

Abstract
In this work, electron-induced site-specific formation of neutral π-type aminyl radicals (RNH•) and their reactions with pyrimidine nucleoside analogs azidolabeled at various positions in the sugar moiety, e.g., at 2′-, 3′-, 4′-, and 5′- sites along with a model compound 3-azido-1-propanol (3AZPrOH), were investigated. Electron paramagnetic resonance (EPR) studies confirmed the site and mechanism of RNH• formation via dissociative electron attachment-mediated loss of N2 and subsequent facile protonation from the solvent employing 15N-labeled azido group, deuterations at specific sites in the sugar and base, and changing the solvent from H2O to D2O. Reactions of RNH• were investigated employing EPR by warming these samples from 77 K to ca. 170 K. RNH• at a primary carbon site (5′-azido-2′,5′-dideoxyuridine, 3AZPrOH) facilely converted to a σ-type iminyl radical (R=N•) via a bimolecular H-atom abstraction forming an α-azidoalkyl radical. RNH• when at a secondary carbon site (e.g., 2′-azido-2′-deoxyuridine) underwent bimolecular electrophilic addition to the C5=C6 double bond of a proximate pyrimidine base. Finally, RNH• at tertiary alkyl carbon (4′-azidocytidine) underwent little reaction. These results show the influence of stereochemical and electronic environment on RNH• reactivity and allow the selection of those azidonucleosides that would be most effective in augmenting cellular radiation damage.
Keywords: EPR; azidonucleosides; prehydrated electron; dissociative electron attachment; aminyl radical; iminyl radical; radical addition to double bond; H-atom abstraction; C-centered radicals; nature of the carbon (primary, secondary, and tertiary) linked to the azide group; nature of the carbon (primary, secondary, and tertiary) linked to the aminyl radical
Graphical Abstract
Introduction:
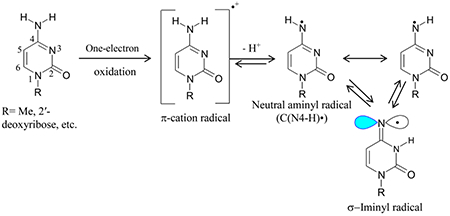
Neutral aminyl radicals (RNH•) are π-type radicals that are important in chemical synthesis and in biology.1–6 RNH• plays an important role in the prototropic equilibria of DNA-base π-cation radicals (such as, guanine cation radical (G•+), cytosine cation radical (C•+), adenine cation radical (A•+)) that are involved in charge (hole) transfer processes followed by localization of damage.4,6 Moreover, •OH addition to the C4=C5 double bond in purine bases (G, A) followed by water elimination lead to the same RNH• species (G(N1-H)•, A(N6-H)•) that are formed via deprotonation of G•+ and A•+.4,6 it is well-established that RNH• undergo a wide variety of reactions that are summarized below:
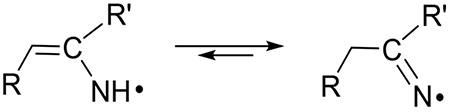
(A). Tautomerization:
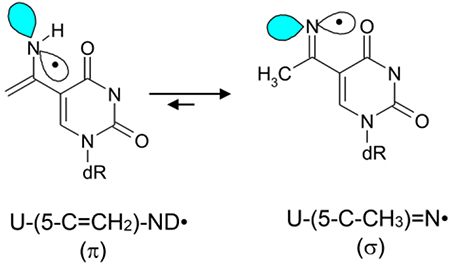
π-Type RNH• is shown to undergo tautomerization to the σ-type R=N•.
 |
(1) |
For example, electron paramagnetic resonance (EPR) studies have established that π-type aminyl radical (U-(5-C=CH2)-NH•) formed from 5-(1-azidovinyl) modified 2′-deoxyuridine, facilely tautomerizes to the σ-type iminyl radical, U-(5-C-CH3)=N• (reaction 1a).7
 |
(1a) |
Also, the deprotonated cytosine π-cation radical (C(N4-H)•) tautomerizes facilely to the σ-iminyl radical (reaction 1b).17
 |
(1b) |
(B). Intermolecular H-atom abstraction generating C-centered radicals:
It is well-established that RNH• can abstract H-atoms from weaker CX-H bonds via a bimolecular pathway to form C-centered radicals.1–16 For example, RNH• generated from AZT undergo bimolecular H-atom abstractions from the –CH3 group at C5 of thymine base forming the allylic radical, dUCH2• (ca. 55%), and from the C5′-site of a proximate AZT producing the strand break precursor radical, C5′•.8
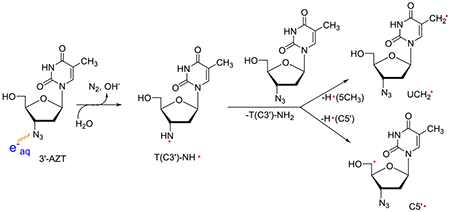 |
(2) |
In addition, intermolecular H-atom abstraction by RNH• from a proximate compound (e.g., 5-azidomethyl-2′-deoxyuridine) having an α-azidoalkyl moiety leads to the formation of an α-azidoalkyl radical which undergoes a facile conversion to the σ-type R′=N•7:
| (2a) |
(C). Transposition of radical site via intramolecular H-atom transfer, e.g., 1,2 shift, 1,5 shift:
Pulse radiolysis of amino acids at ambient temperature presented evidence of 1,2 H-atom shift in the •OH mediated formation of oxidizing radicals in amino acids, for example, aminyl radical (•NH-CH2-CO2−) from glycine rearranges to NH-•CH2-CO2− by intramolecular 1,2 H-shift (reaction 3).22,23
| (3) |
EPR studies have presented evidence of facile and intramolecular 1,5 H-shift in RNH• in lyxofuranoside leading to the formation of C5• radical.9
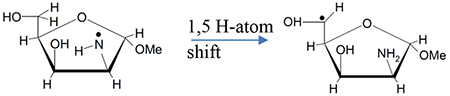 |
(3a) |
(D). β-elimination:
Pulse radiolysis has shown that the oxidizing aminyl radical (e.g., •NH-CH2-CO2¯) generated by the reaction of •OH with an amino acid (e.g., glycine) mediated leads to the formation of CO2•¯ (reaction 4).22,23
| (4) |
(E). Intramolecular cyclization:
Dialkylaminyl radicals [R=H, Me; R′= H, Me (reaction 5)] are shown to undergo intramolecular cyclization reactions leading to the formation of C-centered radicals.10,16
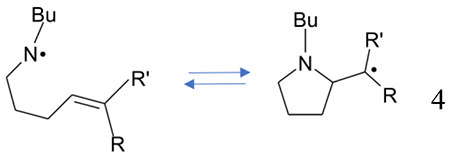 |
(5) |
RNH• radical is generated by one-electron oxidation of primary amines followed by deprotonation of the primary amine cation radical (RNH2•+).4,17–34 Thus, RNH• becomes the Brønsted base, RNH2•+ is the conjugate acid, and these two species (RNH• and RNH2•+) are in prototropic equilibrium if R is a delocalized structure (see for example, reaction 1b). In addition, the formation of RNH• via photodissociation11–15 and via •OH employing pulse radiolysis4,22,23 have been reported.
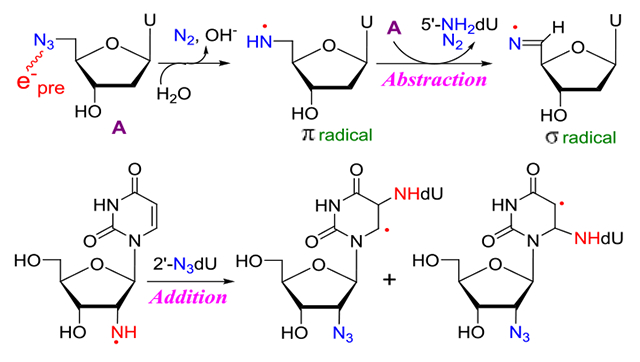
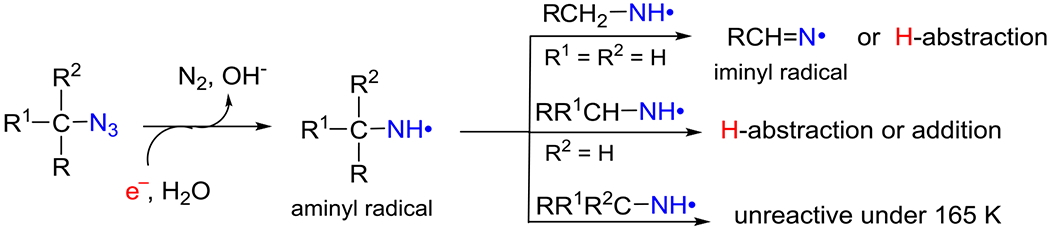
We have developed a novel method of site-specific electron-mediated RNH• generation employing azidonucleosides (reaction 2).6–9 Our work has established that radiation-produced prehydrated electron (epre−)-mediated dissociative electron attachment to azidonucleosides (RN3) in homogeneous supercooled glassy systems at 77 K,9 at first, leads to the formation of highly basic and unstable nitrene anion radical (RN•−) at 77 K via spontaneous loss of N2 from the azidonucleoside anion radical (RN3•−). Subsequent facile protonation of RN•− produces RNH•. Neither RN3•− nor RN•− intermediates were detected by using electron paramagnetic resonance (EPR) spectroscopy even at 77 K, and only the EPR spectra of RNH• were observed.6–9 So far, investigations from our laboratory have presented evidence of reactions 1 to 3a. Also, our work has shown that reaction mechanisms of RNH• depend on the type of compounds (azidomethyl vs. azidovinyl)6,7 and conformation of the sugar (lyxofuranose vs. 2′-deoxyribofuranose)9.
In this work, we have employed uracil and cytosine nucleosides with the azido group substituted at the 2′ (2′-azido-2′-deoxycytidine (2′-AZdC, 1), 2′-azido-2′-deoxyuridine (2′-AZdU, 2)), 3′ (3′-azido-2′,3′-dideoxyuridine (3′-AZ-2′,3′-ddU, 3)), 4′ (4′-azidocytidine (4′-AZC, 4)) and 5′ (5′-azido-2′,5′-dideoxyuridine (5′-AZ-2′,5′-ddU, 5), 5′-azido-5′-deoxythymidine (5′-AZT, 6)) positions. An azido-substituted alcohol (3-azido-1-propanol (3AZPrOH, 7)), in addition, has also been investigated as a model of azido group linked to the primary carbon. Thus, the azido group is attached to the primary carbon, 5′ (5, 6) and 3AZPrOH (7), to the secondary carbon (2′ and 3′), and to the tertiary carbon (4′) site. RNH• formation via attachment of radiation-produced prehydrated electrons to these compounds in γ-irradiated aqueous glassy (7.5 M LiCl) systems and subsequent reactions of RNH• are investigated employing EPR spectroscopy. A set of [2H]-labeled 2 (C2′, C3′, C4′, C5 and C6) isotopomers and of [15N]-azido labeled 2 have led us to unambiguously assign that in all compounds including the isotopically substituted ones, the predominant site of electron attachment is the azido group and to characterize the resultant RNH• and its subsequent reactions. The experimentally obtained hyperfine coupling constant (HFCC) values of RNH• have been compared with the corresponding theoretically calculated values for EPR spectral assignment (Table 1). Similar to our earlier work on the reaction of RNH• from the azido substitution to a primary carbon at C5 of 5-azidomethyl-2′-deoxyuridine (AmdU),7 a bimolecular conversion of RNH• attached to a primary carbon site (5′, 5, 6) to the σ-type iminyl radical (R=N•) involving an α-azidoalkyl radical as intermediate was observed. RNH• from 7 underwent this conversion as well. Thus, our earlier results from AmdU,7 our results from 5, 6, and 7 in this worked have established the general nature of this RNH• to R=N• bimolecular conversion, i.e., this conversion happens in any azido compound in which the azido group is linked to a primary carbon. Bimolecular electrophilic addition of RNH• attached to the secondary carbon site to the C5=C6 double bond of a proximate pyrimidine (uracil and cytosine but not thymine) base is observed. Finally, a little reaction was observed in the glassy system from RNH• at tertiary alkyl carbon. Our results presented in this work demonstrate that the stereochemical and electronic environment of azido substitution at specific sites in the sugar moiety significantly affect the reactivity of RNH•. Azidonucleosides have also been shown to augment radiation damage in various types of cancer cells. 7,35–39 In addition, 2′-azido-2′-deoxyuridine (2) 5′-phosphate is shown to be a potent inhibitor of ribonucleotide reductases following a radical chemistry-based inactivation pathway.40–42 Therefore, the results presented in this work in conjunction with our own work on azido substitution at C5 in the pyrimidine base7 provide the basis to choose the type of azidonucleoside that will be suitable to augment significant radiation damage to tumor cells thereby improving the efficacy of tumor radiochemotherapy.
Table 1.
Hyperfine Coupling Constants (HFCC) and g-values of radicals reported in this work
| Compound/Type/ Radical | Hyperfine couplings (G) | g-value (exp) | ||
|---|---|---|---|---|
| Site(nucleus) | Exp | Theory | ||
| π/T(5′-CH2)-NH• a (5′-AZT) | β-protons, (H5′, H5″) | Sum 91a | g‖ = 2.0020a, g⊥=2.0043a | |
| N5′-H (α-proton) | (-, -, −30)a | (−39.8, 0.4, −26.7)a | ||
| N5′-coupling | (0, 0, 43)a | (0, 0, 41.8) G a | ||
| π /U-5-CH2-NH• b (AmdU) 5/π/dU-5′-CH2-NH• |
β-protons, (H5, H5)b β-protons, (H5′, H5″) (for 5) |
Sum 93.5b Sum 90.5 |
---------------------- | g‖ = 2.0020, g⊥=2.0043 |
| N5-H (α-proton) b N5′-H (α-proton) |
(-, -, −30)a | |||
| N5/N5′-coupling | (0, 0, 42.5) | |||
| α-azidoalkyl radical5/π/dU-5′-CH•-N3 | 1αH 1αH 1N |
------------- | (−10.97, −15.03,−5.09) (−14.46,−20.64,−6.67) (10.5, −0.5, −1.32)c |
2.0038 yy 2.0021 zz 2.0043 xx |
| σ U-5-CH=N• b | β-proton, (H5) b N5-coupling b |
82 b (0, 0, 36.5) b |
(71.8, 78.0,72.8) (−4.2, 0, 37.7)b |
g‖ = 2.0016, g⊥=2.0040 |
| 5 σ dR-5′-CH=N• | β-proton, (H5′) 5 N5′-coupling |
82 (0, 0, 36.5) |
(72.9, 78.7, 73.0)d (−4.44, −0.9, 36.6)d |
|
| 1 π dC(C2′)-ND• (π C(C2′)-ND•) 2 π dU(C2′)-ND• (π U(C2′)-ND•) |
N2′-coupling | (0, 0, 40.5) | -------------------------- | g‖=2.0020, g⊥=2.0043 |
| C2′-H (β-proton) | 51.5 | |||
| N2′-H (α-proton) | (*,*,−28) e | |||
| 3 π 2′,3′-ddU(C3′)-ND• | N3′-coupling | (0, 0, 37.5) e | ------------------------- | g‖=2.0020, g⊥=2.0043 |
| C3′-H (β-proton) | 41 e | |||
| N3′-H (α-proton) | (*,*,−28) e | |||
| 4 π C(C4′)-ND• | N4′-coupling | (0, 0, 37.5) e | -------- | g‖=2.0020, g⊥=2.0043 |
| N4′-H (α-proton) | (*,*,−28) e | |||
Taken from reference 8.
Taken from reference 7.
Employing B3LYP/6-31G** method, structure of 5′-CH•-N3 was optimized and HFCCs of the fully optimized structure of 5′-CH•-N3 was calculated (see SI). Linewidth = 3.5 G, and a mixed line shape (Lorentzian/Gaussian =1) was employed to simulate the experimental spectrum.
Employing B3LYP/6-31G** method, structures of 5′-CH=N• of U-5-CH=N• were fully optimized and HFCCs of these optimized structures were calculated (see SI). Linewidths as (7, 5, 5) G and line shape with a mixed Lorentzian/Gaussian = 1 were employed to simulate the experimental spectrum.
Taken from reference 8. For simulation, we used linewidth as 10 G, and line shape with a mixed Lorentzian/Gaussian = 1.
Experimental:
The 2′-azido-2′-deoxycytidine (2′-AZdC, 1)43 and 4′-azidocytidine (4′-AZC, 4)44 were prepared by slight modifications of the literature protocols. The synthesis and spectroscopic characterization of 2′-azido-2′-deoxyuridine (2′-AZdU, 2) and its deuterated derivatives with deuterations at specific sites of the sugar moiety, e.g., 2′-D-2′-AZdU ([2′-D]-2), 3′-D-2′-AZdU ([3′-D]-2), and 4′-D-2′-AZdU ([4′-D]-2), or at the base, 5,6-D,D-2′-AZdU ([5,6-D,D]-2) and 15N incorporated azido group in 2 are described in the main manuscript (Schemes 1 and 2). The general experimental methodologies and synthesis of all other synthetic intermediates (Schemes S1–S2) are described in SI section. Azido compounds 3′-AZ-2′,3′-ddU (3), 5′-AZ-2′,5′-ddU (5), 5′-AZT (6), 3AZPrOH (7), and 3′-AZT are commercially available. Sodium azide 15N labeled (97%) in the terminal position and deuterium-labeled uracil were purchased from Cambridge Isotope Laboratories.
Scheme 1.
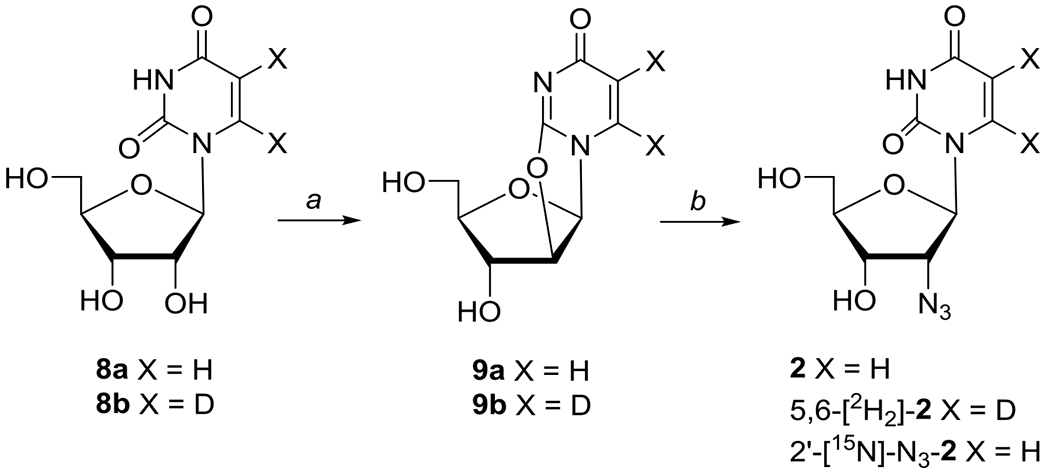
Synthesis of 2′-azido-2′-deoxyuridine 2 and its 5,6-[2H2]-2 and 2′-[15N]-N3-2 isotopes.
Reagents: (a) (PhO)2CO/NaHCO3/DMF; (b) NaN3 or [15N]-NaN3/BzOH/HMPA
Scheme 2.
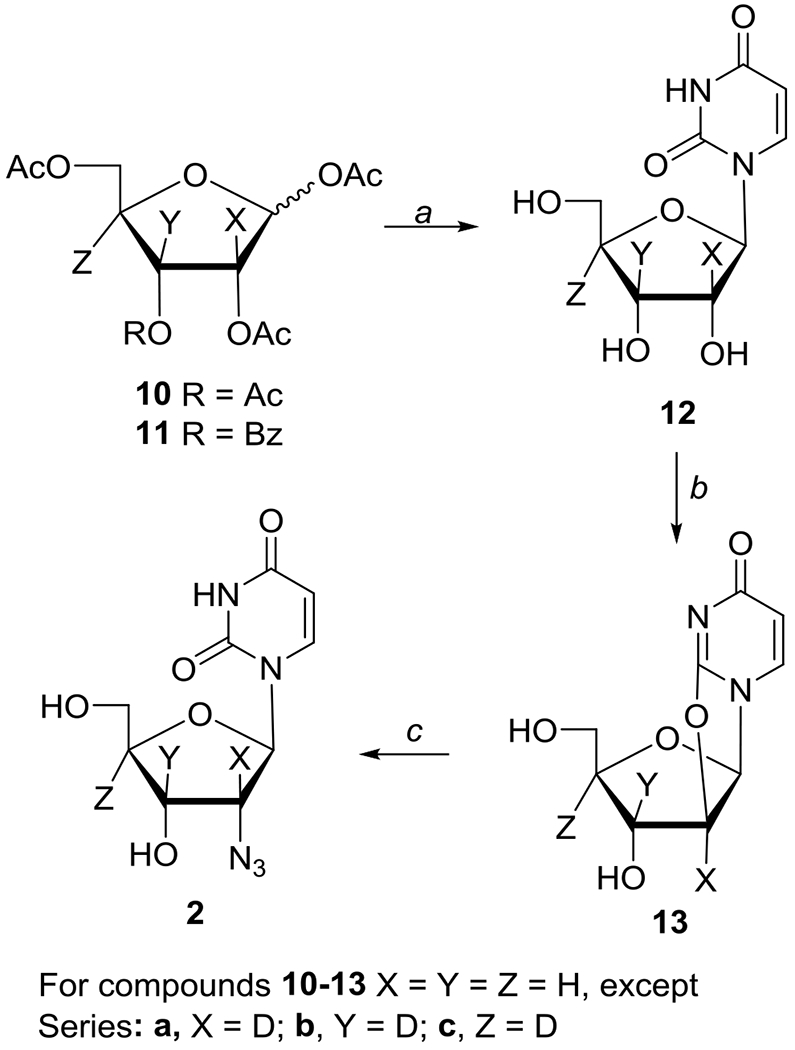
Synthesis of ribose deuterated 2′-azido-2′-deoxyuridine isotopes 2′-[2H]-2 (X = D), 3′-[2H]-2 (Y = D), and 4′-[2H]-2 (Z = D).
Reagents: (a) (i) uracil/HMDS/TMSSiCl, (ii) TMSOTf/MeCN, (iii) NH3/MeOH; (b) (PhO)2CO/NaHCO3/DMF; (c) NaN3/BzOH/HMPA
For EPR studies, methods of preparation of homogeneous glassy samples of 1 to 7, [2′-D]-2, [3′-D]-2), [4′-D]-2), [5,6-D,D]-2, and 15N incorporated azido group in 2 in 7.5 M LiCl/D2O or 7.5 M LiCl/H2O, γ-irradiation and storage of these glassy samples including the stepwise annealing of these glassy samples are described in the Supporting Information. EPR spectrometer, experimental set-up (field calibration employing Fremy’s salt, microwave power, etc.) for recording the EPR spectra at 77 K, and methods of theoretical calculations are described in the Supporting Information.
Results:
(A). Synthesis:
The 2′-azido-2′-deoxyuridine 2 (2′-AZdU) and its selectively labeled 5,6-[2H2]-2, , 2′-[2H]-2, 3′-[2H]-2, and 4′-[2H]-2 analogues were prepared by conversion of uridine 8a and the selectively deuterated uridine analogues 8b and 12a-c to 2,2′-O-anhydrouridine derivatives 9a, 9b and 13a-c employing Hampton and Nichol methodology.45 Subsequent ring-opening with azide anion [generated from trimethylsilylazide (TMSN3)/LiF and tetramethylethylenediamine (TMEDA) or NaN3 in hexamethylphosphoramide (HMPA) in the presence of benzoic acid] gave desired 2′-azido-2′-deoxyuridine 246,47 and its deuterium-labeled analogues in uracil 5,6-[2H2]-2 (Scheme 1) or ribose (2′-[2H]-2, 3′-[2H]-2, and 4′-[2H]-2; Scheme 2) moieties. Ring-opening of 9a with [15N]-NaN3 provided 2′-[15N]-azido-labeled 2 (Scheme 1).48 The selectively deuterated ribofuranose precursors 2-[2H]-10a, 3-[2H]-10b (Schemes S1–S2), and 4-[2H]-11c were prepared following literature methodologies and the details of their synthesis as well as their coupling with uracil to give uridines 12a-c and their subsequent conversion to 2,2′-O-anhydro precursors 13a-c are described in SI section.
2′-Azido-2′-deoxyuridine (2).
The stirred solution of 9a (30 mg, 0.13 mmol; see SI section for the preparation of 9a) and NaN3 (60 mg, 0.93 mmol) in HMPA (0.5 mL) was heated at 150°C. After 30 min, benzoic acid (16 mg, 0.13 mmol) was added and heating was continued for another 15 min. The cooled reaction mixture was diluted with H2O and washed with CHCl3. The organic layer was back-extracted with H2O. The combined aqueous layers was concentrated and purified on silica gel column (EtOAc/MeOH, 9:1) or preparative RP-HPLC (H2O/MeCN, 6:1) to afford 246,47 (20 mg, 56%): 1H NMR (D2O) δ 3.70 (dd, J = 4.3, 12.8 Hz, 1, H5″), 3.82 (dd, J = 2.8, 12.8 Hz, 1, H5′), 4.00 (“ddd”, J = 2.9, 4.2, 6.0 Hz, 1, H4′), 4.22 (“dd”, J = 4.5, 5.7 Hz, 1, H2′), 4.35 (t, J = 5.9 Hz, 1, H3′), 5.77 (d, J = 8.1 Hz, 1, H5), 5.80 (d, J = 4.4 Hz, 1, H1′), 7.75 (d, J = 8.1 Hz, 1, H6); MS (ESI−) m/z 268 (MH−).
2′-[15N]-N3-2′-Azido-2′-deoxyuridine (2′-[15N]-N3-2).
Treatment of 9a (20 mg, 0.09 mmol) with [15N]-NaN3 (40.5 mg, 0.61 mmol; >97% 15N) as described above gave 2′-[15N]-N3-248 (8 mg, 33%) with identical physical, chemical, and spectroscopical properties (1H NMR) as 2 except for MS (ESI−) m/z 269 (M-H−).
5,6-[2H2]-2′-Azido-2′-deoxyuridine (5,6-[2H2]-2).
Azidation of 9b (18 mg, 0.08 mmol; see SI section for the preparation of 9a) as described above afforded 5,6-[2H2]-2 (11 mg, 52%) with identical physical, chemical, and spectroscopical properties as 2 except for the disappearance of H6 peak at 7.75 ppm and the presence of the residual peak (~ 30%) for H5 at 5.77 ppm (s): MS (ESI−) m/z 270 (MH−).
2′-[2H]-2′-Azido-2′-deoxyuridine (2′-[2H]-2).
Azidation of 13a (22 mg, 0.10 mmol) as described above afforded 2′-[2H]-2 (7 mg, 27%) with identical spectroscopical properties as 2 except for the disappearance of H2′ at 4.22 ppm and simplification of proton splitting for H1′ at 5.80 ppm (s) and H3′ at 4.35 ppm (d, J = 6.0 Hz); MS (ESI−) m/z 269 (MH−).
3′-[2H]-2′-Azido-2′-deoxyuridine (3′-[2H]-2).
Azidation of 13b (20 mg, 0.09 mmol) as described above afforded 3′-[2H]-2 (15 mg, 63%) with identical spectroscopical properties as 2 except for the disappearance of H3′peak at 4.35 ppm and simplification of proton splitting for H2′ at 4.22 ppm (d, J = 4.5 Hz) and H4′ at 4.00 ppm (“dd”, J = 2.9, 4.2 Hz): MS (ESI−) m/z 269 (MH−).
4′-[2H]-2′-Azido-2′-deoxyuridine (4′-[2H]-2).
Azidation of 13c (25 mg, 0.11 mmol) as described above afforded 4′-[2H]-2 (20 mg, 67%) with identical spectroscopical properties as 2 except for the disappearance of H4′ peak at 4.00 ppm and simplification of proton splitting for H3′ at 4.35 ppm (d, J = 5.8 Hz), H5′ at 3.82 ppm (d, J = 12.8 Hz), and H5″ at 3.70 ppm (d, J = 12.8 Hz): MS (ESI−) m/z 269 (MH−).
(B). EPR studies:
B.1. Characterization of RNH•:
Characterization of the RNH• formed from glassy samples of 2′-azido-2′-deoxyuridine (2′-AZdU, 2) is presented here as an example. From our previous assignments of RNH•6–9 (for example, T(C2′)-14ND• in 3′-AZT,8) the major HFCC values in RNH• from 2 (dU(C2′)-14ND• and its corresponding 15ND• analog (dU(C2′)-15ND•)) are expected from three sources – the axially symmetric anisotropic HFCC from the azide N, the isotropic β-proton coupling owing to H2′, and the αH/aD anisotropic hyperfine coupling from the exchangeable NH/ND.
B.1.1. Assignment of the axially symmetric anisotropic HFCC due to the aminyl N:
The 77 K EPR spectrum (green) of RNH• (U(C2′)-14ND•) formed via γ-radiation-induced (absorbed dose = 500 Gy at 77 K) electron attachment in aqueous glassy (7.5 M LiCl/D2O (pH ca. 5)) sample of 2 (1 mg/mL) after visible illumination by using a photoflood lamp at 77 K for 15 minutes is presented in Figure 1A. The 77 K visible illumination was carried out following our work on T(C2′)-14ND•) in 3′-AZT8; 77 K visible illumination removed line components of uracil anion radical by photoejection of the excess electron. The spectrum in Figure 1A shows a large central doublet due to an isotropic β-proton HFCC value of H2′ at 51.5 G, a triplet from an axially symmetric anisotropic aminyl 14N (spin =1) HFCC (Azz (A‖) = 40.5 G, A⊥ =Axx = Ayy = 0 G), along with g‖ = 2.0020 and g⊥=2.0043 (see also Figure 2A and Table 1). The anisotropic 14N HFCC and the g-values agree with those of aminyl radicals reported in the literature.7–9,17–29 Based on these results, the green spectrum in Figure 1A is assigned to U(C2′)-14ND•.
Figure 1.
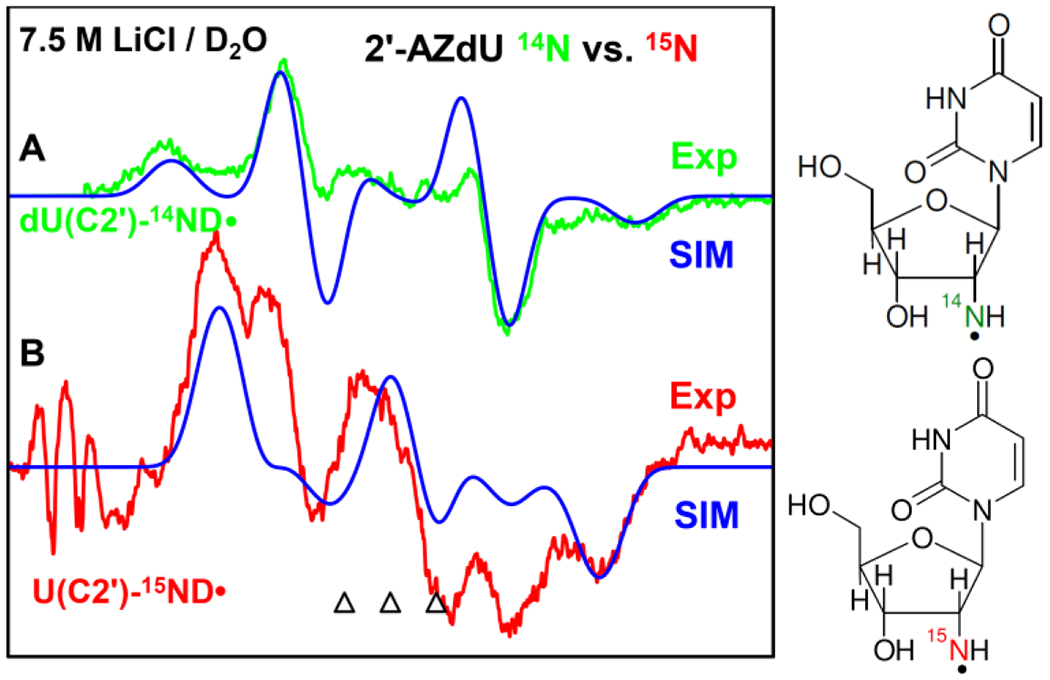
77 K EPR spectrum of RNH• (U(C2′)-14ND•) formed via electron-induced attachment in aqueous glassy (7.5 M LiCl/D2O) sample of (A) unlabeled 2, green color, and of (B) 15N labeled 2, red color after visible illumination of each sample by using a photoflood lamp at 77 K for 15 minutes to remove the uracil anion radical by photoejection of the excess electron. The simulated spectra (blue) (for simulation parameters, see text) are superimposed on the top of the each experimentally recorded spectrum. The three reference markers (open triangles) in this Figure and in other Figures show the position of Fremy’s salt resonance with the central marker at g = 2.0056. Each of these markers is separated from each other by 13.09 G.
Figure 2.
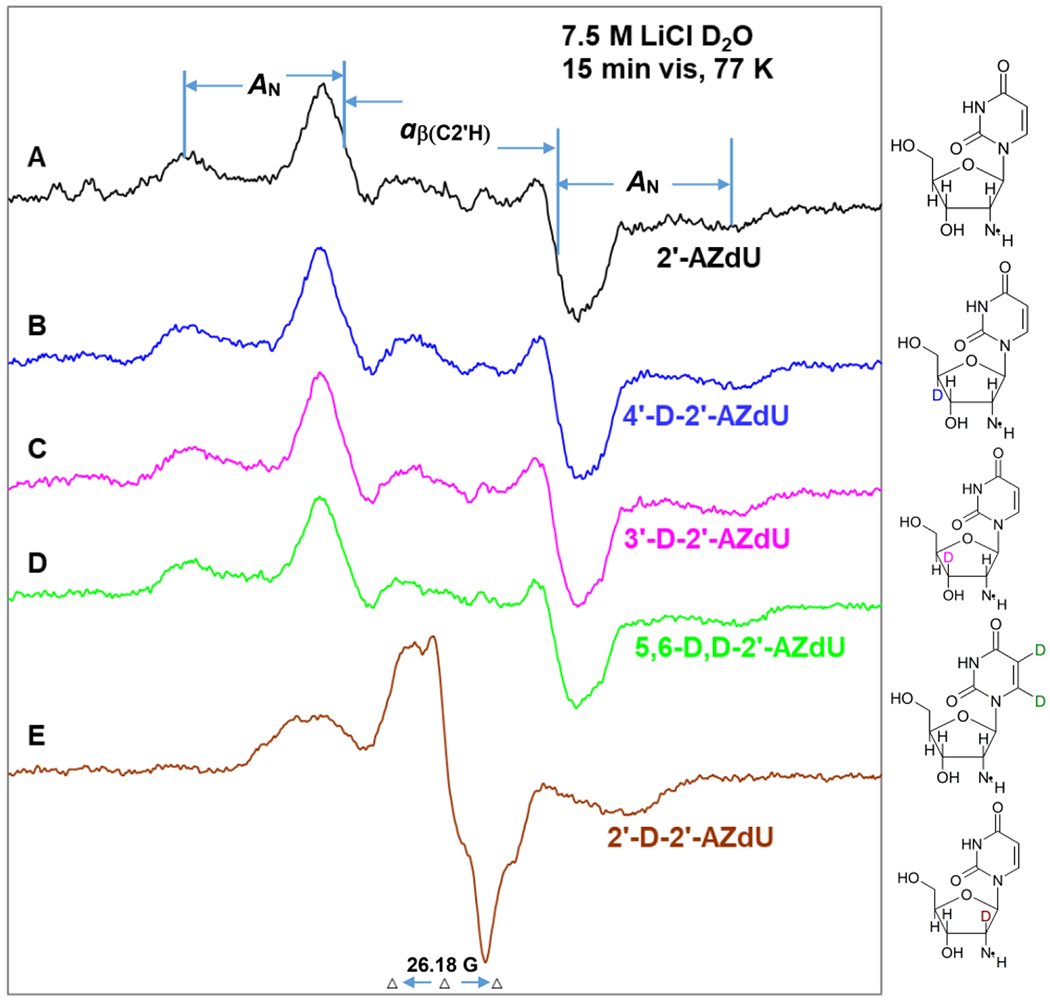
EPR spectra of the aminyl radicals (dU(C2′)-ND•) found in matched samples (1 mg/mL of each compound) via radiation-produced (absorbed dose = 500 Gy at 77 K) one-electron addition at 77 K of the homogeneous glassy solution of 7.5 M LiCl in D2O after visible illumination of each sample by using a photoflood lamp at 77 K for 15 minutes to remove the uracil anion radical by photoejection of the excess electron of (A) 2′-AZdU (2), (B) 4′-D-2′-AZdU ([4′-D]-2), (C) 3′-D-2′-AZdU ([3′-D]-2), (D) 5,6-D,D-2′-AZdU ([5,6-D,D]-2), and (E) 2′-D-2′-AZdU ([2′-D]-2). The extent of β-hyperfine coupling by H2′-atom and the anisotropic nitrogen hyperfine coupling, A‖ in U(C2′)-ND• of 2′-AZdU are represented by stick diagrams.
Based on our previous work on 1-Me-(C2)-15ND• from methyl 2-Azido-2-deoxy-β-D-ribofuranoside,9 experiments were carried out using a matched sample of 15N incorporated 2 (where the N-atoms in the 2′-azido group of 2 were 15N labeled) to make this assignment of U(C2′)-14ND• unequivocal; the results are presented in Figure 1B. Analyses of the experimentally recorded spectrum (red) in Figure 1B shows the large central doublet due to an isotropic β-proton HFCC value of H2′ at 51.5 G and g‖ = 2.0020 and g⊥=2.0043 (same as the green spectrum in Figure 1A). The red spectrum in Figure 1B also shows line components due to an axially symmetric anisotropic aminyl 15N (spin = ½) HFCC (Azz = 56.9 G, Axx = Ayy = 0 G). Following our assignment of the green spectrum in Figure 1A to U(C2′)-14ND•, the red spectrum in Figure 1B is assigned to U(C2′)-15ND•. 15N (spin = ½) couplings are found to be 1.404 times the 14N (spin = 1) and this is equal to the gyromagnetic ratio of 15N/14N.9,18 We note here that each experimentally recorded spectrum shown in Figures 1A and 1B was obtained after subtraction of the line components of Cl2•− spectrum following our previous works on the isolation of 77 K RNH• spectrum.7–9,49–51 A small residual of the Cl2•− subtraction is found on the low field end of the red spectrum in Figure 1B. This does not belong to the experimental U(C2′)-15ND• spectrum (red). Employing these HFCC and g-values, the U(C2′)-14ND• (green, Figure 1A) spectrum and U(C2′)-15ND• (red, Figure 1B) spectrum were simulated employing parameters: anisotropic α-D (N-D) HFCC (4.5, 0, 6) G,7–9 and a mixed Lorentzian/Gaussian (1/1) linewidth of 10 G. The simulated spectra (blue) matched nicely with corresponding experimentally recorded spectra in Figures 1A and B. These results provided unequivocal assignment of the axially symmetric anisotropic N HFCC of RNH•.
B.1.2. Sugar deuteration studies establish that the isotropic β-hydrogen HFCC in dU(C2′)-ND• originates from H2′:
To investigate the origin of β-hydrogen coupling, we have employed matched homogeneous glassy samples (1 mg/ mL of each compound) of 2 and its derivatives with deuterium substitution at specific sites in the deoxyribose sugar (at 2′ ([2′-D]-2), 3′ ([3′-D]-2), 4′ ([4′-D]-2)) and at the C5,C6 sites of uracil base moiety ([5,6-D,D]-2)). EPR spectra of dU(C2′)-ND• formed in these samples via γ-radiation-induced (absorbed dose ca. 500 Gy at 77 K) electron attachment after visible illumination by using a photoflood lamp at 77 K for 15 minutes are shown in Figure 2. Each experimentally recorded spectrum shown in Figure 2 was obtained after subtraction of the line components of Cl2•− spectrum following our previous works on the isolation of 77 K RNH• spectrum.7–9,49–51 Analyses of the spectra presented in Figures 2A to 2E establish that the EPR spectrum of dU(C2′)-ND• is affected only by deuteration at 2′ (spectrum in Figure 2E). The total hyperfine splitting, lineshape, and center of the spectra remain unchanged in spectra presented in Figures 2A to 2D. Following our analyses of the green spectrum in Figure 1(A), spectra presented in Figures 2A to 2D show a large central doublet due to an isotropic β-proton HFCC value of H2′ at 51.5 G, a triplet from an axially symmetric anisotropic aminyl N HFCC (Azz (A‖) = 40.5 G, A⊥ =Axx = Ayy = 0 G), along with g‖ = 2.0020 and g⊥=2.0043.
The collapse of the ca. 51.5 G doublet due to deuteration at 2′ ([2′-D]-2) in spectrum 2E establishes unequivocally the presence of β-H2′ HFCC in dU(C2′)-ND•. All other analogs of 2 having deuteration at the C5 and C6 positions in the uracil ring, or at positions other than C2′ in the sugar moiety show this doublet in dU(C2′)-ND• spectrum.
B.1.3. Studies in D2O vs. H2O establish the αH-atom anisotropic hyperfine coupling of the exchangeable N-H proton in dU(C2′)-ND• (or, dU(C2′)-NH•):
EPR spectral studies of radiation-produced electron attachment to matched samples of 2 in H2O glasses (7.5 M LiCl/H2O) were performed and compared with the results obtained in D2O glasses (7.5 M LiCl/D2O, Figure 1).
These results are shown in Figure 3 below. Comparison of the linewidth of the green-colored spectrum (in D2O) shown in Figure 3A with that of the red-colored spectrum (in H2O, Figure 3B) clearly establishes that the central doublet observed in the red-colored spectrum in Figure 3B is due to an extra proton hyperfine coupling in the aminyl radical in H2O (U(C2′)-NH•) that is present in the H2O glasses and is missing in the D2O glasses (dU(C2′)-ND•, green color) as it is an exchangeable proton. Comparison of these two spectra clearly shows the A‖ (i.e., the Azz) component ca. 22 G due to the α-N-H proton of the aminyl radical in H2O (U(C2′)-NH•) (red color) is lost in the D2O glasses (green color) because a deuteron coupling is only 15% (1/6.514) that of a proton in the same environment.
Figure 3.
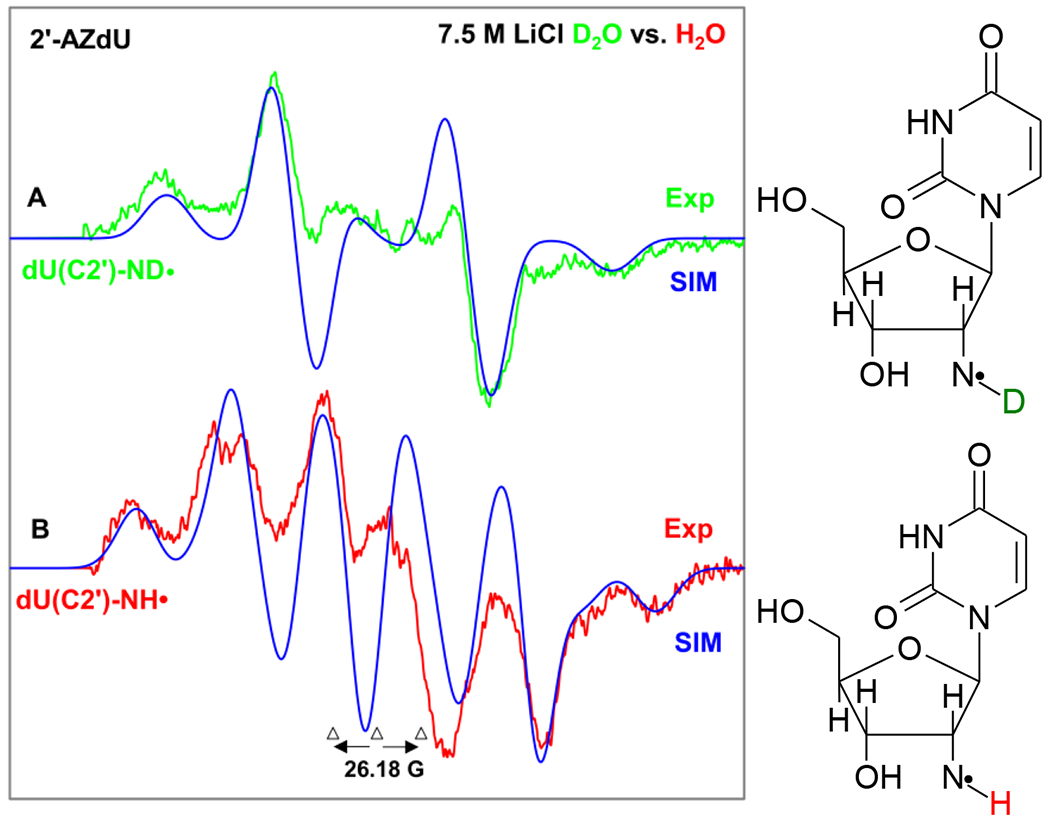
EPR spectrum of aminyl radical formed in matched samples (1 mg/mL of each compound) via radiation-produced (absorbed dose = 500 Gy at 77 K) one-electron addition by γ-irradiation of 2 (A) dU(C2′)-ND• (green color) in 7.5 M LiCl (D2O), (B) dU(C2′)-NH• (red color) in 7.5 M LiCl (H2O) color after visible illumination of each sample by using a photoflood lamp at 77 K for 15 minutes to remove the uracil anion radical by photoejection of the excess electron. Figure 3(A) is the same one shown in Figures 1A and 2A. The simulated spectra (blue) are superimposed on the top of the experimentally recorded spectra for comparison. Each experimentally recorded spectrum shown in Figure 2 was obtained after subtraction of the line components of Cl2•− spectrum following our previous works on the isolation of 77 K RNH• spectrum.7–9,49–51
Therefore, the results presented in Figures 1–3 clearly establish the electron-induced site-specific formation of the π-RND•/π-RNH• (dU(C2′)-ND•) /dU(C2′)-NH•) from 2 and its various deuterated derivatives [2′-D]-2, [3′-D]-2, [4′-D]-2 and [5,6-D,D]-2 (scheme 3). These results further confirm that the HFCC values due to the axially symmetric anisotropic 14N, the isotropic β-proton (H2′), and the anisotropic αH/αD of the exchangeable NH/ND contribute to the total hyperfine splitting of dU(C2′)-ND•.
Scheme 3.
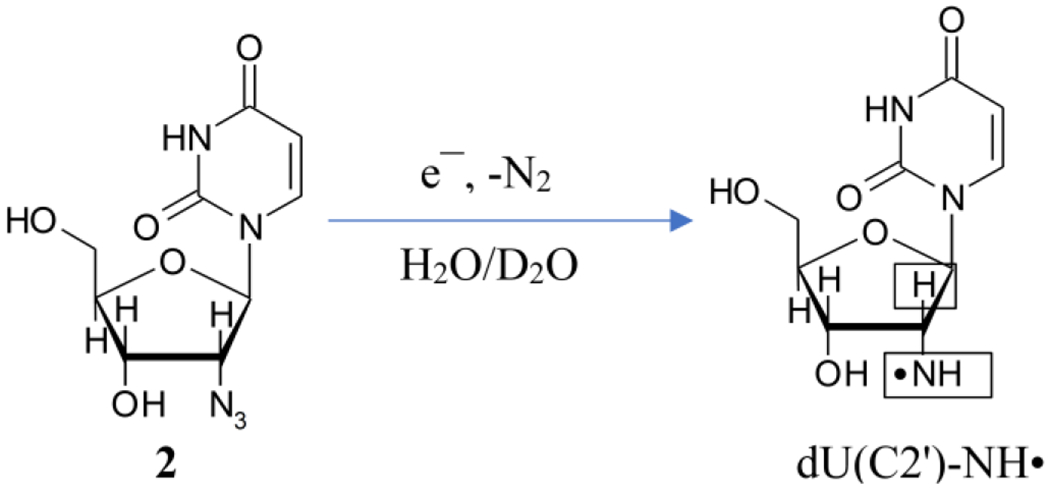
Radiation-produced electron-mediated site-specific formation of dU(C2′)-NH• from 2. Boxes in the structure of dU(C2′)-ND• indicate those nuclei with substantial HFCC values. These couplings arise from the axially symmetric anisotropic azide N, the isotropic β-proton (H2′), and the anisotropic αH/αD of the exchangeable NH/ND.
We note here that these results along with our previous results on radiation-produced prehydrated electron-mediated formation of RNH• and their reactions in 5-azidopyrimidines,7 AZT derivatives,8 azidopentoses,9 reported predominant formation of phenylnitrene anion radical due to gas phase low energy electron (LEE, 0 to 1 eV) attachment to phenylazide.8,52,53 Light-induced reduction of aromatic azides to amines in the presence of CdS and CdSe nanoparticles8,54 showed the facile and site-specific formation of RNH• from a localized RN3•− (Scheme 3). In addition, pulse radiolysis carried out in aqueous solution of 3′-AZT at ambient temperature reports that the solvated (aqueous) electron reacts with 3′-AZT with a diffusion-controlled rate (k = 1.9 x 1010 M−1s−1)55 thereby supporting the reaction mechanism and its facile nature represented in Scheme 3.
B.2. Reactions of aminyl radicals:
B.2.1. RNH• formed at a primary carbon site (5′-azido-2′, 5′-dideoxyuridine (5′-AZ-2′,5′-ddU, 5)) converts bimolecularly to the σ-type iminyl radical (R=N•) involving an α-azidoalkyl radical intermediate:
Figure 4 presents the EPR spectra (red) showing the formation of RNH• by prehydrated electron attachment7–9,50 to 5 (5 mg/mL) and the subsequent reactions of RNH• in supercooled homogeneous glassy (7.5 M LiCl/D2O) solutions. Electrons were produced by gamma radiation (absorbed dose = 500 Gy at 77 K). These spectra are overlapped on our recently published spectra (blue) of radiation-produced electron-induced formation of π-type RNH• and its reactions in 5-azidomethyl 2′-deoxyuridine (AmdU) samples.7 Note that all the EPR spectra presented in Figure 4 and in subsequent Figures are obtained after subtraction of line components due to the expected low field resonance from the matrix radical, Cl2•−, that are produced by scavenging of radiation-produced holes (reactions 6 and 7) .7–9,49–51
Figure 4.
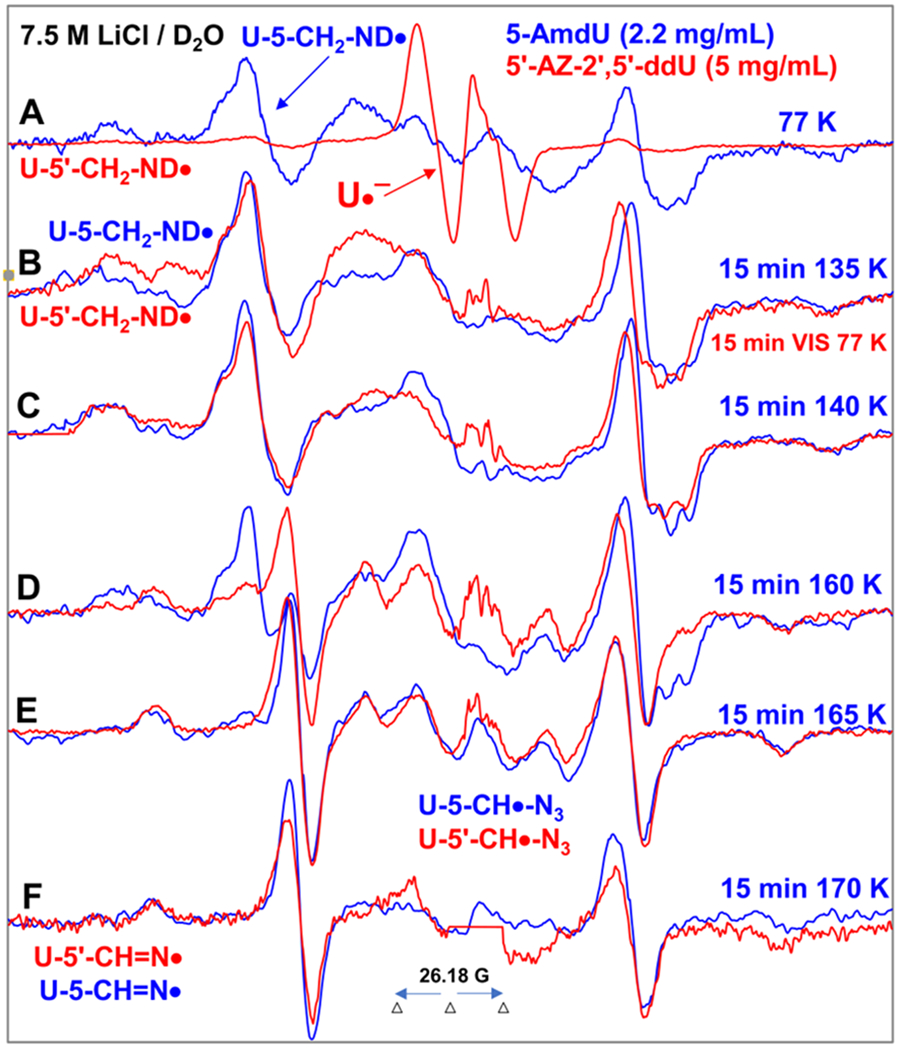
Spectra (A-D) in red were obtained from samples of 5′-AZ-2′,5′-ddU (5) ([5] = 5 mg/mL) after subtraction of the Cl2•− spectrum7–9,49 from the individual experimentally recorded spectrum. (A) EPR spectrum (red) after radiation-produced prehydrated one-electron addition to the same sample of 5 at 77 K in 7.5 M LiCl/D2O. Spectrum (B, red) was obtained after 15 min photoexcitation by a photoflood lamp at 77 K. Spectra (C, red) to (F, red) were obtained via stepwise annealing of the sample for 15 min at 140, 160, 165, and 170 K. All spectra were recorded at 77 K. Sum of two isotropic β-proton couplings is assigned to the central doublet (ca. 90.5 G) found in red spectra (A) to (D); the large central doublet (ca. 82 G) in spectra (D to F, red) is due to one isotropic β-proton coupling. Whereas, the wings in spectra (A) to (F) show the Azz components of the anisotropic nitrogen hyperfine coupling. All spectra were recorded at 77 K. The experimental spectra from AmdU from ref. 7 (2.2 mg/mL, blue) are superimposed in (A) to (F) for comparison.
| (6) |
| (7) |
Figure 4A presents the EPR spectrum (red) of the radicals formed in the sample of 5 at 77 K. The center of spectrum in Figure 4A shows the doublet27–29 expected for U•−. Furthermore, a comparison of this red spectrum with the already reported spectrum (blue) of the neutral π-aminyl radical, U-5-CH2-ND• from AmdU samples at 77 K7 shows that red spectrum has additional line components that match nicely with those due to U-5-CH2-ND•. Therefore, we assign these line components to the neutral π-aminyl radical, U-5′-CH2-ND• in the red spectrum in Figure 4A.
Following our work on AZT,8 photoejection of the electron from the U•− via visible photoexcitation at 77 K results in the red spectrum shown in Figure 4B. Comparison of the red spectrum in Figure 4B with the corresponding one in Figure 4A shows that the central doublet due to U•− is absent in the red spectrum in Figure 4B. Therefore, we conclude that the electron has been completely photoejected from U•− to form U and has reacted with parent 5 to produce additional U-5′-CH2-ND•. Total hyperfine splitting of the red spectra in Figures 4A and 4B is ca. 175.5 G (see Figure S1) in comparison to that (178.5 G) found in the blue spectra in Figures 4A and 4B. Moreover, U-5′-CH2-ND• spectrum (red, Figure 4B) from 5 is very similar to the reported spectrum7 of U-5-CH2-ND• (blue, Figure 4B) from AmdU obtained via annealing in the dark at ca. 135 K for 15 min. Thus, the hyperfine structures of the red spectrum in Figure 4B are due to a single axially symmetric anisotropic nitrogen with hyperfine coupling constant (HFCC) value as (42.5, 0, 0) G along with values of g‖ (2.0020) and g⊥ (2.0043) that are typical of a neutral aminyl radical nitrogen (Table 1). This is manifested by the excellent agreement of these N HFCC, g‖, and g⊥ values with the corresponding nitrogen HFCC, g‖, and g⊥ values of T(5′-CH2)-NH• from 5′-AZT8 and of U-5-CH2-NH• from AmdU7.
The spectra (red, Figures 4A and 4B) of U-5′-CH2-ND• show a broad doublet of ca. 90.5 G (see Figure S1) and this is very similar to the published spectra of (ca. 93.5 G) U-5-CH2-NH• (blue, Figure 4A and 4B) from AmdU7 and of (ca. 91 G) T(5′-CH2)-NH• from 5′-AZT (6)8. As for these similar structures, the radical-site p-orbital on the aminyl-N at C5′-CH2 in U-5′-CH2-ND• strongly couples with two 5′-CH2- protons that are attached to C5′ and gives rise to this broad doublet spectrum. These results show that substitution of –CH2-N3 moiety either at the sugar of 5 or at the uracil base of AmdU7 yields similar π-aminyl radical (U-5′-CH2-ND• vs. U-5-CH2-ND•) with near-identical EPR spectra.
Taking U•− spectrum from the literature27–29 and U-5′-CH2-ND• spectrum (red, Figure 4B) as benchmarks, analysis of the red spectrum in Figure 4A shows that this spectrum is a cohort of spectra due to two radicals, i.e., U-5′-CH2-ND• (ca. 80%) and U•− (ca. 20%). Thus, the predominant site of electron capture in azido compounds used in this work and in our previous works6–9 is the azido group; thus, the azido group is even more electron affinic than uracil – the most electron affinic nucleic acid base. These results, in agreement with our previous results,6–9 further establish the facile formation of site-specific neutral π-aminyl radical at 77 K via radiation-produced electron mediated addition followed by N2 removal via dissociative electron attachment mechanism and subsequent protonation from the surrounding solvent (Schemes 1 and 2).
Progressive annealing of the sample of 5 from ca. 140 K to 160 K has resulted in the spectra (red) that are presented in Figures 4C and 4D (also see Figure S1). The red spectrum in Figure 4C is found to be very similar to that (blue) already reported for AmdU under equivalent conditions7. From the similarities (overall hyperfine splitting, the center of the spectra, and lineshape) found in the red spectra with those in the blue spectra in Figures 4B and 4C, the red spectra in Figures 4B and 4C are assigned to π-aminyl radical (U-5′-CH2-ND•). In addition, at the right of both red and blue spectra in Figures 4B to 4D, the splitting of large broad resonances into three-line components is observed. This, according to our reported analysis of the AmdU sample,7 is due to unresolved deuterium hyperfine couplings.
The broad line component found at the center of the red spectra in Figures 4B and 4C splits into various line components in Figure 4D as found previously for spectra (blue) of AmdU samples.7 Following the similarities in the blue and red spectra in Figure 4D and following the assignment of these line components in the blue spectrum in Figure 4D to the C-centered α-azidoalkyl radical U-5-CH•-N3 formed via H-abstraction by U-5-CH2-ND• from a proximate parent, AmdU7, the corresponding line components at the center of the red spectrum in Figure 4E is assigned to the C-centered α-azidoalkyl radical U-5′-CH•-N3 formed via H-abstraction by U-5′-CH2-ND• from a proximate parent, 5 (Scheme 4).
Scheme 4.
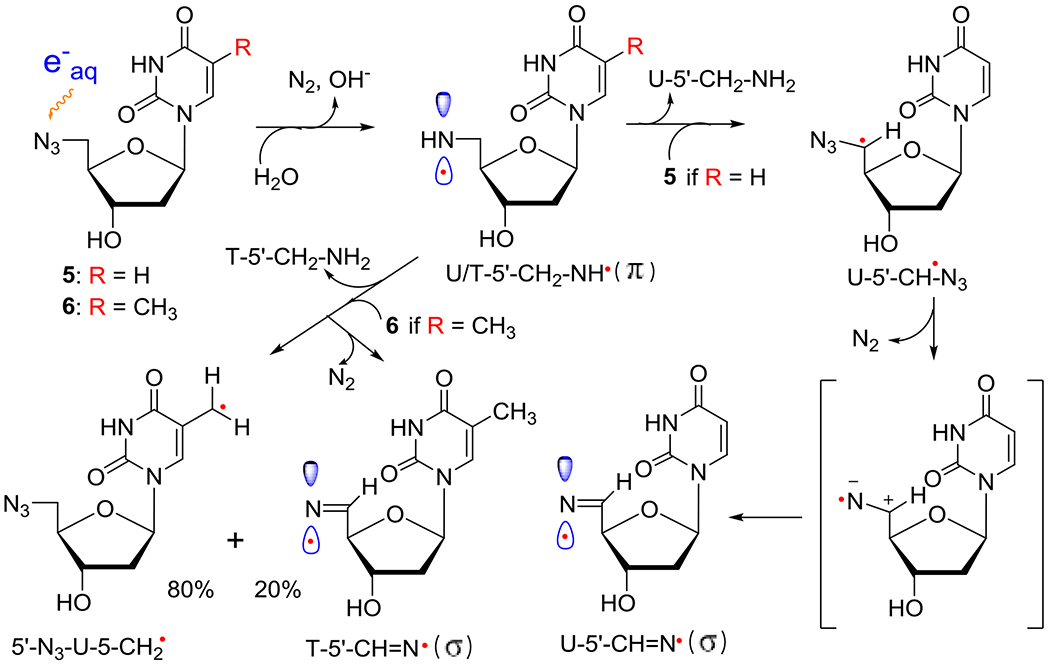
Formation of neutral π-aminyl radical, RNH• from 5′-AZ-2′,5′-ddU (5) and 5′-AZT (6) and its subsequent bimolecular reactions with 5 or 6, respectively. H-atom abstraction from the CH3 group at C5 in 6 by RNH• prevails (ca. 80%) in competition to the H-atom abstraction by the α-azidoalkyl radical, U-5′-CH•-N3, from 5′-CH2 (ca. 20%) in 6 forming the σ-iminyl radical (R=N•). As the allylic radical, U-5-CH2•, is known to undergo either dimerization or addition to the >C=C< double bond,4,33,34 we expect that the allylic radical from 6, 5′-N3-U-5-CH2•, would undergo similar reactions. However, owing to the absence of the CH3 group in 5, H-atom abstraction by the α-azidoalkyl radical, U-5′-CH•-N3, from 5′-CH2 is the only reaction taking place, leading to the facile formation of σ-iminyl radical (R=N•).
Progressive annealing of the sample of 5 from ca. 165 K to 170 K lead to the spectra (red) in Figures 4E and 4F. From the similarities of our reported spectra of AmdU samples7 to the red spectra from 5, it is apparent that apart from the new line components due to C-centered α-azidoalkyl radical U-5′-CH•-N3 at the center, the second species begins to be formed at the wings of the spectrum in Figure 4D and becomes the only radical whose spectra are observed in Figure 4F (see Figure S2).
The total width of the red and blue spectra in Figures 4E and 4F are found to be identical (ca. 155 G) and wings show line components arising from an axially symmetric anisotropic nitrogen HFCCs, A‖, of ca. 36.5 G, A⊥ = 0 and g‖ = 2.0016, g⊥=2.0040. The large doublet in both the red and blue spectra in Figure 4F results from a single β-proton coupling of ca. 82 G. Therefore, following our assignment to the blue spectrum in Figure 4F to the σ-iminyl radical, U-5-CH=N• from AmdU7, we assign the red spectrum in Figure 4F also to σ-iminyl radical, U-5′-CH=N• (For its assignment, see Table 1 and SI section). These results show that the reactive π-U-5′-CH2-ND•, abstracts H-atom from the parent (5) to form the α-azidoalkyl radical intermediate, U-5′-CH•-N3. The U-5′-CH•-N3 promptly undergoes a unimolecular β-N2 elimination from the azide group to produce the thermodynamically more stable σ-U-5′-CH=N• (Scheme 4 and Figure 8). Based on these assignments, we conclude that the red spectrum in Figure 4E is a cohort of spectra due to U-5′-CH•-N3 and U-σ−5′-CH=N• whereas the cohort of spectra due to U-5-CH•-N3 and σ-U-5-CH=N• explains the blue spectrum in Figure 4E. In addition, it is evident from these assignments that the blue spectrum in Figure 4D is a cohort of spectra due to U-5-CH2-ND•, U-5-CH•-N3 and σ-U-5-CH=N•.
Figure 8.
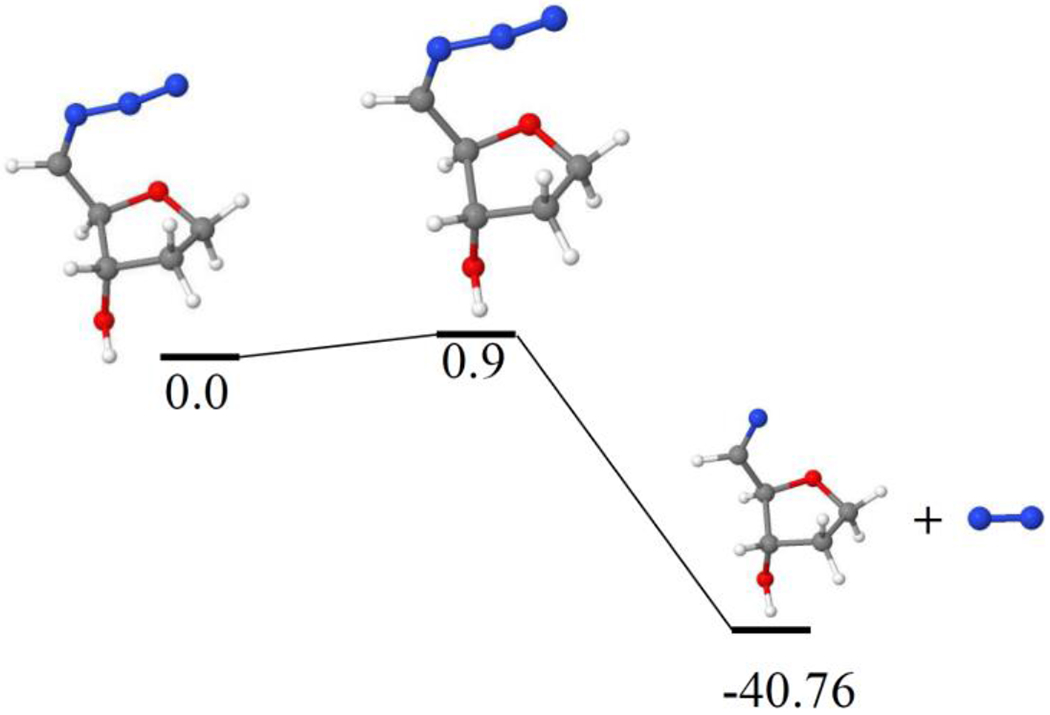
Dissociation of (5′-CH•)-N3 to 5′-CH=N• and N2 in the gas phase with a small activation barrier of only 0.9 kcal/mol. The geometries of these radicals were optimized and relative energies (kcal/mol) were calculated by B3LYP/6-31G** method.
As expected, because of the lack of an exchangeable proton at the iminyl nitrogen of U-5′-CH=N•, the EPR spectrum of this radical was the same in 7.5 M LiCl/D2O and 7.5 M LiCl/H2O (Figure S3), and also in 7.5 M LiBr/H2O (Figure S4). These results add further support to our assignment of spectra in Figure 4F to the σ iminyl radical, U-5′-CH=N•. They also confirm that irrespective of solvent (D2O or H2O) and of the glass (LiCl or LiBr), the formation of σ-U-5′-CH=N• from π-U-5′-CH2-ND• involves the α-azidoalkyl radical intermediate, U-5′-CH•-N3.
Studies employing samples of 5 with different concentrations (0.5 mg/mL and 5 mg/mL) showed that signal-to-noise ratios of EPR spectra increase substantially with a rise in concentration of 5 (Figure S5). These results point out that the C-centered α-azidoalkyl radical, U-5′-CH•-N3, is formed as an intermediate via a bimolecular H-atom abstraction by the π-U-5′-CH2-ND• from a proximate 5 (Scheme 4).
Our reported work (Figures 4D and 5 in reference 8) on 5′-AZT (6; methylazide group at sugar) showed a small observable conversion of π-RNH• to σ-R=N• or σ-T-5′-CH=N• (ca. 20%, see Figure S6). It was shown that the H-atom abstraction reaction from the sterically accessible methyl group at C5 of thymine base by π-T-5′-CH2-NH• predominates (ca. 80%) in 6 leading to the formation of allylic radical, dU-CH2• (Scheme 4). Based on our new finding of the σ-type iminyl radical spectrum from AmdU7 and from 5, this analysis accounts for the central multiline pattern from a superposition of the UCH2• and T-5′-CH=N• spectra (see Figure S6). Therefore, we now conclude that this assignment corrects our earlier assignment of C3′• as the final radical spectrum to the spectra shown in Figures 4D and 5 from 6 in reference 8.
EPR investigation of the prehydrated electron attachment to 3AZPrOH (7), a simple model of the azido substitution at the primary carbon site, also showed σ-iminyl radical formation via the α-azidoalkyl radical intermediate (Figure S7) thereby proving the general character of the azido substitution at any primary carbon site which leads to the facile conversion of π-RNH• to σ-R=N• via the α-azidoalkyl radical intermediate.
B.3. RNH• formed at a secondary carbon site:
B.3.1. Nature of the base (e.g., cytosine in 2′-AZdC (1) or uracil in 2′-AZdU (2)) does not affect the formation of aminyl radicals and their subsequent reactions:
γ-Irradiation-mediated π-RNH• observed in matched homogeneous glassy samples (native pD ca. 5) of 2′-AZdU (black, 2) and 2′-AZdC (blue, 1) are chosen as models of π-RNH• produced at secondary carbons and subsequent reactions of these π-RNH• are presented in Figure 5.
Figure 5.
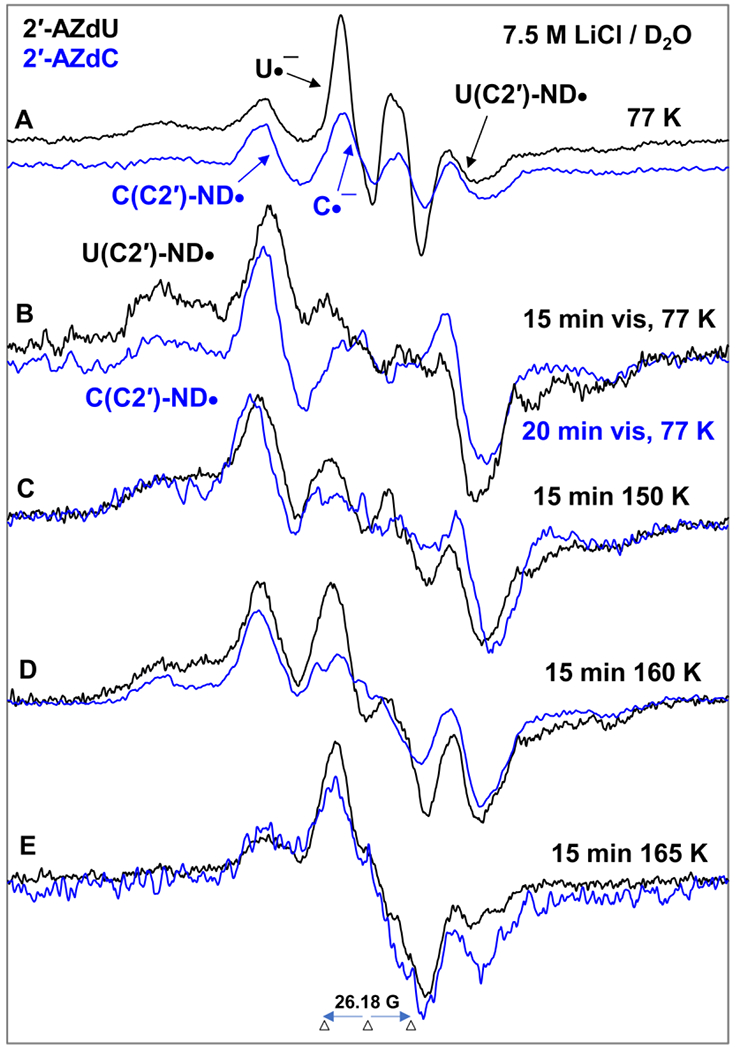
EPR spectra of matched samples of 2′-AZdU (2) (black) and 2′-AZdC (1) (blue) at the native pD (ca. 5) of the homogeneous glassy solution of 7.5 M LiCl in D2O after subtraction of the Cl2•− spectrum7–9,49 from the individual experimentally recorded spectrum. (A) After γ-irradiation (absorbed dose = 500 Gy) produced one-electron addition at 77 K. (B) After visible illumination of samples employing a photoflood lamp at 77 K for 15 to 20 minutes. The central doublet in both spectra due to the uracil anion radical (U•−) and cytosine anion radical (C•−) are removed by photoejection of the excess electron. Spectra (C) to (E) are obtained after subsequent annealing for ca. 15 min in the dark from ca. 150 to ca. 165 K. All EPR spectra shown in Figures A to E were recorded at 77 K. See also SI Figure S8 for additional data.
Figure 5 shows that EPR spectral characteristics (e.g., overall hyperfine splitting, g-value at the center, lineshape) obtained from the samples of 1 and 2 are quite identical. Hence, we conclude that the formation of π-RNH• and their subsequent reactions are similar in these samples and are not affected by the base (C (1) or U (2)).
Following spectral assignments shown in Figures 4A, the central doublets in Figure 5A are assigned to the uracil anion radical (U•−, black) and cytosine anion radical (C•−, blue).
Based on dU(C2′)-ND• assignment (section B.1) and on the spectral similarities observed in Figure 5A and 5B, the blue spectrum in Figure 5B is assigned to dC(C2′)-ND•. Therefore, dC(C2′)-ND• shows a large central doublet due to an isotropic β HFCC value of 2′-H (ca. 51.5 G), wings line component due to an axially symmetric anisotropic 14N-atom HFCC (Azz = 40.5 G, Axx = Ayy = 0 G), g‖ = 2.0020 and g⊥=2.0043 (section B.1, Table 1).
Comparison of spectra in Figure 5B with those of Figure 5A shows that the central doublet owing to U•− (black) or C•− (blue) is absent in the spectra presented in Figure 5B. This result demonstrates that the excess electron is completely photo-ejected either from U•− (black) and from C•− (blue) via photo-excitation at 77 K and reacts with unreacted parent 1 and 2 also at 77 K to produce additional π-RNH• (dU(C2′)-ND• (or, dC(C2′)-ND•)).
Based on these assignments, spectra shown in Figure 5A are found to be a cohort of U•− (black) or C•− (blue) (ca. 20%) and π-RNH• (dU(C2′)-ND•, black (or, dC(C2′)-ND•, blue)) (ca. 80%) (see Figures 4A and 4B).
The spectra shown in Figures 5C to 5E and Figure S8 are due to subsequent reactions of dU(C2′)-ND• or, dC(C2′)-ND• due to progressive annealing up to 165 K that caused softening of the glass and migration of these radicals.
The spectra presented in Figure 5E is due to the final radical formed via bimolecular reaction of either dU(C2′)-ND• (or, dC(C2′)-ND•) with a proximate unreacted parent 2 or 1. EPR spectral studies carried out using matched samples of 2 in 7.5 M LiCl/D2O and in 7.5 M LiCl/H2O glasses (Figure 3 (π-RNH• identification) and Figure 5E, Figure S9 (final radical spectra)) establish that reaction of dU(C2′)-ND• (or, dC(C2′)-ND•) leads to a C-centered radical formation as there is no observable difference in the spectra in D2O and in H2O glasses.
There are two possible assignments of this C-centered radical. (A) The radical site could either be at the C5- or C6 site in uridine (2) or in cytosine (1) via the addition of π-RNH• (dU(C2′)-ND• or dC(C2′)-ND•) to the C6 or C5-site of the C5=C6 double bond. Alternatively, (B) π-RNH• being a good H-atom abstractor 6–9 (also, see the introduction and section B.2.1), it might cause H-atom abstraction from the 5′-site of 1 or 2 and the resulted C5′• has two possibilities: (i) it can be in a conformation which has an anisotropic α-H coupling at C5′ and owing to a significant isotropic β-H coupling at C4′24 ; (ii) it can undergo ring-opening reaction leading to an open-chain conformation of C4′• (similar to our results in azidolyxofuranoside9) with an anisotropic α-H coupling at C4′ and owing to a significant isotropic β-H coupling at C3′.
As samples of 1 or 2 lead to similar final radical spectra (Figure 5, S8, and S9), EPR studies were performed using deuterated derivatives of 2 with deuterations at various specific positions of the sugar moiety and at the base ([2′-D]-2, [3′-D]-2, [4′-D]-2 and [5,6-D,D]-2 (section B.1.2 and Figure 2) to obtain an unequivocal assignment of the final radical species found in 1 or 2. EPR spectra of non-deuterated parent 2 vis-à-vis deuterated at various positions at the sugar moiety (Figures S10 and S11) show no observable difference. These results establish that deuterations at the sugar moiety do not help in radical assignment and rule out the possibility (B) (vide supra). However, the collapse of the central part of the spectrum obtained after annealing at ca. 167 K for 1h in the dark due to deuteration at C5 and C6 of the uracil base of 2 ([5,6-D,D]-2) (Figure S12) suggests the formation of the C-centered radical via electrophilic addition of π-RNH• (dU(C2′)-ND•) to the C5=C6 double bond of the uracil base in 2 (Scheme 5 and Scheme S1). Due to spectral similarities observed from samples of 1 and 2 (Figure 5), formation of the similar C-centered radical occurs in 1 via the same pathway (scheme 5). These results also establish that uracil or cytosine base does not affect the formation and subsequent reactions of π-RNH•.
Scheme 5.
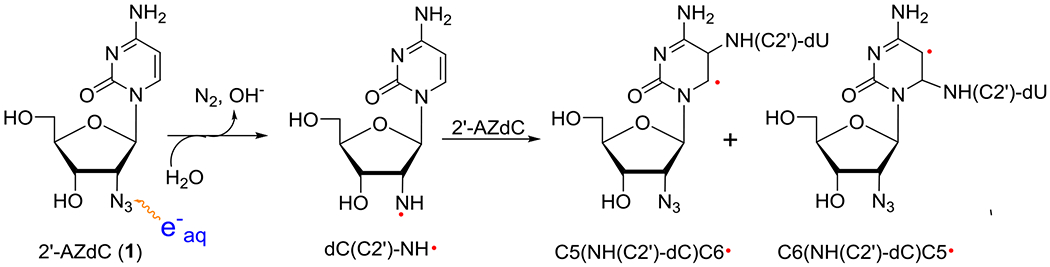
Electrophilic addition of π-RNH• (dC(C2′)-ND•) to the C5=C6 double bond of the cytosine base in 1. The corresponding scheme for electrophilic addition of π-RNH• (dU(C2′)-ND•) to the C5=C6 double bond of the uracil base in 2 is shown in scheme S3.
B.3.2. 3′-azido-2′,3′-dideoxyuridine (3′-AZ-2′,3′-ddU, 3):
Since C3′ in 2′-deoxyribose ring is also a secondary carbon in which the azido group can be attached, EPR studies are performed employing 3′-AZ-2′,3′-ddU, 3; these results (black spectra) are subsequently compared with those obtained from 2 (blue spectra) and are presented in Figure 6.
Figure 6.
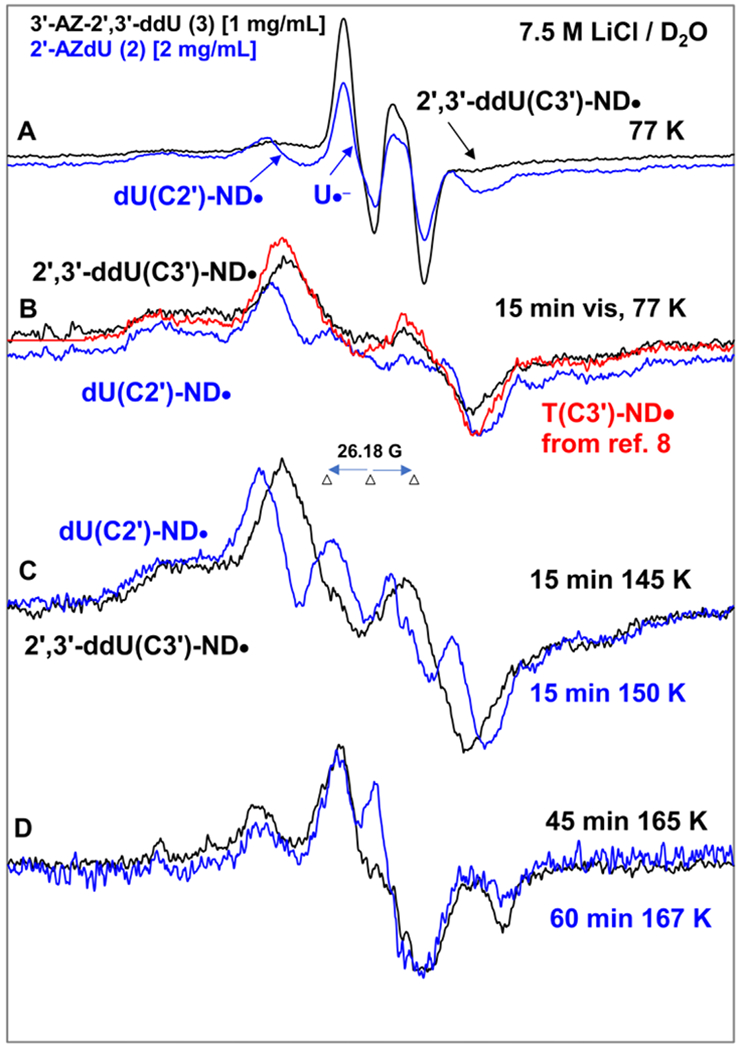
EPR spectra of samples of 2′-AZdU (2) [2 mg/mL] (blue) and 3′-AZ-2′,3′-ddU (3) 1 mg/mL] (black) at the native pD (ca. 5) of the homogeneous glassy solution of 7.5 M LiCl in D2O after subtraction of the Cl2•− spectrum7–9,49 from the individual experimentally recorded spectrum. (A) After γ-irradiation (absorbed dose = 500 Gy) produced one-electron addition at 77 K. (B) After visible illumination of samples employing a photoflood lamp at 77 K for 15 minutes. The central doublet in both spectra due to the uracil anion radical (U•−) and cytosine anion radical (C•−) are removed by photo-ejection of the excess electron. The corresponding data of 3′-AZT sample is taken from reference 8. (C) Spectra obtained after subsequent annealing for ca. 15 min in the dark from ca. 145 to ca. 150 K. (D) Spectra obtained after subsequent annealing for ca. 45 to 60 min in the dark from ca. 165 K.
All EPR spectra shown in Figures A to D were recorded at 77 K.
Following EPR spectral results presented in Figures 4A, 4B, 5A and 5B, we assign the black 77 K spectrum presented in Figure 6A obtained after γ-irradiation as the cohort of spectra due to π-RNH• (2′,3′ddU(C3′)-ND•, ca. 80%) and U•− (ca. 20%). After 15 min photoexcitation at 77 K by a photoflood lamp, the excess electron is completely photo-ejected from U•− leading to only π-RNH• (2′,3′-ddU(C3′)-ND•, black spectrum in Figure 6B) similar to our findings shown in Figures 4B and 5B. As the azido group is at 3′-site in both 3′-AZT and 3, our reported spectrum (pink) of T(C3′)-ND• from 3′-AZT8 is superimposed on the black spectrum of 2′,3′-ddU(C3′)-ND•. EPR characteristics (total hyperfine splitting, g-value at the center, lineshape) of these two spectra match nicely and further support our assignment of the black spectrum in Figure 6B to 2′,3′-ddU(C3′)-ND•. Therefore, β(C3′-H) HFCC, Azz, Axx, Axx values of the axially symmetric anisotropic N, g‖ and g⊥ values of 2′,3′-ddU(C3′)-ND• are similar to those of T(C3′)-ND•, i.e., for 2′,3′-ddU(C3′)-ND• β(C3′-H) HFCC = ca. 41 G, Azz = 37.5 G, Axx = Ayy = 0 G, g‖ = 2.0020 and g⊥=2.0043.
Subsequent annealing of this sample from 3 at ca. 145 K and at ca. 165 K for 15 min in the dark led to the black spectra in Figures 6C and 6D. The black spectrum in Figure 6C, owing to its similarities to the black spectrum in Figure 6B is assigned to 2′,3′-ddU(C3′)-ND•. Owing to the similarities in the blue spectra in Figures 6B and 6C and in Figures 6C and 5B, the blue spectrum in Figure 6C is assigned to dU(C2′)-ND• (section B.3.1).
A nice match observed between the blue and black spectra shown in Figure 6D suggests the formation of the radical species due to electrophilic addition of 2′,3′-ddU(C3′)-ND• to the C5=C6 double bond of the uracil base of a proximate parent, 3. This reaction is similar to those observed in the case of π-RNH• formed from another secondary carbon site (2′) in 1, 2 and their derivatives (Scheme 5 and Scheme S1).
Results presented in sections B.3.1 and B.3.2 establish that unlike azidopyrimidine nucleosides in which the azido group is attached to a primary carbon (-CH2N3) (section B.2) , no observable line components due to σ-R=N• are found in azidopyrimidine nucleosides in which the azido group is attached to a secondary carbon (>CH-N3). In addition, our earlier work on π-RNH• on a secondary carbon site (T(C3′)-ND•/T(C3′)-NH•) formed from electron addition to 3′-AZT show predominant formation of the allylic UCH2• via H-atom abstraction from the -CH3 group at C5 of thymine base.8
B.4. RNH• formed at a tertiary carbon site:
Since 4′ site is a tertiary carbon site in ribose (RNA) and in 2′-deoxyribose (DNA), 4′-azidocytidine (4′-AZ-C, 4) has been chosen as the model to investigate π-RNH• formation at tertiary carbon site and reactions of these π-RNH•. These results are compared with the formation of π-RNH• from 1 and its reactions (Figure 5). These results are presented in Figure 7.
Figure 7.
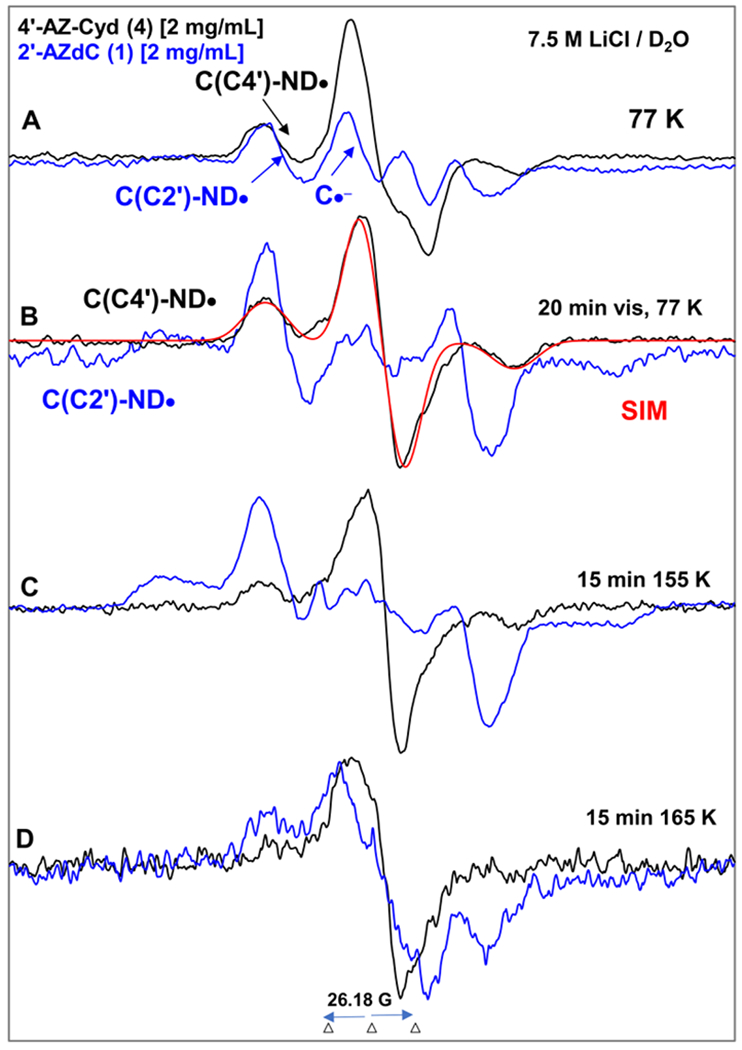
EPR spectra of matched samples of 4′-AZ-C (4, black) and 2′-AZdC (blue,1 also shown in Figure 5) at the native pD (ca. 5) of the homogeneous glassy solution of 7.5 M LiCl in D2O after subtraction of the Cl2•− spectrum7–9,49 from the individual experimentally recorded spectrum. (A) After γ-irradiation (absorbed dose = 500 Gy) produced one-electron addition at 77 K. (B) After visible illumination of samples employing a photoflood lamp at 77 K for 20 minutes. The central doublet in both spectra due to the cytosine anion radical (C•−) is removed by photoejection of the excess electron. The simulated spectrum of C(C4′)-ND• is also shown (for simulation parameters, see text). Spectra (C) and (D) are obtained after subsequent annealing for 15 min in the dark at ca.155 K and at ca. 165 K. All EPR spectra shown in Figures A to D were recorded at 77 K.
Following EPR spectral results presented in Figures 5A, 5B, 6A and 6B, we assign the black 77 K spectrum presented in Figure 7A obtained after γ-irradiation as the cohort of spectra due to π-RNH• (C(C4′)-ND•, ca. 85%) and C•− (ca. 15%). After 15 min photoexcitation at 77 K by a photoflood lamp, the excess electron is completely photo-ejected from C•− leading to only π-RNH• (C(C4′)-ND•, black spectrum in Figure 7B) similar to our findings shown in Figures 5B and 6B. Comparison of the black spectrum of C(C4′)-ND• presented in Figures 7B with those presented in Figures 4, 5, and 6 clearly demonstrates that C(C4′)-ND• does not have any isotropic β-H HFCC owing to the attachment of aminyl radical site N to the tertiary carbon (C4′). This spectrum has been simulated by employing axially symmetric anisotropic 14N HFCC (37.5, 0, 0) G, anisotropic α-D (N-D) (4.5, 0, 6) G, a mixed Lorentzian/Gaussian (1/1) linewidth of 10 G, along with g-values of (2.0020, 2.0043, 2.0043). The simulated spectrum (red) of C(C4′)-ND• obtained by employing these EPR parameters match the experimental spectrum (black) well shown in Figure 7B.
Subsequent annealing of this sample from 4 at ca. 145 K and at ca. 165 K for 15 min in the dark led to the black spectra in Figures 7C and 7D. The black spectrum in Figure 7C, owing to its similarities to the black spectrum in Figure 7B is assigned to C(C4′)-ND•. The blue spectrum in Figure 7C is assigned to dC(C2′)-ND•.
The black spectrum in Figure 7D represents the decay of C(C4′)-ND•. On the other hand, the blue spectrum in Figure 7D is of the radical species due to electrophilic addition of dC(C2′)-ND• to the C5=C6 double bond of the uracil base of a proximate parent, 1 (Scheme 5). Thus, a comparison of spectra in Figure 7D suggests that π-RNH• attached to a tertiary alkyl carbon (generated from 4) could be less reactive than π-RNH• attached to primary and secondary alkyl carbons (Scheme 6).
Scheme 6.
Schematic representation of different types of reactions undergone by π-RNH• attached to a primary, or a secondary, or a tertiary alkyl carbon in azidopyrimidine nucleosides.
(C). Theoretical calculations:
We have calculated the forward barrier for dissociation of the abasic 5-α-azidoalkyl radical, dR-5′-CH•-N3, to the σ iminyl radical, dR-5′-CH=N• and N2 in the gas phase employing dR-5′-CH•-N3 as a model (Figure 8 and section B.2) to reduce the computational cost. The potential energy surface (PES) for the dissociation of dR-5′-CH•-N3 → dR-5′-CH=N• + N2 was calculated in the gas phase using the B3LYP/6-31G** method, see Figure 8. The relative energies (kcal/mol) of barrier height and products with respect to the reactant were calculated from the total SCF energy of each fragment shown in Figure 8. We have used the B3LYP/6-31G** optimized geometry of 5′-CH•-N3 to obtain its HFCCs for accounting of the center of the blue spectrum in Figures 4D and 4E. Employing this method, we have also calculated the HFCCs of other aminyl and iminyl radicals reported in this work.
The B3LYP/6-31G** level of theory predicts a forward barrier of 0.9 kcal/mol which is easily accessible even at the low-temperature range of our EPR experiments. The overall reaction is also predicted to be highly favorable with an energy change of −40.8 kcal/mol. The very similar EPR spectral results from samples of 5, 6 (this work) and AmdU7 (Figure 4 and section B.2) lead us to conclude that the barriers for conversions of U-5-CH•-N3 to σ-U-5-CH=N•, dU-5′-CH•-N3 to σ-dU-5′-CH=N• would be similar to that calculated for dR-5′-CH•-N3 (Table 1).
Discussion/Conclusion:
Our radiation-chemical results demonstrate that:
Unequivocal identification of aminyl and iminyl radicals: Application of 15N isotopic substitution, suitable deuterations at the sugar moiety along with the collapse of the exchangeable α-N-H anisotropic HFCC by changing the solvent from H2O to D2O lead to the unequivocal assignment of electron-mediated site-specific formation of π-RNH•. These isotopic substitution methods also aided the identification of subsequent radical species, e.g., iminyl radicals.
-
Stereochemical structure and electronic environment affect the formation and reactivity of various types of π-RNH• generated from azidopyrimidine nucleosides: Despite different electronic and chemical properties π U-5′-CH2-ND• from 5 (methylazide group in a sugar), π HOCH2-CH2-CH2-ND• from 7 (methylazide group in an alcohol), and π U-5-CH2-ND• from AmdU (methylazide group at the C5 position of the uracil base) have similar spectral features and undergo analogous reactions, i.e., a facile bimolecular H-atom abstraction to form α-azidoalkyl radicals which unimolecularly and facilely convert to σ-type R=N•.
However, electron addition to azido group attached to a secondary carbon site (2′) in 1 and 2 produce π dC(C2′)ND• and π dU(C2′)ND• respectively with very similar radical conformations and these radicals undergo electrophilic addition to C5=C6 double bond of a proximate base (C (1) or U (2)). Moreover, 2′,3′-ddU(C3′)-ND• formed via electron addition to an azido group attached to the 3′-secondary carbon site in the sugar moiety also undergoes electrophilic addition to the C5=C6 double bond of a proximate base (U (3)). Thus while the azido group (and hence the π-RNH• radical) attached to a primary carbon site results in its subsequent facile conversion to σ-type R=N•, our EPR spectral results establish that these reactions are not observed in the case of the π-RNH• radical attached to a secondary carbon site (Scheme 4 vs. Scheme 5) which instead undergoes ring addition. In principle, following the mechanism represented in Scheme 4, π-RNH• generated at a secondary carbon site might lead to an α-azidoalkyl radical in the sugar ring via bimolecular H-atom abstraction from a proximate unreacted parent molecule. However, restricted conformations of the cyclic ring in the transition state likely prevent the formation of iminyl radical in these cases and force the observed electrophilic addition (i.e., addition to the C5=C6 double bond) pathway.
Because of steric hindrance (i.e., due to the crowded transition state) and stabilization due to hyperconjugation, the π-RNH• attached to a tertiary carbon site exhibits lower reactivity in our glassy system at low temperature. Thus, these studies provide the free radical mechanistic basis for the various types of reactions (H-atom abstraction by π-RNH• and addition of π-RNH• to C=C double bond) that were postulated from product analyses studies by Wagner et al.12
Implications as radiosensitizers in radiochemotherapy of cancer: Recently, π-RNH• from AmdU (π-RNH• attached to a primary carbon site) have been shown to significantly augment radiation damage in EMT6 breast cancer cells.7 EPR spectral results presented in this work under anoxic conditions predict that π-RNH• attached to a primary or a secondary carbon site would be more effective in augmenting radiation damage to hypoxic cancer cells than π-RNH• attached to a tertiary carbon site owing to its lower reactivity. Thus, these studies have exhibited the potential to select those azidoDNA-models as adjuvants that could significantly improve cancer radiotherapeutic efficacy by causing higher hypoxic cancer cell killing.
These results are of fundamental importance in the chemistry of aminyl radicals which have widespread applications in synthesis, in environmental degradation of pharmaceuticals, in free radical-mediated damage to DNA and proteins, etc.
Supplementary Material
Acknowledgement:
MDS, AA and AK thank the National Cancer Institute of the National Institutes of Health (Grant RO1CA045424) for support. AA and MDS thank REF, CBR at OU for support. AA thanks the National Science Foundation under Grant No. CHE-1920110. TPD is grateful to the FIU Graduate School for her Doctoral Evidence Acquisition fellowship and Kaufmann Foundation for support and SFW to NIGMS/NCI (SC1CA138176) grant for support.
Footnotes
Supporting Information for publication: Synthetic procedures for the preparation and characterization of the deuterium labeled nucleosides and their sugar precursors (Scheme S1–S2), additional EPR spectra (Figures S1–S12), Scheme S3, and supportive theoretical calculation data. This information is available free of charge via the internet at http://pubs.acs.org/.
Conflicts of interest
The authors declare that there are no conflicts.
References:
- 1.Danen WC; Neugebauer FA Aminyl Free Radicals. Angew. Chem. Int. Ed 1975, 14, 783–789. [Google Scholar]
- 2.Rodriguez MA In Light-Induced Iminyl Radicals: Generation and Synthetic Applications, InTech: 2012; pp 265–282. [Google Scholar]
- 3.Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centered Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- 4.von Sonntag C Free-Radical-Induced DNA Damage and Its Repair; Springer-Verlag: Berlin, Heidelberg, 2006. [Google Scholar]
- 5.Minozzi M; Nnni D; Spagnolo P From Azides to Nitrogen-Centered Radicals: Applications of Azide Radical Chemistry to Organic Synthesis. Chem. Eur. J 2009, 15, 7830–7840. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A; Becker D; Adhikary A; Sevilla MD Reaction of Electrons with DNA: Radiation Damage to Radiosensitization. Int. J. Mol. Sci 2019, 20, 3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen Z; Peng J; Tuttle P; Ren Y; Garcia C; Debnath D; Rishi S; Hanson C; Ward S; Kumar A et al. Electron-mediated Aminyl and Iminyl Radicals from C5-Azido-Modified Pyrimidine Nucleosides Augment Radiation Damage to Cancer Cells. Org. Lett 2018, 20, 7400–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adhikary A; Khanduri D; Pottiboyina V; Rice CT; Sevilla MD Formation of Aminyl Radicals on Electron Attachment to AZT: Abstraction from the Sugar Phosphate Backbone vs. One-Electron Oxidation of Guanine. J. Phys. Chem. B 2010, 114, 9289–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mudgal M; Rishi S; Lumpuy DA; Curran KA; Varley KL; Sobczak AJ; Dang TP; Sulimoff N; Kumar A; Sevilla MD et al. Prehydrated One-electron Attachment to Azido-modified Pentofuranoses: Aminyl Radical Formation, Rapid H-atom Transfer and Subsequent Ring Opening. J. Phys. Chem. B 2017, 121, 4968–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musa OM; Horner JH; Shahin H; Newcomb M A Kinetic Scale for Dialkylaminyl Radical Reactions. J. Am. Chem. Soc 1996, 118, 3862–3868. [Google Scholar]
- 11.Hwu JR; Lin CC; Chuang SH; King KY; Su T-R; Tsay S-C Aminyl and Iminyl Radicals From Arylhydrazones in The Photo-induced DNA Cleavage. Bioorg. Med. Chem 2004, 12, 2509–2515. [DOI] [PubMed] [Google Scholar]
- 12.Kuttappan-Nair V; Samson-Thibault F; Wagner JR Generation of 2′-Deoxyadenosine N6-Aminyl Radicals from the Photolysis of Phenylhydrazone Derivatives. Chem. Res. Toxicol 2010, 23, 48–54. [DOI] [PubMed] [Google Scholar]
- 13.Zheng L; Griesser M; Pratt DA; Greenberg MM Aminyl Radical Generation via Tandem Norrish Type I Photocleavage, β-Fragmentation: Independent Generation and Reactivity of the 2′-Deoxyadenosin-N6-yl Radical. J. Org. Chem 2017, 82, 3571–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun H; Zheng L; Yang K; Greenberg MM Positional Dependence of DNA Hole Transfer Efficiency in Nucleosome Core Particles. J. Am. Chem. Soc 2019, 141, 10154–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng L; Lin L; Qu K; Adhikary A; Sevilla MD; Greenberg MM Independent Photochemical Generation and Reactivity of Nitrogen-Centered Purine Nucleoside Radicals From Hydrazines. Org. Lett 2017, 19, 6444–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newcomb M Radical Kinetics and Clocks In Encyclopedia of Radicals in Chemistry, Biology and Materials, Chatgilialoglu C.; Struder A, Eds. John Wiley & Sons Ltd.: Chichester, UK, 2012; pp 107–124. [Google Scholar]
- 17.Adhikary A; Kumar A; Bishop CT; Wiegand TJ; Hindi RM; Adhikary A; Sevilla MD π-Radical to σ-Radical Tautomerization in One-Electron-Oxidized 1-Methylcytosine and Its Analogs. J. Phys. Chem. B 2015, 119, 11496–11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adhikary A; Kumar A; Becker D; Sevilla MD The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J. Phys. Chem. B 2006, 110, 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adhikary A; Khanduri D; Sevilla MD Direct Observation of the Protonation State and Hole Localization Site in DNA-oligomers. J. Am. Chem. Soc 2009, 131, 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adhikary A; Kumar A; Munafo SA; Khanduri D; Sevilla MD Prototropic Equilibria in DNA Containing One-electron Oxidized GC: Intra-duplex vs. Duplex to Solvent Deprotonation. Phys. Chem. Chem. Phys 2010, 12, 5353–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adhikary A; Kumar A; Khanduri D; Sevilla MD The Effect of Base Stacking on the Acid-base Properties of the Adenine Cation Radical [A•+] in solution: ESR and DFT studies. J. Am. Chem. Soc 2008, 130, 10282–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonifačić M; Armstrong DA; Carmichael I; Asmus K-D β-Fragmentation and Other Reactions Involving Aminyl Radicals from Amino Acids. J. Phys. Chem. B 2000, 104, 643–649. [Google Scholar]
- 23.Bonifačić M; Sÿtefanić I; Hug GL; Armstrong DA; Asmus K-D Glycine Decarboxylation: The Free Radical Mechanism. J. Am. Chem. Soc 1998, 120, 9930–9940. [Google Scholar]
- 24.Adhikary A; Khanduri D; Kumar A; Sevilla MD Photoexcitation of Adenine Cation Radical [A•+] in the Near UV-vis Region Produces Sugar Radicals in Adenosine and in Its Nucleotides. J. Phys. Chem. B 2008, 112, 15844–15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banyasz A; Ketola T-M; Muñoz-Losa A; Rishi S; Adhikary A; Sevilla MD; Martinez-Fernandez L; Improta R; Markovitsi D. UV-Induced Adenine Radicals Induced in DNA A-Tracts: Spectral and Dynamical Characterization. J. Phys. Chem. Lett 2016, 7, 3949–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adhikary A; Becker D; Sevilla MD Electron Spin Resonance of Radicals in Irradiated DNA In Applications of EPR in Radiation Research; Lund A, Shiotani M, Eds.; Springer International Publishing: Heidelberg, New York, London, 2014; pp 299–352. [Google Scholar]
- 27.Bernhard WA Radical Reaction Pathways Initiated by Direct Energy Deposition in DNA by Ionizing Radiation. Wiley Ser. React. Intermed. Chem. Biol 2009, 2, 41–68. [Google Scholar]
- 28.Close D From the Primary Radiation Induced Radicals in DNA Constituents to Strand Breaks: Low Temperature EPR/ENDOR Studies In Radiation Induced Molecular Phenomena in Nucleic Acids; Shukla M, Leszczynski J, Eds.; Springer: Netherlands, 2008; Vol. 5, pp 493–529. [Google Scholar]
- 29.Sagstuen E; Hole EO In Radiation produced radicals; Brustolon M, Giamello E, Eds.; John Wiley & Sons, Inc.: NJ, 2009; pp 325–382. [Google Scholar]
- 30.Steenken S Purine Bases, Nucleosides, and Nucleotides: Aqueous Solution Redox Chemistry and Transformation Reactions of Their Radical Cations and e− and OH Adducts. Chem. Rev 1989, 89, 503–520. [Google Scholar]
- 31.Steenken S Electron-Transfer-Induced Acidity/Basicity and Reactivity Changes of Purine and Pyrimidine Bases. Consequences of Redox Processes for DNA Base Pairs. Free Radical Res. Commun 1992, 16, 349–379. [DOI] [PubMed] [Google Scholar]
- 32.O’Neill P; Davies SE Pulse Radiolytic Study of the Interaction of SO4•− With Deoxynucleosides. Possible Implications for Direct Energy Deposition. Int. J. Radiat. Biol 1987, 52, 577–587. [DOI] [PubMed] [Google Scholar]
- 33.Wagner JR; Cadet J Oxidation Reactions of Cytosine DNA Components by Hydroxyl Radical and One-electron Oxidants in Aerated Aqueous Solutions. Acc. Chem. Res 2010, 43, 564–571. [DOI] [PubMed] [Google Scholar]
- 34.Burrows CJ; Muller JG Oxidative Nucleobase Modifications Leading to Strand Scission. Chem. Rev 1998, 98, 1109–1151. [DOI] [PubMed] [Google Scholar]
- 35.Jagetia GC; Aruna R Correlation of Micronuclei-induction With the Cell Survival in HeLa Cells Treated With A Base Analogue, Azidothymidine (AZT) Before Exposure to Different Doses of Gamma-radiation. Toxicology Lett 2003, 139, 33–43. [DOI] [PubMed] [Google Scholar]
- 36.Coucke PA; Cottin E; Decosterd LA Simultaneous Alteration of De Novo and Salvage Pathway to the Deoxynucleoside Triphosphate Pool by (E)-2′-Deoxy-(fluoromethylene)cytidine (fmdc) and Zidovudine (AZT) Results in Increased Radiosensitivity In Vitro. Acta Oncologica 2007, 46, 612–620. [DOI] [PubMed] [Google Scholar]
- 37.Liao Z-K; Zhou F-X; Luo Z-G; Zhang W-J; Jie X; Bao J; Han G; Zhang M-S; Xie C-H; Zhou Y-F. Radio-activation of hTERT Promoter in Larynx Squamous Carcinoma Cells: An ‘Indirected-activator’ Strategy in Radio-gene-therapy. Oncology Reports 2008, 19, 281–286. [PubMed] [Google Scholar]
- 38.Zhou F-X; Liao Z-K; Dai J; Jie X; Xie C-H; Luo Z-G; Liu S-Q; Zhou Y-F. Radiosensitization Effect of Zidovudine on Human Malignant Glioma Cells. Biochem. Biophys. Res. Commun 2007, 354, 351–356. [DOI] [PubMed] [Google Scholar]
- 39.Westphal EM; Blackstock W; Feng W; Israel B; Kenney SC Activation of Lytic Epstein-Barr Virus (EBV) Infection by Radiation and Sodium Butyrate In Vitro and In Vivo: A Potential Method for Treating EBV-positive Malignancies. Cancer Res. 2000, 60, 5781–5788. [PubMed] [Google Scholar]
- 40.van der Donk WA; Stubbe J; Gerfen GJ; Bellew BF; Griffin RG, EPR Investigations of the Inactivation of E. coli Ribonucleotide Reductase With 2′-Azido-2′-deoxyuridine 5′-Diphosphate: Evidence For the Involvement of the Thiyl Radical of C225-R1. J. Am. Chem. Soc 1995, 117, 8908–8916. [Google Scholar]
- 41.Fritscher J; Artin E; Wnuk S; Bar G; Robblee JH; Kacprzak S; Kaupp M; Griffin RG; Bennati M; Stubbe J, Structure of the Nitrogen-Centered Radical Formed During Inactivation of E. coli Ribonucleotide Reductase by 2′-Azido-2′-deoxyuridine-5′-diphosphate: Trapping of the 3′-Ketonucleotide. J. Am. Chem. Soc 2005, 127, 7729–7738. [DOI] [PubMed] [Google Scholar]
- 42.Wnuk SF; Mudgal MM; Nowak I; Robins MJ, Model Substrate/Inactivation Reactions for MoaA and Ribonucleotide Reductases: Loss of Bromo, Chloro, or Tosylate Groups from C2 of 1,5-Dideoxyhomoribofuranoses upon Generation of an α-Oxy Radical at C3. Molecules 2020, 25, 2539–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fauster K; Hartl M; Santner T; Aigner M; Kreutz C; Bister K; Ennifar E; Micura R 2′-Azido RNA, A Versatile Tool for Chemical Biology: Synthesis, X-ray Structure, siRNA Applications, Click Labeling. ACS. Chem. Biol 2012, 7, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maag H; Rydzewski RM; McRoberts MJ; Crawford-Ruth D; Verheyden JPH; Prisbe EJ Synthesis and Anti-HIV Activity of 4′-Azido- and 4′-Methoxynucleosides. J. Med. Chem 1992, 35, 1440–1451. [DOI] [PubMed] [Google Scholar]
- 45.Hampton A; Nichol AW, Nucleotides. V. Purine Ribonucleoside 2′,3′-Cyclic Carbonates. Preparation and Use for the Synthesis of 5′-Monosubstituted Nucleosides. Biochemistry 1966, 5, 2076–2082. [DOI] [PubMed] [Google Scholar]
- 46.Wnuk SF; Chowdhury SM; Garcia PI Jr.; Robins MJ Stereo-defined Synthesis of O3′-Labeled Uracil Nucleosides. 3′-[17O]-2′-Azido-2′-deoxyuridine 5′-Diphosphate as a Probe for the Mechanism of Inactivation of Ribonucleotide Reductases. J. Org. Chem 2002, 67, 1816–1819. [DOI] [PubMed] [Google Scholar]
- 47.Li H; Jiang Z; Wang X; Zheng C Synthesis of 2′-Amino-2′-deoxyuridine Modified by 1,8-Naphthalimide. Synth. Commun 2006, 36, 1933–1940. [Google Scholar]
- 48.Gai XS; Fenlon EE; Brewer SH A Sensitive Multispectroscopic Probe for Nucleic Acids. J. Phys. Chem. B 2010, 114, 7958–7966. [DOI] [PubMed] [Google Scholar]
- 49.Khanduri D; Adhikary A; Sevilla MD Highly Oxidizing Excited States of One-Electron-Oxidized Guanine in DNA: Wavelength and pH Dependence. J. Am. Chem. Soc 2011, 133, 4527–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrovici A; Adhikary A; Kumar A; Sevilla MD Presolvated Electron Reaction with Methylacetoacetate: Electron Localization, Proton-Deuteron Exchange, and H-atom Abstraction. Molecules 2014, 19, 13486–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaczmarek R; Ward S; Debnath D; Jacobs T; Stark AD; Korczyński D; Kumar A; Sevilla MD; Denisov SA; Shcherbakov V et al. One Way Traffic: Base-to-backbone Hole Transfer in Nucleoside Phosphorodithioates. Chem. Eur. J 2020, 26, 9495–9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDonald RN; Chowdhury AK; Setser DW Gas-Phase Generation of Phenylnitrene Anion Radical-Proton Affinity and ΔH°f of PhN−• and Its Clustering with ROH Molecules. J. Am. Chem. Soc 1981, 103, 6599–6603. [Google Scholar]
- 53.Živanov S; Ibanescu BC; Paech M; Poffet M; Baettig P; Sergenton A-C; Grimme S; Allan M Dissociative Electron Attachment and Electron Energy-loss Spectra of Phenyl Azide. J. Phys. B: At. Mol. Opt. Phys 2007, 40, 101–109. [Google Scholar]
- 54.Warrier M; Lo MKF; Monbouquette H; Garcia-Garibay M Photocatalytic reduction of aromatic azides to amines using CdS and CdSe nanoparticles. Photochem. Photobiol. Sci 2004, 3, 859–863. [DOI] [PubMed] [Google Scholar]
- 55.Joshi R; Adhikari S; Mukherjee T Redox reactions of 3′-azido-3′-deoxythymidine (AZT) and interaction of its •OH-derived radical with bilirubin and riboflavin: A pulse radiolysis study. Res. Chem. Intermed 2001, 27, 623–634. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.