

Abstract
Background
Residual disease of glioblastoma (GBM) causes recurrence. However, targeting residual cells has failed, due to their inaccessibility and our lack of understanding of their survival mechanisms to radiation therapy. Here we deciphered a residual cell–specific survival mechanism essential for GBM relapse.
Methods
Therapy resistant residual (RR) cells were captured from primary patient samples and cell line models mimicking clinical scenario of radiation resistance. Molecular signaling of resistance in RR cells was identified using RNA sequencing, genetic and pharmacological perturbations, overexpression systems, and molecular and biochemical assays. Findings were validated in patient samples and an orthotopic mouse model.
Results
RR cells form more aggressive tumors than the parental cells in an orthotopic mouse model. Upon radiation-induced damage, RR cells preferentially activated a nonhomologous end joining (NHEJ) repair pathway, upregulating Ku80 and Artemis while downregulating meiotic recombination 11 (Mre11) at protein but not RNA levels. Mechanistically, RR cells upregulate the Su(var)3-9/enhancer-of-zeste/trithorax (SET) domain and mariner transposase fusion gene (SETMAR), mediating high levels of H3K36me2 and global euchromatization. High H3K36me2 leads to efficiently recruiting NHEJ proteins. Conditional knockdown of SETMAR in RR cells induced irreversible senescence partly mediated by reduced H3K36me2. RR cells expressing mutant H3K36A could not retain Ku80 at double-strand breaks, thus compromising NHEJ repair, leading to apoptosis and abrogation of tumorigenicity in vitro and in vivo. Pharmacological inhibition of the NHEJ pathway phenocopied H3K36 mutation effect, confirming dependency of RR cells on the NHEJ pathway for their survival.
Conclusions
We demonstrate that the SETMAR-NHEJ regulatory axis is essential for the survival of clinically relevant radiation RR cells, abrogation of which prevents recurrence in GBM.
Keywords: glioblastoma, NHEJ, radiation resistance, residual disease, SETMAR
Key Points.
Radiation RR GBM cells cause recurrence and poor patient outcome.
Higher NHEJ via increased Ku80 recruitment at double-strand breaks mediates residual cell survival.
SETMAR‒H3K36me2‒NHEJ axis inhibition eliminates residual GBM preventing recurrence.
Importance of the Study.
GBM recurrence remains a clinical challenge due to poor knowledge of survival strategies of therapy refractory RR cells. We developed a radiation resistant cellular model from clinically relevant patient samples that recapitulate clinical scenarios of GBM resistance and recurrence. Here, we demonstrate that post radiotherapy temporal recruitment of NHEJ repair protein Ku80 via SETMAR mediated H3K36me2 is indispensable for DNA repair and survival of RR cells. Accordingly, conditional knockdown of SETMAR specifically in RR cells reduces Ku80 recruitment leading to irreversible senescence of these cells. Furthermore, H3.3K36A mutation or inhibition of the NHEJ repair pathway induces complete death of RR cells in vitro and in vivo in an orthotopic mouse model. This study demonstrates the dependency of therapy refractory RR disease cells on the NHEJ repair pathway and identifies SETMAR and the NHEJ pathway as molecular targets for selective elimination of residual disease, thus preventing GBM relapse.
Glioblastoma (GBM) is a highly malignant brain tumor associated with poor overall survival contributed by the treatment refractory cells. Lack of accessibility of these cells from tumors has prevented our understanding of their survival mechanisms. Using cellular radiation resistant models, we showed that in heterogeneous GBM, a subpopulation of inherently resistant residual (RR) cells survives radiation exposure by overexpressing multiple survival pathways and is significantly associated with poor patient survival.1–4 DNA double-strand breaks (DSBs) are the most toxic lesion generated by ionizing radiation used in cancer treatment5,6 and are inevitably repaired to maintain chromosomal integrity and prevent cell death. DSB repair is brought about by 2 major pathways: homologous recombination (HR) and nonhomologous end joining (NHEJ). Compared with the error-prone NHEJ,7 HR repairs DNA in an error-free manner predominantly in S and G2 phases of the cell cycle.8
Chromatin plays an important role in DNA repair by controlling accessibility of DNA repair factors to the DNA lesions. Histone methylations including H3K4me2, H3K36me2, H3K79me2, H3K9me2, and H3K27me2 have been associated with the transcription of active euchromatin, transcriptional repression, DNA repair (via NHEJ pathway), and recombination.9–13 However, the above-mentioned modulations of DNA repair are very context dependent. Therefore, here we aimed to investigate the regulation of DSB repair in radiation resistant cells of GBM. We demonstrate the dependency of RR cells on the NHEJ repair pathway. Mechanistically, we show that following radiation exposure, RR cells undergo global chromatin decompaction mediated by SETMAR, a histone methyltransferase that induces histone methylations H3K36me2 and H3K4me2 to facilitate efficient NHEJ repair via enhanced recruitment of NHEJ repair proteins. Mutant H3K36 and pharmacological inhibition of the NHEJ pathway induces apoptosis, while genetic knockdown of Su(var)3-9/enhancer-of-zeste/ trithorax (SET) domain and mariner transposase fusion gene (SETMAR) causes irreversible senescence in RR cells, thus preventing recurrence in GBM.
Materials and Methods
Detailed methods are provided in the Supplementary Material.
Statistical Methods
In Figures 1–6, all data are represented as means ± standard error means (SEMs). P-value of ≤0.05 in a paired 2-sided nonparametric t-test was used to test for statistically significant differences. Results in bar and line graphs are the composite data from 3 independent experiments (mean ± SEM); **P ≤ 0.01; ***P ≤ 0.001.
Fig. 1.

Residual cells show euchromatization and tumorigenic in vivo. (A) Schematic representation of the in vitro cellular model developed using primary brain tumor samples and cell lines. (B) CT images (upper panel) of mouse brain orthotopically injected with parent and RR cells. 3D reconstruction (lower panel) of the CT images. Graph represents quantitation of tumor volume as calculated from CT images from mice (n = 3). (C) Immunofluorescence of gamma-H2AX (green) and nuclei counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) in U87MG and SF268 cells at different time points as indicated post irradiation. NR, non-radiated. Graphs represent % of cells with foci from each population (scale bar = 5 µm). (D) Transmission electron micrograph of control and RR cells of U87MG and SF268 showing euchromatin and heterochromatin (dark staining). Bar graph quantifies % heterochromatin from electron microscope images. (E) Immunofluorescence of HP1 alpha (green) in parent and RR cells of indicated cell lines. Nuclei were counter stained with DAPI (blue). Scale bar 5 μm. Bar graph shows mean fluorescence intensity of HP1 alpha staining in at least 50 cells. Intensities were calculated by ImageJ software. All data are represented as means ± SEMs. P-value of ≤0.05 in a paired 2-sided nonparametric t-test was used to test for statistically significant differences. Results in bar graph are the composite data from 3 independent experiments (mean ± SEM); *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Fig. 2.

Residual cells show preferential recruitment and activation of NHEJ repair. (A) Representative immunofluorescence images of parent and RR cells stained for Ku80 and pBRCA1 and nuclei counterstained with DAPI in U87MG, SF268, and primary cultures of patient samples (PS). Graph represents % of cells with Ku80 and pBRCA1 foci from each sample as indicated. (B) Western blot for pBRCA1, total BRCA1, and Ku80 in parent and RR cells of indicated cell lines and patient samples. Actin was used as loading control. Graphs shows the densitometric analysis for Ku80 and pBRCA1 proteins in parent and RR cells lines. (C) Western blot for NBS1, Mre11, and Artemis in parent and RR cells of U87MG and SF268 cell lines. Actin was used as loading control. Graphs shows the densitometric analysis for NBS1, Mre11, and Artemis proteins in parent and RR cells lines. (D) HR and NHEJ vector reactivation assay performed in parent and RR cells of U87MG and SF268 cells. Upper panel shows the transfection of HR and NHEJ vectors. Flow cytometry for the percentage of GFP and TdRed cells 72 h post transfection. Percentage of dual positive and GFP positive cells is indicated in the respective quadrants. (Scale bar = 5 µm.) All data are represented as means ± SEMs. P-value of ≤0.05 in a paired 2-sided nonparametric t-test was used to test for statistically significant differences. Results in bar graph are the composite data from 3 independent experiments (mean ± SEM); *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Fig. 3.

Residual cells show high levels of H3K36me2 and upregulation of SETMAR. (A) Western blot for histone modifications (H3K36me2, H3K4me2, H3K9me2, H3K27me2, and H4K79me2) in parent and RR cells of U87MG and SF268 cell lines. Total H4 was used as loading control. Graphs show the results of Wilcoxon matched-pair signed rank test between parent and RR population of respective cell lines for H3K36me2, H3K4me2, H3K9me2, H3K27me2, and H3K79me2. (B) Western blot for H3K36me2, H3K4me2 in parent, and RR cells of patient samples (PS1–PS8). Total H4 was used as loading control. Graphs shows the results of Wilcoxon matched-pair signed rank test between parent and RR population of respective primary cell lines for H3K36me2 and H3K4me2. (C, D) Graphs represent fold change differences in the expression of SETMAR and NSD1 transcripts in RR cells as compared with the corresponding parent cells of U87MG and SF268 and patient samples (PS1–PS8) determined using quantitative PCR analysis. All data are represented as means ± SEMs. P-value of ≤0.05 in a paired 2-sided nonparametric t-test was used to test for statistically significant differences. Results in bar graphs (C–D) are the composite data from 3 independent experiments (mean ± SEM); *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Fig. 4.

SETMAR knockdown induce premature senescence in residual cells. (A–D) Line graphs show the growth of parent and RR cells of U87MG and SF268 cells stably transfected with scrambled (Scr), shRNA, or siRNA against SETMAR. Results in the line graph are the composite data from 3 independent experiments. (E, F) Senescence-associated β-galactosidase staining at days 12 and 20 of the RR cells with shRNA and days 10 and 14 of the RR cells treated with siRNA against SETMAR. (G) Western blot for H3K36me2 in parent and RR cells of U87MG and SF268 treated with scrambled, sh1, and siRNA against SETMAR. Total H3 was used as loading control. (H) Representative immunofluorescence images of parent and RR cells treated with scrambled, shRNA, and siRNA against SETMAR stained for Ku80 (green) and nuclei counterstained with DAPI (blue). Graphs represent the quantitation of immunofluorescence of Ku80. Scale bar 10 μm. Results in the bar graph are the composite data from 3 independent experiments.
Fig. 5.

H3K36me2 is essential for the recruitment of NHEJ protein Ku80. (A) Western blot for H3K36me2 in parent and RR cells of U87MG and SF268 with wild-type (wt) H3K36 or mutant (mt) H3K36A. Total H3 was used as loading control. (B) Immunofluorescence staining for H3K36me2 (green) in parent and RR cells of U87MG and SF268 with wt H3K36 or mt H3K36A. Nuclei were counterstained with DAPI (blue). (C, D) Western blot for Ku80, pBRCA1 and total BRCA1 in parent cells at different time points (as indicated) post irradiation and in RR cells of U87MG and SF268 with wt H3K36 or mt H3K36A. Actin was used as loading control. (E, F) Line graphs show the growth of wt and mt cells of U87MG and SF268 cells before and after irradiation. Results in line graph are the composite data from 3 independent experiments. (G) CT images of mice brain with wt or mt U87MG cells. Scale bar 10 μm. Scale bar 5 μm.
Fig. 6.

Inhibition of the NHEJ repair pathway abolishes the tumorigenic potential of residual cells in vitro and in vivo. (A, B) Immunofluorescence for Ku80 and pBRCA1 (green) in parent cells at different time points (as indicated) post irradiation and in RR cells of U87MG and SF268 with wild-type (wt) H3K36 or mutant (mt) H3K36A. Nuclei were counterstained with DAPI (blue). Graphs show the quantitation of immunofluorescence data where the number of cells with foci (Ku80 or BRCA1) are plotted against the time as indicated. (C, D) Representative images of clonogenic assay performed with parent and RR cells of U87MG and SF268 treated with NHEJ inhibitor (NU7026). Bar graph shows the quantitation of the clonogenic assay where survival fractions of different cells with different treatments as indicated are plotted. (E) Representative bioluminescence images after orthotopic injection of U87MG-Luciferase labeled parent (P) and RR cells with vehicle control (DMSO) and NHEJ inhibitor (NU7026). Mice were images at the indicated times post injection. (F) Line graph represents bioluminescence signal at day 13 and day 26 post injection in mice injected with parent and RR cells with DMSO and NU7026. At least n = 3 mice were used per group. (G) Model showing new insights into the complex crosstalk between pathways governing survival and proliferation, senescence, and apoptosis in the innately radiation RR GBM cells. Scale bar 5 μm. All data are represented as means ± SEMs. P-value of ≤0.05 in a paired 2-sided nonparametric t-test was used to test for statistically significant differences. Results in bar graph are the composite data from 3 independent experiments (mean ± SEM); *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Results
Residual Cells Display Residual DNA Damage and Euchromatization
Here, we captured RR cells from 8 primary cultures developed from naïve GBM patient samples, as shown in Figure 1A and reported previously,1 and 2 cell lines (U87MG and SF268). Alpha thalassemia/mental retardation syndrome X-linked (ATRX), p53, and O6-methylguanine-DNA methyltransferase promoter methylation status of cell lines14–17 and patient samples are given in Supplementary Table 2. Similar results were obtained with fractionated dose of radiation (Supplementary Figure 1A–D) and when the cells were cultured as glioma spheres (Supplementary Figure 2A–C). Using the orthotopic mouse model, we observed that the RR cells from U87MG and patient derived culture display higher tumorigenic potential than the parental cells as determined by the tumor volume calculated from the microCT images taken at days 32 and 13, respectively, post injection (Figure 1B and Supplementary Figure 2D). RR cells not only showed tumorigenic potential in vivo, they formed more aggressive tumors with higher volume (41 ± 7.09 mm3) compared with the parent cells (20.83 ± 6.043 mm3) (Figure 1B). Further, we wanted to confirm if the residual cells escape radiation-induced DNA damage or are able to survive the damage. As expected, residual cells showed significantly high DNA damage as assessed by gamma–H2A histone family member X positivity and comet assay (Figure 1C and Supplementary Figure 3). Interestingly, electron microscopy revealed increased euchromatin in RR cells, which was further confirmed by reduction in the intensity of heterochromatin protein 1 (HP1) alpha immunostaining in RR cells compared with the parent population (Figure 1D, E).
RR Cells Undergo NHEJ Driven DSB Repair
Since the chromatin organization is known to influence DNA repair pathways by regulating the recruitment of various DNA repair proteins, we examined the expression and recruitment of Ku80 (NHEJ) and phosphorylated breast cancer 1 (pBRCA1) (HR) proteins in the RR cells. The radiation resistant cells generated from cell lines and patient-derived primary cultures (n = 5) demonstrated higher recruitment of Ku80 compared with pBRCA1 (Figure 2A). When checked for the total protein levels, interestingly RR cells from U87MG, SF268, and patient samples also exhibited higher protein levels of Ku80 compared with pBRCA1 (Figure 2B). Additionally, Artemis, another NHEJ repair pathway protein, was also upregulated while meiotic recombination 11 (Mre11) (HR pathway protein) was downregulated in RR cells compared with parent cells and no differential expression of Nijmegen breakage syndrome 1 was observed (Figure 2C), indicating that RR cells preferentially upregulate NHEJ proteins. In an independent experiment, we performed total RNA sequencing of RR cells from U87MG, SF268, and patient samples 1 and 2 (PS1, PS2). An unbiased analysis of transcripts of DNA repair pathway genes showed no particular pattern or preference for differential expression of NHEJ or HR pathway genes in RR cells, suggesting that it is at the protein levels that the DNA damage response (DDR) repair proteins were regulated (Supplementary Figure 4). These results indicate that RR cells preferentially stabilize NHEJ but not HR pathway proteins for the repair of their DSBs.
To ascertain that higher expression and recruitment of NHEJ protein translates into higher NHEJ repair efficiency in RR cells, we measured HR and NHEJ activity in vivo using I-SceI based green fluorescent protein (GFP) assay.18 Average NHEJ repair efficiency in U87MG parent was 31.42%, while in RR cells it was 51.09%. In SF268 parent cells, it was observed to be 30.74%, while in RR cells it was 53.74%. No significant difference in the HR repair efficiency was observed in parent and RR cells, which was in the range of 25–33%. Indeed, we observed that RR cells showed enhanced NHEJ repair compared with the HR pathway (Figure 2D).
RR Cells Show Higher H3K36me2 and H3K4me2 Modifications
We then wanted to understand what could influence the DDR pathway choice in residual cells. Histone methylations prominently influence the choice of NHEJ or HR repair pathway as well as chromatin architecture.19,20 Thus, we screened histone methylations (H3K36me2, H3K4me2, H3K9me2, H3K27me2, and H3K79me2) with reported DDR association.21,22 Data from parent and RR cells of cell lines revealed that H3K36me2 and H3K4me2 were upregulated in RR cells (Figure 3A). Interestingly, these histone marks are involved in mediating euchromatization, which was also observed in RR cells (Figure 1D). Furthermore, H3K9me2 levels were lower in RR cells, a histone mark that interacts with HP1 to induce heterochromatin, again corroborating our earlier findings of RR cell euchromatization. H3K27me2 remained unchanged, while H3K79me2 showed variable results. Since we found enhanced H3K36me2 and H3K4me2 in the RR cells, we went ahead to examine the status of these modifications in the RR cells generated from primary patient cultures (N = 8). As shown in Figure 3B, indeed, 6 out of the 8 patient samples (75%) showed higher H3K36me2, while dimethylation of H3K4 was observed to be high in 4/8 samples (50%). We also evaluated expression of some of the histone trimethylations involved in DDR such as H3K4me3, H3K9me3, H3K27me3, H3K79me3, and H3K36me3 in the parent and RR cells of U87MG and SF268 cells. However, no significant differences were seen among these histone marks (Supplementary Figure 4B).
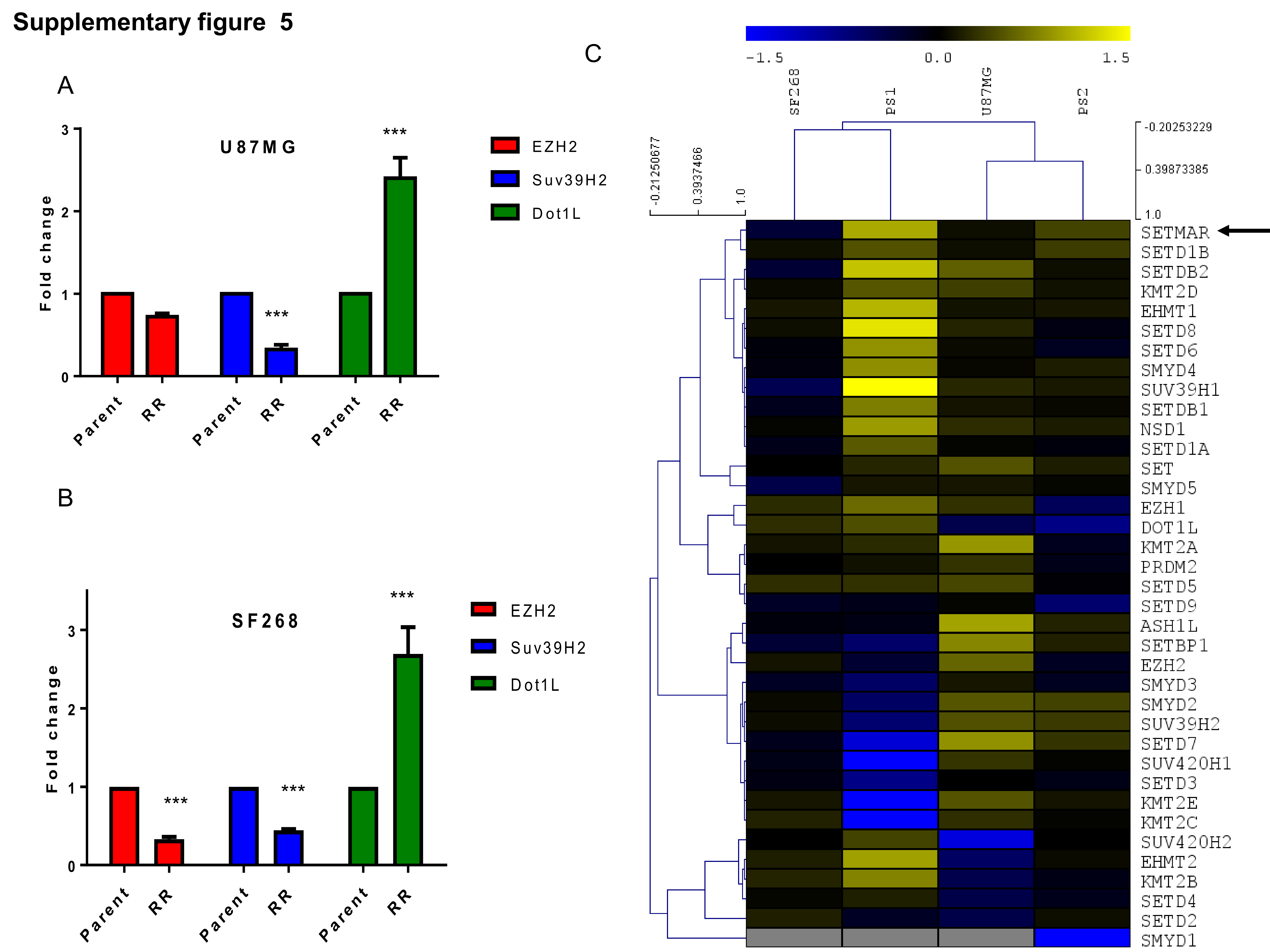
Independently, we also screened parent and RR cells from cell lines and 8 primary cultures of patient samples for the expression of methyltransferases that mediate H3K36 and H3K4 dimethylations (SETMAR and nuclear receptor binding SET domain protein 1 [NSD1]). Transcript levels of SETMAR as determined using quantitative real-time PCR were found to be significantly upregulated in 7/10 RR cells (Figure 3C), correlating with the expression of H3K36me2 (Figure 3B), while the transcript levels of NSD1 were upregulated in 2/10 samples (Figure 3D) compared with the parent cells. We also analyzed the expression of other methyltransferases (enhancer of zeste homolog 2 [EZH2], suppressor of variegation 3-9 homolog 2 [SUV39H2], and disruptor of telomeric silencing 1-like [DoT1L]). Of these, EZH2 and SUV39H2 were downregulated in the RR cell lines. Although DoT1L transcripts were significantly upregulated, the corresponding differential change in the levels of major histone modification (H3K79) mediated by DoT1L (Supplementary Figure 4A and B) was not observed. Furthermore, among all the epigenetic factors probed in the transcriptome dataset, 2-way unsupervised clustering based on Pearson coefficient criteria identified SETMAR as the top-ranked gene based on the average upregulation among PS1 and PS2 samples (Supplementary Figure 5C).
SETMAR Knockdown Induces Premature Senescence in RR Cells
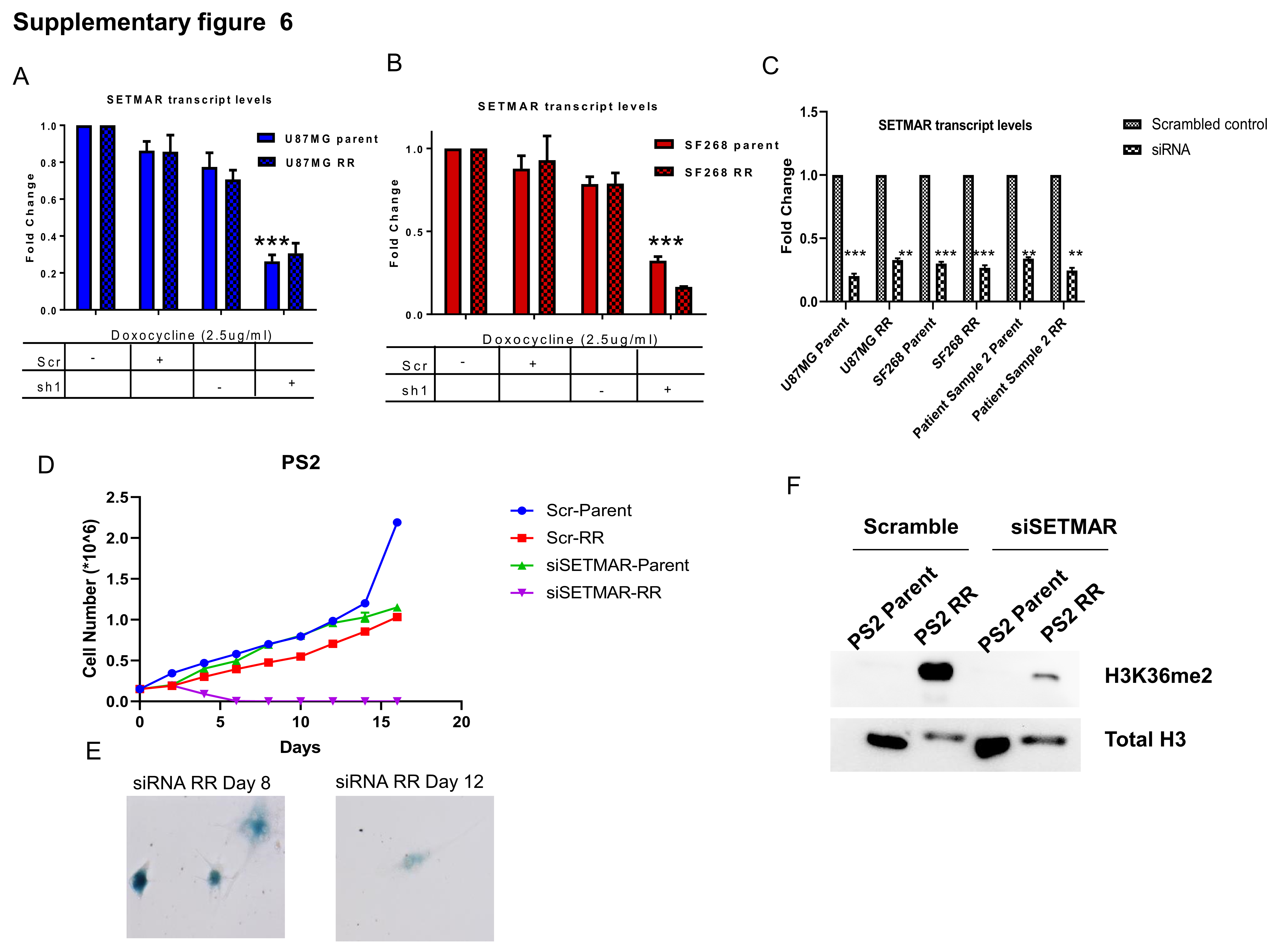
Since significant number of samples overexpressed SETMAR transcripts and corresponding histone modification of H3K36me2 in RR cells, we wanted to investigate the role of SETMAR and H3K36me2 in RR cells. For this, we adopted 2 strategies: (i) knockdown of SETMAR in RR cells and (ii) mutation of lysine 36 residue to alanine in H3.3. SETMAR was knocked down in U87MG and SF268 by short hairpin (sh)RNAs (sh1) (cloned under the tetracycline-inducible promoter) and mixture of 3 small interfering (si)RNAs. Upon induction with 2.5 µg/mL doxycycline, sh1, showed 70–76% and 82–90% reduction of SETMAR transcripts and siRNA showed about 70% reduction in SETMAR transcripts in parent and RR cells, respectively (Supplementary Figure 6A–C). As shown in Figure 4A, B, shRNA (sh1) or siRNA (mixture of 3 siRNAs) mediated knockdown of SETMAR initially induced cell death of RR cells; however, eventually (12 days post induction of shRNA and 10-day post siRNA treatment) the cells ceased to grow and became senescent (Figure 4E, F). Importantly, SETMAR knockdown did not alter the growth pattern of parent cells but specifically halted the proliferation of RR cells, highlighting the dependency of RR cells on SETMAR to escape from senescence. A significant reduction in the H3K36me2 was observed in SETMAR knockdown cells (Figure 4G). Similar results were found in primary cultures of patient sample (Supplementary Figure 6D–F). Since SETMAR mediated H3K36me2 modification promotes DSB repair via the NHEJ pathway,23 and we also observed the NHEJ pathway preference in RR cells, we asked whether SETMAR is involved in aiding the NHEJ pathway in the radiation RR cells of GBM. For this, we examined Ku80 recruitment in RR cells with SETMAR knockdown and found, indeed, that SETMAR knockdown significantly reduced Ku80 recruitment (Figure 4H), demonstrating that SETMAR knockdown reduces H3K36me2 levels and recruitment of NHEJ proteins, and highlighting that SETMAR knockdown is sufficient to render RR cells senescent, so as not to induce apoptosis.
H3K36me2 Is Essential for Ku80 Retention and Residual Cell Survival
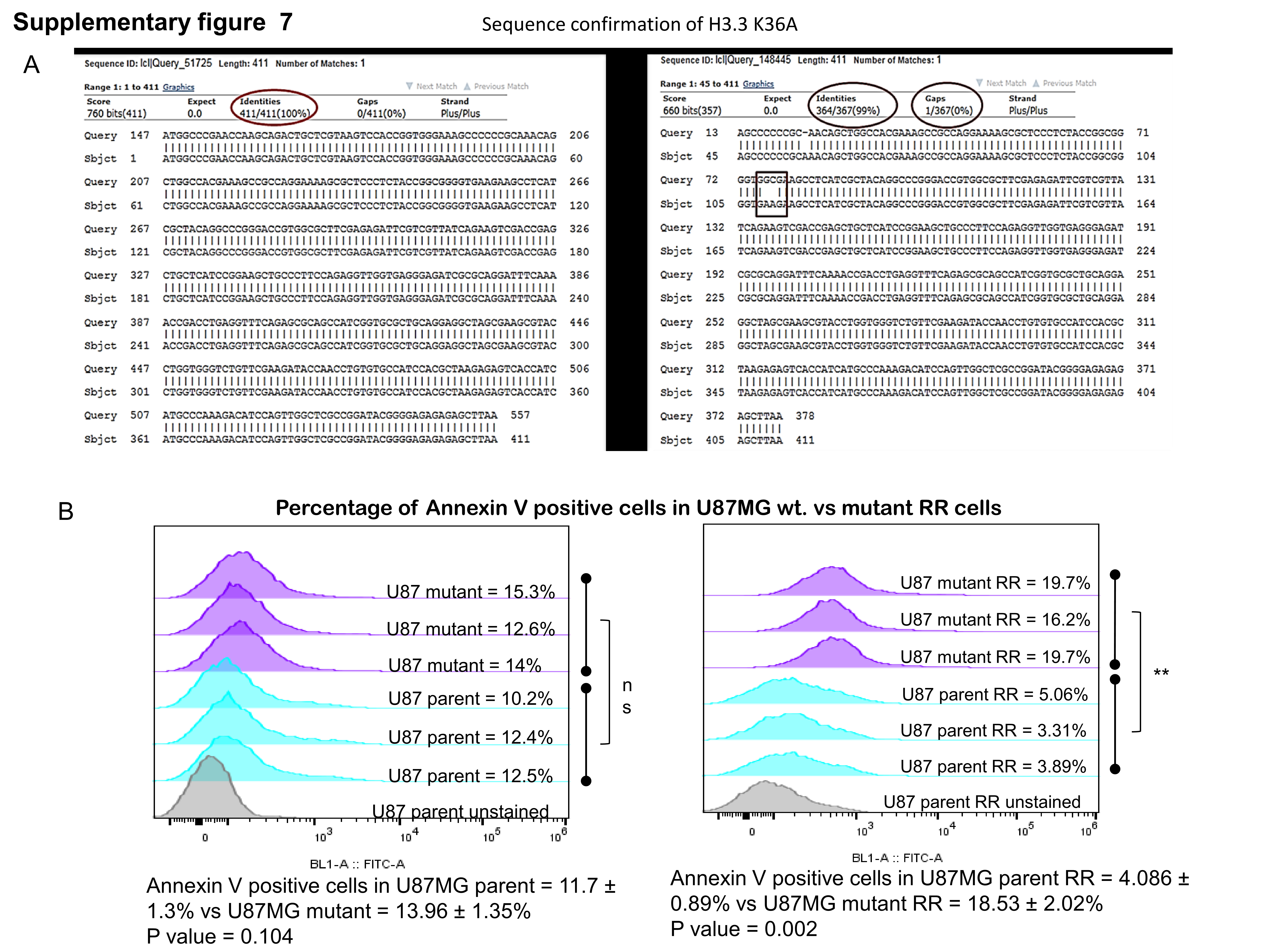
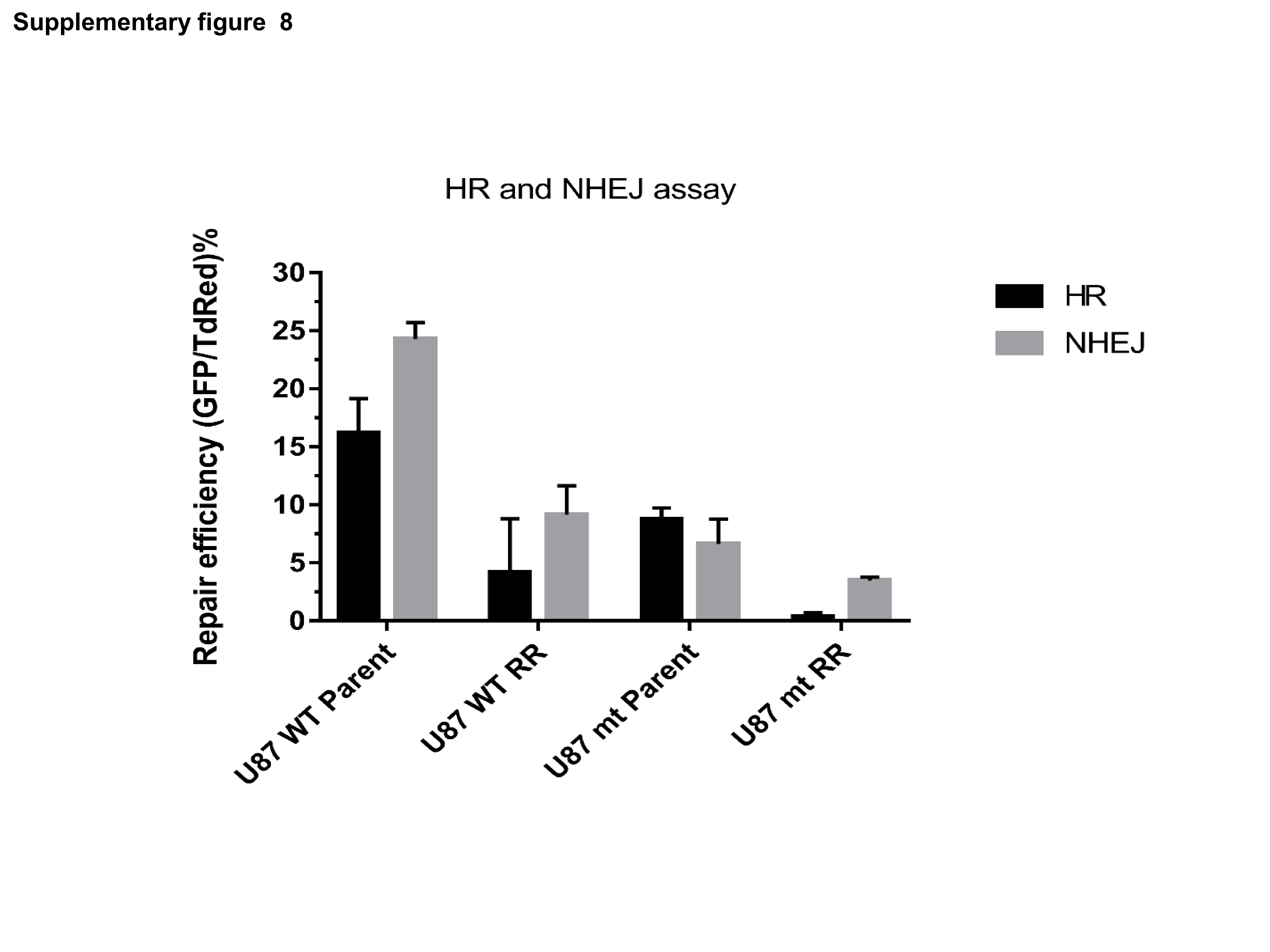
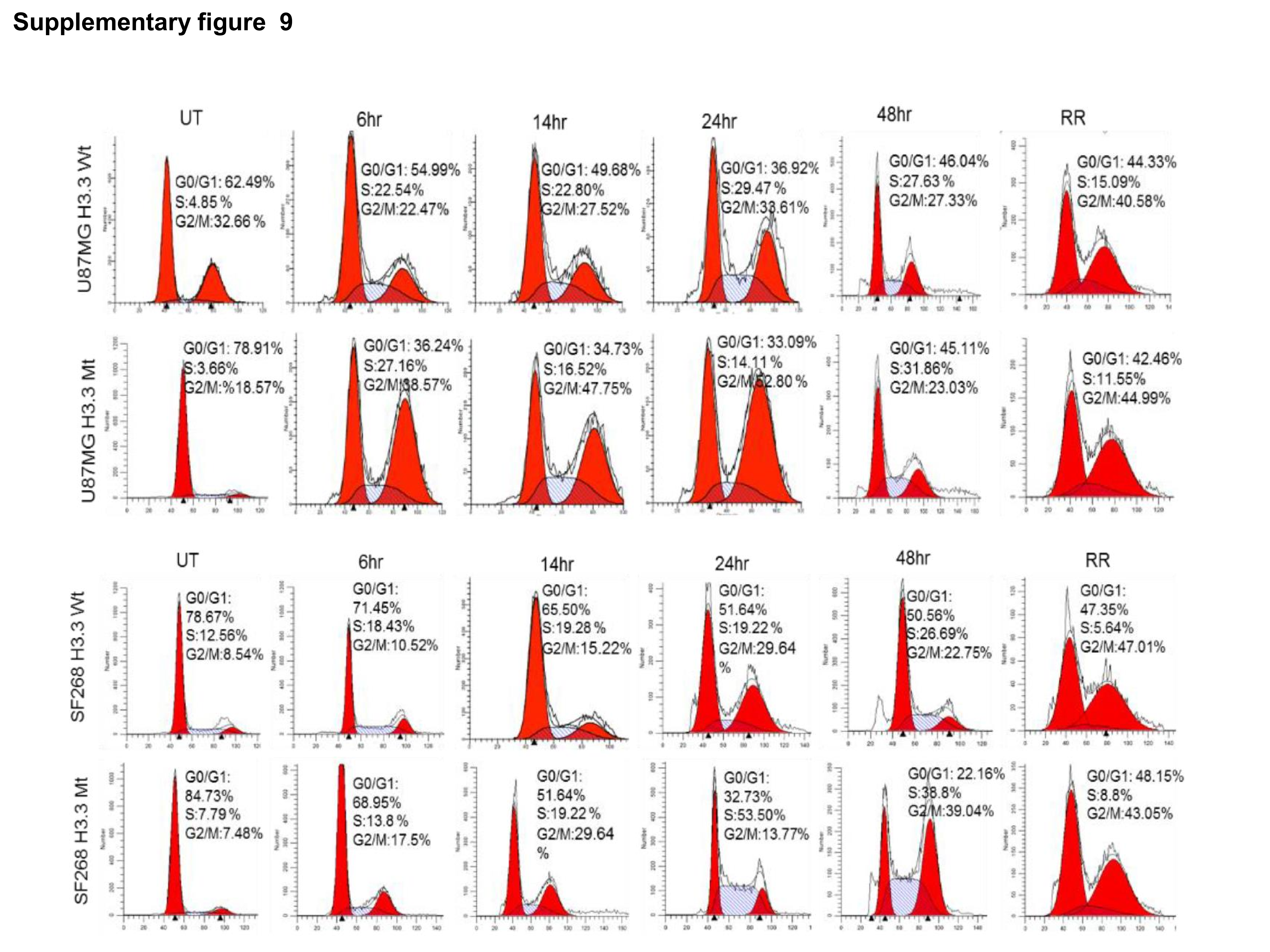
To understand the direct effect of H3K36me2, we mutated H3K36 to alanine (Supplementary Figure 7A) and transfected U87MG and SF268 cells with construct. Western blot analysis showed that RR cells with mutant H3K36 showed no or very low levels of H3K36me2 modification, which was further confirmed by immunofluorescence for H3K36me2 (Figure 5A, B). Unexpectedly, the parent cells expressing mutant H3.3 showed reduced expression of Ku80 compared with cells expressing wild type H3.3, suggesting H3K36me2 involvement in regulation of Ku80 protein levels in the cells with high stress; however, these results require further investigation. Accordingly, there was marginal expression of Ku80 in RR cells expressing H3.3K36 mutation. Furthermore, compared with the wild-type cells, cells expressing mutant H3.3 exhibited loss of total BRCA1 expression at later time points post radiation (Figure 5C, D). Accordingly, in RR cells expressing mutant H3.3 compared with wild-type H3.3, there was reduced NHEJ or HR activity as assessed by NHEJ and HR vectors (Supplementary Figure 8). DNA repair protein levels are cell cycle dependent, therefore we analyzed cell cycle distribution of cells expressing mutant H3. We found similar distribution of cells in S and G2-M population in both wild-type and mutant expressing cells (Supplementary Figure 9), ruling out the cell cycle changes as the cause of differential protein levels in wild-type and mutant cells. The observed differences could be due to the continuous apoptosis in the mutant cells or regulation via H3K36me2; however, this requires experimental confirmation.
H3K36me2 has been shown to be essential for the recruitment of Ku80.23 In order to examine this, parent cells were treated with a lethal dose of ionizing radiation (IR) and the recruitment of Ku80 as well as pBRCA1 was determined at different time points till the generation of RR cells. No difference was seen in the initial (till 14 h post radiation) recruitment of Ku80 in H3.3 wild-type and H3.3 mutant expressing cells. However, at a later time point post radiation, the retention of Ku80 was affected specifically in mutant H3.3 expressing cells. Further, no difference in pBRCA1 recruitment kinetics was observed in mutant and wild-type expressing cells at DSBs (Figure 6A, B). Together, these data demonstrate that H3K36me2 is required for retention of Ku80 at the site of DSBs generated post radiation. Since the H3.3 mutant cells could not recruit DDR proteins at DSBs, we examined the growth kinetics of the non-irradiated and irradiated wild and mutant H3.3 expressing cells. As shown in Figure 5E, F, cells expressing H3K36A showed reduced cell proliferation. To understand if the RR cells with mutant H3.3 were undergoing apoptosis, annexin V staining was performed in RR cells of U87MG cell with wild-type and mutant H3.3. As seen from Supplementary Figure 7B, mutant RR cells showed significant increase in apoptosis compared with cells with wild-type H3.3. Furthermore, orthotopic injections of wild-type and cells containing mutant H3K36A in mouse brain showed that the wild-type cells formed large tumors at day 26 post injection, while no tumors were seen with the cells expressing mutant H3K36 (Figure 5G), thus confirming that H3K36me2 is required for survival and relapse of residual cell disease in vitro and in vivo.
We also analyzed the levels of SETMAR transcripts, H3K36me2 modification, and recruitment of Ku80 before and after irradiation in the newly diagnosed primary (PS A and PS B) and recurrent (RS A and RS B) patient samples. Although there was increase in the SETMAR transcript levels and H3K36me2 modification post irradiation in primary and recurrent samples, the differences were not significant (Supplementary Figure 10A, B). We did not find any differences in the Ku80 recruitment in primary and recurrent cells post irradiation (Supplementary Figure 10C).
Residual Cells Dependent on NHEJ Repair for Survival and Relapse In Vitro and In Vivo
We reasoned if the apoptosis induced in H3K36 mutant cells was due to impaired NHEJ repair, then the inhibition of the NHEJ repair pathway should phenocopy the effect of H3K36 mutation in GBM cells. To test this hypothesis, we determined the clonogenic capacity of RR cells treated with 10 μM NHEJ inhibitor (NU7026). Similar to mutant RR cells, clonal ability of RR was completely abolished with NHEJ inhibitor (Figure 6C, D). To confirm the effect of NU7026 treatment on the tumorigenic potential of RR cells in vivo, we treated U87MG luciferase-expressing cells with the NU7026 inhibitor (10 µM) for 8 days following the formation of the RR population. Appropriate controls (untreated, vehicle treated, radiation alone, and inhibitor alone treated cells) were taken at the same time. Cells were injected intracranially in nude mice and tumor growth was monitored by bioluminescence. As seen from the graph, tumorigenicity of parent cells treated with vehicle (dimethyl sulfoxide [DMSO]) and NU7026 was not significantly different compared with the untreated parent cells (Figure 6E, F). Also, untreated and vehicle (DMSO) treated RR cells showed similar tumorigenicity. However, RR cells treated with NU7026 showed significantly reduced tumors, confirming the dependency of RR cells to survive and form tumors but not of parent cells on the NHEJ repair pathway in vivo.
Discussion
GBM is the most common and lethal primary brain tumor. Despite multimodal therapy, recurrence remains inevitable with the dismal median survival of 14.5 months. Although clinical importance of the residual disease is known, molecular mechanisms leading to their survival and recurrence remain elusive. This is primarily due to inaccessibility of residual disease cells as biopsies are not available post therapy. To overcome this problem and to study the residual disease we have previously developed a cellular model of therapy resistance and recurrence from fresh primary naïve GBM patient samples and cell lines that recapitulate the clinical scenario of disease recurrence, thus providing us with the opportunity to capture residual cells.
We analyzed residual cells generated from primary cultures of patient samples and cell lines after subjecting them to radiation as previously reported.1 As expected, residual cells had significantly high levels of DNA DSBs. However, importantly, unlike the bulk tumor cells, residual cells not only survived DNA damage but recurred in vitro as well as in vivo. We thus wanted to understand how residual cells repair their DSBs. Several reports suggest heterochromatin architecture inhibits DNA repair processes,24,25 thus we hypothesized that global chromatin decompaction would make damaged DNA sites accessible to repair proteins. Indeed, RR cells exhibit global decompaction and higher recruit of NHEJ protein Ku80 compared with HR protein BRCA1. The NHEJ pathway is shown to be a highly deleterious mode of DNA repair process that enhances genomic instability, eventually leading to carcinogenesis.26 Indeed, in multiple cancer types, NHEJ factors are observed to be altered.27–30 Additionally, overexpression or enhanced NHEJ repair activity has been associated with therapy resistance, including GBM.31–34
Furthermore, these cells displayed high levels of H3K36 and H3K4 dimethylation and overexpression of their corresponding histone methylase enzyme—SETMAR. These findings were specific to radiation and not a generic stress response as shown in Supplementary Figure 11 . Temozolomide treatment (10 μM) alone showed delayed cell growth in U87MG and PS1, but the effects were significant only with radiation alone and temozolomide + radiation. These combinations also showed significantly higher number of multinucleated giant cell formation (Supplementary Figure 11A–D). Further, only IR alone or temozolomide + IR treatment enhanced SETMAR transcript levels; H3K36me2, Ku80, and, as expected, pBRCA1 were not activated even after the combination treatment (Supplementary Figure 11E–H).
High H3K36me2 is shown to initiate oncogenic programming influencing myeloma pathogenesis35 and is associated with poorly differentiated squamous cell carcinoma of the head and neck.36 SETMAR is overexpressed in acute myeloid leukemia and breast cancer, mediating resistance to the DNA damaging drug etoposide and adriamycin, respectively.37,38 Additionally, SETMAR mediates dimethylation of H3K36 adjacent to DSB site, recruiting and retaining Ku and Mre11-Rad50-NBS1 complex at DSBs leading to enhanced DSB repair via the NHEJ pathway.23 Accordingly, upon SETMAR knockdown in RR cells, we observed significant reduction in H3K36me2 levels. However, complete loss of H3K36me2 was not seen possible because of incomplete SETMAR knockdown or the redundancy of other methyltransferases with similar function. Furthermore, knockdown of SETMAR in RR cells leads to reduced Ku80 recruitment and induction of senescence. It has been previously shown that functions of DNA repair proteins are controlled by epigenetic processes including DNA methylation and histone modifications.39 Importantly, SETMAR knockdown did not affect cell proliferation of parent cells, highlighting the dependency of residual cells on SETMAR for DNA repair and regrowth. Loss of H3K36me2 in H3K36A-expressing cells completely abolished the growth capacity of residual cells, causing apoptosis in vitro and in vivo similar to the effect observed with NHEJ inhibitor.
This is the first study providing insights into the crosstalk between pathways governing proliferation, senescence, and apoptosis in the innately radiation resistant residual GBM cells (Figure 6G). We show that radiation RR disease cells but not bulk GBM cells preferentially undergo NHEJ repair, regulated by the SETMAR-mediated H3K36me2. Importantly, we demonstrate that targeting SETMAR and the NHEJ repair pathway can prevent GBM recurrence by inducing premature senescence and apoptosis, respectively, in the RR cells. Thus, drugs targeting SETMAR may enable specific eradication of residual disease cells without impacting normal cells.
Funding
This work was supported by Department of Biotechnology (BT/PR4020/MED/30/792/2012) to S.D. E.K, J.N., and S.S. are CSIR fellows. A.G. is Department of Science and Technology-Science and Engineering Research Board National Post Doctoral Fellow, (PDF/2016/00158). S.V.M. is a DBT fellow.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
HR and NHEJ reporter constructs were a kind gift from Dr Vera Gorbunova (University of Rochester, New York). We also thank Ketaki Patkar for her help with immunohistochemistry.
Conflict of interest statement. The authors declare no potential conflicts of interest.
Authorship statement. S.D and E.K conceptualized and designed experiments. E.K, J.N, S.S, P.I, A.G, N.G, S. Desai, S.V.M, A.A, M.K, D.S, and R.T performed experiments. S.D and E.K analyzed data. A.D and A.M provided reagents. S.D, E.K. prepared manuscript. S.D supervised research.
References
- 1. Kaur E, Rajendra J, Jadhav S, et al. Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis. 2015;36(6):685–695. [DOI] [PubMed] [Google Scholar]
- 2. Kaur E, Sahu A, Hole AR, et al. Unique spectral markers discern recurrent Glioblastoma cells from heterogeneous parent population. Sci Rep. 2016;6:26538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rajendra J, Datta KK, Ud Din Farooqee SB, et al. Enhanced proteasomal activity is essential for long term survival and recurrence of innately radiation resistant residual glioblastoma cells. Oncotarget. 2018;9(45):27667–27681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaur E, Goda JS, Ghorai A, et al. Molecular features unique to glioblastoma radiation resistant residual cells may affect patient outcome - a short report. Cell Oncol (Dordr). 2019;42(1):107–116. [DOI] [PubMed] [Google Scholar]
- 5. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4): 497–510. [DOI] [PubMed] [Google Scholar]
- 6. Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol. 2014;6(9):a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. [DOI] [PubMed] [Google Scholar]
- 9. Conrad T, Akhtar A. Dosage compensation in Drosophila melanogaster: epigenetic fine-tuning of chromosome-wide transcription. Nat Rev. Genet. 2011;13(2):123–134. [DOI] [PubMed] [Google Scholar]
- 10. Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev. Genet. 2011;13(2):97–109. [DOI] [PubMed] [Google Scholar]
- 11. Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25(13):1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferrari KJ, Scelfo A, Jammula S, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. 2014;53(1):49–62. [DOI] [PubMed] [Google Scholar]
- 13. Peng JC, Karpen GH. Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS Genet. 2009;5(3):e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Chen JX, Liu YH, You C, Mao Q. Mutant TP53 enhances the resistance of glioblastoma cells to temozolomide by up-regulating O(6)-methylguanine DNA-methyltransferase. Neurol Sci. 2013;34(8):1421–1428. [DOI] [PubMed] [Google Scholar]
- 15. Li F, Deng Z, Zhang L, et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 2019;38(19):e96659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mashima T, Oh-hara T, Sato S, et al. p53-defective tumors with a functional apoptosome-mediated pathway: a new therapeutic target. J Natl Cancer Inst. 2005;97(10):765–777. [DOI] [PubMed] [Google Scholar]
- 17. Gaspar N, Marshall L, Perryman L, et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010;70(22):9243–9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp. 2010;2002:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faucher D, Wellinger RJ. Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Gen. 2010;6(8) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J Cell Biol. 2017;216(7):1959–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chatterjee S, Senapati P, Kundu TK. Post-translational modifications of lysine in DNA-damage repair. Essays Biochem. 2012;52:93–111. [DOI] [PubMed] [Google Scholar]
- 22. Hunt CR, Ramnarain D, Horikoshi N, et al. Histone modifications and DNA double-strand break repair after exposure to ionizing radiations. Radiat Res. 2013;179(4):383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fnu S, Williamson EA, De Haro LP, et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci U S A. 2011;108(2):540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kruhlak MJ, Celeste A, Dellaire G, et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172(6):823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murga M, Jaco I, Fan Y, et al. Global chromatin compaction limits the strength of the DNA damage response. J Cell Biol. 2007;178(7):1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Srivastava M, Raghavan SC. DNA double-strand break repair inhibitors as cancer therapeutics. Chem Biol. 2015;22(1):17–29. [DOI] [PubMed] [Google Scholar]
- 27. Beskow C, Skikuniene J, Holgersson A, et al. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br J Cancer. 2009;101(5):816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pucci S, Mazzarelli P, Rabitti C, et al. Tumor specific modulation of KU70/80 DNA binding activity in breast and bladder human tumor biopsies. Oncogene. 2001;20(6):739–747. [DOI] [PubMed] [Google Scholar]
- 29. Shintani S, Mihara M, Li C, et al. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003;94(10):894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sirzén F, Nilsson A, Zhivotovsky B, Lewensohn R. DNA-dependent protein kinase content and activity in lung carcinoma cell lines: correlation with intrinsic radiosensitivity. Eur J Cancer. 1999;35(1):111–116. [DOI] [PubMed] [Google Scholar]
- 31. Zhang T, Chai J, Chi L. Induction Of XLF And 53BP1 expression is associated with temozolomide resistance in glioblastoma cells. Onco Targets Ther. 2019;12:10139–10151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst). 2004;3(8-9):909–918. [DOI] [PubMed] [Google Scholar]
- 33. Frit P, Canitrot Y, Muller C, et al. Cross-resistance to ionizing radiation in a murine leukemic cell line resistant to cis-dichlorodiammineplatinum(II): role of Ku autoantigen. Mol Pharmacol. 1999;56(1):141–146. [DOI] [PubMed] [Google Scholar]
- 34. Yang S, Wang XQ. XLF-mediated NHEJ activity in hepatocellular carcinoma therapy resistance. BMC Cancer. 2017;17(1):344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kuo AJ, Cheung P, Chen K, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44(4):609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saloura V, Cho HS, Kiyotani K, et al. WHSC1 promotes oncogenesis through regulation of NIMA-related kinase-7 in squamous cell carcinoma of the head and neck. Mol Cancer Res. 2015;13(2):293–304. [DOI] [PubMed] [Google Scholar]
- 37. Wray J, Williamson EA, Sheema S, et al. Metnase mediates chromosome decatenation in acute leukemia cells. Blood. 2009;114(9):1852–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wray J, Williamson EA, Royce M, et al. Metnase mediates resistance to topoisomerase II inhibitors in breast cancer cells. PLoS One. 2009;4(4):e5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Salunkhe S, Mishra SV, Nair J, et al. Inhibition of novel GCN5-ATM axis restricts the onset of acquired drug resistance in leukemia. Int J Cancer. 2018;142(10):2175–2185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.