

Abstract
Behçet’s disease (BD) is presumably an autoinflammatory disease of unknown etiology for which several animal models have been described over the years. Agents and methods used for the development of these models have ranged from the herpes simplex type one virus (hsv-1) pathogen to the use of transgenic mice. Other models have also been used to investigate a possible autoimmune component. Each model possesses its own unique set of benefits and shortcomings, with no one model fully being able to recapitulate the disease phenotype. Here, we review the proposed models and provide commentary on their effectiveness and usefulness in studying the disease.
Keywords: Behçet’s disease (BD), rare disease, Behçet’s disease animal models, autoimmune disease, herpes simplex virus type 1, human leukocyte antigen
1. INTRODUCTION
Behçet’s disease (BD) is a rare and debilitating illness first described by the Turkish dermatologist Hulusi Behçet, in 1937, years before he was to become its namesake [1]. The symptoms associated with this condition had been described in ancient writings long before the characteristics of the disease were established. BD is an inflammatory condition [2] of unknown etiology [3], which can lead to vasculitis in both large and small blood vessels throughout the body [4]. It is a multisystemic syndrome that can manifest itself in several different ways, leading to varied clinical presentation in affected patients, which makes it more difficult to definitively diagnose patients [5]. In addition to the most commonly observed symptoms, viz., recurrent oral contagious aphthosis, genital ulcers, and ocular abnormalities, patients may also present with skin and gastrointestinal lesions [6], arterial aneurysms [7], arthritis [8], and neurological symptoms [9].
Although classified as a rare disease in the Western world, with one study reporting as few as 5.2 cases per 100,000 in a geographically defined population in the USA [10], the disease is far more common in countries along the Old Silk Route [11]. In Turkey, the disease prevalence ranges from 20 to 420 cases per 100,000, while Iran and Japan [12] report 80 and 16 cases per 100,000, respectively. Genome-wide association studies have been performed by several groups in an attempt to uncover specific genes that may be associated with increased susceptibility to BD [12–17]. These studies included populations from China, Europe, Japan, Iran, Korea, Turkey, and the Middle East. Over 20 genes have been found to be associated with increased disease susceptibility, with the human leukocyte antigen gene encoding B*51 (HLA-B*51) showing the strongest association across multiple populations.
Despite its strong association with BD, HLA-B*51 cannot be used as a singular genetic determinant for disease development, since the presence of HLA-B*51, alone, does not always lead to disease. Also, only a fraction of BD patients is HLA-B*51 positive [18]. BD is most likely caused by a combination of genetic and environmental factors, including pathogens and environmental pollutants. Although it remains to be proven, there is evidence to suggest the involvement of both herpes simplex virus type 1 (HSV-1) [19, 20], as well as members of the Streptococcus genus [21–25] in BD pathogenesis.
There is currently no cure for BD, however, individual symptoms can be treated with preexisting drugs. Attempts made to develop animal models have only been partially successful, with the HSV-1 mouse model showing the greatest ability to recapitulate some symptoms of the BD phenotype [26]. Herein, we review current BD models, offering commentary on the effectiveness of each model for studying the disease.
2. HLA-B*51 MODEL
The human leukocyte antigen complex (HLA) is analogous to the mouse major histocompatibility complex (MHC), which plays a role in differentiating self from non-self-antigens [27]. These membrane proteins are necessary for the presentation of antigen-derived peptide fragments and play important roles in immune regulation [28]. Various HLA alleles have been linked to different autoimmune conditions, some of which include HLA-DRB1 (rheumatoid arthritis) [29], HLA-DR-DQ (type 1 diabetes and celiac disease) [30], and HLA-C*06:02 (psoriasis) [31].
Several HLA alleles have also been studied and found to either increase susceptibility to BD or to have a protective effect in certain populations. Of these alleles, HLA-B*51 is perhaps the most well-studied, and the consensus is that it increases susceptibility to BD across multiple populations [32–38]. One investigation [39] reported no association between HLA-B*51 and BD in a cohort of Irish patients of Caucasian descent. However, it was reported that increased frequencies of four other HLA-B alleles appeared in BD patients as compared to the general population. These alleles included B*57, B*35, B*15 and B*27. It is important to note that the sample size was rather small (20 patients) and additional studies spanning a wider region would be needed to validate these findings [39]. Indeed, another study [40] using DNA from a cohort of 56 western European Caucasians with BD and 90 Caucasian controls, uncovered the opposite result. It is most likely that BD results from a combination of genetic and environmental factors such as HLA-B*51, along with pathogens or chemical elements, such as environmental pollutants. Indeed, a case of BD was reported, which arose 2 years after a 32-year-old Caucasian woman moved from her home in Finland to Japan [41]. She had no familial history of BD. This lends credence to the theory that environmental interactions made possible by certain conditions that exist in countries along the Old Silk Road, where the disease is most common, support BD development.
Various other HLA genes have been found to be associated with an increased susceptibility to BD. One example is HLA-B*15 in Iranians [42], Egyptians [43], and Moroccans [44]. However, one study reported this allele to be protective in Saudi Arabians, but additional larger-scale investigations are needed to confirm this association [45]. Other population associations have been made, specifically, HLA-A*26 and HLA-A*31 in Saudi Arabians [45], HLA-B*57 in Spaniards [46, 47], HLA Class-I A*24, -A*68, and -B*42 in Egyptian patients [43], HLA-A*26 in Northeast Asians [48, 49], and HLA-A*02:07, A*26:01, and A*30:04 in Korean patients [50]. On the other hand, various other HLA alleles have been associated with a more protective effect against BD, including HLA Class-I A*03 and B*52 in Egyptian patients [43] and HLA-B*35 in Spaniards [47].
Variations in HLA genotypes have also been shown to play a role in clinical BD manifestation as well as response to certain drugs. One study showed that the genetic differences in the HLA complex partly affected the response of two different groups to infliximab therapy, namely, Japanese patients diagnosed with Behçet’s uveitis and genotyped as HLA-A*2601 and HLA-B*5101, respectively [51]. The presence of different subtypes of HLA-B*51 has been shown to result in different clinical manifestations of BD. One study [52] in Turkish patients showed that those patients who had papulopustular lesions were less likely to carry the HLA-B*5109 subtype, whereas the HLA-B*5103 subtype was seen at higher frequencies in those with central nervous system involvement. This led the authors to conclude that HLA-B*5109 could be protective against the development of papulopustular lesions in BD while HLA-B*5103 could have the opposite effect in the development of neuro-Behçet’s disease (NBD) development in BD patients. Another study in Korean BD patients showed that while an earlier disease onset was observed in patients carrying HLA-B*51, it appeared to be protective against the development of neurological and gastrointestinal symptoms [53]. In a report on the influence of HLA-B*51 on clinical manifestations in Japanese BD patients, a negative association between HLA-B*51 and gastrointestinal involvement in BD was validated in Japanese patients [54]. It was also found that HLA-B*51-positive patients were at lower risk for genital ulcerations but higher risk for ocular lesions. Other studies have also been done to study the clinical effects of different HLA alleles on patients with BD [49, 50].
The relationship between HLA-B*51 and neutrophil hyperfunction in patients with BD has been explored [55]. To assess abnormalities in the functioning of neutrophils, the levels of superoxide production were measured in peripheral blood neutrophils collected from both humans and HLA-B*51 transgenic mice. Experiments were conducted in a HLA-B*51 transgenic mouse model, utilizing N-Formylmethionyl-leucyl-phenylalanine (fMLP), phorbol myristate acetate (PMA), or opsonized zymosan for neutrophil induction. This model was selected and generated based on observations made in healthy patients when compared to patients with BD and other individuals carrying HLA-B*51. HLA-B*35 transgenic mice, as well as non-transgenic mice, were included in the experiments. When stimulated with fMLP, neutrophils from HLA-B*51 transgenic mice showed marked increases in superoxide production as opposed to those from HLA-B*35 transgenic mice and non-transgenic mice, in which superoxide production levels were similar to those of unstimulated neutrophils. Contrary to the responses observed using fMLP for neutrophil stimulation, all of the mice tested, both the transgenic lines as well as the non-transgenic line, showed elevated levels of superoxide production when stimulated by PMA or opsonized zymosan [55]. There was no clear evidence to explain why the HLA-B*51 neutrophils were more responsive when induced with fMLP [55].
The HLA-B*51 transgenic mice failed to show signs of clinical or pathologic changes that would indicate the development of active lesions reminiscent of those observed with BD in humans. These results lend credence to the idea that BD is caused by a combination of factors, both genetic and environmental, as opposed to independent factors working in isolation. However, another study concluded that there was no direct correlation between MHC and the development of BD symptoms in an animal model developed using HSV-1 virus [56]. In this study, the authors used 5 different mouse strains, two of which shared a common haplotype, and infected these subjects with HSV-1. Of the 5 lines infected, viz., B10.BR (H-2k), B10.RIII (H-2r), C57BL/6 (H-2b), C3H/He (H-2k), Balb/c (H-2d), only three showed promising results with 40–50% of the infected mice presenting with BD-like symptoms, while only 2% of mice of the other two strains showed BD-like symptoms. Interestingly, B10.BR and C3H/He strains presented different patterns of disease development, 40% and 2%, respectively, despite the fact that they shared a common haplotype. The authors concluded that a mouse strain susceptibility to a certain viral strain may be more important than MHC association when trying to induce BD-like symptoms [56]. However, no final conclusions concerning BD and HLA-B*51 can be made until a study is accomplished using HLA-B*51 transgenic mice in combination with a pathogen suspected to be involved in BD, such as HSV-1. Also, due to obvious differences in the physiology of mice versus humans, the findings cannot be extrapolated from mice to humans. While no clinical disease signs were observed in HLA-B*51 transgenic mice, these mice may still be a useful tool for investigating this enigmatic disease. Genetics may only be part of the causative factors, but by combining this model with a suspected pathogen involved in BD, answers may be provided to many currently unanswered questions.
Major histocompatibility complex class I chain-related gene A and B (MICA and MICB) are both members of a family of non-classical MHC molecules and are located in close proximity to HLA-B [57, 58]. Multiple groups have reported a relationship between MICA and BD, an association, which appears to exist across different populations [57–60]. One study generated MICB transgenic mouse lines to investigate the relationship between MICB expression and BD pathogenesis [58]. However, the results of the experiments suggested a lack of direct association between MICB expression and BD.
3. ALPHA-TROPOMYOSIN MODEL
The existence of an autoimmune component in BD development has been suggested by a number of investigators [25, 61–64]. In one of these studies [64], 179 genes were found to be modified in peripheral blood cells (PBCs) that were collected from ten BD patients that exhibited signs of active disease when compared to ten healthy controls. Among these, increased expression of Th17-related and type I interferon (IFN)-inducible genes were observed. Another study found that mouse filamentous neuronal processes, the likely antigen being neurofilament medium (NF-M) in the brain, retina and scrotal skin, was able to react with sera collected from BD patients [65]. Further analysis of the human form of NF-M revealed structural homology between specific regions of this antigen and bacterial HSP-65, as well as one region of mycobacterial HSP-65, which has been shown to be associated with BD.
An investigation was conducted regarding possible autoantigens toward which antibodies were being produced in patients with BD. Random selection of fifteen patients who had been diagnosed with BD from a rheumatology clinic was accomplished [66]. Sera were then collected from these patients as well as control patients with recurrent aphthous stomatitis and healthy individuals. These sera were then used as the primary antibody source in Western blot experiments. Initially, the serum of one typical patient with active BD was tested against rat tissue lysates originating from several different organs, including vagina, tongue, skin, muscle, heart, thymus, brain, myelin, kidney, lung, and intestine. These samples were then used as an evaluation panel. Extracts from fat and serum were also included in the evaluation panel. Western blot analyses revealed a 37-kDa band which was present in vagina, tongue, skin, muscle, and heart extracts, but not in the other extracts. Employing in-gel digestion and mass spectrometry, the authors were able to identify the 37 kDa band as α-tropomyosin.
Tropomyosin is a coiled-coil type protein found in both muscle and non-muscle cells, which is involved in the modulation of the cytoskeleton [67], as well as heart and skeletal muscle contractions [68]. Tropomyosin binds actin and regulates actin/myosin interactions [69]. This protein is found in both invertebrate and vertebrate species. While tropomyosin is non-allergenic in vertebrates, this is not the case for invertebrates [70]. Tropomyosin from invertebrate species such as crustaceans, mites, mollusks, and cockroaches, is highly allergenic [71]. Of the five major human tropomyosin isoforms described [72], human tropomyosin isoform 5 (hTM5) has been shown to be autoreactive in patients with ulcerative colitis (UC) [73]. Therefore, it is not surprising that this protein is associated with yet another disease that has been proposed to be autoinflammatory.
The sera collected from the other BD patients, as well as the healthy and otherwise diseased (e.g., recurrent aphthous stomatitis patients) controls, were tested against skin and tongue lysate. This revealed three additional BD patients carrying antibodies against α-tropomyosin [66]. Neither the aphthous stomatitis patients nor healthy controls tested positive. The authors also assessed serum reactivity against a panel consisting of extracts of rat skin, bovine α-tropomyosin, human muscle, and recombinant skeletal muscle human α-tropomyosin. Again, the serum used reacted with every protein in the panel, producing a band of the same size /electrophoretic mobility.
To assess the auto-immunogenic potential, Lewis rats were immunized with bovine α-tropomyosin [66]. These immunized rats presented with clinical symptoms indicative of both eye and skin involvement, some of which included thickening of the iris and uvea in the eyes and micro-abscess formation in the epidermis of the skin. In order to examine the possible involvement of anti-tropomyosin T cells, rats were also injected with a T-cell line specific for bovine tropomyosin. This resulted in ocular clinical symptoms, such as thickening and infiltration of the ciliary body and iris. This study was not only able to identify a new possible autoantigen in patients with BD, but also allowed for the development of an animal model which could be useful in studying BD pathogenesis. Immunization of Lewis rats with α-tropomyosin led to the development of two out of three symptoms that form the characteristic BD triad for diagnosis, namely eye and skin involvement. Serum collected from a patient in this study also interacted with vaginal extracts collected from rats, showing reactivity to vaginal tissue. One possible downside in this study is the fact that only a fraction of BD patients tested showed evidence of auto-reactivity toward α-tropomyosin. The authors did however acknowledge this limitation and suggested potential reasons for this. One of these reasons is epitope spreading [66], which refers to the development of antibodies that react or bind to sites other than specific primary disease-causing sites [74, 75]. Another proposed explanation [66] was the possibility of misdiagnosis in patients who were negative for anti-α-tropomyosin antibodies. Symptoms associated with BD are not specific for the disease and without a definitive diagnostic test for patients, misdiagnosis could occur.
Another investigation also used α-tropomyosin to immunize rats. However, this time the focus was on the characterization of the arthritic symptoms generated using this model [76]. Arthritis was observed in 94% of the female and in 93% of the male rats used, while the control mice failed to develop any disease. This study also included a treatment group in which infliximab was administered to a subset of the animals [76]. Infliximab is a monoclonal antibody generated against tumor necrosis factor (TNFα) [77], a proinflammatory cytokine [78]. This antibody is comprised of two variable regions derived from mice that bind TNFα and a constant region of human origin [77]. Rats treated with this drug did not show signs of arthritis [76]. Treatment was administered on days 0, 4, 7, 14, 21, and 28 post-vaccination. This would indicate that TNFα could be a potential target in patients with active BD. However, the use of a mouse/human chimeric antibody, e.g., infliximab, could induce unfavorable immune responses in humans who may react negatively to the murine portion of the antibody. Adalimumab, another TNFα inhibitor in which the non-human portions have been replaced with those of human, may be a more viable option for the treatment of BD [77].
In an overall sense, this model shows great potential for use in clinical drug trials as well as for studies involving BD pathogenesis. While it may not necessarily be representative of the mechanism of disease development in the entire BD population, it may actually be representative of a subset of that community. The etiology of BD remains unclear and multiple routes may exist for disease development, one of which may include auto-immune reactions to α-tropomyosin. One additional study [79] also reported the presence of anti-tropomyosin antibodies in four of eighteen BD patients tested. To more definitively determine whether auto-reactivity against α-tropomyosin is a determinant for BD development, additional large-scale studies using diverse patient cohorts would be necessary [66].
4. HEAT SHOCK PROTEIN MODEL
Heat shock proteins (HSPs) are highly conserved ubiquitous proteins that are present in every organism [80]. They are induced under varying conditions of stress, such as hypoxia [81], chemical stress [82], or hyperthermia [83]. Temperatures constituting a hyperthermic stressor would vary based on the normal temperature of an organism [83]. HSPs can function as molecular chaperones that aid in folding and as facilitators of peptide loading to the MHC [84]. They can also intervene under ischemic conditions to reduce neuronal cell death. These proteins are highly conserved across different species. For example, the bacterial 90 kDa heat shock protein (HSP90), found in Escherichia coli, shows a 50% sequence similarity to its human homolog [85]. Mycobacterial HSP60 shares approximately 60% homology with mammalian HSP60 [86]; and DnaK proteins (bacterial homolog of HSP70 [87]) are 50% identical to eukaryotic proteins at the amino acid level [88]. Due to the high levels of homology across species, the invasion of foreign HSPs can result in the induction of an immune response in the infected host [89]. Consequently, self-HSPs can be attacked by the body’s defense system leading to certain autoimmune conditions, such as rheumatoid arthritis [90], inflammatory bowel diseases [91], and ulcerative colitis. The presence of antibodies against HSP60 in the sera of BD patients suggests the involvement of this protein in the pathogenesis or development of the disease [92, 93].
It was found that uveitis could be induced in Lewis rats immunized with peptide 336–351 of human HSP-60 [94]. Uveitis was observed in 80% of rats immunized with a combination of this peptide and B. pertussis. Rats were also observed for signs of extra-ocular disease but failed to exhibit anything obvious. Histological examination revealed the presence of mononuclear cell infiltration in and around the ciliary body and iris, and a few polymorphonuclear leukocytes were also present. Additional experiments were conducted with the aim of investigating whether the development of uveitis would be hindered by the introduction of the peptide via oral or nasal routes. However, changes to the route of administration failed to prevent the development of uveitis. Iridocyclitis was induced in 42.2% of rats immunized by any route, while only a 4.9% loss of photoreceptors was reported. However, a drastic switch in the induction of iridocyclitis and photoreceptor loss was observed when the peptide was administered sequentially via a combined mucosal-subcutaneous route. Iridocyclitis was only recorded in 25% of rats, while photoreceptor loss was recorded in as many as 40% of rats.
Three different treatment options were also evaluated to determine their effects on uveitis development. Signs of uveitis could be observed in 50% of untreated rats immunized with HSP-60 peptide 336–351 by day 22, with 82% of rats showing symptoms by day 30. In contrast, when treated with CD8 monoclonal antibodies, more than 50% of rats developed uveitis in 11 days (half the original time observed). By day 25, all of these rats had developed uveitis, suggesting the suppression of uveitis development by CD8+ cells. The opposite was observed in rats treated with CD4 monoclonal antibodies. The two different doses of the antibody administered illustrated a dose-dependent response, with only 55.5% of rats developing uveitis by day 30 (from 82.2% untreated) when treated with a low dose and only 25% developing uveitis by day 30 when treated with the high dose. CD4+ cells, therefore, appear to incite disease development. The third treatment tested was IL-4. The cohort undergoing this regimen showed a substantial decrease in disease occurrence, from 68% to only 30.4%. This is not surprising since IL-4 is a Th2 cytokine and Th2 cytokines have been shown to be present at lower concentrations in patients with BD [95]. However, the authors [94] were not able to find significant differences in the mRNA levels of Th1 and Th2 cytokines between rats that were immunized with peptide and those that were not. There were also no significant differences in the mRNA levels of these cytokines in rats that developed uveitis and those that did not.
Altogether, this HSP animal model can be used as a BD model for several different reasons. HSPs share a high level of homology across species and infection with bacterial and mycobacterial HSPs have been implicated in BD development. This would, therefore, imitate at least one form of disease acquisition and progression. Another advantage of this model is one factor that may initially appear as a downside to using it, i.e., the fact that rats develop no extra-ocular symptoms. The use of single symptom models might be useful to observe or study the mechanisms involved with each symptom in isolation. Another advantage of using this model arises from the fact that rats respond positively to treatments. However, it is interesting to note that rats were unable to show any differences in the Th1 versus Th2 cytokine profiles. Further studies utilizing the model may be necessary to determine whether a timing issue exists, as suggested, or something entirely different [94]. In addition, HSPs could be viable therapeutics in the treatment of uveitis associated with BD. As ironic as it may seem, clinical studies [96] have also shown the utility of HSPs in the treatment of certain autoimmune conditions. The proposed logic for this restored tolerance is rooted in three different aspects, which include the high level of evolutionary conservation of HSPs, the fact that HSPs most frequently provide ligands for MHC class II molecules in the cytosol and nucleus, and that inflammatory stress leads to an upregulation of HSPs in tissues. Further studies could expand on this model and therapeutic options by introducing an HSP treatment regimen to evaluate the responses of the animals.
5. S ANTIGEN MODEL
Uveitis is one of the major symptoms observed in patients with BD and refers to inflammation of the pigmented layer of the eye comprising the choroid, iris, and ciliary body (uvea) [97]. Several animal models of experimental uveitis have been described [98], some of which include immunization with interphotoreceptor retinoid-binding protein (IRBP) in BIO.A [99], BlO.S (8R), A, and MRL (+/+) mice [100], recoverin in Lewis rats [101], and retinal soluble antigen or S-Ag in SJL/J X AKR, and AKR mice [100]. S-Ag is a 48 kD protein, which is found in the outer segments of rod cells in the retina [102, 103]. This protein shares a high degree of homology among mammals [104] and induces uveitis upon immunization in mice [100].
The development of an experimental autoimmune uveitis model (EAU) using Lewis rats injected with S-Ag has been reported [105]. Here, the authors also explored uveitopathogenicity of different peptides originating from two different viral proteins with sequences somewhat similar to that of S-Ag. Female Lewis rats were immunized with bovine S-Ag, or with peptides from one of four different sources: peptide M (main site present in S-Ag capable of inducing uveitis [106]), hepatitis B virus, murine leukemia virus, and potato proteinase. The rats were immunized a second time two weeks later. EAU was recorded in rats immunized with bovine S-Ag (60%), peptide M (80%), and the hepatitis B virus peptide (60%), with symptoms developing between 16 and 25 days. Retinal detachment, accompanied by an accumulation of cellular exudate, was observed in the subretinal space. In addition, the authors reported vasodilation of the iris and posterior iris synechiae. Inflammatory cell infiltration was also observed in the vitreous and anterior chamber. However, a progressive reduction in inflammatory activity was observed a week after EAU onset.
Further studies were conducted with the aim of examining the stimulation of lymphocytic proliferation of cells derived from the lymph nodes of rats immunized with different viral peptides, or peptide M. These cells were stimulated with either peptide M or viral peptides. All of the peptides showed the ability to induce lymphocytic cell proliferation, whether immunized with peptide M or viral peptides. It is important to note that although the peptides used for the rat immunizations only shared as little as 3–4 consecutive amino acids, this was still sufficient to elicit a cross-reactive response. A mechanism of molecular mimicry that may be induced by a viral infection was proposed. Infection with the viral agent may then lead to the induction or sensitization of mononuclear cells that may potentially cross-react with host proteins [105]. This mechanism is similar to the autoimmune response, which was proposed for HSP and lends credence to the theory that BD is a disease of autoinflammatory origin.
An earlier study investigated the role of superoxide in ocular inflammation using two different animal models [107]. The first model was an EAU model generated using S-Ag in female Hartley strain guinea pigs. Clinical evidence of EAU was observed approximately two weeks after injection. The second model was a reverse passive type endophthalmitis model generated via bovine serum albumin (BSA) immunization of albino rabbits. Concentrated anti-BSA serum was injected into the vitreous of naïve rabbits three hours before intravenously administering a 3% BSA injection. In both models, one eye was treated with superoxide dismutase (SOD). Significantly higher levels of superoxide were recorded in neutrophils from guinea pigs with EAU as compared to the healthy controls. Superoxide levels in healthy versus diseased rabbits were not reported. While treatment with SOD did not prevent inflammation in either of the models, a reduction in the severity of inflammation was observed. The results of this study led the authors to conclude that superoxide plays a role in tissue damage, although additional factors may be contributing to the level of cell damage in the inflamed tissues.
All in all, this model appears to be useful for the study of uveitis, which is one of the major symptoms associated with BD. While it may be most appropriate to utilize a model that recapitulates the disease as fully as possible, single symptom models may also provide the added advantage of studying specific pathways involved in each symptom. This would allow an assessment of similarities and differences between pathways involved in the presentation of each symptom. As BD appears to be the result of a combination of both genetic and environmental factors, it may be useful to regenerate this model using transgenic rats or even mice of different genetic backgrounds, encoding some of the genes most commonly associated with BD. Another useful addition would be an investigation into the upregulation and downregulation of specific Th1- and Th2-associated cytokines. As it is likely that BD is caused by different combinations of stimuli, diversity among animal models is crucial. However, it is not only important to determine the underlying mechanisms that give rise to each BD phenotype but also to evaluate the cellular, molecular, and immunological features that unite the various disease presentations. The increased superoxide production recorded in the HLA-B*51 transgenic mice which was also observed in the second S-Ag model (guinea pig model), unfortunately, occurred in the absence of any clinical symptoms [55]. The fact that this was observed in animals expressing a gene associated with BD development, as well as the S-Ag EAU model, leads to the belief that superoxide overproduction may play a significant role in the development of the disease/disease pathogenesis. The fact that both models showed an increase in superoxide levels while presenting with different clinical phenotypes lends credence to the theory that cell damage is further exacerbated by factors other than superoxide [107]. It may prove important to conduct additional investigations into increased superoxide production in other single symptom models.
6. HUMAN SERA MODEL
The discovery of antibodies to tropomyosin and HSP in the sera of patients with BD has been discussed earlier [66, 92, 93]. Additional evidence suggests that autoimmune responses to retinal antigens may play a role in uveitis pathogenesis, a phenotype that is also observed with BD [108, 109]. These antigens include IRBP and S-Ag, both of which were also discussed in earlier sections [109]. Another investigation [110] reported the development of an NBD (neuro-Behçet’s disease) animal model using antibodies present in the sera of patients with NBD. These sera showed the ability to react with membrane antigens of neuronal axonal processes (neuropils).
The pathogenic effects of the antibodies were studied both in vivo and in vitro. For the in vivo studies, sera collected from seven different NBD patients were pooled along with the sera of seven healthy controls. The sera were depleted of IgG before administering each of the four treatment options into the lateral ventricles of male Sprague-Dawley rats. The four treatment options included NBD IgG-depleted sera, healthy IgG-depleted sera, purified IgG from healthy subjects, and purified IgG from NBD subjects; PBS (phosphate-buffered saline) was used as a fifth treatment and control. The authors performed a range of behavioral assessments on the rats, including locomotor activity, anxiety-like behaviors, and passive avoidance learning and memory tasks. The results of these assessments showed a significant reduction in the locomotor activity of rats injected with NBD IgG but not in any of the other treatment groups. However, the other behavioral assessments yielded no significant differences among the treatment groups tested. The results of in vitro experiments conducted with SH-SY5Y cells implied that NBD IgG induced neuronal cell death by the enhancement of apoptosis [110].
This model could prove to be very useful due to the fact that the “infectious agent” was obtained from actual BD patients showing neurological symptoms. Serum IgG, but not IgG depleted sera, was shown to induce both neurotoxic effects in vitro and a decrease in locomotor activity in vivo [110], lending further credence to the idea that BD is an autoinflammatory disease.
7. HERPES SIMPLEX VIRUS TYPE-1 MODEL
Patients with BD commonly present with a number of recrudescent symptoms, including oral aphthous ulcers, genital ulcers, uveitis, and skin lesions [111]. These symptoms are similar to some of those observed in patients infected with HSV-1 [112]. Additional results showed significantly elevated levels of anti-HSV-1 antibodies in the sera of patients with BD as compared to controls [113]. Furthermore, HSV DNA has been detected in both the saliva [114] and skin lesions (one patient) [115] of BD patients. Overall, an HSV model aimed at recapitulating the phenotypes associated with BD would appear logical and attractive. A previous study reported the findings of such a model using HSV-1 in the Institute of Cancer Research (ICR) mice [116].
In this model, mice were inoculated with the KOS strain of HSV-1 via the ear and observed for four weeks [116]. After the first month, they were inoculated once again and observed for an additional four months. Three different outcomes were observed with the infected mice: a) approximately one third died, b) another third showed BD-like symptoms, while c) the final third showed a single symptom or none at all. For a mouse to be considered to exhibit BD-like symptoms, the presence of two recorded symptoms would be necessary. Observed symptoms included skin ulcers (17.1% of mice), eyes syndromes (11.6%), hair loss (10.1%), genital ulcers (5.8%), bullae (3.5%), arthritis (1.6%), gastrointestinal (GI) symptoms (1.6%), and oral ulcers (1.2%). The polymerase chain reaction (PCR) also revealed the presence of HSV DNA sequences in skin lesions and in the GI tract; HSV DNA was not detected in healthy skin. Histological studies revealed the accumulation of inflammatory cells around blood vessels, a sign of vasculitis.
This model, at first glance, appears very attractive, given the fact that it mirrors so many of the symptoms observed in patients with BD. Nonetheless, given the low penetrance rates for some of these symptoms, it would be necessary to use very large sample sizes for studies using this model. For this investigation, a total of 258 mice were used, approximately one-third of which died and another third of which were considered healthy, leaving only the final third to be classified as BD-like. However, this model could be very useful in studies where the researchers are not limited in the number of mice needed to be used [116]. Another similar study [56] examined the relationship between MHC haplotype and the development of BD-like symptoms. These symptoms were induced in inbred mouse strains with different MHC haplotypes. This report showed large differences in the presentation of BD-like symptoms among the different mouse strains, with as much as 50% of one strain showing BD-like symptoms and as little as 2% of another. It is, therefore important to consider the benefits of using one mouse strain over another.
Another important factor to consider would be the genetic and environmental elements involved in developing such an animal model. It appears very likely that BD results not just from an infection with a single pathogen or just one or multiple genetic influences, but rather from a combination of these factors as well as environmental factors. The environmental conditions in which mice are held, or in which their food/water is prepared, could have a significant impact on response to an infectious agent such as HSV-1. In addition, inbred mice, even those having originated from the same source, may, over time, alter critical genetic elements that impact susceptibility to different illnesses. This is underscored by an observation (unpublished results) made by our group using C57BL/6 mice (to reproduce this HSV-1 model), where there appeared to be variation in frequency or even the types of symptoms displayed.
The HSV-1 model, nonetheless, is by far the most common model utilized for BD animal studies. A modified version of this HSV-1 model was used to study Galectin-9 (Gal9) as a possible BD-like treatment and found that it was able to reduce proinflammatory cytokine levels and lead to a decreased severity score in the mice that were treated [117]. Another study [118] conducted also utilized this model to study a BD therapeutic, the target this time being TNFα. Here, a small interfering RNA (siRNA) against TNFα was utilized to negatively alter over-expression and hence treat chronic inflammation. They also compared the efficacy of their siRNAs to that of two previously developed drugs-infliximab and etanercept. Not only did the siRNA treatment appear to decrease BD-like symptoms in a larger number of mice, it also appeared to lead to an improvement of symptoms in a shorter period of time than infliximab. Other studies utilized this model to look at the effects of TNFα inhibition [119], Interleukin-6 (IL-6) inhibition [120], vitamin D administration [121], famciclovir (an antiviral agent) [122], polyinosinic:polycytidylic acid (Poly I:C) administration (Poly I:C is an immunostimulant that induces interleukin-15 (IL-15) and IL-15 receptor alpha) [123], irradiation [124], gemcitabine [125], microRNA- 21 (miRNA-21) inhibition [126], classical versus alternative macrophage activation [127], T cell immunoglobulin mucin 1 and 4 (TIM-1 and 4) [128], thalidomide [129] and N-acetyl galactosamine-4-SO4 (GalNAc) [130] on BD-like mice. This model has also been used in several other works, most of which have been inflammation-based.
8. AUTHOR’S INSIGHTS ON THE TOPIC
BD remains a rather elusive disease, leaving many questions unanswered. One of the important first steps in elucidating the etiology of the disease is the successful development of a reliable and reproducible animal model. Due to the strong likelihood of diverse contributing elements, the development of relevant models would be compulsory if we are to understand the underlying mechanisms involved in the development and pathogenesis of BD. This article has reviewed the development of different BD models, each having its own benefits and limitations.
BD can manifest differently in individual patients [131], and so there may be different mechanisms involved in the development of individual symptoms. The use of single-symptom models, such as the uveitis model, would be useful for studying individual pathways or mechanisms involved in specific symptom development. However, such a model may not be as appropriate for a more inclusive study of BD. Although the HLA-B*51 mouse model failed to show any clinical symptoms, this model could prove very useful if coupled with another model such as the HSP or, even better, the HSV-1 model. Again, it is most likely that BD results from a combination of both genetic and environmental/pathogenic factors (Fig. 1), and so a model depicting this route of disease development may prove most promising. The HSV-1 model, alone, appears to have been the most successful, but given the fact that the model relies solely on a pathogenic or viral factor, there is a need for improvement. With two-thirds of the world having HSV-1 [132], it is very unlikely that this virus, even if shown to be a contributing factor, could be the major cause of BD.
Fig. (1).
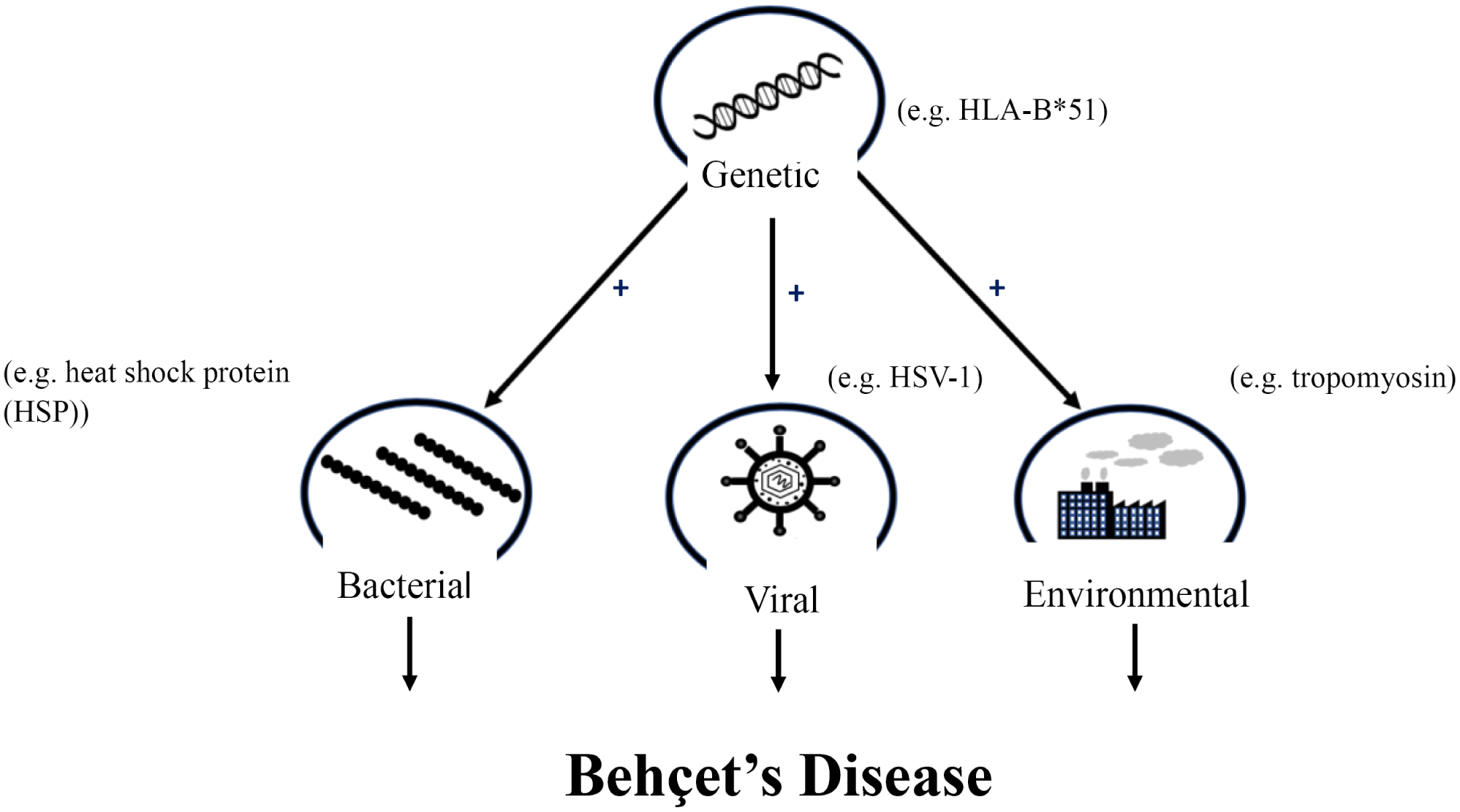
Summary of proposed mechanism of Behçet’s disease development.
In conclusion, several BD animal models have been described, each having their strengths and weaknesses (Table 1). The selected model will depend on the focus of the study. The best model to mirror the disease entirely would most likely be a cross between the HLA-B*51 model and the HSV-1, or even the HSP model as this disease most likely results from a combination of both genetic and environmental/pathogenic factors.
Table 1.
Major advantages and disadvantages of individual BD models.
| Model | Major Advantage | Major Disadvantage |
|---|---|---|
| HLA-B*51 [55] |
Transgenic mouse model developed being gene associated with BD | No clinical BD symptoms |
| Alpha-tropomyosin [66] |
Model developed using antibodies found in BD patients | Alpha-tropomyosin antibody only found in a small subset of BD patients |
| Heat shock protein [94] |
Models involvement of a protein that has already been shown to be associated with BD | No significant differences in the mRNA levels of Th1 and Th2 cytokines in immunized animals vs. those that were not immunized. |
| S antigen (S-Ag) [107] [105] |
Successfully induced EAU in guinea pigs with increased superoxide production (uveitis and increased superoxide production have been observed with BD patients) EAU also successfully induced in Lewis rats immunized with both S-Ag and Hepatitis B virus peptides implying autoreactive mechanisms activated by a viral infection |
May not be useful for more comprehensive studies. |
| Human sera [110] |
Model developed using antibodies found in BD patients | Sera possibly containing heterogeneous mixture of antibodies against different targets and not actual antigens were used; therefore, one cannot be certain that changes observed were induced solely or even partly by antibodies against neuropil antigens. |
| Herpes simplex virus type 1 [116] |
Best able to recapitulate BD phenotype | Low penetrance rates necessitate the use of large numbers of animals. |
ACKNOWLEDGEMENTS
The authors would also like to thank Dr. Kritika Singh for her assistance in the preparation of Figure 1.
FUNDING
We gratefully acknowledge financial support from the Warren Center for Drug Discovery, the University of Notre Dame Behcet’s Disease Research Fund, and a grant from the W. M. Keck Center for Transgene Research.
Footnotes
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- [1].Tan SY, Poole PS. Hulusi Behçet (1889–1948): Passion for dermatology. Singapore Med J 2016; 57(7): 408–9. 10.11622/smedj.2016123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Soomro AH, Khan E, Noori S, Lone MA, Syal Z, Sheikh S. Assessment of Cytokine Release against Oral Mucosal Cell Line Culture (TR146) Stimulated by Neutrophil Elastase Associated with Behcet’s Disease. Int J Dent 2019; 20196095628 10.1155/2019/6095628 [DOI] [PMC free article] [PubMed]
- [3].Shahram F, Kazemi J, Mahmoudi M, Jadali Z. Single Nucleotide Polymorphisms of FCRL3 in Iranian Patients with Behcet’s Disease. Iran J Public Health 2019; 48(6): 1133–9. [PMC free article] [PubMed] [Google Scholar]
- [4].Villiger RA, Stefanski AL, Grobéty V, Adler S, Villiger PM. Behçet’s syndrome: clinical presentation and prevalence in Switzerland. Swiss Med Wkly 2019; 149w20072 10.4414/smw.2019.20072 [DOI] [PubMed] [Google Scholar]
- [5].Talarico R, Elefante E, Parma A, Taponeco F, Simoncini T, Mosca M. Sexual dysfunction in Behçet’s syndrome. Rheumatol Int 2019; ••• 10.1007/s00296-019-04455-w [DOI] [PubMed]
- [6].Furuya MY, Temmoku J, Fujita Y, et al. Vasculo-Behçet disease complicated by conversion disorder diagnosed with 18F-fluorodeoxy-glucose positron emission tomography combined with computed tomography (PET/CT). Fukushima J Med Sci 2019; 65(2): 55–60. 10.5387/fms.2019-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Petrushkin H, Norman PJ, Lougee E, et al. KIR3DL1/S1 Allotypes Contribute Differentially to the Development of Behçet Disease. J Immunol 2019; 203(6): 1629–35. 10.4049/jimmunol.1801178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Islam SMS, Byun H-O, Choi B, Sohn S. Inhibition of CD83 Alleviates Systemic Inflammation in Herpes Simplex Virus Type 1-Induced Behçet’s Disease Model Mouse. Mediators Inflamm 2019; 20195761392 10.1155/2019/5761392 [DOI] [PMC free article] [PubMed]
- [9].Kim SW, Kim TG, Oh J, et al. Clinical and Radiographic Characteristics of Neuro-Behçet’s Disease in South Korea. J Clin Neurol 2019; 15(4): 429–37. 10.3988/jcn.2019.15.4.429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Calamia KT, Wilson FC, Icen M, Crowson CS, Gabriel SE, Kremers HM. Epidemiology and clinical characteristics of Behçet’s disease in the US: a population-based study. Arthritis Rheum 2009; 61(5): 600–4. 10.1002/art.24423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Davatchi F, Chams-Davatchi C, Shams H, et al. Behcet’s disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol 2017; 13(1): 57–65. 10.1080/1744666X.2016.1205486 [DOI] [PubMed] [Google Scholar]
- [12].Kirino Y, Nakajima H. Clinical and Genetic Aspects of Behçet’s Disease in Japan. Intern Med 2019; 58(9): 1199–207. 10.2169/internalmedicine.2035-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kappen JH, Medina-Gomez C, van Hagen PM, et al. Genome-wide association study in an admixed case series reveals IL12A as a new candidate in Behçet disease. PLoS One 2015; 10(3): e0119085-. 10.1371/journal.pone.0119085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fei Y, Webb R, Cobb BL, Direskeneli H, Saruhan-Direskeneli G, Sawalha AH. Identification of novel genetic susceptibility loci for Behçet’s disease using a genome-wide association study. Arthritis Res Ther 2009; 11(3): R66–6. 10.1186/ar2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kirino Y, Bertsias G, Ishigatsubo Y, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat Genet 2013; 45(2): 202–7. 10.1038/ng.2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu P, Du L, Hou S, et al. Association of LACC1, CEBPB-PTPN1, RIPK2 and ADO-EGR2 with ocular Behcet’s disease in a Chinese Han population. Br J Ophthalmol 2018; 102(9): 1308–14. 10.1136/bjophthalmol-2017-311753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim SW, Jung YS, Ahn JB, et al. Identification of genetic susceptibility loci for intestinal Behçet’s disease. Sci Rep 2017; 7: 39850 10.1038/srep39850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shenavandeh S, Jahanshahi KA, Aflaki E, Tavassoli A. Frequency of HLA-B5, HLA-B51 and HLA-B27 in patients with idiopathic uveitis and Behçet’s disease: a case-control study. Reumatologia 2018; 56(2): 67–72. 10.5114/reum.2018.75516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sohn S, Lee E-S, Kwon HJ, Lee SI, Bang D, Lee S. Expression of Th2 cytokines decreases the development of and improves Behçet’s disease-like symptoms induced by herpes simplex virus in mice. J Infect Dis 2001; 183(8): 1180–6. 10.1086/319681 [DOI] [PubMed] [Google Scholar]
- [20].Zheng Z, Sohn S, Ahn KJ, Bang D, Cho SB. Serum reactivity against herpes simplex virus type 1 UL48 protein in Behçet’s disease patients and a Behçet’s disease-like mouse model. Acta Derm Venereol 2015; 95(8): 952–8. 10.2340/00015555-2127 [DOI] [PubMed] [Google Scholar]
- [21].Kaneko KNF, Togashi A, Nomura E. Behcet’s Disease and Related Diseases -Immune Reactions to Oralstreptococci in Their Pathogenesis. J Dermatological Res 2016; 1(3): 41–50. 10.17554/j.issn.2413-8223.2016.01.12 [DOI] [Google Scholar]
- [22].Galeone M, Colucci R, D’Erme AM, Moretti S, Lotti T. Potential Infectious Etiology of Behçet’s Disease. Pathol Res Int 2012; 2012595380 10.1155/2012/595380 [DOI] [PMC free article] [PubMed]
- [23].Rozin AP Is Behcet’s syndrome associated with infection? Ann Rheum Dis 2005; 64(3): 513–5. [PMC free article] [PubMed] [Google Scholar]
- [24].Kaneko F, Oyama N, Nishibu A. Streptococcal infection in the pathogenesis of Behçet’s disease and clinical effects of minocycline on the disease symptoms. Yonsei Med J 1997; 38(6): 444–54. 10.3349/ymj.1997.38.6.444 [DOI] [PubMed] [Google Scholar]
- [25].Lule S, Colpak AI, Balci-Peynircioglu B, et al. Behçet Disease serum is immunoreactive to neurofilament medium which share common epitopes to bacterial HSP-65, a putative trigger. J Autoimmun 2017; 84: 87–96. 10.1016/j.jaut.2017.08.002 [DOI] [PubMed] [Google Scholar]
- [26].Sohn S, Lee ES, Bang D. Learning from HSV-infected mice as a model of Behçet’s disease. Clin Exp Rheumatol 2012; 30(3)(Suppl. 72): S96–S103. [PubMed] [Google Scholar]
- [27].Nakamura T, Shirouzu T, Nakata K, Yoshimura N, Ushigome H. The Role of Major Histocompatibility Complex in Organ Transplantation-Donor Specific Anti-Major Histocompatibility Complex Antibodies Analysis Goes to the Next Stage. Int J Mol Sci 2019; 20(18): 4544 10.3390/ijms20184544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bodis G, Toth V, Schwarting A. Role of Human Leukocyte Antigens (HLA) in Autoimmune Diseases. Rheumatol Ther 2018; 5(1): 5–20. 10.1007/s40744-018-0100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].van Drongelen V, Holoshitz J. Human Leukocyte Antigen-Disease Associations in Rheumatoid Arthritis. Rheum Dis Clin North Am 2017; 43(3): 363–76. 10.1016/j.rdc.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Type Goodwin G. 1 Diabetes Mellitus and Celiac Disease: Distinct Autoimmune Disorders That Share Common Pathogenic Mechanisms. Horm Res Paediatr 2019; 92(5): 285–92. 10.1159/000503142 [DOI] [PubMed] [Google Scholar]
- [31].Prinz JC. Melanocytes: Target Cells of an HLA-C*06:02-Restricted Autoimmune Response in Psoriasis. J Invest Dermatol 2017; 137(10): 2053–8. 10.1016/j.jid.2017.05.023 [DOI] [PubMed] [Google Scholar]
- [32].Carapito R, Shahram F, Michel S, et al. On the genetics of the Silk Route: association analysis of HLA, IL10, and IL23R-IL12RB2 regions with Behçet’s disease in an Iranian population. Immunogenetics 2015; 67(5–6): 289–93. 10.1007/s00251-015-0841-6 [DOI] [PubMed] [Google Scholar]
- [33].Sakly K, Maatouk M, Hammami S, et al. HLA-G 14 bp insertion/deletion polymorphism and its association with sHLA-G levels in Behçet’s disease Tunisian patients. Hum Immunol 2016; 77(1): 90–5. 10.1016/j.humimm.2015.10.016 [DOI] [PubMed] [Google Scholar]
- [34].Hamzaoui A, Houman MH, Massouadia M, et al. Contribution of Hla-B51 in the susceptibility and specific clinical features of Behcet’s disease in Tunisian patients. Eur J Intern Med 2012; 23(4): 347–9. 10.1016/j.ejim.2011.12.011 [DOI] [PubMed] [Google Scholar]
- [35].Kaburaki T, Takamoto M, Numaga J, et al. Genetic association of HLA-A*2601 with ocular Behçet’s disease in Japanese patients. Clin Exp Rheumatol 2010; 28(4)(Suppl. 60): S39–44. [PubMed] [Google Scholar]
- [36].Kongkaew S, Yotmanee P, Rungrotmongkol T, et al. Molecular dynamics simulation reveals the selective binding of human leukocyte antigen alleles associated with behçet’s disease. PLoS One 2015; 10(9)e0135575 10.1371/journal.pone.0135575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Muruganandam M, Rolle NA, Sibbitt WL Jr, et al. Characteristics of Behcet’s Disease in the American Southwest. Semin Arthritis Rheum 2019; 49(2): 296–302. 10.1016/j.semarthrit.2019.03.003 [DOI] [PubMed] [Google Scholar]
- [38].Castaño-Núñez Á, Montes-Cano MA, García-Lozano JR, et al. Association of Functional Polymorphisms of KIR3DL1/DS1 With Behçet’s Disease. Front Immunol 2019; 10: 2755 10.3389/fimmu.2019.02755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Adeeb F, Ugwoke A, Stack AG, Fraser AD. Associations of HLA-B alleles with Behçet’s disease in Ireland. Clin Exp Rheumatol 2017; 35(2)(Suppl. 104): 22–3. [PubMed] [Google Scholar]
- [40].Hughes EH, Collins RW, Kondeatis E, et al. Associations of major histocompatibility complex class I chain-related molecule polymorphisms with Behcet’s disease in Caucasian patients. Tissue Antigens 2005; 66(3): 195–9. 10.1111/j.1399-0039.2005.00465.x [DOI] [PubMed] [Google Scholar]
- [41].Kimura T, Asano Y, Yamamoto M, Sugaya M, Sato S. Development of Behçet’s disease in a Caucasian with human leukocyte antigen B51 after immigration to Japan. J Dermatol 2011; 38(6): 581–4. 10.1111/j.1346-8138.2010.01125.x [DOI] [PubMed] [Google Scholar]
- [42].Xavier JM, Davatchi F, Abade O, et al. Characterization of the major histocompatibility complex locus association with Behçet’s disease in Iran. Arthritis Res Ther 2015; 17: 81 10.1186/s13075-015-0585-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Elfishawi MM, Elgengehy F, Mossallam G, et al. HLA Class I in Egyptian patients with Behçet’s disease: new association with susceptibility, protection, presentation and severity of manifestations. Immunol Invest 2019; 48(2): 121–9. 10.1080/08820139.2018.1517364 [DOI] [PubMed] [Google Scholar]
- [44].Choukri F, Chakib A, Himmich H, Hüe S, Caillat-Zucman S. HLA-B* 51 and B*15 alleles confer predisposition to Behçet’s disease in Moroccan patients. Hum Immunol 2001; 62(2): 180–5. 10.1016/S0198-8859(00)00249-4 [DOI] [PubMed] [Google Scholar]
- [45].Al-Okaily F, Al-Rashidi S, Al-Balawi M, Mustafa M, Arfin M, Al-Asmari A. Genetic association of HLA-A*26,-A*31, and-B*51 with Behcet’s disease in Saudi patients. Clin Med Insights Arthritis Musculoskelet Disord 2016; 9: 167–73. 10.4137/CMAMD.S39879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ortiz-Fernández L, Carmona FD, Montes-Cano MA, et al. Genetic analysis with the immunochip platform in Behçet disease. Identification of residues associated in the HLA class I region and new susceptibility loci. PLoS One 2016; 11(8)e0161305 10.1371/journal.pone.0161305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Montes-Cano MA, Conde-Jaldón M, García-Lozano JR, et al. HLA and non-HLA genes in Behçet’s disease: a multicentric study in the Spanish population. Arthritis Res Ther 2013; 15(5): R145 10.1186/ar4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakamura J, Meguro A, Ishii G, et al. The association analysis between HLA-A*26 and Behçet’s disease. Sci Rep 2019; 9(1): 4426 10.1038/s41598-019-40824-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kato H, Takeuchi M, Horita N, et al. HLA-A26 is a risk factor for Behçet’s disease ocular lesions. Mod Rheumatol 2020; •••: 1–5. 10.1080/14397595.2019.1705538 [DOI] [PubMed] [Google Scholar]
- [50].Kang EH, Kim JY, Takeuchi F, et al. Associations between the HLA-A polymorphism and the clinical manifestations of Behcet’s disease. Arthritis Res Ther 2011; 13(2): R49 10.1186/ar3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kuroyanagi K, Sakai T, Kohno H, et al. Association between the major histocompatibility complex and clinical response to infliximab therapy in patients with Behçet uveitis. Jpn J Ophthalmol 2015; 59(6): 401–8. 10.1007/s10384-015-0404-2 [DOI] [PubMed] [Google Scholar]
- [52].Demirseren DD, Ceylan GG, Akoglu G, et al. HLA-B51 subtypes in Turkish patients with Behçet’s disease and their correlation with clinical manifestations. Genet Mol Res 2014; 13(3): 4788–96. 10.4238/2014.July.2.8 [DOI] [PubMed] [Google Scholar]
- [53].Ryu HJ, Seo MR, Choi HJ, Baek HJ. Clinical phenotypes of Korean patients with Behcet disease according to gender, age at onset, and HLA-B51. Korean J Intern Med (Korean Assoc Intern Med) 2018; 33(5): 1025–31. 10.3904/kjim.2016.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mizuki Y, Horita N, Horie Y, et al. The influence of HLA-B51 on clinical manifestations among Japanese patients with Behçet’s disease: A nationwide survey. Mod Rheumatol 2019; •••: 1–7. 10.1080/14397595.2019.1649103 [DOI] [PubMed] [Google Scholar]
- [55].Takeno M, Kariyone A, Yamashita N, et al. Excessive function of peripheral blood neutrophils from patients with Behçet’s disease and from HLA-B51 transgenic mice. Arthritis Rheum 1995; 38(3): 426–33. 10.1002/art.1780380321 [DOI] [PubMed] [Google Scholar]
- [56].Sohn S, Lee ES, Lee S. The correlation of MHC haplotype and development of Behçet’s disease-like symptoms induced by herpes simplex virus in several inbred mouse strains. J Dermatol Sci 2001; 26(3): 173–81. 10.1016/S0923-1811(01)00086-X [DOI] [PubMed] [Google Scholar]
- [57].Marin MLC, Savioli CR, Yamamoto JH, Kalil J, Goldberg AC. MICA polymorphism in a sample of the São Paulo population, Brazil. Eur J Immunogenet 2004; 31(2): 63–71. 10.1111/j.1365-2370.2004.00446.x [DOI] [PubMed] [Google Scholar]
- [58].Nomura E, Sato M, Suemizu H, et al. Hyperkeratosis and leukocytosis in transgenic mice carrying MHC class I chain-related gene B (MICB). Tissue Antigens 2003; 61(4): 300–7. 10.1034/j.1399-0039.2003.00014.x [DOI] [PubMed] [Google Scholar]
- [59].Zhang J, Liao D, Yang L, Hou S. Association between Functional MICA-TM and Behcet’s Disease: A Systematic Review and Meta-analysis. Sci Rep 2016; 6: 21033 10.1038/srep21033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yabuki K, Mizuki N, Ota M, et al. Association of MICA gene and HLA-B*5101 with Behçet’s disease in Greece. Invest Ophthalmol Vis Sci 1999; 40(9): 1921–6. [PubMed] [Google Scholar]
- [61].Çolpak AI, Özdemir YG, Kalyoncu U. The presence of autoantibodies against vascular and nervous tissue in sera from patients with neuro-behçet’s disease. Noropsikiyatri Ars 2014. [DOI] [PMC free article] [PubMed]
- [62].Cebeci F, Onsun N, Pekdemir A, Uras AR, Kayataş K. Thyroid autoimmunity and Behçet’s disease: is there a significant association? ScientificWorldJournal 2013; 2013956837 10.1155/2013/956837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hussain M, Xiao J, Zhang Y, Chen P, Du H. Identification of hnRNP C1/C2 as an Autoantigen in Patients with Behcet’s Disease Iran J Immunol 2018; 15(2): 133–41. [PubMed] [Google Scholar]
- [64].Puccetti A, Fiore PF, Pelosi A, et al. Gene expression profiling in behcet’s disease indicates an autoimmune component in the pathogenesis of the disease and opens new avenues for targeted therapy. J Immunol Res 2018; 20184246965 10.1155/2018/4246965 [DOI] [PMC free article] [PubMed]
- [65].Taşçi B, Direskeneli H, Serdaroǵlu P, Akman-Demir G, Eraksoy M, Saruhan-Direskeneli G. Humoral immune response to mycobacterial heat shock protein (hsp)65 in the cerebrospinal fluid of neuro-Behçet patients. Clin Exp Immunol 1998; 113(1): 100–4. 10.1046/j.1365-2249.1998.00620.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mor F, Weinberger A, Cohen IR. Identification of alpha-tropomyosin as a target self-antigen in Behçet’s syndrome. Eur J Immunol 2002; 32(2): 356–65. [DOI] [PubMed] [Google Scholar]
- [67].Hitchcock-DeGregori SE, Barua B. Tropomyosin structure, function, and interactions: A dynamic regulator. Subcell Biochem 2017; 82: 253–84. 10.1007/978-3-319-49674-0_9 [DOI] [PubMed] [Google Scholar]
- [68].Gunning PW, Hardeman EC, Lappalainen P, Mulvihill DP. Tropomyosin - master regulator of actin filament function in the cytoskeleton. J Cell Sci 2015; 128(16): 2965–74. 10.1242/jcs.172502 [DOI] [PubMed] [Google Scholar]
- [69].Khaitlina SY. Chapter Seven - Tropomyosin as a Regulator of Actin Dynamics K W B T-I R of C and Jeon MB, Ed. Academic Press; 2015; 318: pp. 255–91. [DOI] [PubMed] [Google Scholar]
- [70].Mikita CP, Padlan EA. Why is there a greater incidence of allergy to the tropomyosin of certain animals than to that of others? Med Hypotheses 2007; 69(5): 1070–3. 10.1016/j.mehy.2006.12.060 [DOI] [PubMed] [Google Scholar]
- [71].Faber MA, Pascal M, El Kharbouchi O, et al. Shellfish allergens: tropomyosin and beyond. Allergy 2017; 72(6): 842–8. 10.1111/all.13115 [DOI] [PubMed] [Google Scholar]
- [72].Mirza ZK, Sastri B, Lin JJC, Amenta PS, Das KM. Autoimmunity against human tropomyosin isoforms in ulcerative colitis: localization of specific human tropomyosin isoforms in the intestine and extraintestinal organs. Inflamm Bowel Dis 2006; 12(11): 1036–43. 10.1097/01.mib.0000231573.65935.67 [DOI] [PubMed] [Google Scholar]
- [73].Kovvali G, Das KM. Molecular mimicry may contribute to pathogenesis of ulcerative colitis. FEBS Lett 2005; 579(11): 2261–6. 10.1016/j.febslet.2005.02.073 [DOI] [PubMed] [Google Scholar]
- [74].Powell AM, Black MM. Epitope spreading: protection from pathogens, but propagation of autoimmunity? Clin Exp Dermatol 2001; 26(5): 427–33. 10.1046/j.1365-2230.2001.00852.x [DOI] [PubMed] [Google Scholar]
- [75].Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol 2002; 2(2): 85–95. 10.1038/nri724 [DOI] [PubMed] [Google Scholar]
- [76].Baharav E, Mor F, Halpern M, Quintana F, Weinberger A. Tropomyosin-induced arthritis in rats. Clin Exp Rheumatol 2007; 25(4)(Suppl. 45): S86–92. [PubMed] [Google Scholar]
- [77].Jackson JM. TNF- α inhibitors. Dermatol Ther 2007; 20(4): 251–64. 10.1111/j.1529-8019.2007.00138.x [DOI] [PubMed] [Google Scholar]
- [78].Umare V, Pradhan V, Nadkar M, et al. Effect of proinflammatory cytokines (IL-6, TNF-α, and IL-1β) on clinical manifestations in Indian SLE patients. Mediators Inflamm 2014; 2014385297 10.1155/2014/385297 [DOI] [PMC free article] [PubMed]
- [79].Mahesh SP, Li Z, Buggage R, et al. Alpha tropomyosin as a self-antigen in patients with Behçet’s disease. Clin Exp Immunol 2005; 140(2): 368–75. 10.1111/j.1365-2249.2005.02760.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li Z, Srivastava P. Heat-Shock Proteins. Curr Protoc Immunol 2003; ••• 10.1002/0471142735.ima01ts58 [DOI] [PubMed]
- [81].Miller DJ, Fort PE. Heat shock proteins regulatory role in neurodevelopment. Front Neurosci 2018; 12: 821 10.3389/fnins.2018.00821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Beere HM. “The stress of dying”: the role of heat shock proteins in the regulation of apoptosis. J Cell Sci 2004; 117(Pt 13): 2641–51. 10.1242/jcs.01284 [DOI] [PubMed] [Google Scholar]
- [83].Candido EPM. Heat Shock ProteinsS Brenner and J H B T-E of Miller G, Eds. New York: Academic Press; 2001; pp. 914–5. 10.1006/rwgn.2001.0588 [DOI] [Google Scholar]
- [84].Dukay B, Csoboz B, Tóth ME. Heat-shock proteins in neuroinflammation. Front Pharmacol 2019; 10: 920 10.3389/fphar.2019.00920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Maleki F, Khosravi A, Nasser A, Taghinejad H, Azizian M. Bacterial heat shock protein activity. J Clin Diagn Res 2016; 10(3): BE01–3. 10.7860/JCDR/2016/14568.7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zügel U, Kaufmann SHE. Role of heat shock proteins in protection from and pathogenesis of infectious diseases. Clin Microbiol Rev 1999; 12(1): 19–39. 10.1128/CMR.12.1.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Singh B, Gupta RS. Conserved inserts in the Hsp60 (GroEL) and Hsp70 (DnaK) proteins are essential for cellular growth. Mol Genet Genomics 2009; 281(4): 361–73. 10.1007/s00438-008-0417-3 [DOI] [PubMed] [Google Scholar]
- [88].Urban-Chmiel R, Dec M, Puchalski A, Wernicki A. Characterization of heat-shock proteins in Escherichia coli strains under thermal stress in vitro. J Med Microbiol 2013; 62(Pt 12): 1897–901. 10.1099/jmm.0.064857-0 [DOI] [PubMed] [Google Scholar]
- [89].Moudgil KD, Thompson SJ, Geraci F, De Paepe B, Shoenfeld Y. Heat-shock proteins in autoimmunity. Autoimmune Dis 2013; 2013621417 10.1155/2013/621417 [DOI] [PMC free article] [PubMed]
- [90].Koliński T, Marek-Trzonkowska N, Trzonkowski P, Siebert J. Heat shock proteins (HSPs) in the homeostasis of regulatory T cells (Tregs). Cent Eur J Immunol 2016; 41(3): 317–23. 10.5114/ceji.2016.63133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kasperkiewicz M, Tukaj S, Gembicki AJ, et al. Evidence for a role of autoantibodies to heat shock protein 60, 70, and 90 in patients with dermatitis herpetiformis. Cell Stress Chaperones 2014; 19(6): 837–43. 10.1007/s12192-014-0507-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ghasemi Y, Dabbagh F, Rasoul-Amini S, Borhani Haghighi A, Morowvat MH. The possible role of HSPs on Behçet’s disease: a bioinformatic approach. Comput Biol Med 2012; 42(11): 1079–85. 10.1016/j.compbiomed.2012.08.009 [DOI] [PubMed] [Google Scholar]
- [93].Direskeneli H Innate and Adaptive Responses to Heat Shock Proteins in Behcet’s Disease. Genet Res Int 2013; 2013249157 10.1155/2013/249157 [DOI] [PMC free article] [PubMed]
- [94].Hu W, Hasan A, Wilson A, et al. Experimental mucosal induction of uveitis with the 60-kDa heat shock protein-derived peptide 336–351. Eur J Immunol 1998; 28(8): 2444–55. [DOI] [PubMed] [Google Scholar]
- [95].Shahram F, Nikoopour E, Rezaei N, et al. Association of interleukin-2, interleukin-4 and transforming growth factor-beta gene polymorphisms with Behcet’s disease. Clin Exp Rheumatol 2011; 29(4)(Suppl. 67): S28–31. [PubMed] [Google Scholar]
- [96].van Eden W, Jansen MAA, Ludwig I, van Kooten P, van der Zee R, Broere F. The enigma of heat shock proteins in immune tolerance. Front Immunol 2017; 8: 1599 10.3389/fimmu.2017.01599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Petris CK, Almony A. Ophthalmic manifestations of rheumatologic disease: diagnosis and management. Mo Med 2012; 109(1): 53–8. [PMC free article] [PubMed] [Google Scholar]
- [98].Bansal S, Barathi VA, Iwata D, Agrawal R. Experimental autoimmune uveitis and other animal models of uveitis: An update. Indian J Ophthalmol 2015; 63(3): 211–8. 10.4103/0301-4738.156914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chan CC, Caspi RR, Ni M, et al. Pathology of experimental autoimmune uveoretinitis in mice. J Autoimmun 1990; 3(3): 247–55. 10.1016/0896-8411(90)90144-H [DOI] [PubMed] [Google Scholar]
- [100].Caspi RR, Roberge FG, Chan CC, et al. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol 1988; 140(5): 1490–5. [PubMed] [Google Scholar]
- [101].Gery I, Chanaud NP III, Anglade E. Recoverin is highly uveitogenic in Lewis rats. Invest Ophthalmol Vis Sci 1994; 35(8): 3342–5. [PubMed] [Google Scholar]
- [102].Pfister C, et al. Retinal S antigen identified as the 48K protein regulating light-dependent phosphodiesterase in rods Science (80- ) 1985. 10.1126/science.2988124 [DOI] [PubMed]
- [103].de Smet MD, Bitar G, Mainigi S, Nussenblatt RB. Human S-antigen determinant recognition in uveitis. Invest Ophthalmol Vis Sci 2001; 42(13): 3233–8. [PubMed] [Google Scholar]
- [104].Petty RE, Hunt DWC, Rollins DF, Schroeder M-L, Puterman ML. Immunity to soluble retinal antigen in patients with uveitis accompanying juvenile rheumatoid arthritis. Arthritis Rheum 1987; 30(3): 287–93. 10.1002/art.1780300307 [DOI] [PubMed] [Google Scholar]
- [105].Hamzaoui K, Boussen E, Gorgi Y, Ouertani A, Ayed K. Molecular mimicry between S-Antigen and viral peptides | Mimetisme moleculaire entre l’antigene-s et les peptides viraux. Tunis Med 1999. [PubMed]
- [106].Yamamoto JH, Minami M, Inaba G, Masuda K, Mochizuki M. Cellular autoimmunity to retinal specific antigens in patients with Behçet’s disease. Br J Ophthalmol 1993; 77(9): 584–9. 10.1136/bjo.77.9.584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].YAMADA M. SHICHI H, YUASA T, TANOUCHI Y, and MIMURA Y, “Superoxide in ocular inflammation: Human and experimental uveitis. Adv Free Radic Biol Med 1986; ••• 10.1016/s8755-9668(86)80005-9 [DOI] [PubMed]
- [108].Yamamoto JH, Fujino Y, Lin C, Nieda M, Juji T, Masuda K. S-antigen specific T cell clones from a patient with Behçet’s disease. Br J Ophthalmol 1994; 78(12): 927–32. 10.1136/bjo.78.12.927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Takeuchi M, Usui Y, Okunuki Y, et al. Immune responses to interphotoreceptor retinoid-binding protein and S-antigen in Behcet’s patients with uveitis. Invest Ophthalmol Vis Sci 2010; 51(6): 3067–75. 10.1167/iovs.09-4313 [DOI] [PubMed] [Google Scholar]
- [110].Erdağ E, Şahin C, Küçükali Cİ, et al. Effects of in vivo and in vitro administration of neuro-Behcet’s disease IgG. Neurol Sci 2017; 38(5): 833–43. 10.1007/s10072-017-2856-2 [DOI] [PubMed] [Google Scholar]
- [111].Sakane T, Takeno M, Suzuki N, Inaba G. Behçet’s disease. N Engl J Med 1999; 341(17): 1284–91. 10.1056/NEJM199910213411707 [DOI] [PubMed] [Google Scholar]
- [112].Tognarelli EI, Palomino TF, Corrales N, Bueno SM, Kalergis AM, González PA. Herpes simplex virus evasion of early host antiviral responses. Front Cell Infect Microbiol 2019; 9: 127 10.3389/fcimb.2019.00127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Hamza M, Elleuch M, Slim A, Hamzaoui K, Ayed K. Antibodies to herpes simplex virus in patients with Behçet’s disease. Clin Rheumatol 1990; 9(4): 498–500. 10.1007/BF02030511 [DOI] [PubMed] [Google Scholar]
- [114].Lee S, Bang D, Cho YH, Lee ES, Sohn S. Polymerase chain reaction reveals herpes simplex virus DNA in saliva of patients with Behçet’s disease. Arch Dermatol Res 1996; 288(4): 179–83. 10.1007/BF02505221 [DOI] [PubMed] [Google Scholar]
- [115].Tojo M, Zheng X, Yanagihori H, et al. Detection of herpes virus genomes in skin lesions from patients with Behçet’s disease and other related inflammatory diseases. Acta Derm Venereol 2003; 83(2): 124–7. 10.1080/00015550310007472 [DOI] [PubMed] [Google Scholar]
- [116].Sohn S, Lee ES, Bang D, Lee S. Behçet’s disease-like symptoms induced by the Herpes simplex virus in ICR mice. Eur J Dermatol 1998; 8(1): 21–3. [PubMed] [Google Scholar]
- [117].Shim JA, Park S, Lee ES, Niki T, Hirashima M, Sohn S. Galectin-9 ameliorates herpes simplex virus-induced inflammation through apoptosis. Immunobiology 2012; 217(6): 657–66. 10.1016/j.imbio.2011.11.002 [DOI] [PubMed] [Google Scholar]
- [118].Choi B, Hwang Y, Kwon HJ, et al. Tumor necrosis factor alpha small interfering RNA decreases herpes simplex virus-induced inflammation in a mouse model. J Dermatol Sci 2008; 52(2): 87–97. 10.1016/j.jdermsci.2008.05.001 [DOI] [PubMed] [Google Scholar]
- [119].Choi B, Kim J, Lee ES, Bang D, Sohn S. Synthesized pyridine compound derivatives decreased TNF alpha and adhesion molecules and ameliorated HSV-induced inflammation in a mouse model. Eur J Pharmacol 2011; 657(1–3): 167–72. 10.1016/j.ejphar.2011.01.062 [DOI] [PubMed] [Google Scholar]
- [120].Shim J, Byun HO, Lee YD, Lee ES, Sohn S. Interleukin-6 small interfering RNA improved the herpes simplex virus-induced systemic inflammation in vivo Behcet’s disease-like mouse model. Gene Ther 2009; 16(3): 415–25. 10.1038/gt.2008.180 [DOI] [PubMed] [Google Scholar]
- [121].Choi B, Lee ES, Sohn S. Vitamin D3 ameliorates herpes simplex virus-induced Behçet’s disease-like inflammation in a mouse model through down-regulation of Toll-like receptors. Clin Exp Rheumatol 2011; 29(4)(Suppl. 67): S13–9. [PubMed] [Google Scholar]
- [122].Sohn S, Bang D, Lee ES, Kwon HJ, Lee SI, Lee S. Experimental studies on the antiviral agent famciclovir in Behçet’s disease symptoms in ICR mice. Br J Dermatol 2001; 145(5): 799–804. 10.1046/j.1365-2133.2001.04498.x [DOI] [PubMed] [Google Scholar]
- [123].Choi J, Lee ES, Choi B, Sohn S. Therapeutic potency of Poly I:C in HSV-induced inflammation through up-regulation of IL-15 receptor alpha. Immunobiology 2013; 218(9): 1119–30. 10.1016/j.imbio.2013.03.005 [DOI] [PubMed] [Google Scholar]
- [124].Kang S, Lee ES, Choi B, et al. Effects of irradiation on cytokine production in a mouse model of Behçet’s disease. Clin Exp Rheumatol 2009; 27(1): 54–63. [PubMed] [Google Scholar]
- [125].Sohn S, Lutz M, Kwon HJ, Konwalinka G, Lee S, Schirmer M. Therapeutic effects of gemcitabine on cutaneous manifestations in an Adamantiades-Behçet’s disease-like mouse model. Exp Dermatol 2004; 13(10): 630–4. 10.1111/j.0906-6705.2004.00210.x [DOI] [PubMed] [Google Scholar]
- [126].Choi B, Kim HA, Suh CH, Byun HO, Jung JY, Sohn S. The relevance of miRNA-21 in HSV-induced inflammation in a mouse model. Int J Mol Sci 2015; 16(4): 7413–27. 10.3390/ijms16047413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Anower AKMM, Shim JA, Choi B, Kwon HJ, Sohn S. The role of classical and alternative macrophages in the immunopathogenesis of herpes simplex virus-induced inflammation in a mouse model. J Dermatol Sci 2014; 73(3): 198–208. 10.1016/j.jdermsci.2013.11.001 [DOI] [PubMed] [Google Scholar]
- [128].Shim JA, Lee ES, Choi B, Sohn S. The role of T cell immunoglobulin mucin domains 1 and 4 in a herpes simplex virus-induced Behçet’s disease mouse model. Mediators Inflamm 2013; 2013903948 10.1155/2013/903948 [DOI] [PMC free article] [PubMed]
- [129].Lee ES, Kim YA, Kwon HJ, Bang D, Lee S, Sohn S. Thalidomide upregulates macrophage inflammatory protein-1α in a herpes simplex virus-induced Behçet’s disease-like animal model. Arch Dermatol Res 2004; 296(4): 175–81. 10.1007/s00403-004-0498-8 [DOI] [PubMed] [Google Scholar]
- [130].Choi B, Sayeed HM, Islam SMS, Sohn S. Role of N-acetyl galactosamine-4-SO4, a ligand of CD206 in HSV-induced mouse model of Behçet’s disease. Eur J Pharmacol 2017; 813: 42–9. 10.1016/j.ejphar.2017.07.022 [DOI] [PubMed] [Google Scholar]
- [131].Kokturk A Clinical and pathological manifestations with differential diagnosis in Behçet’s disease. Pathol Res Int 2012; 2012690390 10.1155/2012/690390 [DOI] [PMC free article] [PubMed]
- [132].Koganti R, Yadavalli T, Shukla D. Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms 2019; 7(10): 429 10.3390/microorganisms7100429 [DOI] [PMC free article] [PubMed] [Google Scholar]