

Abstract
The mammalian immune system can tune its inflammatory response to the threat level posed by an invading pathogen. It is well established that the host utilizes numerous ‘patterns of pathogenicity’, such as microbial growth, invasion, and viability, to achieve this tuning during bacterial infections. This review discusses how this notion fits during fungal infection, particularly regarding Aspergillus fumigatus infection. Moreover, how the environmental niches filled by A. fumigatus may drive the evolution of the fungal traits responsible for inducing the strain-specific inflammatory responses that have been experimentally observed will be discussed. Moving forward understanding the mechanisms of the fungal strain-specific inflammatory response due to the initial interactions with the host innate immune system will be essential for enhancing our therapeutic options for the treatment of invasive fungal infections.
Keywords: invasive aspergillosis, invasive fungal infections, Aspergillus fumigatus, fungal pathogenesis, fungal immunology, Innate immunity, inflammation
INTRODUCTION
It is estimated that approximately 2 million cases of life-threatening invasive fungal infections are reported worldwide each year [1]. While hundreds of species exist within fungal genera, only a handful have been shown to cause invasive mycoses in humans. The rapid rise in cases of life-threatening invasive fungal infections during the second half of the 20th century can be attributed to an increase in immunocompromised patients due to the advent of myeloablative chemotherapy, glucocorticoid usage for organ transplantation, and the AIDS pandemic. Moreover, the number of invasive fungal infection cases is predicted to continue to rise due to the sustained growth of the immunocompromised patient populations and the ongoing emergence of drug resistance [1]. However, not all immunocompromised patients develop invasive fungal infections, polymorphisms in numerous innate immune sensing and signaling pathways alter the susceptibility of transplant patients to developing invasive fungal disease (reviewed in [2], [3] and [4]). Additionally, patients with primary immunodeficiencies in the NADPH oxidase complex, STAT3 signaling pathway, CARD9 signaling pathway, leukocyte adhesion deficiencies, and those with severe congenital neutropenia have been shown to be predisposed to developing invasive fungal infections (reviewed in [2] and [5]).
These polymorphisms and primary immunodeficiencies in the Innate immune sensing pathways demonstrate the importance of innate immunity in maintaining resistance against fungal pathogens, including Aspergillus fumigatus. Experimental studies using mouse models have demonstrated a number of these innate immune sensing and signaling pathways are essential for coordinating the inflammatory response to A. fumigatus. Alveolar macrophages are critical for killing inhaled conidia and contribute to the initial antifungal inflammatory response [6–8], although others have shown them to be dispensable [9]. In zebrafish, tissue macrophages are essential for maintaining resistance against only certain A. fumigatus strains, which might explain the discrepancy in the murine studies [10]. Lung-infiltrating CCR2+ inflammatory monocytes are also critical in establishing the inflammatory milieu following A. fumigatus challenge [11, 12]. These cells serve as critical sources of IFNα [13] and IL-1α [14], which regulate the recruitment and activation of neutrophils whose timely recruitment are essential for maintaining host resistance [15]. CCR2+ inflammatory monocytes also regulate recruitment of plasmacytoid dendritic cells who maintain resistance against A. fumigatus through the enhancement of neutrophil antifungal functions [16]. In addition to orchestrating the inflammatory cytokine response, CCR2+ inflammatory monocytes can differentiate into monocyte-derived dendritic cells which are potent antifungal killers themselves [11]. The interplay of macrophage and neutrophil-dependent inflammatory responses is further highlighted in a zebrafish model where macrophages controls slow growing A. fumigatus strains, while neutrophils are critical for controlling rapidly growing strains [10]. Thus, the host can initiate a well-organized innate immune response necessary for the maintenance of host resistance against A. fumigatus.
In this review, we discuss the importance of sensing the growth, viability, and danger posed by these fungal pathogens to tune the inflammatory response to the threat posed by a given isolate. We highlight the importance of environmental niche evolution in the ability of A. fumigatus isolates to drive stress resistance needed for virulence in a host and how this intersects with the induction of the innate immune response. Overall, the balance between these host-pathogen interactions are essential for driving appropriately tuned immune responses to maintain host resistance against fungal pathogens without too much collateral tissue pathology.
STRAIN VARIABLITY: IMPACTS ON PATHOGENTICITY AND IMMUNITY
A. fumigatus is ubiquitously found throughout the environment, flourishing in a wide variety of environmental niches. Each of these environmental niches present their own unique microbial and metabolic pressures. However, the most advantageous traits for surviving in these different environmental niches -- temperature tolerance, UV resistance, protease repertoire, hypoxic tolerance, resistance to amoeba predators, toxin production for inter-microbial competition -- may or may not reflect the pressures found within the mammalian lung.
Studies comparing A. fumigatus isolates have demonstrated that differences in metabolic and stress adaptation impact fungal pathogenesis, as well as the host inflammatory response. In experimental models of invasive aspergillosis, typically, one of two clinical isolates are commonly utilized: Af293 [17] or CEA10 [18]. Af293 is less virulent than CEA10 in immune competent mice [14, 19], immune competent zebrafish [10], and immunosuppressed mice [20]. Importantly, this phenomenon is not restricted to these two laboratory strains of A. fumigatus as a number of clinical and environmental isolates demonstrate a full range of virulence in mouse models [14, 20]. Interestingly, these differences in virulence are not seen in the chemotherapeutic (i.e. neutropenic) murine model of invasive aspergillosis [20]; thus, immunological pressure by neutrophils and metabolic changes within the respiratory environment likely drive the observed virulence differences.
One major pressure found in the lungs is tissue hypoxia which is most prevalent in the immunosuppressed model of invasive aspergillosis [21]. Hypoxic fitness of A. fumigatus isolates correlates with virulence in the immunosuppressed model of invasive aspergillosis [20]. Further experimental support for this correlation is provided by the experimental evolution of the Af293 strain, which has a poor fitness in hypoxic environments, resulting in a novel strain that had increased hypoxic fitness and was significantly more virulence than the parental Af293 strain in the immunosuppressed model of invasive aspergillosis [20]. The genetic basis underlying this phenotypic evolution is dependent on the hrmA gene [22]. Specifically, the hypoxia evolved variant of hrmA results in a transcriptional rewiring of the hypoxic response such that the strain is primed for rapid growth in low oxygen driving increased virulence in the immunosuppressed model of invasive aspergillosis. Moreover, hrmA variants had altered biofilm architecture and cell wall composition, which results in more robust inflammation. The more robust inflammation likely drives strain-specific induction of lung hypoxia due to the increased neutrophil accumulation. In addition to fungal adaptation to hypoxia, host hypoxia signaling in myeloid cells, through the transcription factor HIF-1α, is critical for murine survival in CEA10 [19, 23] but not Af293 infections [19]. Thus, not only does hypoxia impact the fungal biology and virulence but, also, alters the host inflammatory response in a strain-specific manner.
A second major pressure found in the lungs is the limited nutrient availability within the airways. A consistent phenotype is that more virulent strains of A. fumigatus are more rapidly able to undergo conidial germination both in vitro and in vivo [10, 14, 24]. The molecular mechanism behind why certain isolates of A. fumigatus initiate germination more rapidly within the nutrient limited environment of the airways remains unresolved. However, it is apparent that strains which undergo more rapid conidial germination drive a more robust and broader inflammatory response [14, 19]. Not only is the utilization of nutrients likely critical to initiate fungal germination and growth, but the controlled use of carbon sources through CreA-mediated carbon catabolite repression is critical within the hypoxic environment of established infection for disease progression. Specifically, a creA-null mutant is unable to thrive in the low oxygen environment within the host, due to its inability to shift to a more hypoxic-based glycolytic metabolism [25]. Thus, the ability to utilize the limited nutrients found in the lung environment is critical for the growth throughout A. fumigatus infection.
Another major pressure found in the lungs is the host innate immune cells. Typically, A. fumigatus spores are rapidly engulfed and killed by host phagocytes through both oxidative and non-oxidative mechanisms [7]. In the natural environment A. fumigatus may be preyed upon by amoeba. Some of the interactions between A. fumigatus with amoebas resemble those occurring between A. fumigatus and phagocytes, which has led to a hypothesis that amoeboid predation may drive the evolution of animal virulence determinants in the fungi [26, 27]. One such virulence determinant could be 1,8-dihydroxynapthalene (DHN) melanin. Spores of A. fumigatus lacking DHN melanin are ingested at higher rates by macrophages [28] and the amoeba Dictyostelium discoideum [29]. Moreover, DHN melanin inhibits phagolysosome maturation and acidification which significantly impairs conidial killing by macrophages [30, 31]. In addition to DHN melanin, secondary metabolites are key determinants in the interactions between A. fumigatus and amoebas. Specific examples include gliotoxin which is important in killing D. discoideum [29] or fumagillin which inhibits Entamoeba histolytica growth [32], while these secondary metabolites can inhibit NADPH oxidase, innate immunity signaling hubs, or drive epithelial cell damage in mammalian systems [33–36]. In addition to gliotoxin and fumagillin, A. fumigatus express a wide array of secondary metabolites that could have critical roles in regulation the host-pathogens and microbe-environment interactions of A. fumigatus [37].
Heterogeneity within A. fumigatus due to its evolution within its natural environmental niche likely drives the virulence heterogeneity observed in mice. Additionally, nutritional conditions in which spore development occurs appears to be critical in the regulation of the conidial development program, dormancy, and cell wall architecture which could influence germination rates, cell wall carbohydrate exposure and interactions with host phagocytes [38]. Thus, much remains to be understood about the heterogeneity among A. fumigatus isolates and how that impacts the host inflammatory response.
PHAGOCYTOSIS AND ANTI-FUNGAL KILLING OF FUNGAL PATHOGENS
A. fumigatus enters the body through the inhalation of airborne resting conidia. Resting conidia of A. fumigatus are tightly covered by a hydrophobic layer of rodlet proteins. Rodlets conceal the fungal polysaccharide-rich cell wall which is composed of an outer DHN melanin layer, a core layer composed of β−1,3-linked glucans, mannoproteins, and galactomannan, and an inner layer of chitin (Figure 1a–c) (reviewed by [39]). The rodlets are tightly packed limiting the exposure of the polysaccharide-rich cell wall components limiting activation of the innate immune response [40]. Recently, the CcpA protein was also found in the outer proteinaceous layer of the resting spores. Resting spores lacking CcpA maintained normal cell surface structure of the rodlet layer but had enhanced immunostimulatory potential [41]. The fungal lectin FleA is also found in the outer proteinaceous layer of the resting spore and is a ligand for fucosylated glycoproteins that are found in lung mucins and is critical for the recognition and uptake of resting conidia by macrophages [42].
Figure 1. Phagosome maturation and the patterns of pathogenesis for regulating the innate immune response induced against Aspergillus fumigatus.
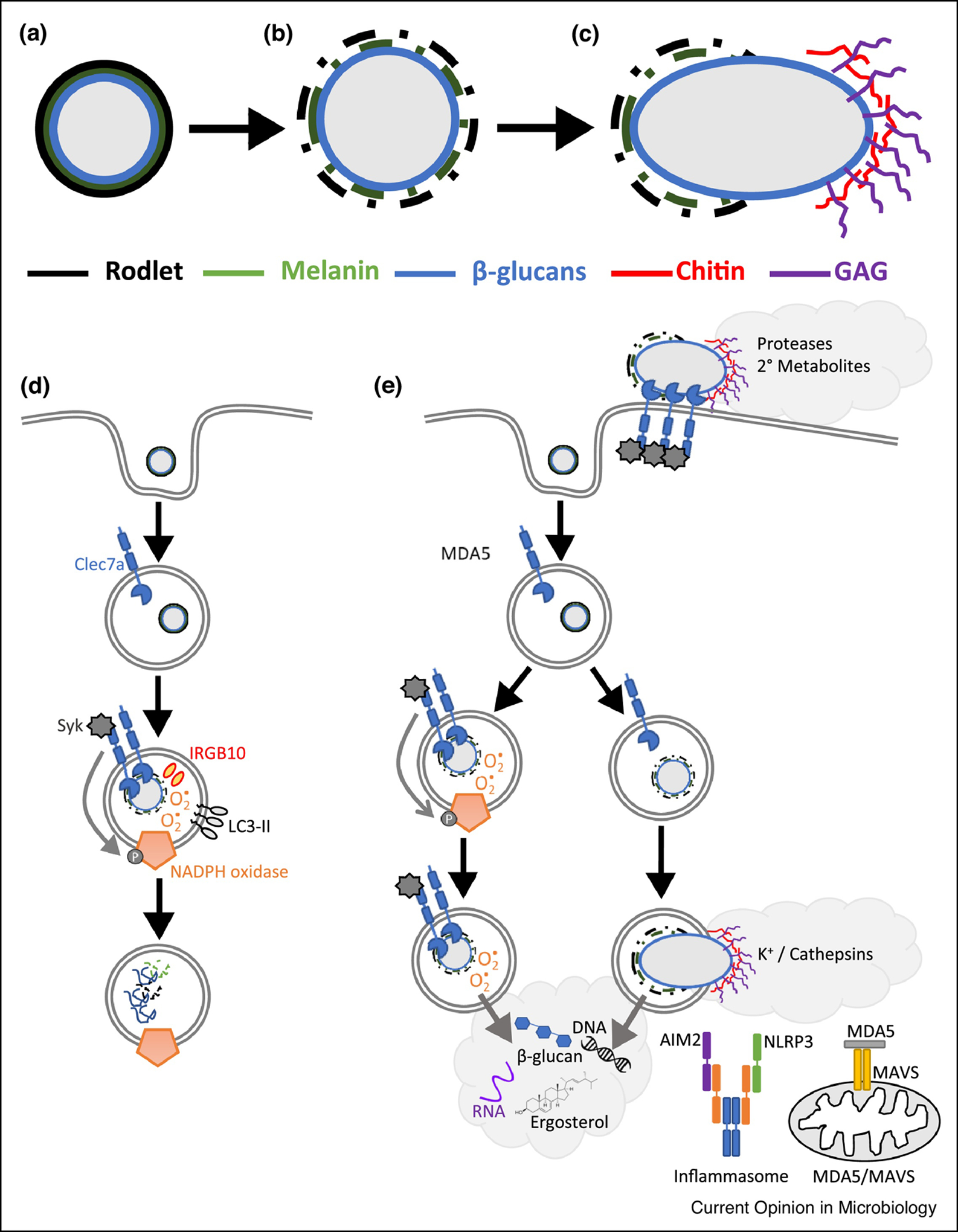
Immune sensing of A. fumigatus is highly dependent on the interplay between the changes in the fungal cell wall as the Aspergillus grows and the host innate immune cells and its phagosome. (a) Resting conidia of A. fumigatus have tightly packed rodlet hydrophobin and melanin layers, which limit the exposure of the core cell wall carbohydrates. (b) Upon osmotic swelling the rodlet hydrophobin and melanin layers are broken down exposing the underlying core carbohydrates, predominately β−1,3-glucans. (c) Upon germ tube emergence further changes occur, particularly the expression and integration of galactosaminogalactan on the outer surface of the cell wall. (d) During phagosome maturation A. fumigatus spores that were engulfed can undergo conidial germination and stripping of both the hydrophobin rodlet and DHN melanin layers resulting in the exposure of the β−1,3-linked glucans that drive Dectin 1 signaling. Activation of Dectin1 results in NADPH oxidase activation and enhanced LC3-associated phagocytosis for the optimal killing of A. fumigatus. (e) For sensing the patterns of pathogenesis during fungal infection, several key events can be sensed by the host innate immune cells including cell surface recognition of germlings, phagosome rupture, or the release of fungal components into the cytosol. Release of fungal components, such as fungal DNA, β−1,3-glucans, and ergosterol or fungal RNA, drive the activation of the inflammasome for IL-1β release or Mda5/MAVS-dependent IFN production, respectively.
Upon fungal spore uptake, the phagosome undergoes a highly regulated process of maturation to mediate antifungal killing (Figure 1d). Upon phagocytosis, conidial germination and stripping of both the rodlet and DHN melanin layers exposes the β−1,3-linked glucans within the cell wall [43]. Exposure of β−1,3-linked glucans results in the engagement of Dectin-1 driving Syk-dependent signaling which is necessary for phagosome maturation [43] and reprogramming macrophage metabolism to glycolysis which is necessary for antifungal immunity [44]. Dectin-1/Syk signaling results in the phosphorylation of the p47phox protein, which drives NADPH oxidase complex formation and activation [43]. Upon activation the NADPH oxidase complex generates reactive oxygen species (ROS) which is not only necessary for antifungal killing within the phagosome, but also enhances the incorporation of LC3 into the phagosomal membrane [43]. LC3-associated phagocytosis is essential for mediating host antifungal immunity [45]. Interestingly, corticosteroid treatment can dampen LC3-associated phagocytosis which could contribute to the immunosuppressive environment in those patients ultimately making them susceptible to invasive aspergillosis [46].
It has been well established that DHN melanin in A. fumigatus mediates resistance against phagocytic killing. The role of DHN melanin in protecting the fungal spores is two-pronged: 1) directly protecting the spores from the damaging effects of ROS [47] and 2) impairing the LC3-associated phagosome maturation process [43]. In this latter role DHN melanin appears to sequester phagosomal Ca2+ thereby preventing calmodulin-dependent signaling and phagosome maturation [48]. Ca2+-dependent signaling through calcineurin and NFAT downstream of TLR9 activation in the phagosome through a MyD88-independent, but Btk-dependent signaling pathway is critical in antifungal immunity against A. fumigatus in mice [49]. The importance of Btk signaling in preventing invasive fungal infections in humans is highlighted by the observation that cancer patients treated with ibrutinib are at increased risk of developing these infections [50–53]. Interestingly, β−1,3-linked glucan polysaccharide activation of Dectin-1/Syk signaling is necessary for the recruitment of TLR9 to phagosomes containing fungal spores and the induction of an interferon (IFN)-dependent transcriptional signature [54]. LC3-associated phagocytosis directly enhances IFNα production downstream of CpG-induced TLR9 activation through the direct interaction of LC3 with IKKα [55]. Both type I and type III IFNs are critical in regulating the protective innate antifungal immune response after A. fumigatus challenge [13]. Much more work on the cross-talk between phagosome localized PRRs and LC3-associated phagocytosis is needed to understand the induction of antifungal killing and inflammation in the context of A. fumigatus infection, as therapeutic targeting of these pathways can modulate disease outcomes [56, 57].
SENSING OF PATTERNS OF PATHOGENESIS AND VITA-PAMPs ENHANCE THE INFLAMMATORY RESPONSE TO BACTERIAL PATHOGENS
Mammalian hosts must be able to distinguish between commensal and pathogenic interactions given their microbiota. This has led to an explosion of research into how innate immune sensing pathways can distinguish these two outcomes. Vance, Isberg, and Portnoy have proposed a theory based on “patterns of pathogenesis”, which include: 1) microbial growth, 2) cytosolic access, 3) disruption of host cytosolic function [58]. Microbial growth would be the result of the sum of microbial replication and microbial cell death. It has been well established that live microbes induce stronger and more diverse immune responses than dead microbes. This observation led Blander and colleagues to propose the hypothesis that certain PAMPs may acts at markers of vitality (vita-PAMPs). Vita-PAMPs that have been identified during bacterial infections include microbial RNA [59] and cyclic-di-adenosine monophosphate [60], which can drive enhanced type I interferon and IL-1β inflammation leading to more robust protective immunity. Similarly, cytosolic access of microbial products or microbial effectors leads to the activation of numerous different inflammasomes (reviewed by [61]). Some of these microbial effectors are critical for altering cytosolic cytoskeleton proteins critical in cellular function and phagocytosis, which can be sensed by guard proteins like pyrin [62]. Overall, the mammalian innate immune system is well designed to assess the risk of each microbial insult and tune the overall inflammatory response in such a manner to deal with the pathogenic insult.
THE FUNGAL-MENTALS OF PATHOGENESIS FOR PREVENTING INVASIVE FUNGAL INFECTIONS
The patterns of pathogenesis framework is likely critical in understanding how the mammalian innate immune response is tuned to the threat posed by fungal species as well, given the emergence of the importance of the host mycobiome in the intestinal tract [63], as well as the fact that the respiratory tract is continually exposed to ubiquitous environmental fungal spores in the air. The former is critical in regards to Candida infections as it has recently been demonstrated by Hohl and colleagues that the bloodstream Candida spp. isolate from invasive candidiasis patients who had undergone allogeneic hematopoietic cell transplant originated in the intestinal mycobiome [64]. In agreement with the patterns of pathogenesis hypothesis both the interferon response [4, 13, 65] and inflammasome-dependent IL-1β production [66–71] are critical for maintaining host resistance against fungal pathogens. Thus, we will now highlight the role of fungal patterns of pathogenesis in tuning the inflammatory response to fungal pathogens, particularly focused on A. fumigatus.
Sensing of A. fumigatus growth.
One of the key features of A. fumigatus growth that can be sensed by the innate immune system is the observed changes in the fungal cell wall (Figure 1e). Sensing of these changes in the Aspergillus cell wall have been shown to be critical in regulating the antifungal immune response. For example, exposure of the β−1,3-glucans upon swelling by A. fumigatus conidia is recognized by Dectin-1 (Clec7a) [72, 73]. Additionally, Dectin-2 (Clec4n) can recognize both swollen conidia and germlings of A. fumigatus [74, 75]. While the exact fungal ligand driving Dectin-2 activation by A. fumigatus remains unknown; in other fungi, cell wall mannans and high mannose structures have been shown to drive Dectin-2 activation [76, 77]. Both Dectin-1 and Dectin-2 therefore enable the host to sense the early growth of A. fumigatus directly upon conidial swelling. The hyphal-specific galactosaminogalactan carbohydrate can also modulate the inflammatory response [78–80], but how galactosaminogalactan interacts with the immune system remains unknown. Moreover, germlings/hyphae from A. fumigatus are known to secrete numerous hydrolytic enzymes (proteases and lipases) and secondary metabolites, which likely influence the inflammatory response. Our laboratory has shown that A. fumigatus germlings drive much stronger and broader inflammation, in particular driving IL-1α and LTB4 secretion resulting in increased neutrophil recruitment [14, 19]. The exact fungal triggers of this increased inflammatory response are unknown. Thus, the biochemical changes in the cell wall and metabolic status are important rheostats for the inflammatory response against A. fumigatus.
Inflammasome sensing and IL-1β secretion.
During fungal infections inflammasome activation and IL-1β secretion are critical for host antifungal immunity. Specific to A. fumigatus, both the NLRP3 and AIM2 inflammasomes can be activated [66]. β−1,3-glucans are critical in the priming and activation of the NLRP3 inflammasome [71, 81]. Moreover, ROS and K+ efflux may contribute to the activation of NLRP3 [68, 71]. The activation of the AIM2 inflammasome is driven by fungal DNA [66]. This brings about an interesting paradox whereby the fungal ligands for both the NLRP3 and AIM2 inflammasomes are typically found within the phagosome, but the receptors are localized to the cytosol of the cell; thus, how do these ligand get transferred into the cytosol for detection by these inflammasome sensors? Recently, Kanneganti and colleagues demonstrated that IRGB10 induces damage to A. fumigatus hyphae to enhance the release of β−1,3-glucans from the fungal cell wall driving NLRP3 [81], but the exact mechanism by which this ligand gets transferred to the cytosol remain unresolved.
In other fungal systems it has been shown that phagosome rupture by hyphal growth can play an important role in NLRP3 pyroptosis and IL-1β release [70, 82, 83]. Analogously, challenging bone marrow-derived macrophages with A. fumigatus germlings results in greater IL-1α and IL-1β secretion compared to spores and these germlings had to be alive to elicit this response [14]. More recent work using several mutant collections demonstrate that hyphal growth is not absolutely required for NLRP3-depednent pyroptosis and IL-1β secretion [82, 84–87]. One important pathway identified using these mutant collections is the ergosterol biosynthetic enzymes [85, 86]. Moreover, ergosterol-containing liposomes were sufficient for the induction of NLRP3 inflammasome-dependent IL-1β secretion [86]. Our laboratory has also observed NLRP3 inflammasome activation using ergosterol crystals (unpublished observation). Thus, multiple mechanisms appear to exist in which fungal-derived ligands necessary for inflammasome activation can escape the phagosome and drive IL-1β secretion, which was dependent on metabolically active fungi surviving within the phagosome.
Regulation of the interferon response.
Studies in mice have shown that both type I and type III interferons (IFNs) are crucial for maintaining host resistance against A. fumigatus [13]. In this model, upon A. fumigatus challenge type I IFNs are rapidly produced by CCR2+ monocytes, which promotes type III IFN signaling, expression, and induction of optimal antifungal activity of neutrophils [13]. However, the fungal triggers and host receptors responsible for initiating the IFN response following A. fumigatus challenge remains largely unresolved. Our recent work has described an essential role for Mda5 in maintaining host resistance against A. fumigatus [88]. Mda5 is a cytosolic receptor for double-stranded RNA and is critical for the induction of antiviral IFN responses [89, 90]. We found that double-stranded RNA from A. fumigatus was sufficient for Mda5 activation and that the type I IFN response was only partially dependent on Mda5, while the type III IFN response was entirely dependent on Mda5 [88]. Thus, other innate immune sensing receptors likely participate in inducing the type I IFN response, such as Dectin 1 and TLR9 [54, 91].
Similarly following Candida albicans infection IFNAR-depending signaling is essential for host resistance [4, 65, 92]. Following C. albicans challenge, both fungal nucleic acids [92, 93] and β−1,3-glucans [65] are involved in the induction of the type I IFN response. Induction of IFN-β by Dectin-1 requires Syk-dependent activation of Irf5 [65], while type I IFN induction by fungal nucleic acids is partially dependent on TLR7 and TLR9 [92, 93]. Additionally, in humans polymorphisms in the IFIH1 gene are associated with altered risks for developing systemic Candida infections [94]. However, Ifnb expression in murine bone marrow derived macrophages was largely independent of Mda5 [94]. Thus, both type I and type III interferons are critical in maintaining host resistance against fungal pathogens, but much remains unknown about the initiation of these cytokines during fungal infection.
CONCLUDING REMARKS
As highlighted in this review, A. fumigatus induces inflammatory responses in mice that are tightly tuned to the virulence of the infecting fungal isolate. These differences in virulence appear to be due to the ability of an isolate to adapt to the stresses found in the infected lung, such as hypoxia, nutrient availability, and engulfment by host phagocytes. The ability of a strain to adapt or not to the stresses imposed within the mammalian lungs will result in changes in a strains metabolic activity, ability to survive, and ability to grow. These changes in the fungi are interrogated by the host innate immune system to maintain resistance against the fungal pathogen while hopefully minimizing collateral tissue damage. By understanding the regulation of these processes by both the fungi and mammalian immune response we should be able to develop better therapeutic avenues for these difficult to treat invasive fungal pathogens.
HIGHLIGHTS.
Host-fungal interactions tune the innate inflammatory response to maintain host resistance against distinct fungal isolates
Fungal growth, viability, phagosome escape, and cell wall changes are critical ‘patterns of pathogenesis’ during fungal infection
Environmental evolution and growth conditions drive isolate-specific virulence traits
ACKNOWLEDGEMENTS
Thank you to members of the Obar laboratory who helped shaped the ideas for this review article. This work was supported by NIH grants R01-AI139133 and R21-AI152019.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Brown GD, et al. , Hidden killers: human fungal infections. Sci Transl Med, 2012. 4(165): p. 165rv13. [DOI] [PubMed] [Google Scholar]
- 2.Wojtowicz A, et al. , IL1B and DEFB1 Polymorphisms Increase Susceptibility to Invasive Mold Infection After Solid-Organ Transplantation. J Infect Dis, 2015. 211(10): p. 1646–57. [DOI] [PubMed] [Google Scholar]
- 3.Romani L, Immunity to fungal infections. Nat Rev Immunol, 2011. 11(4): p. 275–88. [DOI] [PubMed] [Google Scholar]
- 4.Smeekens SP, et al. , Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat Commun, 2013. 4: p. 1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lanternier F, et al. , Primary immunodeficiencies underlying fungal infections. Curr Opin Pediatr, 2013. 25(6): p. 736–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia S, et al. , Rapid host defense against Aspergillus fumigatus involves alveolar macrophages with a predominance of alternatively activated phenotype. PLoS One, 2011. 6(1): p. e15943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jhingran A, et al. , Tracing conidial fate and measuring host cell antifungal activity using a reporter of microbial viability in the lung. Cell Rep, 2012. 2(6): p. 1762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubourdeau M, et al. , Interaction of Aspergillus fumigatus with the alveolar macrophage. Med Mycol, 2006. 44(Supplement_1): p. S213–S217. [DOI] [PubMed] [Google Scholar]
- 9.Mircescu MM, et al. , Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J Infect Dis, 2009. 200(4): p. 647–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **10.Rosowski EE, et al. , Macrophages inhibit Aspergillus fumigatus germination and neutrophil-mediated fungal killing. PLoS Pathog, 2018. 14(8): p. e1007229. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper using the zebrafish model to demonstrate that macrophages and neutrophils play different roles in regulating growth and germination of Aspergillus fumigatus spores and hyphae. Moreover, the importance of each cell in maintain host resistance dependents on the germinate rate of each strain.
- 11.Espinosa V, et al. , Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog, 2014. 10(2): p. e1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hohl TM, et al. , Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe, 2009. 6(5): p. 470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **13.Espinosa V, et al. , Type III interferon is a critical regulator of innate antifungal immunity Science Immunology, 2017. 2(16): p. eaan5357. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper highlights the importance of both type I and type III interferons in regulating the antifungal neutrophil response following A. fumigatus challenge through the enhancement of ROS prodcution by neutrophils.
- *14.Caffrey-Carr AK, et al. , IL-1alpha is Critical for Resistance Against Highly Virulent Aspergillus fumigatus Isolates. Infect Immun, 2017. 85(12): p. e00661–17. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that Aspergillus fumigatus germlings are more inlfammatory than fungal spores. Moreover, they induced a distinct inflammatory response that is necessary for maintaining host resistance against the germling form.
- 15.Bonnett CR, et al. , Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus Conidia. Infect Immun, 2006. 74(12): p. 6528–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **16.Guo Y, et al. , During Aspergillus Infection, Monocyte-Derived DCs, Neutrophils, and Plasmacytoid DCs Enhance Innate Immune Defense through CXCR3-Dependent Crosstalk. Cell Host Microbe, 2020: p. doi: 10.1016/j.chom.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper highlights the important triprate cross-talk between monocyte-derived dendritic cells, plasmacytoid dendritic cells, and neutrophils for the maintainence of host resistance against Aspergillus fumigatus. Monocyte-dervived dendritic cells and neutrophils are important sources of CXCL9/10, which recruit plasmacytoid denritic cells to the site of infection. There the plasmacytoid dendritic cells enhance antifungal effector functions of the neutrophils.
- 17.Nierman WC, et al. , Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature, 2005. 438(7071): p. 1151–6. [DOI] [PubMed] [Google Scholar]
- 18.Fedorova ND, et al. , Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet, 2008. 4(4): p. e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caffrey-Carr AK, et al. , Host-Derived Leukotriene B4 Is Critical for Resistance against Invasive Pulmonary Aspergillosis. Front Immunol, 2017. 8: p. 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *20.Kowalski CH, et al. , Heterogeneity among Isolates Reveals that Fitness in Low Oxygen Correlates with Aspergillus fumigatus Virulence. MBio, 2016. 7(5): p.:e01515–16. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that differences in the hypoxic fitness of A. fumigatus isolates correlate with in vivo virulences in the corticosteroid-induced invasive aspergillosis model in mice.
- 21.Grahl N, et al. , In vivo hypoxia and a fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary aspergillosis. PLoS Pathog, 2011. 7(7): p. e1002145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kowalski CH, et al. , Fungal biofilm morphology impacts hypoxia fitness and disease progression. Nat Microbiol, 2019. 4(12): p. 2430–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shepardson KM, et al. , Myeloid derived hypoxia inducible factor 1-alpha is required for protection against pulmonary Aspergillus fumigatus infection. PLoS Pathog, 2014. 10(9): p. e1004378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knox BP, et al. , Characterization of Aspergillus fumigatus Isolates from Air and Surfaces of the International Space Station. mSphere, 2016. 1(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beattie SR, et al. , Filamentous fungal carbon catabolite repression supports metabolic plasticity and stress responses essential for disease progression. PLoS Pathog, 2017. 13(4): p. e1006340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casadevall A, et al. , The ‘Amoeboid Predator-Fungal Animal Virulence’ Hypothesis. J Fungi (Basel), 2019. 5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novohradska S, Ferling I, and Hillmann F, Exploring Virulence Determinants of Filamentous Fungal Pathogens through Interactions with Soil Amoebae. Front Cell Infect Microbiol, 2017. 7: p. 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luther K, et al. , Phagocytosis of Aspergillus fumigatus conidia by murine macrophages involves recognition by the dectin-1 beta-glucan receptor and Toll-like receptor 2. Cell Microbiol, 2007. 9(2): p. 368–81. [DOI] [PubMed] [Google Scholar]
- 29.Hillmann F, et al. , Virulence determinants of the human pathogenic fungus Aspergillus fumigatus protect against soil amoeba predation. Environ Microbiol, 2015. 17(8): p. 2858–69. [DOI] [PubMed] [Google Scholar]
- 30.Thywissen A, et al. , Conidial Dihydroxynaphthalene Melanin of the Human Pathogenic Fungus Aspergillus fumigatus Interferes with the Host Endocytosis Pathway. Front Microbiol, 2011. 2: p. 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jahn B, et al. , PKSP-dependent reduction of phagolysosome fusion and intracellular kill of Aspergillus fumigatus conidia by human monocyte-derived macrophages. Cell Microbiol, 2002. 4(12): p. 793–803. [DOI] [PubMed] [Google Scholar]
- 32.Arico-Muendel C, et al. , Antiparasitic activities of novel, orally available fumagillin analogs. Bioorg Med Chem Lett, 2009. 19(17): p. 5128–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guruceaga X, et al. , A possible role for fumagillin in cellular damage during host infection by Aspergillus fumigatus. Virulence, 2018. 9(1): p. 1548–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pahl HL, et al. , The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-kappaB. J Exp Med, 1996. 183(4): p. 1829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlam D, et al. , Gliotoxin Suppresses Macrophage Immune Function by Subverting Phosphatidylinositol 3,4,5-Trisphosphate Homeostasis. mBio, 2016. 7(2): p. e02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsunawaki S, et al. , Fungal metabolite gliotoxin inhibits assembly of the human respiratory burst NADPH oxidase. Infect Immun, 2004. 72(6): p. 3373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raffa N and Keller NP, A call to arms: Mustering secondary metabolites for success and survival of an opportunistic pathogen. PLoS Pathog, 2019. 15(4): p. e1007606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bleichrodt RJ, et al. , Cell Wall Composition Heterogeneity between Single Cells in Aspergillus fumigatus Leads to Heterogeneous Behavior during Antifungal Treatment and Phagocytosis. mBio, 2020. 11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latge JP and Chamilos G, Aspergillus fumigatus and Aspergillosis in 2019. Clin Microbiol Rev, 2019. 33(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aimanianda V, et al. , Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature, 2009. 460(7259): p. 1117–21. [DOI] [PubMed] [Google Scholar]
- 41.Voltersen V, et al. , Proteome Analysis Reveals the Conidial Surface Protein CcpA Essential for Virulence of the Pathogenic Fungus Aspergillus fumigatus. mBio, 2018. 9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kerr SC, et al. , FleA Expression in Aspergillus fumigatus Is Recognized by Fucosylated Structures on Mucins and Macrophages to Prevent Lung Infection. PLoS Pathog, 2016. 12(4): p. e1005555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **43.Akoumianaki T, et al. , Aspergillus Cell Wall Melanin Blocks LC3-Associated Phagocytosis to Promote Pathogenicity. Cell Host Microbe, 2016. 19(1): p. 79–90. [DOI] [PubMed] [Google Scholar]; This paper confirms that LC3-mediated phagocytosis is necessary for host resistance against A. fumigatus in mice. Importantly, this paper extends those findings demonstrating that melanin promotes virulence by inhibiting LC3-mediated phagocytosis which results in a dampen ROS response.
- 44.Goncalves SM, et al. , Phagosomal removal of fungal melanin reprograms macrophage metabolism to promote antifungal immunity. Nat Commun, 2020. 11(1): p. 2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *45.Martinez J, et al. , Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol, 2015. 17(7): p. 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; This paper was the first to demonstrate that LC3-associated phagocytos was essential for maintaining host resistance agasinst A. fumigatus in mice.
- 46.Kyrmizi I, et al. , Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J Immunol, 2013. 191(3): p. 1287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugareva V, et al. , Characterisation of the laccase-encoding gene abr2 of the dihydroxynaphthalene-like melanin gene cluster of Aspergillus fumigatus. Arch Microbiol, 2006. 186(5): p. 345–55. [DOI] [PubMed] [Google Scholar]
- 48.Kyrmizi I, et al. , Calcium sequestration by fungal melanin inhibits calcium-calmodulin signalling to prevent LC3-associated phagocytosis. Nat Microbiol, 2018. [DOI] [PubMed] [Google Scholar]
- 49.Herbst S, et al. , Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway coordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med, 2015. 7(3): p. 240–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghez D, et al. , Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood, 2018. 131(17): p. 1955–1959. [DOI] [PubMed] [Google Scholar]
- 51.Lionakis MS, et al. , Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell, 2017. 31(6): p. 833–843 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruchlemer R, et al. , Ibrutinib-associated invasive fungal diseases in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: An observational study. Mycoses, 2019. 62(12): p. 1140–1147. [DOI] [PubMed] [Google Scholar]
- 53.Zarakas MA, et al. , Fungal Infections with Ibrutinib and Other Small-Molecule Kinase Inhibitors. Curr Fungal Infect Rep, 2019. 13(3): p. 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khan NS, et al. , Dectin-1 Controls TLR9 Trafficking to Phagosomes Containing beta-1,3 Glucan. J Immunol, 2016. 196(5): p. 2249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayashi K, Taura M, and Iwasaki A, The interaction between IKKalpha and LC3 promotes type I interferon production through the TLR9-containing LAPosome. Sci Signal, 2018. 11(528). [DOI] [PMC free article] [PubMed] [Google Scholar]
- *56.de Luca A, et al. , IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A, 2014. 111(9): p. 3526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *57.Seyedmousavi S, et al. , Exogenous Stimulation of Type I Interferon Protects Mice with Chronic Granulomatous Disease from Aspergillosis through Early Recruitment of Host-Protective Neutrophils into the Lung. MBio, 2018. 9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]; References 56 & 57 together demonstrate the altering the inflammatory milieu in CGD mice with invasive aspergillosis can ameriolate the course of invasive aspergillosis disease.
- 58.Vance RE, Isberg RR, and Portnoy DA, Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe, 2009. 6(1): p. 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sander LE, et al. , Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature, 2011. 474(7351): p. 385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moretti J, et al. , STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell, 2017. 171(4): p. 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Christgen S, Place DE, and Kanneganti TD, Toward targeting inflammasomes: insights into their regulation and activation. Cell Res, 2020. 30(4): p. 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung LK, et al. , The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome. Cell Host Microbe, 2016. 20(3): p. 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iliev ID, et al. , Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science, 2012. 336(6086): p. 1314–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhai B, et al. , High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat Med, 2020. 26(1): p. 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.del Fresno C, et al. , Interferon-beta production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity, 2013. 38(6): p. 1176–86. [DOI] [PubMed] [Google Scholar]
- 66.Karki R, et al. , Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe, 2015. 17(3): p. 357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moretti S, et al. , IL-37 inhibits inflammasome activation and disease severity in murine aspergillosis. PLoS Pathog, 2014. 10(11): p. e1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Said-Sadier N, et al. , Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One, 2010. 5(4): p. e10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Joly S, et al. , Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol, 2009. 183(6): p. 3578–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hise AG, et al. , An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe, 2009. 5(5): p. 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gross O, et al. , Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature, 2009. 459(7245): p. 433–6. [DOI] [PubMed] [Google Scholar]
- 72.Hohl TM, et al. , Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog, 2005. 1(3): p. e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steele C, et al. , The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog, 2005. 1(4): p. e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goyal S, et al. , The Interaction of Human Pathogenic Fungi With C-Type Lectin Receptors. Front Immunol, 2018. 9: p. 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Loures FV, et al. , Recognition of Aspergillus fumigatus hyphae by human plasmacytoid dendritic cells is mediated by dectin-2 and results in formation of extracellular traps. PLoS Pathog, 2015. 11(2): p. e1004643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saijo S, et al. , Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity, 2010. 32(5): p. 681–91. [DOI] [PubMed] [Google Scholar]
- 77.McGreal EP, et al. , The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology, 2006. 16(5): p. 422–30. [DOI] [PubMed] [Google Scholar]
- 78.Gresnigt MS, et al. , A polysaccharide virulence factor from Aspergillus fumigatus elicits anti-inflammatory effects through induction of Interleukin-1 receptor antagonist. PLoS Pathog, 2014. 10(3): p. e1003936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee MJ, et al. , The Fungal Exopolysaccharide Galactosaminogalactan Mediates Virulence by Enhancing Resistance to Neutrophil Extracellular Traps. PLoS Pathog, 2015. 11(10): p. e1005187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gravelat FN, et al. , Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta-glucan from the immune system. PLoS Pathog, 2013. 9(8): p. e1003575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *81.Briard B, et al. , Fungal ligands released by innate immune effectors promote inflammasome activation during Aspergillus fumigatus infection. Nat Microbiol, 2019. 4(2): p. 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that IRGB10 is necessary for the relases of NLRP3 ligands from the fungal wall and their release from the phagosome to the cytosol for the activation of the NLRP3 inflammation.
- 82.Wellington M, et al. , Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot Cell, 2014. 13(2): p. 329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Uwamahoro N, et al. , The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. MBio, 2014. 5(2): p. e00003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O’Meara TR, et al. , Global analysis of fungal morphology exposes mechanisms of host cell escape. Nat Commun, 2015. 6: p. 6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Meara TR, et al. , Mapping the Hsp90 Genetic Network Reveals Ergosterol Biosynthesis and Phosphatidylinositol-4-Kinase Signaling as Core Circuitry Governing Cellular Stress. PLoS Genet, 2016. 12(6): p. e1006142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koselny K, et al. , A Genome-Wide Screen of Deletion Mutants in the Filamentous Saccharomyces cerevisiae Background Identifies Ergosterol as a Direct Trigger of Macrophage Pyroptosis. mBio, 2018. 9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O’Meara TR, et al. , High-Throughput Screening Identifies Genes Required for Candida albicans Induction of Macrophage Pyroptosis. mBio, 2018. 9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang X, et al. , MDA5 is an essential vita-PAMP sensor necessary for host resistance against Aspergillus fumigatus. bioRxiv, 2020: p. 2020.07.06.182154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pichlmair A, et al. , Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol, 2009. 83(20): p. 10761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kato H, et al. , Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature, 2006. 441(7089): p. 101–5. [DOI] [PubMed] [Google Scholar]
- 91.Dutta O, et al. , Dectin-1 Promotes Type I and III Interferon Expression to Support Optimal Antifungal Immunity in the Lung. Front Cell Infect Microbiol, 2020. 10: p. 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Biondo C, et al. , Recognition of yeast nucleic acids triggers a host-protective type I interferon response. Eur J Immunol, 2011. 41(7): p. 1969–79. [DOI] [PubMed] [Google Scholar]
- 93.Biondo C, et al. , Recognition of fungal RNA by TLR7 has a nonredundant role in host defense against experimental candidiasis. Eur J Immunol, 2012. 42(10): p. 2632–43. [DOI] [PubMed] [Google Scholar]
- 94.Jaeger M, et al. , The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur J Clin Microbiol Infect Dis, 2015. 34(5): p. 963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]