic T cells by inducing perforin- and granzyme-associated apoptosis, or by decreasing Wnt and Yap signaling (Hippo pathway) in tumor cells. Abbreviations: GPC3, glypican 3; CAR-T cells, chimeric antigen receptor T-cells.
2.2.1.2. Antibody targeting of GPC3
The effectiveness of anti-GPC3 antibodies has also been studied in the treatment of HCC. In a recent study using a xenograft mouse model inoculated with 107 Huh-7 and HepG2 tumor cells, decreased growth was observed in the antibody treated group compared with the controls.51 GC33 is a monoclonal antibody (mAb) against C-terminal 30-kDa fragment of human GPC3. In one study, GC33 mAb therapy was found to suppress tumor development in ectopic and orthotopic GPC3-positive HCC xenograft models.52 In addition, a double-blind, phase II trial utilizing a humanized GC33 mAb therapy was conducted in 185 patients with advanced HCC, who had failed to respond to sorafenib (Trial identifier: NCT01507168). In this study, 125 patients received the antibody while another 60 patients were given a placebo. Although GC33 mAb did not show an overall clinical benefit in this previously treated population, data did suggest that a higher dose of GC33, or selection of patients with high tumor GPC3 or high expression of CD16 on peripheral immune cells, may offer prolonged progression free survival and overall survival.53
In addition to mAbs targeting the GPC3 core protein, HS20, a human mAb recognizing the GPC3 heparin sulfate (HS) chains, has also been studied. GPC3-HS chains appear to play a key role in Wnt/β-catenin signaling pathway and hepatocyte growth factor binding/c-Met activation. The data from two studies found that the HS20 mAb can impair GPC3-Wnt3a interaction and suppress hepatocyte growth factor-associated cell migration leading to suppressing cell proliferation.54,55
Bispecific antibodies can simultaneously bind two separate unique antigens and present another potential therapeutic approach. Unfortunately, such antibodies may have some undesirable and challenging characteristics, such as unwanted aggregations, poor structural stability, and short serum half-life. To overcome these issues, a novel form of T-cell redirecting antibody has been designed specific for membrane proteoglycans GPC3 of HCC, as well as the T-cell-specific antigen CD3. This has been found to have thermo-stability characteristics similar to general IgG-like bispecific antibodies and was shown to significantly inhibit tumor growth in a murine xenograft model utilizing a subcutaneous injection of Huh-7 cells.56
2.2.1.3. GPC3-derived peptide vaccine
Peptide vaccines have also been a potential alternative method for generating an effective anti-tumor immune reaction. In one recent phase 1 study, a GPC3 peptide vaccination was given to 33 patients with advanced HCC. The primary and secondary endpoints of safety and immune response were met. One patient showed a partial response while 19 others were found to have disease stability for a limited period.57 A second phase II trial utilized the same vaccine to assess the rates of recurrence in patients undergoing surgical resection or thermal ablation. The best outcomes were observed at one-year among those patients who had GPC3-positive cancer and who received surgical resection.58
2.2.2. Gal-1
Galectins are glycan-binding proteins that bind specifically to β-galactoside proteins and have a known role in the promotion of inflammation and dampening the T cell-mediated immune response. Galectin-1, −3, and −9 can regulate immune cells and modulate tumor cell growth.59 Gal-1 has attracted much attention because of its general roles in tumor progression, migration, and angiogenesis. The natural adhesive characteristic of Gal-1 has been demonstrated in clinical settings.59 In human cancers, such as HCC, Gal-1 is overexpressed, a fact that has been exploited in devising cancer treatments. The overexpression of Gal-1 in HCC has been postulated to trigger epithelial-mesenchymal transition, thereby making the tumor cells resistant to sorafenib by inducing the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signaling pathway.60
2.2.2.1. Small molecule inhibition of Gal-1
OTX008 is a small-molecule and selective inhibitor of Gal-1. OTX008 has been studied in a clinical phase 1 trial of patients with solid tumors that have failed standard treatments. Although it was found to effectively reduce serum Gal-1 levels, overall it was not well tolerated and had dose-limiting toxicities (Trial identifier: NCT01724320).61 Other approaches have also been considered to reduce the expression of Gal-1. The tumor suppressor microRNA-22 (miR-22), has been shown to reduce the expression of Gal-1 as well as Cyclin-A2 and several other protein deacetylases.62,63 Interestingly, researchers comparing miR-22 expression in HCC versus normal tissues found miR-22 to be significantly lower in HCC tumor tissues.64–67 Under expression of miR-22 leads to Gal-1 overexpression in HCC. It has been shown that the expression of Gal-1 and retention in endoplasmic reticulum 1 (RER1), a Golgi transmembrane protein, has a significant positive correlation with its oncogenic effect; while miR-22 was negatively correlated with Gal-1 and its oncogenic effect.68 In the same study, the combination therapy of OTX008 and sorafenib showed more effectiveness in comparison with sorafenib administration alone.68
3. Impact of epigenetic drugs and microbiome on immunotherapy
3.1. Epigenetic drugs in HCC
Cancer cells use epigenetic mechanisms, such as DNA methyltransferase (DNMTs) enzymes-mediated hypermethylation and histone deacetylases (HDACs)-mediated histone modification. Preclinical investigations on HCC have considered DNMT and HDAC inhibitors. An HDAC inhibitor, belinostat was found to improve the effect of anti-CTLA4 checkpoint inhibitor as demonstrated in mouse models utilizing subcutaneously inoculated Hepa129 cells. Belinostat, in combination with an anti-CTLA4 checkpoint inhibitor, could reduce tumor size and prolong survival time compared with belinostat alone. Such an improved effect was accompanied by increased tumor infiltration of M1 macrophages and reduced Tregs.12
The DNMT inhibitor 5-azacytidine can induce the expression of neoantigens on HCC cells. Compared with single agent treatments, when 5-azacytidine was used in combination with anti-PD-L1, the tumor size was further reduced and was noted to be accompanied by an increased T-lymphocyte infiltration in a mouse model. Such findings have revealed epigenetic modulation as a new approach for improving HCC immunotherapy.69
Recently, it has been proven that the epigenetic modificatory enhancer of zeste homolog 2 (EZH2) can suppress the expression of PD-L1 in hepatoma cells. The authors also showed negative correlations between EZH2 and PD-L1 expression in clinical samples from HCC patients. More investigations are needed to accredit these findings to evaluate EZH2 as s potential therapeutic target for HCC immunotherapy.70
3.2. Microbiome
Regardless of the etiology, through mechanisms of the gut-liver axis, liver injury can change the composition of the gut microbiome and their metabolites, thereby affecting host signaling.71 In general, HCC patients have a gut microbiome profile characterized by increased pro-inflammatory bacteria and a commensurate decrease in short-chain fatty acid-producing bacteria.72–76 Depleting gut microbes using antibiotics, such as vancomycin, can lower the abundance of microorganisms (e.g., Clostridium) that convert the primary bile acids into the secondary bile acids. The primary bile acids are known to increase the expression of the membrane-bound chemokine and mediator of innate immunity, CXCL16. Increased CXCL16 expression leads to activated CXCR6+ natural killer T (NKT) cells which inhibit growth of both B16 and EL4 tumor cells (Fig. 3).77 Although encouraging, whether such observations in animal models will translate into those that are clinically relevant in humans has yet to be determined.
Fig. 3. Antibiotics lowered the abundance of Clostridium which leads to increased ratio of the primary to the secondary bile acids.
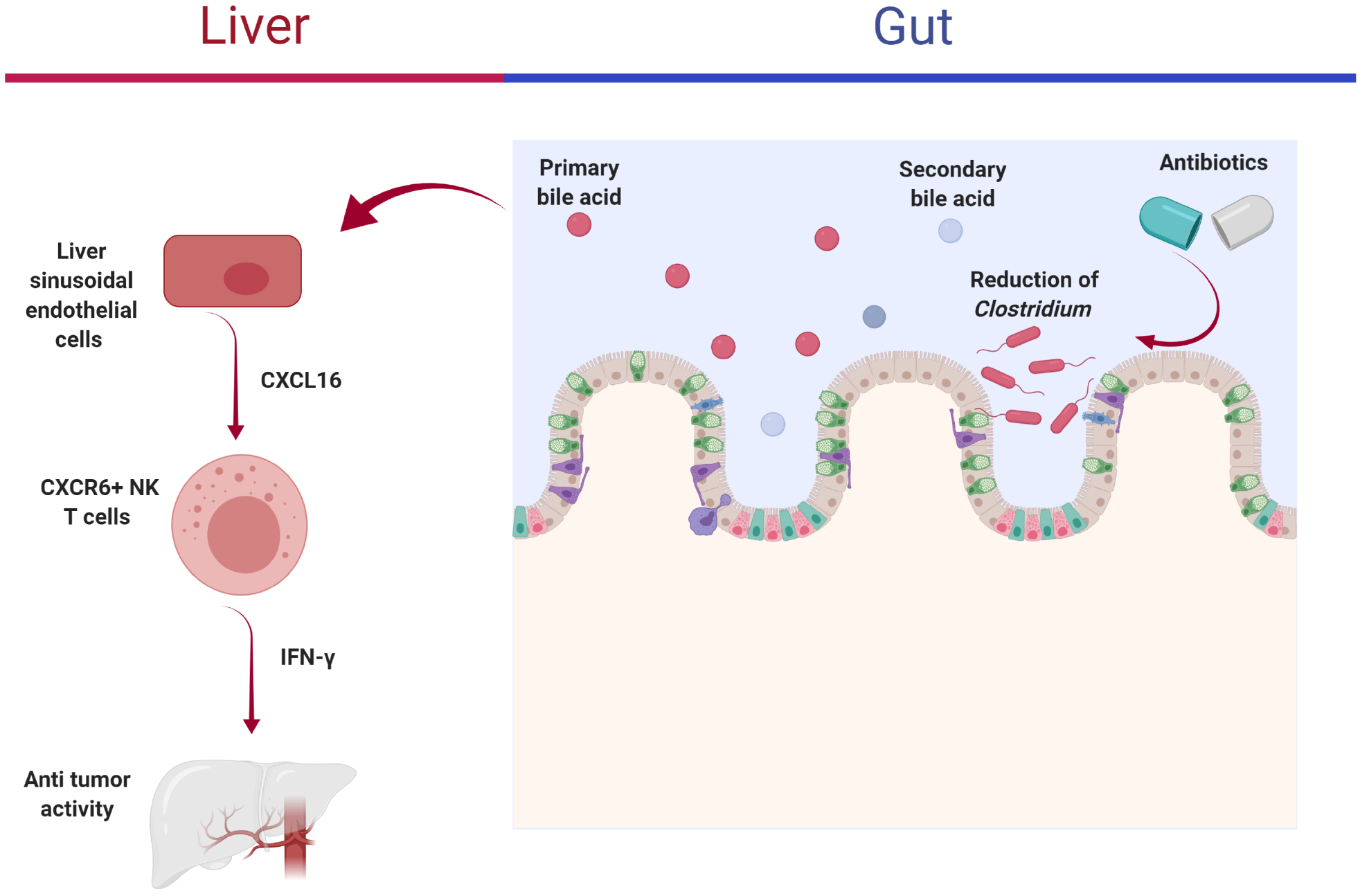
The primary bile acids increased CXCL16 expression produced by liver sinusoidal endothelial cells. Increased CXCL16 leads to activated CXCR6+ NKT cells, which have anti-tumor activity via IFN-γ production.77 Abbreviations: NKT, natural killer T; IFN, interferon.
Zheng et al.18 recently studied a small sample of patients with HCC refractory to sorafenib and their response to anti-PD-1 antibody immunotherapy. Patients were classified as responders (complete response, partial response, or stable) or non-responders based on imaging using response evaluation criteria in solid tumors (RECIST). Fecal samples were collected at intervals. In this study, non-responders had increased Proteobacteria from the third week, which became dominant by week twelve. However, responders had enriched Akkermansia muciniphila and Ruminococcaceae spp. These results suggest that the gut microbiome can affect the outcome of anti-PD-1 immunotherapy.18
Hepatic cirrhosis is often an underlying condition in HCC patients. Cirrhosis is associated with an extreme dysbiosis, which, in some circumstances, can contribute to drug resistance. It is therefore reasonable to speculate that modulating the gut microbiome very likely has a greater impact on the treatment of HCC as compared with other common tumors.78 The combination of antibiotic therapy (e.g., vancomycin) with immune checkpoint blockade has been used to study the effect of the gut microbiota in HCC treatment (Trial identifier: NCT03785210).78 Clearly, this is an area of exciting potential in the study of HCC treatment. The main HCC immunotherapy approaches of this review article are summarized in Table 1.
Table 1.
The main HCC immunotherapy approaches that are summarized in this review article.
| Trials or Drugs | Outcomes | Reference |
|---|---|---|
| CheckMate 040 (nivolumab) and KEYNOTE-224 (pembrolizumab) | Acceptable safety profile and tolerability | 21,22 |
| KEYNOTE-240 (pembrolizumab) | Acceptable co-primary endpoints of progression-free survival and overall survival, but statistical significance was not reached | 23 |
| CheckMate-459 (nivolumab vs. sorafenib) | Improved in overall survival, but statistical significance was not reached | 24 |
| Inhibitors of Poliovirus rececptor-related-1 | Increased ratio of cytotoxic to regulatory T-cells, reduced tumor growth and prolonged survival observed in mice | 33 |
| Sunitinib malate | Reduced tumor burden greater than monotherapy in female C57BL/6 mice | 34 |
| Listeria-based HCC vaccine | Eliminated T-cell inhibitory signals in mice | 35 |
| GPC3-28BBZ and CAR (hYP7)-T cells | Eradicated or decreased GPC3-positive HCC cell populations in mice model | 47, 50 |
| GC33 (mAb for GPC3 C-terminal) | Suppressed tumor development in mice | 53 |
| HS20 (mAb for GPC3 heparin sulfate chain) | Impaired GPC3-Wnt3a interaction in mice | 54, 55 |
| GPC3 bispecific antibodies | Inhibited tumor growth in a murine xenograft model | 56 |
| GPC3-derived Peptide Vaccine | Phase 1 study: Acceptable primary and secondary endpoints of safety and immune response Phase 2 study: The best outcomes were observed among patients who had received surgical resection | 57, 58 |
| OTX008, Galectin-1 inhibitor | Reduced serum Gal-1 levels, but it was not well tolerated and had dose-limiting toxicities in phase 1 trial | 61 |
| Belinostat | In combination with an anti-CTLA4 checkpoint inhibitor could reduce tumor size and prolong survival time in mice | 12 |
| 5-azacytidine | The tumor size was reduced, which was accompanied by an increased T-lymphocyte infiltration in mice | 69 |
Abbreviations: HCC, hepatocellular carcinoma; GPC3, glypican 3; CAR-T cells, chimeric antigen receptor T-cells; CTLA-4, cytotoxic T-lymphocyte-associated antigen-4; Gal-1, galectin-1.
4. Conclusions
Immunotherapy only leads to less than 20% clinical responses. The immune-suppressive TME is a main barrier for a successful anti-tumor activity through immunotherapeutic treatments. The weaknesses of the current immunotherapy approaches should be balanced using combination therapy, epigenetic medications or manipulation of the microbiome. Future effective approaches will likely include combinations of various immunotherapeutic approaches for eliciting a successful anti-cancer reaction, chemotherapy/checkpoint inhibitors and a balancing of the immune-suppressive TME. New investigations on the gut microbiome, especially those focusing on fecal microbiota transplantation, will likely assist in the development of new paradigms and personalized treatments to enhance immunotherapy of HCC.79
Acknowledgements
This study was supported by grants funded by the National Institutes of Health U01CA179582 and R01CA222490.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is publishedin its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no conflict of interest.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391: 1301–1314. [DOI] [PubMed] [Google Scholar]
- 3.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35–S50. [DOI] [PubMed] [Google Scholar]
- 4.Thomas MB, Jaffe D, Choti MM, et al. Hepatocellular carcinoma: consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2010;28:3994–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eatrides J, Wang E, Kothari N, Kim R. Role of systemic therapy and future directions for hepatocellular carcinoma. Cancer Control. 2017;24: 1073274817729243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. [DOI] [PubMed] [Google Scholar]
- 7.Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology. 2018;68:723–750. [DOI] [PubMed] [Google Scholar]
- 8.Pardee AD, Butterfield LH. Immunotherapy of hepatocellular carcinoma: unique challenges and clinical opportunities. Oncoimmunology. 2012;1:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tagliamonte M, Mauriello A, Cavalluzzo B, et al. Tackling hepatocellular carcinoma with individual or combinatorial immunotherapy approaches. Cancer Lett. 2020;473:25–32. [DOI] [PubMed] [Google Scholar]
- 10.Nakano S, Eso Y, Okada H, Takai A, Takahashi K, Seno H. Recent advances in immunotherapy for hepatocellular carcinoma. Cancers (Basel). 2020;12:775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Khoueiry A The promise of immunotherapy in the treatment of hepatocellular carcinoma. Am Soc Clin Oncol Educ Book. 2017;37:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Llopiz D, Ruiz M, Villanueva L, et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol Immunother. 2019;68:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359: 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. [DOI] [PubMed] [Google Scholar]
- 16.Shui L, Yang X, Li J, Yi C, Sun Q, Zhu H. Gut Microbiome as a Potential Factor for Modulating Resistance to Cancer Immunotherapy. Front Immunol. 2019;10:2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vetizou M, Pitt JM, Daillere R, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng Y, Wang T, Tu X, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hato T, Goyal L, Greten TF, Duda DG, Zhu AX. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology. 2014;60:1776–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immunol. 2015;15:45–56. [DOI] [PubMed] [Google Scholar]
- 21.El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940–52. [DOI] [PubMed] [Google Scholar]
- 23.Merck Provides Update on KEYNOTE-240, a Phase 3 Study of KEYTRUDA® (pembrolizumab) in Previously Treated Patients with Advanced Hepatocellular Carcinoma. [press release]. 2019.
- 24.Bristol-Myers Squibb Announces Results from CheckMate-459 Study Evaluating Opdivo (nivolumab) as a First-Line Treatment for Patients with Unresectable Hepatocellular Carcinoma [press release]. 2019.
- 25.Jiang Y, Zhao X, Fu J, Wang H. Progress and challenges in precise treatment of tumors with PD-1/PD-L1 blockade. Front Immunol. 2020;11:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894–1905. [DOI] [PubMed] [Google Scholar]
- 27.Lee HW, Cho KJ, Park JY. Current status and future direction of immunotherapy in hepatocellular carcinoma: what do the data suggest? Immune Netw. 2020;20:e(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh P, Toom S, Avula A, Kumar V, Rahma OE. The immune modulation effect of locoregional therapies and its potential synergy with immunotherapy in hepatocellular carcinoma. J Hepatocell Carcinoma. 2020;7:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson AC. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res. 2014;2:393–398. [DOI] [PubMed] [Google Scholar]
- 30.Han Y, Yang Y, Chen Z, et al. Human hepatocellular carcinoma-infiltrating CD4+CD69+Foxp3− regulatory T cell suppresses T cell response via membrane-bound TGF-beta1. J Mol Med (Berl). 2014;92:539–550. [DOI] [PubMed] [Google Scholar]
- 31.Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol Lett. 2013;150:116–122. [DOI] [PubMed] [Google Scholar]
- 32.Johnston MP, Khakoo SI. Immunotherapy for hepatocellular carcinoma: current and future. World J Gastroenterol. 2019;25:2977–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kung-Chun Chiu D, Wai-Hin Yuen V, Wing-Sum Cheu J, et al. Hepatocellular carcinoma cells upregulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology. 2020;159:609–623. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Zhan M, Quan YY, et al. Modulating the tumor immune microenvironment with sunitinib malate supports the rationale for combined treatment with immunotherapy. Int Immunopharmacol. 2020;81:106227. [DOI] [PubMed] [Google Scholar]
- 35.Xu G, Feng D, Yao Y, et al. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene. 2020;39:1429–1444. [DOI] [PubMed] [Google Scholar]
- 36.Baruch EN, Berg AL, Besser MJ, Schachter J, Markel G. Adoptive T cell therapy: an overview of obstacles and opportunities. Cancer. 2017;123:2154–2162. [DOI] [PubMed] [Google Scholar]
- 37.Cai XR, Li X, Lin JX, et al. Autologous transplantation of cytokine-induced killer cells as an adjuvant therapy for hepatocellular carcinoma in Asia: an update meta-analysis and systematic review. Oncotarget. 2017;8:31318–31328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caraballo Galva LD, Cai L, Shao Y, He Y. Engineering T cells for immunotherapy of primary human hepatocellular carcinoma. J Genet Genomics. 2020;47:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lukasik Z, Elewaut D, Venken K. MAIT cells come to the rescue in cancer immunotherapy? Cancers (Basel). 2020;12:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu L, Jiang J, Zhan M, et al. Targeting neoantigens in hepatocellular carcinoma for immunotherapy: A futile strategy? Hepatology. 2020. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat Med. 2019;25:1488–1499. [DOI] [PubMed] [Google Scholar]
- 42.Guo M, Zhang H, Zheng J, Liu Y. Glypican-3: a new target for diagnosis and treatment of hepatocellular carcinoma. J Cancer. 2020;11:2008–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou F, Shang W, Yu X, Tian J. Glypican-3: a promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med Res Rev. 2018;38:741–767. [DOI] [PubMed] [Google Scholar]
- 44.Baumhoer D, Tornillo L, Stadlmann S, Roncalli M, Diamantis EK, Terracciano LM. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: a tissue microarray analysis of 4,387 tissue samples. Am J Clin Pathol. 2008;129:899–906. [DOI] [PubMed] [Google Scholar]
- 45.Dargel C, Bassani-Sternberg M, Hasreiter J, et al. T cells engineered to express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology. 2015;149:1042–1052. [DOI] [PubMed] [Google Scholar]
- 46.Zhu ZW, Friess H, Wang L, et al. Enhanced glypican-3 expression differentiates the majority of hepatocellular carcinomas from benign hepatic disorders. Gut. 2001;48:558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao H, Li K, Tu H, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20:6418–6428. [DOI] [PubMed] [Google Scholar]
- 48.Zhai B, Shi D, Gao H, et al. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC). J Clin Oncol. 2017;35:3049. [Google Scholar]
- 49.Wang Y, Wang Y, Mu H, Liu T, Chen X, Shen Z. Enhanced specific antitumor immunity of dendritic cells transduced with the glypican 3 gene and co-cultured with cytokine-induced killer cells against hepatocellular carcinoma cells. Mol Med Rep. 2015;11:3361–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li D, Li N, Zhang YF, et al. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. 2020;158:2250–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng M, Gao W, Wang R, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2013;110:E1083–E1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishiguro T, Sugimoto M, Kinoshita Y, et al. Anti-glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res. 2008;68:9832–9838. [DOI] [PubMed] [Google Scholar]
- 53.Abou-Alfa GK, Puig O, Daniele B, et al. Randomized phase II placebo controlled study of codrituzumab in previously treated patients with advanced hepatocellular carcinoma. J Hepatol. 2016;65:289–295. [DOI] [PubMed] [Google Scholar]
- 54.Gao W, Kim H, Feng M, et al. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology. 2014;60:576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao W, Kim H, Ho M. Human monoclonal antibody targeting the heparan sulfate chains of glypican-3 inhibits HGF-mediated migration and motility of hepatocellular carcinoma cells. PLoS One. 2015;10:e0137664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu L, Yang X, Huang N, et al. A novel targeted GPC3/CD3 bispecific antibody for the treatment hepatocellular carcinoma. Cancer Biol Ther. 2020;21:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sawada Y, Yoshikawa T, Nobuoka D, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18:3686–3696. [DOI] [PubMed] [Google Scholar]
- 58.Sawada Y, Yoshikawa T, Ofuji K, et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology. 2016;5:e1129483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chou FC, Chen HY, Kuo CC, Sytwu HK. Role of galectins in tumors and in clinical immunotherapy. Int J Mol Sci. 2018;19:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang PF, Li KS, Shen YH, et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016;7:e(2201). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delord JP, Awada A, Raymond E, et al. A first-in-man Phase I study of the galectin-1 (gal-1) inhibitor OTX008 given subcutaneously as a single agent to patients with advanced solid tumors. Mol Cancer Ther. 2013;12:A72. [Google Scholar]
- 62.Hu Y, French SW, Chau T, et al. RARbeta acts as both an upstream regulator and downstream effector of miR-22, which epigenetically regulates NUR77 to induce apoptosis of colon cancer cells. FASEB J. 2019;33:2314–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang F, Hu Y, Liu HX, Wan YJ. MiR-22-silenced cyclin A expression in colon and liver cancer cells is regulated by bile acid receptor. J Biol Chem. 2015;290:6507–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo LJ, Zhang LP, Duan CY, et al. The inhibition role of miR-22 in hepatocellular carcinoma cell migration and invasion via targeting CD147. Cancer Cell Int. 2017;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Yang Y, Yang T, et al. MicroRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br J Cancer. 2010;103:1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, Rohatgi AP, Wan YJ. Retinoic acid and microRNA. Methods Enzymol. 2020;637:283–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang L, Wang YS, Mugiyanto E, Chang WC, Wan YJ. MiR-22 as a Metabolic Silencer and Liver Tumor Suppressor. Liver Res. 2020;4:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leung Z, Ko FCF, Tey SK, et al. Galectin-1 promotes hepatocellular carcinoma and the combined therapeutic effect of OTX008 galectin-1 inhibitor and sorafenib in tumor cells. J Exp Clin Cancer Res. 2019;38:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hong YK, Li Y, Pandit H, et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol. 2019;336:66–74. [DOI] [PubMed] [Google Scholar]
- 70.Xiao G, Jin LL, Liu CQ, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. 2019;7:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keenan BP, Fong L, Kelley RK. Immunotherapy in hepatocellular carcinoma: the complex interface between inflammation, fibrosis, and the immune response. J Immunother Cancer. 2019;7:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Q, Li F, Zhuang Y, et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ni J, Huang R, Zhou H, et al. Analysis of the relationship between the degree of dysbiosis in gut microbiota and prognosis at different stages of primary hepatocellular carcinoma. Front Microbiol. 2019;10:1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pinero F, Vazquez M, Bare P, et al. A different gut microbiome linked to inflammation found in cirrhotic patients with and without hepatocellular carcinoma. Ann Hepatol. 2019;18:480–487. [DOI] [PubMed] [Google Scholar]
- 75.Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. 2019;69:107–120. [DOI] [PubMed] [Google Scholar]
- 76.Ren Z, Li A, Jiang J, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68:1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma C, Han M, Heinrich B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwabe RF, Greten TF. Gut microbiome in HCC-mechanisms, diagnosis and therapy. J Hepatol. 2020;72:230–238. [DOI] [PubMed] [Google Scholar]
- 79.Zhou A, Tang L, Zeng S, Lei Y, Yang S, Tang B. Gut microbiota: a new piece in understanding hepatocarcinogenesis. Cancer Lett. 2020;474:15–22. [DOI] [PubMed] [Google Scholar]