

ABSTRACT
Genome editing via homology-directed repair (HDR) has made possible precise and deliberate modifications to gene sequences. CRISPR/Cas9-mediated HDR is the simplest means to carry this out. However, technical challenges remain to improve efficiency and broaden applicability to any genetic background of Drosophila melanogaster as well as to other Drosophila species. To address these issues, we developed a two-stage marker-assisted strategy in which embryos are injected with RNPs and pre-screened using T7EI. Using sgRNA in complex with recombinant Cas9 protein, we assayed each sgRNA for genome-cutting efficiency. We then conducted HDR using sgRNAs that efficiently cut target genes and the application of a transformation marker that generates RNAi against eyes absent. This allows for screening based on eye morphology rather than colour. These new tools can be used to make a single change or a series of allelic substitutions in a region of interest, or to create additional genetic tools such as balancer chromosomes.
KEYWORDS: CRISPR, gene editing, Drosophila, homology dependent repair
Introduction
Genome editing by CRISPR/Cas9 has transformed research and development in the life and health sciences [1]. Although use of CRISPR has expanded beyond the original genome editing capabilities [2–4], genome editing remains a very popular application. The Cas9 endonuclease associates with a single guide RNA (sgRNA), and the complex localizes to DNA sequences in the genome by following simple DNA:sgRNA base-pairing rules. The complex then introduces a double-strand break (DSB) in the DNA, triggering repair of the broken ends. If there is available a separate DNA template that contains sequences homologous to the regions flanking the DSB, then homology-directed repair (HDR) can result in incorporation of the repair template into the genomic DNA. The repair template can be a homologous chromosome or an exogenous donor DNA molecule. Exogenous templates come in one of two forms: a single-stranded oligonucleotide or a double-stranded DNA plasmid. Plasmids can be much larger than oligonucleotides, allowing for modifications to be made at a greater distance from the DSB. In the absence of a repair template, non-homologous end joining (NHEJ) ligates the broken ends, resulting in stochastic insertions and deletions (indels) at the break site. Although NHEJ-mediated genome editing is useful for gene disruption, HDR affords precise and programmable alterations in genome sequence.
The adaptation of CRISPR/Cas9 editing to Drosophila research occurred shortly after its invention [5–7]. The first-generation of methods used either expression plasmids or RNAs coding for Cas9 and sgRNA. Subsequently, homemade Cas9 protein complexed with tracrRNA and crRNA was shown to work in Drosophila [8]. Additionally, a number of germline-specific Cas9 transgenic lines were generated [9–13], and these have been widely used by Drosophila researchers. Typically, a plasmid encoding a sgRNA driven by RNA polymerase III transcription is injected into Cas9 transgenic embryos. This approach greatly increases the efficiency of germline editing events. It has been particularly beneficial for the development of CRISPR/Cas9-induced HDR in Drosophila [6,10,11,14,15].
CRISPR-induced HDR for genome editing is not straightforward because of two issues. First, computational design of a targeting sgRNA does not predict the efficiency of cleavage, which varies considerably at different target sites [16]. This could be due to many reasons such as secondary structure within the sgRNA, stability of the sgRNA-DNA duplex, or accessibility of the target sequence within the context of chromatin. Therefore, editing of cultured cells often relies on multiple sgRNAs targeting one gene, as a way to ‘cover all bases’ [16]. This approach has also been developed for Drosophila, in which multiplexed sgRNAs are expressed from one vector [17]. The second challenge for editing by HDR is that the individuals who have inherited the desired edits must be identified. This challenge exists because HDR resolution of DSBs is much rarer than NHEJ repair, and so the vast majority of individuals have not been edited. Furthermore, imprecise HDR can occur to create undesired edits. For Drosophila, molecular screening methods such as PCR and sequencing are time-consuming because G1 progeny of injected G0 flies must be individually assayed for precise HDR events.
As an alternative screening method, a visible transformation marker gene can be incorporated into the repair template plasmid [18]. The marker gene is placed between the right and left homology arms used for template-driven repair. This affords rapid screening of G1 animals without their sacrifice. Use of such a selection scheme faces several challenges after transformants are identified. First, imprecise HDR events involving crossover repair at the site of a DSB are frequent occurrences [19]. There, the repair template plasmid backbone is incorporated into the genome, and such events are scored positive using a transformation marker. To identify such events, HDR repair template plasmids can contain a mini-white gene in their backbones (https://flycrispr.org/; Addgene 80801), a modification inspired by recombinase-mediated cassette exchange (RMCE) vectors [20]. The presence of white+ in the plasmid backbone allows for a counter selection against the integration of the whole plasmid when incorporated into a white mutant background, ensuring that only the DNA between the homology arms integrates. However, the general utility of the white marker is limited by the necessity of using it in a white− genetic background and by the large size of its coding and control sequences. A second challenge is the presence of the marker gene at the site of editing. If the goal of editing is to determine the effects of precise base changes, then the marker gene must be removed prior to phenotypic analysis. Although φC31-mediated RMCE and FLP-FRT have been used to excise an HDR marker gene [15,21], they leave scars in the form of ectopic sequences at the excision site. The PiggyBac transposase has been harnessed to excise a 3xP3-DsRed marker gene from the edited site, and this approach has the benefit of leaving no sequence scar at the excision site (https://flycrispr.org/). An alternative scarless approach involves integration of the marker gene at the edited site, followed by a second round of HDR that replaces the marker gene with the desired edits [20,22]. Although reversion events are easily scored, the overall process requires two rounds of CRISPR/Cas9 injections and screening.
In summary, many developments have improved the scope and efficacy of genome editing in Drosophila. However, several impediments still remain until editing becomes as straightforward and efficient as more established genetic technologies in Drosophila. Here, we describe a series of modifications to the HDR-mediated editing procedure that overall enhances the success rate of achieving precise edits. Moreover, these enhancements can be adopted across a broad range of genetic backgrounds in D. melanogaster and even in other Drosophila species. We believe that this new procedure greatly expands the potential use for precise genome editing in Drosophila. The potential to modify one, two, or many more basepairs makes it a powerful tool for testing the phenotypic consequences of changes in genome sequence ranging from single base variants to more substantial differences.
Results
A survey of the existing technologies related to HDR-mediated editing by CRISPR identified several obstacles. These are listed in Figure 1. We systematically describe each obstacle and the method we developed to overcome it.
Figure 1.

Sources of Drosophila CRISPR reagents and their individual pros and cons. (a) Sources of Cas9. Source shaded grey highlights the novel source used in this study. (b) Sources of sgRNA. Since some sgRNAs are inactive for inducing DSBs in vivo, a quality control (QC) test is preferable. Region shaded grey highlights the novel prescreening method to identify active sgRNAs. (c) Sources of donor plasmids to act as repair templates for HDR. This panel only shows plasmids related to the novel plasmid used in this study (shaded grey)
Genome editing by RNP injection
The source of Cas9 used to induce DSBs varies, but the most commonly used source is a transgenic Cas9 specifically expressed in germ cells using regulatory sequences from the genes vasa or nanos (Figure 1(a)). Although it usually induces DSBs with high efficiency, the reliance on a transgenic line limits the genetic background available for G0 founders and often complicates the background of G1 and subsequent generations. Cas9 plasmid and mRNA are less efficient and are more variable if injected. However, DSBs have been demonstrated to occur in cultured human cells using electroporation-mediated delivery of sgRNA and Cas9 in the form of in vitro assembled ribonucleoproteins (RNPs) [23,24]. This approach has also been adapted for generation of NHEJ-induced mutagenesis in Drosophila and Caenorhabditis elegans by microinjection [8,25]. However, the method for Drosophila relied upon homemade Cas9 protein combined with two RNAs rather than the standard sgRNA [8]. Moreover, the efficiency of DSBs using RNPs was not directly compared to the use of Cas9 transgenes. Finally, the utility of RNPs for HDR-mediated genome editing was not tested.
To determine the efficiency of RNPs for Drosophila editing, we assembled sgRNA-Cas9 RNPs by co-incubation of in vitro-synthesized sgRNA with commercially available recombinant Cas9 protein. The sgRNA was generated by T7 polymerase transcription of a synthetic DNA template as described [5]. Preparation was as simple and reproducible as generating sgRNA-expression plasmids (Figure 1(b)). Moreover, the commercially available Cas9 protein is typically affordable.
An RNP solution designed to induce DSBs in the forked gene was injected into 328 white embryos using a previously validated sgRNA that targeted forked coding sequence [17]. We then testcrossed the resulting 52 adults to forked mutants and scored G1 offspring for germline transmission of forked mutations induced by NHEJ. Of 39 crosses that produced G1 offspring, 23 (59.0%) resulted in one or more forked mutant offspring. We compared the efficiency of generating such mutants to the efficiency when the sgRNA alone was delivered into a transgenic vasa-Cas9 line (BDSC #51324). Injection of 331 vasa-Cas9 embryos resulted in 68 adults. When testcrossed to a forked mutant, 54 produced G1 offspring, and 12 (22.2%) of these crosses resulted in one or more forked mutant offspring. Thus, simple assembly and injection of RNPs were 2.5-fold more potent than using transgenic vasa-Cas9 for inducing indel mutations in the forked gene (p = 0.0005, Fisher's exact test). However, this Cas9 transgene has slightly weaker efficiency than some other Cas9 transgenes [11], and thus it will be interesting to see if RNPs using other sgRNAs also outperform this and other transgenes.
sgRNA screening
Clearly, use of RNPs expands the potential for inducing DSBs in any genetic background or even other Drosophila species. However, it has another important benefit. Given that sgRNAs with different target sites exhibit different endonuclease activities in vivo, prescreening the activity of various sgRNAs within the gene of interest would be advantageous before designing an appropriate repair template that fits with the selected sgRNA (Figure 1(b)). At present, prescreening in Drosophila is not commonly performed because of the use of transgenic germline Cas9 lines. Screening G1 or G2 animals constitutes a time-consuming and expensive process. However, injection of assembled RNPs induces DSBs not only in the germline but in the somatic cells of the animal as well. Therefore, we could screen for NHEJ-mediated mutations in G0 animals without having to generate HDR repair template plasmids.
The presence of indels induced by CRISPR can be detected by a T7 endonuclease I (T7EI) assay (Figure 2(a)) [23]. A prior study of RNP-mediated NHEJ mutagenesis in Drosophila found that the T7EI assay was able to detect indels in G0 and G1 adults [8]. We reasoned that if such events could be detected in G0 embryos, then the time to analyse a sgRNA for cutting efficiency could be reduced from 12 to 25 days to 3 days. We injected a minimum of 8 embryos with RNPs, and purified genomic DNA from each individual after 24 hours. A small region surrounding the target site was amplified by PCR from individual DNA. If the RNPs had induced indels in a sizable number of an embryo’s cells, then the amplicons would be a composite of wildtype and mutant DNA duplexes. If the RNPs failed to induce many indels, then the amplicons would be primarily composed of wildtype strands. We then denatured the amplicons from each individual embryo source and hybridized the strands back together. This was followed by a T7EI digestion of the hybridized DNA, and agarose gel electrophoresis to resolve DNA bands. T7EI recognizes and cleaves mismatched DNA which arises from hybridization of wildtype and mutant DNA strands. T7EI cleavage occurs specifically at the mismatch site. Each lane of the gel received the reaction products from one embryo. If there were few or no mismatched heteroduplex DNAs because the sgRNA induced few indels in an embryo, then T7EI cleavage products would not be detected in that lane of the gel. Detection of cleavage products would indicate that a significant number of indel mutations had been induced in that individual embryo (Figure 2(a)). We could then tally the fraction of injected embryos that scored positive by this T7EI assay for each tested sgRNA.
Figure 2.

Screening sgRNAs for cleavage activity in vivo. (a) Schematic of the screening assay. Individual embryos are injected with RNPs composed of a particular sgRNA. Genomic DNA from each embryo is PCR-amplified, and amplicons are denatured and re-annealed. Heteroduplexes with mismatches due to indels in embryonic DNA are cleaved by T7EI enzyme. Gel electrophoresis identifies embryos with detectable cleavage events. (b) PCR products of a target site in the forked gene 892 bp in length were digested by T7EI as indicated. Shown are two representative embryos out of the nine assayed that were injected with forked RNPs. Also shown are two out of the six embryos that were uninjected. The predicted T7EI digest products are 393 and 436 bp (arrows). Although a minority of heteroduplexes derived from an embryo are T7EI-sensitive, they can be detected by this assay. A total of 6 out of 9 injected embryos showed evidence of T7EI sensitivity, whereas 0 out of 6 control embryos showed evidence. This difference is statistically significant (p = 0.0168, Fishers exact test). (c) A T7EI assay was performed using a different sgRNA that targets non-coding DNA. T7EI digestion products of sizes 295 and 502 bp (arrows) would be detected if indels are significantly generated. Since a background band runs at 500 bp, only detection of a band at 295 bp would be diagnostic for the presence of indels. None of the RNP-treated or uninjected control samples exhibit a detectable 295 bp digestion product. In sum, no 295 bp product was detected for 12/12 embryos treated with RNPs and 6/6 control embryos (p > 0.05, Fishers exact test)
We injected RNPs assembled from the forked sgRNA into syncytial embryos. T7EI reactions of amplicon heteroduplexes from nine embryos were run on an agarose gel, revealing that six of the nine samples produced detectable cleavage products (Figure 2(b)). T7EI treatment of PCR amplicons from six uninjected embryos resulted in no detectable cleavage products for the embryos tested. This difference in outcome depending on the sgRNA was statistically significant (p = 0.0168, Fisher's exact test). Thus, injection of RNPs into syncytial embryos is sufficient to induce NHEJ events that are frequent enough to be detected in late-stage embryos. We validated the method by testing 18 other sgRNAs targeting five different genes. For each sgRNA, we injected and analysed a minimum of eight single embryos and analysed control uninjected embryos in parallel. The percentage of embryos with detectable cleavage events depended on the sgRNA, suggesting that sgRNA activity is quite variable. Of the total 19 sgRNAs tested, 5 sgRNAs (26%) failed to yield any embryos with detectable NHEJ events, and were statistically indistinguishable from control uninjected embryos (p > 0.05, Fisher's exact test). An example of one such negative test is shown in Figure 2(c). Note that DNA from the uninjected control samples generated multiple T7EI digestion products. T7E1 is a bacteriophage enzyme that naturally recognizes and resolves Holliday junctions [26]. It also cleaves a range of DNA structures including DNA mismatches. However, resolvases also cleave DNA where there is a long tract of A residues [27], meaning that they will generate background digestion products from DNA substrates if there are A-tracts or other unusual DNA structures present. Indeed, we mapped the cleavage sites of the control DNA in Figure 2(c) to three sites that contained A-tracts that were 18 to 37 bases long. No other A-tracts were present in the DNA substrate. Note that the presence of background digestion products is not a consequence of poor sgRNA efficacy, and such background bands can also be seen in samples with successful sgRNA-induced indels.
We confirmed that one of the failed sgRNAs was inactive for HDR-mediated editing by using it to induce DSBs in Cas9 transgenic lines, accompanied by a repair template plasmid. Injection of ~900 G0 embryos failed to elicit a single HDR event. In contrast, four other sgRNAs that were positive in the T7EI assay and were then used for HDR-mediated genome editing, resulted in successful germline transmission of the desired genome edits. From these experiments, we conclude that a significant fraction of sgRNAs are inactive in Drosophila despite being selected by computational prediction programs. These findings are consistent with studies in cell culture and mammals [16,28].
A broadly applicable repair template vector that incorporates a novel transformation marker
The injection of RNPs clearly provides benefits for rapid selection of active sgRNAs and for broader application to genetically diverse Drosophila backgrounds. However, the potentially broader applications are limited by the use of existing repair template vectors. The scarless 3xP3-DsRed donor vector should work across Drosophila and other insect genera owing to the fact that 3xP3-DsRed can function in many insect species [29]. However, the counter-selection marker mini-white gene only functions in white mutant genetic backgrounds. Thus, counter-selection for imprecise HDR events is not possible when using mini-white in other backgrounds or species with a wildtype white locus.
We have developed an alternative counter-selection marker to be used either independently or in conjunction with the scarless 3xP3-DsRed vector that will work in any genetic background and does not require the use of fluorescent microscopy to score. The counter-selection marker gene is composed of the GMR promoter driving a short hairpin RNA (shRNA) against the eyes absent (eya) gene, taken from the TRiP collection (HMS04515) (Figure 3(a)). Eya is essential for proper compound eye development, and its loss results in small rough eyes [40]. Since the GMR promoter is specifically active in compound eye cells, the eya gene should be knocked down by RNAi and generate small eyes. As proof of principle, we tested the effect of one copy of the GMR-eya(shRNA) marker on the adult eye phenotype and found that it produced a 100% penetrant small-eyed phenotype. Notably, enough residual eye tissue is present in heterozygous flies to allow scoring of white+ or 3XP3 fluorescent eye markers. We tested the general utility of this marker by constructing a new fourth chromosome balancer by insertion of GMR-eya(shRNA) into the gat gene on the fourth chromosome. This balancer, GATeya, has a morphological phenotype more robust and easier to score than existing fourth chromosome balancers, and is also much healthier than the commonly used CiD balancer. Stocks of the new balancer have been deposited at the Bloomington Drosophila Stock Centre (GATeya/CiD, BDSC #90852; GATeya/CrkdsRed, BDSC #90851).
Figure 3.

Identifying precise HDR edits by eye colour. (a) Shown is the transgenic marker for counterselection of imprecise HDR events. The GMR element contains 5 tandem binding sites for the transcription factor Glass fused to the Hsp70 minimal promoter. The transcript contains a shRNA stem-loop followed by an intron from the ftz gene to facilitate transcript stability. After the shRNA is processed by Drosha and Dicer, the guide RNA strand is loaded into RISC. The guide RNA is perfectly complementary to all mRNA isoforms of eya. Shown only is isoform C, and the location of the RNAi target is indicated. (b-g) Eye phenotypes of adults that had been injected with RNPs and the forked HDR donor plasmid. (b,d,f) Eyes visualized with brightfield illumination. (c,e,g) Same eyes visualized for DsRed fluorescence. All eyes are oriented anterior left and dorsal top. Listed above panels are the number of G0 lines that produced G1s with the same phenotypes as the ones shown. (b,c) Adult without apparent HDR event. (d,e) Adult with DsRed expression and no eya RNAi phenotype. (f,g) Adult with DsRed expression and an eya RNAi phenotype
To construct a counter selectable repair template backbone, the GMR-eya(shRNA) marker was inserted into pBlueScript to create the pBS-GMR-eya(shRNA) plasmid (Addgene #157991; Drosophila Genomic Resource Centre #1518). Upon linearization at its multi-cloning site, typically with EcoRV, the pBS-GMR-eya(shRNA) plasmid serves as the backbone for construction of a donor plasmid. Using Gibson assembly, the scarless 3xP3-DsRed transformation marker flanked by 1 kb homology arms and desired genomic sequence modification can be assembled with the pBS-GMR-eya(shRNA) backbone. Thus, precise HDR events can be discriminated from integration of the entire plasmid into the genome by the presence of DsRed fluorescence and the absence of a small eye phenotype. An additional benefit to replacing the mini-white gene with GMR-eya(shRNA) is that mini-white, whose size belies its name, is ~2,800 bp while GMR-eya(shRNA) is only 820 bp. This reduces the donor plasmid size, making it easier to construct repair templates.
We tested the efficacy of the pBS-GMR-eya(shRNA) vector by placing left and right homology arms targeting the forked gene on either side of 3xP3-DsRed. The HDR donor plasmid was designed to induce insertion of 3xP3-DsRed into the coding region of forked, thus disrupting gene function. We injected 461 white embryos with the HDR donor plasmid and Cas9 protein complexed with the forked sgRNA used previously. Of the 50 fertile G0 adults that emerged after injection, three produced G1 offspring that expressed DsRed in eyes that were normal sized (Figure 3(b-e)), while five produced G1 offspring that expressed DsRed in small eya-like eyes (Figure 3(f,g)). Normal-sized eyes with DsRed fluorescence in the G1 offspring indicate precise HDR of the target gene. Nine G1 adults met these criteria. Another nine G1 adults displayed small eyes that expressed DsRed, presumably the result of imprecise HDR events. G1 adults with normal-sized DsRed-expressing eyes were testcrossed to a forked stock, and six crosses generated 100% penetrant forked mutant phenotypes. Sequence analysis showed repair with both homology regions matching the donor plasmid sequence, confirming that we had precisely introduced the DsRed marker in the six lines.
As another test, we targeted the essential gene crk on the fourth chromosome. We established a DsRed positive, non-small-eyed line in which the crk coding sequence was replaced by the 3xP3-DsRed marker, thereby creating the first fluorescently marked fourth chromosome balancer that we are aware of (CrkdsRed, BDSC#90850 and #90851).
Other loci were independently targeted with donor plasmids and sgRNAs, and in four out of the five targeted, some percentage of DsRed-positive G1 adults had small eya-like eyes (Table 1). The per cent ranged from 9% to 49% depending on the target. Overall, including results from the forked editing, an average of 29% of edited germline events were imprecise. Thus, pBS-GMR-eya(shRNA) is an effective and useful counter-selection marker for HDR-mediated editing.
Table 1.
Scoring for precise HDR events using eya(shRNA) counterselection
| HDR Construct | Number injected | Number fertile G0 adults | Number G1 DsRed+ eya+ | Number G1 DsRed+ eya(shRNA) |
|---|---|---|---|---|
| 1 | 342 | 84 | 52 | 5 |
| 2 | 340 | 70 | 27 | 26 |
| 3 | 344 | 95 | 12 | 4 |
| 4 | 295 | 52 | 18 | 0 |
| 5 | 310 | 64 | 14 | 10 |
Discussion
We have developed tools and protocols to implement a two-step genome editing workflow suitable for making precise changes at targeted sites in Drosophila (Figure 4). Detailed protocols for each step in the workflow are available in the Supplementary Information, and plasmid reagents have been deposited in Addgene and the Drosophila Genome Resource Centre (DGRC). Our workflow adds to the available methods that do not leave scars in the genome, such as those that occur when ablating PAM sites or using integrase-mediated excision to remove selectable markers. Injection of Cas9-sgRNA molecular complexes is several fold more efficient at generating DSBs than vasa-Cas9. Injection of RNPs is also well-suited for making changes in other Drosophila species. This feature realizes the potential for genome editing to make changes at a gene’s native locus and in its native genome. For example, the function of sequences that have diverged between two Drosophila species can now be tested in their native context rather than in D. melanogaster, as had been previously done.
Figure 4.
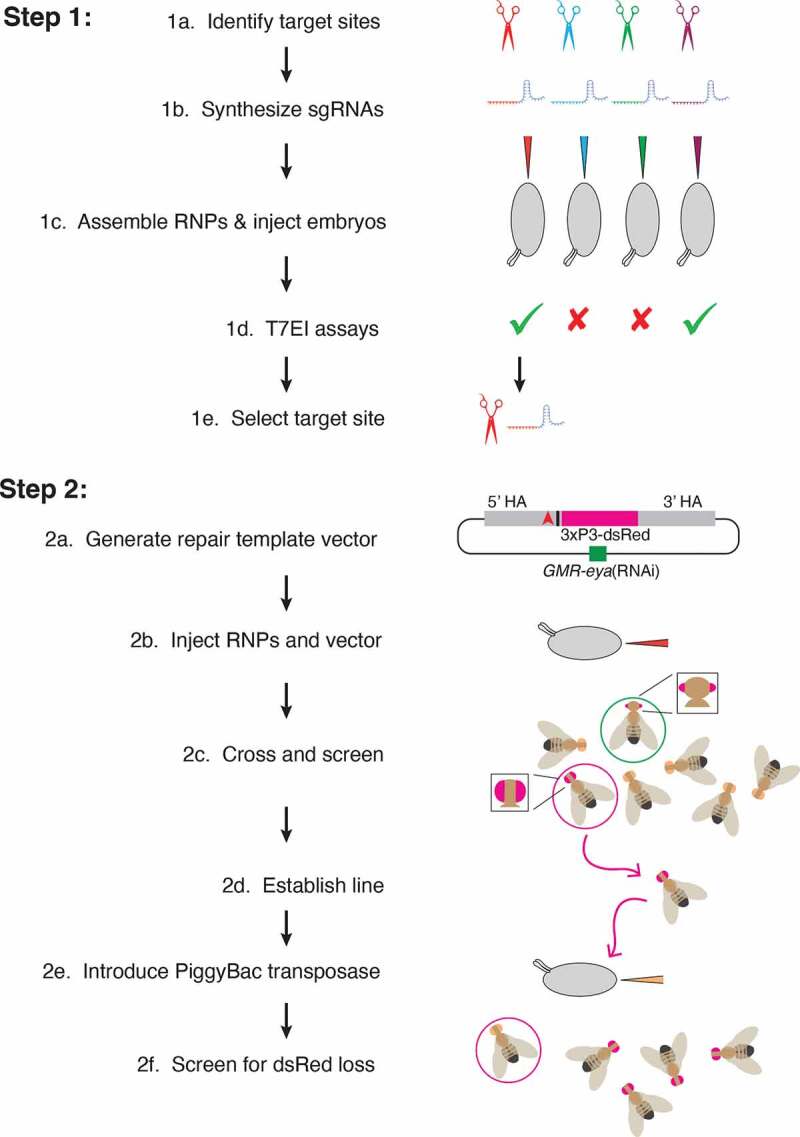
Workflow for two-step genome editing. (1a) Target sites flanking the area to be edited are identified (red, blue, green, purple) using online tools searching for optimal targets and with minimal off-target cleavage. (1b) Sequences from the selected target sites are transcribed in vitro to generate sgRNAs. (1c) Cas9 protein is incubated with sgRNAs before injection into embryos. (1d) Active sgRNAs that cleave embryo DNA are identified by T7 endonuclease I reactions. (1e) One of the active sgRNAs is chosen for genome editing in Step 2. (2a) Homology arms flanking the region of interest are cloned into the pBS-GMR-eya(shRNA) donor plasmid. In this example, the CRISPR target site (red triangle) is 5ʹ to the bases to be edited (black bar). (2b) Embryos are injected with the repair template plasmid and RNPs composed of sgRNA and Cas9 protein. (2c) Adult flies that develop from injected embryos are crossed back to the parental line. G1 progeny are screened for the DsRed marker. Positive G1 animals may have small eyes due to eya(shRNA) but these are not selected (green circle). Only positive G1 animals with normal eyes are selected (red circle). (2d) These are crossed to make purebred lines and molecularly analysed to determine if they contain the desired editing events. (2e) PiggyBac transposase is expressed in the germline, either by a single cross to a transgenic line, or in this example, by embryo injection of a plasmid expressing the transposase. (2f) Since the DsRed marker is dominant, adult flies developing from injected embryos that do not have red fluorescent eyes are then crossed and analysed with molecular tests to determine whether they have precisely excised the marker gene. Only the intended genomic edit remains
The first step in the workflow is the design and testing of candidate sgRNAs. Since sgRNAs vary in their ability to induce DSBs in genomes, it is worthwhile to test a number of sgRNAs for making the desired edit. We found that 26% of tested sgRNAs are inactive in Drosophila embryos. Therefore the first step in the workflow is to rapidly synthesize a few sgRNAs by in vitro transcription, followed by assembly of RNPs with Cas9 protein. A simple PCR-based endonuclease assay then measures the ability of each sgRNA to induce NHEJ indel mutations in injected embryos. This procedure takes at most 3 days and will work in other species besides D. melanogaster. The 3 days effort potentially saves 2 to 3 months of wasted effort to perform HDR using a sgRNA that does not work in vivo. Once a sgRNA is selected, the second step in the workflow is taken. Homology arms, desired genome alterations, and the scarless 3xP3-DsRed marker are Gibson assembled with pBS-GMR-eya(shRNA) such that the arms flank the scarless DsRed marker. This construct is co-injected with matching RNPs into the strain or species of choice, and G1 individuals with red fluorescent eyes are selected. Although it is easier to screen such individuals in a white mutant background, it is not necessary. Red fluorescence can be detected in the adult eyes of white+ D. melanogaster expressing 3xP3-DsRed, although fluorescence is only visible in a small spot of 10 ommatidia (data not shown and [29]). This property of 3xP3-DsRed fluorescence is also observed in other Drosophila species [30]. If compound eye fluorescence is too weak, the adult ocelli or larval Bolwigs organs can also be examined [29,31].
Our method uses a novel marker placed in a pBluescript backbone to select against editing events in which the entire vector has integrated into the genome. The marker uses RNAi to knock down expression of the endogenous eya gene, resulting in a small eye. The eya shRNA is designed to work in any D. melanogaster strain and will work in the closely related D. erecta species. D. simulans, yakuba, and ananassae have only single base variant in the sequence targeted by RNAi, so the backbone can easily be modified via site-directed mutagenesis for these species. Since we have found that 29% of DsRed transformants also have eya(RNAi) phenotypes, our method reduces the laborious molecular characterization of false-positives.
There are added potential functions for the GMR-eya(shRNA) gene in that its insertion into any chromosome renders a simple dominant phenotype that is highly penetrant. Thus, it can be used to mark any balancer chromosome. We have done so for the fourth chromosome balancer, and this new balancer (GATeya) is deposited at the Bloomington Drosophila Stock Centre. It should be possible to use CRISPR/HDR to insert GMR-eya(shRNA) in other balancers with subtle dominant markers, such as TM6,Ubx.
The final step in the workflow is the scarless excision of 3xP3-DsRed from the genome by PiggyBac-mediated transposition. Scarless editing requires the presence of a TTAA motif at the target locus, which adds some restriction to target selection. However, it circumvents the need for a second round of injections because D. melanogaster carrying the marker can be crossed to existing transgenic lines that express the PiggyBac transposase [31]. To apply 3xP3-DsRed excision to species other than D. melanogaster, a subsequent injection step is needed to introduce the PiggyBac transposase. There are appropriate expression plasmids that are freely available [32].
In conclusion, the applicability of our method to many types of experiments in a wide variety of genetic backgrounds makes it a valuable addition to the existing methods and tools for scarless genome editing available to the Drosophila research community.
Materials and methods
A detailed protocol and description of reagents are provided in Supplementary Information.
Statistics were performed using Graphpad software.
Drosophila strains
Drosophila was raised on standard cornmeal-molasses food at room temperature. The vasa-Cas9 strain (BDSC#51324) used for some experiments is from [10]. This transgene is located on the third chromosome and has been shown to have slightly weaker efficiency than some other Cas9 transgenes [11]. Unless otherwise stated, all injections of CRISPR RNPs, sgRNAs, and donor plasmids were into a w1118 strain.
Plasmids
pBS-GMR-eya(shRNA) is derived from pBSII-KS(-), and contains a marker gene GMR-eya(shRNA), which is comprised of the Glass Multimer Reporter (GMR) enhancer driving an eya(shRNA) from the Transgenic RNAi Project (TRiP) [33]. GMR is a synthetic eye-specific enhancer composed of five tandem repeats of a 29 bp element from the Rh1 gene [34]. Tests using GMR-Gal4 to drive UAS-eya(shRNA) from the TRiP lines HMS04515 and HM05716 revealed that HMS04515 produced a stronger eye phenotype than HM05716. Therefore, the eya(shRNA) sequence from HMS04515 was used to construct the GMR-eya(shRNA) marker gene. This shRNA targets all three transcripts of the eya gene. A PCR fragment containing the GMR enhancer and the hsp70 minimal promoter was amplified from pGMR DNA [35] with XhoI and XbaI ends. This fragment was inserted into a version of pKanC5 [36] in which a linker containing XhoI and AvrII sites had been inserted between the BbvI and AscI sites. The GMR cassette was then shuttled into the HMS04515 Valium20 eya(shRNA) plasmid, inserted between the StuI and XbaI sites using Gibson assembly [37]. This replaced the UAS enhancer and upstream Gypsy insulator with the GMR cassette. The ~440 bp Gypsy insulator was eliminated to minimize the final size of the gene. The efficacy of the resulting GMR-eya(shRNA) Valium construct was tested by integrating it into the genome by φC31-mediated recombination [38]. Once validated, the GMR-eya(shRNA) cassette was amplified by PCR from the Valium plasmid, and inserted into the KpnI site of pBSII-KS(-) using Gibson assembly [37]. The resulting pBS-GMR-eya(shRNA) plasmid has been deposited with AddGene (#157991) and the Drosophila Genomics Resource Centre (#1518). Its annotated sequence is found in Supplementary File 1.
To construct an HDR donor plasmid, 3xP3-DsRed and homology arms flanking the targeted region of interest were PCR amplified to generate overlapping regions of homology for cloning into pBS-GMR-eya(shRNA) via Gibson Assembly [37]. The 3xP3-DsRed marker gene from the pScarlessHD-DsRed plasmid (Addgene #64703) was PCR amplified. It is flanked by piggyBac TTAA transposition sites that can be used to cleanly remove the entire marker gene after successful integration of the modification via HDR. The ~1 kb 5′ homology arm was PCR amplified from D. melanogaster with a region homologous to the pBS-GMR-eya(shRNA) vector on the 5′ end. The ~1 kb 3′ homology arm was amplified from D. melanogaster with a region homologous to the pBS-GMR-eya(shRNA) vector on the 3′ end. The PCR amplicons were appended with sequence corresponding to sequence at a native TTAA site in the genome near the sgRNA site (ideally less than 30 bp) or within a TTAA site in the intended modification. A DNA fragment encompassing the sequence modification of interest was synthesized using gBlocks (IDT) with regions appended on both ends to facilitate Gibson assembly. These DNA fragments, the amplicons, and the pBS-GMR-eya(shRNA) vector linearized with EcoRV, were altogether assembled using New England Biolabs (NEB) NEBuilder HiFi DNA Master Mix. This generated the HDR donor plasmid. Inserts into the EcoRV site of pBS-GMR-eya(shRNA) can be sequenced using the custom primer 5ʹ-actgggctcgaggcgatc-3ʹ on one side of the EcoRV site and custom primer 5ʹ-ggcggccgctctagaactag-3ʹ or the standard M13-forward and T7 primers on the other side.
The forked HDR donor plasmid was composed of the following elements:
5ʹ homology arm: X:17,269,009 to X:17,270,001 (Flybase release 6)
3xP3-DsRed cassette: 5ʹ-TTAA-(3xP3-DsRed)-TTAA-3ʹ
forked mutagenesis fragment: This fragment of forked (X:17,270,002 to X:17,270,050) spanned from a TTAA site where 3xP3-DsRed was ligated to the sgRNA cut site. We had to introduce several synonymous mutations in the targeting fragment. There were no nearby TTAA sites, and so we created one with a synonymous mutation that changed a TCAA site (X:17,270,002 to X:17,270,005) in the genome to TTAA. We inactivated the sgRNA cut site in the fragment (X:17,270,034 to 17,270,050) with 2 synonymous mutations, as is common practice. The novel TTAA site is 45 bp from the sgRNA cut site. The fragment sequence with the synonymous mutations underlined is:
5ʹ-TTAAGTTTCTGGTGCTCGAGGCCGGCGGCTCTTTGTACGTCCGTGCTCGT-3ʹ
4) 3ʹ homology arm: X:17,270,051 to X:17,271,035 (FlyBase release 6)
sgRNA and RNP preparations
Candidate sgRNAs were identified using flyCRISPR Optimal Target Finder (http://targetfinder.flycrispr.neuro.brown.edu). High stringency filtering was used, and only NGG PAM sites were considered. Potential off-target sites were minimized to 0 predicted off-target sites. The sgRNA target site sequence was validated in the particular Drosophila strain being injected. This was done by Sanger sequencing the site from the strain’s genomic DNA.
The sgRNAs were synthesized by in vitro transcription using T7 RNA polymerase. Transcription templates were created by PCR using a high fidelity polymerase. PCR amplification used the plasmid pU6-BbsI-chiRNA as template [6]. pU6-BbsI-chiRNA was a gift from M. Harrison, K. O’Connor-Giles and J. Wildonger (RRID:Addgene_45946). The forward primer contained the T7 promoter sequence at the 5ʹ end followed by the sgRNA sequence (without the PAM) and then 19 bases complementary to the plasmid at the 3ʹ end. The complementary site on the plasmid corresponds to the 5ʹ end of the sgRNA scaffold at position 590.
5ʹ-TTAATACGACTCACTATAGG[sgRNA_sequence]GTTTTAGAGCTAGAAATAG-3ʹ
The reverse primer was a 20 base oligonucleotide complementary to the 3ʹ end of the sgRNA scaffold ending at position 669.
5ʹ-AAAAGCACCGACTCGGTGCC-3ʹ
The PCR amplicon was used in a MEGAscript in vitro transcription reaction (ThermoFisher #AM1333) supplemented with 0.5 µL Ribolock RNase inhibitor (ThermoFisher #EO0381) at 37°C overnight. The resulting sgRNA was purified with a Monarch RNA Purification Kit (NEB).
sgRNA-Cas9 RNPs were prepared fresh immediately before injecting embryos. 2.36 µg sgRNA was incubated with 11.9 µg Cas9-NLS recombinant protein (IDT #1081058) in 5 µL of 150 mM KCl. They were incubated at room temperature for 10 min followed by centrifugation at maximum speed for 10 min at room temperature. Supernatant was then loaded into injection needles.
The sequence of the sgRNA synthesized to target the forked gene was: 5ʹ-UUGUACGUCCGUGCACGCGA-3ʹ. It corresponds to X:17,270,034 to 17,270,053.
Drosophila embryo injections
Injections were performed in pre-cellularized embryos without dechorionation using Gompel and Schröder’s method (http://gompel.org/wp-content/uploads/2015/12/Drosophila-transformation-with-chorion.pdf). After injection, any embryos that were skipped during injection due to age or other defects were ruptured with a needle. Then, as much halocarbon oil was removed from the coverslip holding the embryos as possible. For T7EI assays, the coverslip with embryos was placed on an egg-laying plate, and the plate was incubated in a humid chamber at 25°C for 24 hours. For HDR editing, the coverslip with embryos was placed in a standard fly vial.
For injections to induce NHEJ, only freshly prepared sgRNA-Cas9 RNPs were injected. To induce HDR edits, RNPs were assembled as described above except donor plasmid DNA was also added to the reaction. 2.36 µg sgRNA was incubated with 11.9 µg Cas9-NLS recombinant protein (IDT #1081058) plus 0.6 pmoles plasmid DNA in 5 µL of 150 mM KCl. They were incubated at room temperature for 10 min followed by centrifugation at maximum speed for 10 min at room temperature. Supernatant was then loaded into injection needles.
Assay for sgRNA-Cas9 mediated DNA cleavage
Injected embryos were individually extracted using a single-fly genomic prep (https://kumarlab.bio.indiana.edu/7_labresources/protocols/016%20Single%20Fly%20Genomic%20DNA%20Extraction.pdf). L1 larvae or late-stage embryos only were chosen, indicating survival of the injection process. 3–5 µL genomic DNA was used as a template for a 50 µL PCR reaction in which primers were used that bounded the CRISPR target site being assayed. The amplicon was designed to be ~700 – 1200 bp in length with the target site located close to the centre of the amplicon. The PCR product from one embryo was heat-denatured and slowly allowed to re-anneal. 10 µL re-annealed PCR product was digested with 2 units T7 Endonuclease I (NEB #M0302L) in NEBuffer 2 for 60 min at 37°C. Reaction products were run on a 2% (w/v) agarose gel with ethidium bromide and 0.5X TBE. Samples with cleavage products at expected sizes often showed cleavage bands that were very faint.
Genetic screening
To screen for the presence of the GMR-eya(shRNA) marker, we examined G1 adults under standard dissecting microscopes for presence of small rough eyes. Such animals were annotated and then discarded. To screen G1 adults for the DsRed marker, we used a Nikon SMZ 1500 stereoscope equipped with a Fluorescence Illuminator. DsRed expression from the reporter gene used in this study becomes easily detectable in white eyes of adults. If the eyes have normal pigmentation, then the DsRed fluorescence is difficult to visualize except for a spot of ~10 ommatidia within the overall eye. Positive G1 adults were crossed to an appropriate balancer line. From these balancer crosses, siblings with both the DsRed phenotype and the balancer phenotype were crossed to form homozygous or balanced lines.
To precisely excise the DsRed marker, lines were crossed to a stock expressing the piggyBac transposase. The piggyBac transposase transgene is under control of the α-tubulin promoter and is tightly linked to a 3XP3-CFP transgenic marker [39]. This is located on chromosome 2 (Bloomington Stock Centre #32070). The stock also contains third chromosome balancers (MKRS/TM6B,Tb), facilitating tracking of the third chromosomes independent of the piggyBac transposase. Heterozygous offspring were crossed to appropriate balancer strains, and their offspring were screened for absence of DsRed and CFP fluorescence in adult eyes. Excision of DsRed occurs about 10% of the time. Positive animals were again crossed to a balancer strain to establish balanced stocks.
Molecular screening
To confirm integration of the Ds-Red marker gene into the correct genomic location, we used a PCR reaction that amplifies DNA sequence from within the reporter gene sequence to outside the homology region on both the 5′ and 3′ sides of the reporter gene. The amplicons from these PCR reactions were Sanger sequenced to confirm scarless repair at both the target sites and throughout both homology regions. To screen for correct HDR after the DsRed excision, the entire edited locus was amplified via PCR using primers outside the homology regions. The amplicon was Sanger sequenced to confirm the presence of expected sequence edits. All diagnostic PCRs were performed using genomic DNA extracted from single flies following the squish prep protocol (https://kumarlab.bio.indiana.edu/7_labresources/protocols/016%20Single%20Fly%20Genomic%20DNA%20Extraction.pdf).
Gateya and CrkdsRed fourth chromosome balancers
The Gateya balancer fourth chromosome was created by targeting the Gat locus using the two sgRNAs detailed below and an HDR template constructed by Gibson assembly of 3xP3-DsRed flanked by Gat 5ʹ and 3ʹ homology arms in the pBS-GMR-eya(shRNA) backbone. Homologous repair from this template should result in deletion of all but the first 10 amino acids of the Gat open reading frame. Of three DsRed positive lines established from vasa-Cas9 G0 animals (BDSC stock #55821), two were precise repair events (Eya+ eyes) and one was an integration event (eya eyes). An eya line was established from the G0 with the integration event. This line was then crossed to the Piggybac transposase line to excise the DsRed transformation marker. A single F1 male lacking 3xP3-DsRed was used to establish the Gateya chromosome. Neither the nature of the original insertion event nor the molecular nature of the Gateya mutation produced by the dsRed excision has been investigated. The vasa-Cas9, piggyBac transposase, other markers and potential off-target mutations were removed from the background by backcrossing the Gateya chromosome against w1118 for five generations. The Gateya chromosome serves as a fourth chromosome balancer because there is essentially no recombination on the fourth chromosome, the Gateya mutation causes a dominant small-eye phenotype, and it is recessive lethal (1,435 out of 1,435 adults in a stock of Gateya/CrkdsRed were DsRed+, eya RNAi).
The assembled order of the circular Gat/Scarless dsRed HDR template construct was: pBS eya(shRNA) backbone … Gat 5ʹ homology arm … 3xP3-DsRed … Gat 3ʹ homology arm … pBS eya(shRNA) backbone. The sequences of the primers used to create that Gat targeting construct were (Gat sequences underlined):
Gat 5ʹ homology arm forward: 5ʹ – CCGGGCTGCAGGAATTCGATCAGGATCAATAGCCAAGTCGATCT – 3ʹ
Gat 5ʹ homology arm reverse: 5ʹ – CTTTAACGTACGTCACAATATGATTATCTTTCTAGGGTTAAGTCACCATCGCTTGCGGA – 3ʹ
Gat 3ʹ homology arm forward:
5ʹ – GAGCAATATTTCAAGAATGCATGCGTCAATTTTACGCAGACTATCTTTCTAGGGTTAAAGTGGTATGCCAGAAATATCTAG – 3ʹ
Gat 3ʹ homology arm reverse: 5ʹ – CGACGGTATCGATAAGCTTGATCATATTCACTCTTGTGAATAGACAC – 3ʹ
To construct Gat sgRNA plasmids, the following oligonucleotides were annealed and ligated into pU6-BbsI-chiRNA (RRID:Addgene_45946) to create two plasmids that produce sgRNAs targeting the 5ʹ and 3ʹ regions of the Gat gene:
Gat sgRNA1 forward: 5ʹ- CTTCGCCGCAAGCGATGGTGACGG-3ʹ
Gat sgRNA1 reverse: 5ʹ- AAACCCGTCACCATCGCTTGCGGC −3ʹ
Gat sgRNA2 forward 5ʹ- CTTCGTTGTCGTACTTACTTAAAG-3ʹ
Gat sgRNA2 Reverse 5ʹ- AAACCTTTAAGTAAGTACGACAAC-3ʹ
The CrkdsRed fourth chromosome balancer was created by targeting the Crk locus with an HDR template composed of the 3xP3-DsRed marker flanked by Crk 5ʹ and 3ʹ homology arms (see below) in the pBS-GMR-eya(shRNA) backbone. Homologous repair from the HDR template should result in the replacement of almost the entire open reading frame region of the Crk gene with the DsRed gene, from 48 bp 5ʹ of the start codon to 121 bp 5ʹ of the TAA stop codon. Two DsRed Eya+ G1’s were identified from vasa-Cas9 G0 animals. One line was established from a single G1 male to found the CrkdsRed chromosome. The precise nature of the repair event that created the CrkdsRed mutation has not been investigated. The vasa-Cas9 and potential off-target mutations were removed from the background by backcrossing the CrkdsRed chromosome against w1118 for five generations. The CrkdsRed chromosome serves as a fourth chromosome balancer because there is essentially no recombination on the fourth chromosome, the CrkdsRed mutation causes a dominant eye phenotype, and it is recessive lethal.
The assembled order of the HDR template construct is: pBS eya(shRNA) backbone … Crk 5ʹ homology arm … 3xP3-DsRed … Crk 3ʹ homology arm … pBS eya(shRNA) backbone. The sequences of the primers used to create the Crk targeting construct were (Crk sequences underlined):
Crk 5ʹ homology arm forward: 5ʹ – CCGGGCTGCAGGAATTCGATATTTTTGATCCTAGCTTCAAAATCT – 3ʹ
Crk 5ʹ homology arm reverse: 5ʹ – CTTTAACGTACGTCACAATATGATTATCTTTCTAGGGATAAATAGAAATTATGTGATATAATGCAAATATA – 3ʹ
Crk 3ʹ homology arm forward:
5ʹ – GAGCAATATTTCAAGAATGCATGCGTCAATTTTACGCAGACTATCTTTCTAGGGAATTGGAAATAGGTGACATTATTAAAGTCA – 3ʹ
Crk 3ʹ homology arm reverse: 5ʹ – CGACGGTATCGATAAGCTTGATAGAAGCACTAACTAACTATTGATCTAAAGAT – 3ʹ
To construct Crk sgRNA plasmids, the following oligonucleotides were annealed and ligated into pU6-BbsI-chiRNA targeting the 5ʹ and 3ʹ regions of the Crk gene:
Crk sgRNA1 forward: 5ʹ- CTTCGAATTTCTATTTATTTAATC −3ʹ
Crk sgRNA1 reverse: 5ʹ- AAACGAsTTAAATAAATAGAAATTC −3ʹ
Crk sgRNA2 forward 5ʹ- CTTCGGATAAGACTGCATTAAAAT −3ʹ
Crk sgRNA2 Reverse 5ʹ- AAACATTTTAATGCAGTCTTATCC −3’
Supplementary Material
Funding Statement
Fly stocks from the Bloomington Drosophila Stock Center are gratefully appreciated. Plasmids were from the Drosophila Genomics Resource Center. The DGRC is supported by a grant from the National Institutes of Health. Number NIH grant 2P40OD010949 .Funding was provided from the NIH to the authors (F32GM122349, K.G.N.; R01GM108964, G.J.B.; R35GM118144, R.W.C.)
Disclosure statement
The authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Komor AC, Badran AH, Liu DR.. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rees HA, Liu DR.. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19:770–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Klompe SE, Vo PLH, Halpin-Healy TS, et al. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature. 2019;571:219–225. [DOI] [PubMed] [Google Scholar]
- [4].Strecker J, Ladha A, Gardner Z, et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bassett AR, Tibbit C, Ponting CP, et al. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013;4:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gratz SJ, Cummings AM, Nguyen JN, et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yu Z, Ren M, Wang Z, et al. Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics. 2013;195:289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee JS, Kwak S-J, Kim J, et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3 (Bethesda). 2014;4:1291–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kondo S, Ueda R. Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics. 2013;195:715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gratz SJ, Ukken FP, Rubinstein CD, et al. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics. 2014;196:961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Port F, Chen HM, Lee T, et al. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc Natl Acad Sci U S A. 2014;111:E2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sebo ZL, Lee HB, Peng Y, et al. A simplified and efficient germline-specific CRISPR/Cas9 system for Drosophila genomic engineering. Fly (Austin). 2014;8:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xue Z, Ren M, Wu M, et al. Efficient Gene Knock-out and Knock-in with Transgenic Cas9 in Drosophila. G3 (Bethesda). 2014;4:925–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu Z, Chen H, Liu J, et al. Various applications of TALEN- and CRISPR/Cas9-mediated homologous recombination to modify the Drosophila genome. Biol Open. 2014;3:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang X, Koolhaas WH, Schnorrer F. A versatile two-step CRISPR- and RMCE-based strategy for efficient genome engineering in Drosophila. G3 (Bethesda). 2014;4:2409–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Port F, Bullock SL. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat Methods. 2016;13:852–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gratz SJ, Rubinstein CD, Harrison MM, et al. CRISPR-Cas9 genome editing in Drosophila. Curr Protoc Mol Biol. 2015;111:31 32 31–31 32 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bier E, Harrison MM, O’Connor-Giles KM, et al. Advances in Engineering the Fly Genome with the CRISPR-Cas System. Genetics. 2018;208:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li-Kroeger D, Kanca O, Lee P-T, et al. An expanded toolkit for gene tagging based on MiMIC and scarless CRISPR tagging in Drosophila. Elife. 2018;7. DOI: 10.7554/eLife.38709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee PT, Zirin J, Kanca O, et al. A gene-specific T2A-GAL4 library for Drosophila. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lamb AM, Walker EA, Wittkopp PJ. Tools and strategies for scarless allele replacement in Drosophila using CRISPR/Cas9. Fly (Austin). 2017;11:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lin S, Staahl BT, Alla RK, et al. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3:e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liang X, Potter J, Kumar S, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53. [DOI] [PubMed] [Google Scholar]
- [25].Prior H, Jawad AK, MacConnachie L, et al. Rapid and Co-CRISPR-independent genome editing in caenorhabditis elegans. G3 (Bethesda). 2017;7:3693–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Babon JJ, McKenzie M, Cotton RG. The use of resolvases T4 endonuclease VII and T7 endonuclease I in mutation detection. Mol Biotechnol. 2003;23:73–81. [DOI] [PubMed] [Google Scholar]
- [27].Bhattacharyya A, Murchie AIH, von Kitzing E, et al. Model for the interaction of DNA junctions and resolving enzymes. J Mol Biol. 1991;221(4):1191–1207. . [DOI] [PubMed] [Google Scholar]
- [28].Jang DE, Lee JY, Lee JH, et al. Multiple sgRNAs with overlapping sequences enhance CRISPR/Cas9-mediated knock-in efficiency. Exp Mol Med. 2018;50(4):16. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Horn C, Schmid BG, Pogoda FS, et al. Fluorescent transformation markers for insect transgenesis. Insect Biochem Mol Biol. 2002;32:1221–1235. [DOI] [PubMed] [Google Scholar]
- [30].Stern DL, Crocker J, Ding Y, et al. Genetic and transgenic reagents for Drosophila simulans, D. mauritiana, D. yakuba, D. santomea, and D. virilis. G3 (Bethesda). 2017;7:1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Griswold CM, Roebuck J, Anderson RO, et al. A toolkit for transformation and mutagenesis in Drosophila using piggyBac. Drosophila Inf Serv. 2002;85:129–132. [Google Scholar]
- [32].Handler AM, Harrell RA 2nd. Germline transformation of Drosophila melanogaster with the piggyBac transposon vector. Insect Mol Biol. 1999;8:449–457. [DOI] [PubMed] [Google Scholar]
- [33].Ni JQ, Zhou R, Czech B, et al. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods. 2011;8:405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ellis MC, O’Neill EM, Rubin GM. Expression of Drosophila glass protein and evidence for negative regulation of its activity in non-neuronal cells by another DNA-binding protein. Development. 1993;119:855–865. [DOI] [PubMed] [Google Scholar]
- [35].Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. [DOI] [PubMed] [Google Scholar]
- [36].Le T, Yu M, Williams B, et al. CaSpeR5, a family of Drosophila transgenesis and shuttle vectors with improved multiple cloning sites. Biotechniques. 2007;42(164):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gibson DG, Smith HO, Hutchison CA 3rd, et al. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 2010;7:901–903. [DOI] [PubMed] [Google Scholar]
- [38].Venken KJ, He Y, Hoskins RA, et al. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. [DOI] [PubMed] [Google Scholar]
- [39].Horn C, Offen N, Nystedt S, et al. piggyBac-based insertional mutagenesis and enhancer detection as a tool for functional insect genomic. Genetics. 2003;163:647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Silver SJ, Rebay I. Signaling circuitries in development: insights from the retinal determination gene network. Development. 2005;132:3–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.