

Abstract
The structure–activity relationships and hit-to-lead optimization of dual Top1–TDP1 inhibitors in the indenoisoquinoline drug class were investigated. A series of nitrated 7-, 8-, 9-, and 10-hydroxyindenoisoquinolines were synthesized and evaluated. Several compounds displayed potent dual Top1–TDP1 inhibition. The 9-hydroxy series exhibited potencies and cytotoxicities vs Top1 that surpassed those of camptothecin (CPT), the natural alkaloid that is being used as a standard in the Top1-mediated DNA cleavage assay. One member of this series was a more potent Top1 inhibitor at a concentration of 5 nM and produced a more stable ternary drug–DNA–Top1 cleavage complex than CPT.
Graphical Abstract
INTRODUCTION
Human topoisomerase type IB (Top1) is an essential enzyme for various cellular processes as it relaxes DNA supercoils and resolves DNA topology so that genetic information can be accessed, and critical events, including DNA replication and transcription, can take place.1,2 The enzyme cuts a single strand of DNA to form a transient complex that allows supercoils to be dissipated, followed by religation of the broken strand.3 The cleavage complex can be trapped by Top1 poisons that intercalate between the DNA base pairs at the cleavage site and thereby inhibit the religation reaction. Collision of the replication fork with the single-strand break then generates a double-strand break, causing cell death.4 Defective repair explains the therapeutic effect of Top1 inhibitors. DNA damage produced by Top1 inhibitors can be reversed by a group of enzymes that includes tyrosyl-DNA phosphodiesterase I (TDP1), which hydrolyzes the phosphotyrosyl linkage between degraded or denatured Top1 and the DNA at the cleavage site to produce DNA 3′-phosphates and free peptides containing terminal tyrosyl residue.5–8 Polynucleotide kinase phosphatase (PNKP) then hydrolyzes that 3′-phosphate and installs a phosphate on the 5′-end on the other side of the break, and DNA ligase III reseals the DNA.9 Because TDP1 counteracts the action of Top1 inhibitors, it is possible that TDP1 inhibitors could potentiate the cytotoxic effects of Top1 inhibitors in combination anticancer drug therapy.9 The discovery of camptothecin (CPT, 1, Figure 1) as a selective and potent Top1 poison led to the validation of Top1 as an anticancer target,10 and in turn attracted a considerable amount of research effort in the design and synthesis of Top1 inhibitors.11,12 As TDP1 plays an important role in the maintenance of genomic stability13 and is a key enzyme in DNA repair machinery,14–17 TDP1 is also being pursued as a possible anticancer drug target.9,15,18–26
Figure 1.

Structure and activity of camptothecin (1). (a) See Table 1 for the Top1 inhibition grading rubric. (b) The MGM is the mean-graph midpoint value obtained from cytotoxicity testing in the National Cancer Institute’s panel of 60 human cancer cell lines.29
TDP1-dependent repair pathways are normally redundant with other DNA damage response pathways that are often compromised in cancer cells. This provides a reason to expect selective TDP1 inhibitor potentiation of Top1 cytotoxicity in cancer cells vs normal cells. Moreover, checkpoint deficiencies are common in cancer cells,27 and in these cases TDP1 becomes the main repair mechanism for removal of Top1-mediated DNA damage.5,9,28 The goal of the present studies was to design and synthesize potential anticancer agents that would possess dual Top1-TDP1 inhibitory activities and thereby potentially enhance anticancer activity and selectivity.
DESIGN RATIONALE
Two indenoisoquinoline Top1 inhibitors, indotecan (LMP400, 2) and indimitecan (LMP776, 3), are currently in phase I clinical trials at the National Cancer Institute (Figure 2).30,31 These compounds induce persistent Top1-linked DNA breaks, overcome some of the drug resistance issues associated with the camptothecins, and have a long half-life in humans.32 Recently, Cinelli et al. reported the synthesis of some hydroxylated indenoisoquinolines that were utilized as synthetic standards in the metabolism studies of 2 and 3 in human liver microsomes in the presence of NADPH.33 Four of the synthetic compounds were confirmed as the actual metabolites of the two clinical drugs based on matches in molecular masses and retention times with the most abundant peaks of metabolites detected by LC-MS/MS.33 Four other hydroxyindenoisoquinolines 8–11 (Figure 2) were not identified as actual metabolites, but they exhibited attractive Top1 inhibition and antiproliferative activities, especially the two 9-hydroxy-8-methoxy compounds 10 and 11. Through molecular modeling, Cinelli rationalized the lower potencies of their regioisomers (8-hydroxy-9-methoxy, compounds 8 and 9) as a consequence of possible steric and electronic clashes of the 9-methoxy group with the nonscissile DNA strand in the ternary DNA–Top1-inhibitor complex.33 However, independent studies reported by Morrell et al. showed that placing a variety of bulky substituents (e.g., methoxycarbonyl, ethoxy, halogen) at the 9-position did not result in a significant attenuation or loss of Top1 inhibition or antiproliferative activity of indenoisoquinolines when a nitro group was present at the 3-position.34–36 This observation led to the hypothesis that the 3-nitro group exhibits a “steric-rescuing” effect, which is probably facilitated by its hydrogen bonding to the Asn722 residue in the open environment of the cleaved DNA strand, as observed in the hypothetical docking model of compound 5 in the ternary complex (Figure 3),35 thus moving the inhibitors away from the uncleaved DNA strand so that bulky substituents could be tolerated in this crowded area. Because both the 9-hydroxyl group and the 3-nitro group enhance the anti-Top1 potency, the combination of these two features in a single indenoisoquinoline molecule might create exceptionally potent Top1 inhibitors. Although the nitro group is a toxicophore, one of the objectives in the present case was to synthesize very cytotoxic indenoisoquinolines that could be linked to cancer cell-targeting moieties through the phenolic hydroxyl groups, which would make the nitro group more tolerable.
Figure 2.

Indenoisoquinoline Top1 inhibitors and their hydroxylated analogues.
Figure 3.

(top) Proposed hydrogen bonding network of indenoisoquinoline 5 (red) in the DNA–Top1–indenoisoquinoline ternary complex. (bottom) Hypothetical model of the ternary complex derived from the PDB ID 1SC7 crystal structure.35 Hydrogen bonds are shown as yellow lines. The diagram is programmed for wall-eyed (relaxed) viewing.
Morrell et al. reported a series of indenoisoquinolines bearing a 3-nitro group on the A-ring and a 7- or 9-methoxy group on the D-ring (compounds 4–7) as cytotoxic and potent Top1 inhibitors.35,36 The 9-methoxy group was shown to typically improve cytotoxicity at the expense of Top1 inhibition, whereas the 3-nitro group enhanced Top1 inhibition to a greater extent than cytotoxicity.35 Although the importance of the 3-nitro group to the biological activity of this drug class was rationalized in detail by molecular modeling and electrostatic potential calculations,35 explanations of the role of the 9-methoxy group were not completely confirmed. In particular, electrostatic potential modeling revealed charge complementarity between the electron-rich, 9-position oxygen atom and the outer edges of the nonscissile DNA base pairs in the Top1 cleavage complex, while the hypothetical binding model of 5 derived from the high resolution crystal structure of topotecan (PDB ID: 1K4T, in which water molecules were present in the intercalation site) showed a single bridging hydrogen bond (2.78 Å) between a water molecule and the 9-methoxy group (Figure 4).36 From these observations, it was concluded that the optimal place to incorporate a methoxy group on the D-ring was the 9-position (as in compounds 5–7).36 The generally lower potencies of compounds in the 7-methoxy series was rationalized as being the result of the 7-methoxy group having deleterious steric interactions with the lactam side chain, limiting the number of planar conformations (relative to the 9-methoxy group) required to confer Top1 inhibition, and increasing steric repulsion with the DNA base pairs of the nonscissile DNA strand.36 However, Morrell did observe an exception in that the 7-methoxy-3-nitroindenoisoquinoline 4 (Figure 2) was a potent Top1 inhibitor.
Figure 4.

Hypothetical molecular model depicting a water-mediated hydrogen bond from indenoisoquinoline 5 (red) to the backbone of the nonscissile DNA strand in ternary complex derived from the PDB ID 1K4T crystal structure.36 The diagram is programmed for wall-eyed (relaxed) viewing.
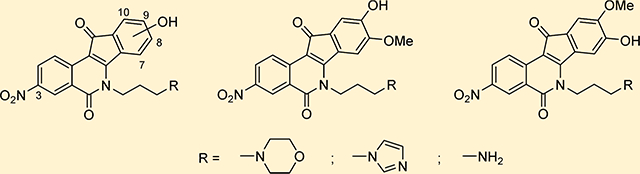
A hydroxyl group could possibly serve as a biologically active replacement for the methoxy group. It might also provide a point of attachment for the synthesis of prodrugs so that the pharmacokinetics could be modulated and optimized. Hydroxylated indenoisoquinolines might also offer an advantage in terms of solubility for intravenous formulation, which has been a concern with this drug class. In the present studies, the syntheses of l8 nitrated 7-, 8-, 9-, and 10-hydroxyindenoisoquinolines bearing a 3-nitro substituent were investigated (Figure 5), all of which are analogues of the highly potent dual Top1–TDP1 inhibitors 4–7,34,37 so as to optimize the dual Top1–TDP1 inhibitory potency and gain more insight into the importance of the location of the hydroxyl group on the overall biological activities of this drug class.
Figure 5.

Proposed Top1–TDP1 inhibitors.
CHEMISTRY
The indenoisoquinoline system can be constructed by several methods: (1) condensation of indenoisochromenone with a primary amine,38 (2) Suzuki–Miyaura cross-coupling reaction followed by ring-closing metathesis,39 and (3) oxidative cyclization of cis acid produced by the condensation of a homophthalic anhydride and a Schiff base.34–36 The latter methodology was chosen in this case to prepare all of the proposed compounds because of the ease in synthesis of intermediates and manipulation of starting materials to obtain indenoisoquinolines bearing the desired substituents.
A. Synthesis of 8-Hydroxy-9-methoxy-3-nitroindenoisoquinolines and 9-Hydroxy-8-methoxy-3-nitroindenoisoquinolines.
The synthesis of nitrated 9-hydroxy-8-methoxyindenoisoquinolines 21, 23, and 25 is depicted in Scheme 1, and the preparation of 8-hydroxy-9-methoxyindenoisoquinolines 32, 34, and 36 is shown in Scheme 2. Commercially available homophthalic acid 12 was nitrated with fuming HNO3 to provide the diacid 13, which underwent dehydration in AcCl to provide anhydride 14. The reactive hydroxyl groups in vanillin (15) and isovanillin (26) were protected with a benzyl group. Benzylvanillin (16) and benzylisovanillin (27) reacted with 3-bromopropylamine hydrobromide to give Schiff bases 17 and 28, which upon condensation with anhydride 14 in CHCl3 furnished cis acids 18 and 29, respectively, in good yields with excellent diastereoselectivities. Treatment of 18 or 29 with SOCl2 (neat) provided a mixture of indenoisoquinoline 19 (from 18) or 30 (from 29) and their regioisomers, which was evident by 1H NMR spectroscopy and consideration of the reaction mechanism (Scheme 3). Isomers 19 and 30 appeared as the major products. The structure assignments were based on the distinct splitting patterns of the D-ring hydrogens in the 1H NMR spectra of the two regioisomers. The very similar Rf values of the regioisomers led to lengthy and difficult purifications by flash column chromatography. The yields of pure 19 and 30 after two chromatographic purifications were low (15–25%) and inconsistent. The low yields may be due to the nitro group activating epimerization to the trans diastereomers, which exist in pseudodiaxial conformations and do not oxidize and cyclize in SOCl2.40,41
Scheme 1.

Synthesis of Nitrated 9-Hydroxy-8-methoxyindenoisoquinolinesa
aReagents and conditions: (a) fuming HNO3, 0–23 °C; (b) AcCl; reflux; (c) BnCl, DMF, IK2CO3, 23 °C; (d) 3-bromopropylamine hydrobromide, Et3N, Na2SO4, CHCl3, 23°C; (e) CHCl3, 0–23 °C; (f) SOCl2, 0–23 °C; (g) morpholine, 1,4-dioxane, 23 °C; (h) aqueous HBr, 70 °C; (i) imidazole, 1,4-dioxane, 70 °C; (j) NaN3, DMSO, 23 °C; (k) (i) P(OEt)3, benzene, reflux, (ii) aqueous HBr, 70 °C.
Scheme 2.

Synthesis of Nitrated 8-Hydroxy-9-methoxymdenoisoquinolmesa
aReagents and conditions: (a) BnCl, DMF, K2CO3, 23 °C; (b) 3-bromopropylamine hydrobromide, Et3N, Na2SO4, CHCl3, 23 °C; (c) anhydride 14, CHCl3, 0–23 °C; (d) SOCl2, 0 to 23 °C; (e) morpholine, 1,4-dioxane, 23 °C; (f) aqueous HBr, 70 °C; (g) imidazole,1,4-dioxane, 70 °C; (h) NaN3, DMSO, 23 °C; (i) (i) P(OEt)3, benzene, reflux, (ii) aqueous HBr, 70 °C.
Scheme 3.

Formation of Two Regioisomers in the Cyclization of Acid Chloride Intermediates
Treatment of cis acids 18 and 29 with SOCl2 alone resulted in conversion to the acid chlorides, dehydrogenation, and intramolecular Friedel–Crafts cyclization to provide the aromatic indenoisoquinoline systems. Purification of crude 19 or 30 by column chromatography (SiO2) caused decomposition of the products, and therefore the products were used directly in the next step without further purification.
Displacement of the terminal bromide in 19 or 30 with morpholine or imidazole in 1,4-dioxane, or azide in DMSO, yielded the benzyl-protected compounds 20, 22, and 24 (from 19), or 31, 33, and 35 (from 30), respectively. Many attempts were made to optimize the yields of these SN2 reactions. For morpholine derivatives 20 and 31, the best yields (44–52%) were obtained when the starting materials 19 or 30 were stirred with morpholine for 16 h at room temperature. In contrast, the syntheses of imidazole derivatives 22 and 33 required constant heating at 70 °C for 16 h, and the yields (36–40%) were slightly lower than those of morpholine compounds (44–52%). Column chromatographic purification of the crude mixtures provided analytically pure products with much ease as compared to the separation of the bromides 19 or 30 from their regioisomers. These results supported the direct use of the crude bromides 19 and 30 from the previous step.
Many attempts were made to achieve the debenzylation of the protected compounds 20, 22, 24, 31, 33, and 35. 1,4-Cyclohexadiene has been shown to be an effective hydrogen donor in catalytic transfer hydrogenation, a method that was utilized to remove N-benzyloxycarbonyl, benzyl ester, and benzyl ether protecting groups in peptide chemistry.42 Disappointingly, attempted implementation of this method on the benzyl-protected morpholine compound 31 in various solvents (CHCl3, EtOH, or glacial AcOH) at room temperature was not successful in cleaving the benzyl group. In all cases, starting material was recovered. Attempted debenzylation with HBr in acetic acid (33 wt %) not only removed the benzyl group in 31, but a subsequent Fisher esterification of the resulting phenol 32 also occurred to provide the acetate 37 as the major product while the desired phenol 32 was only obtained in minor amount (Scheme 4).
Scheme 4.

Debenzylation of 31 with HBr in AcOH (33 wt %)
Treatment of the benzyl-protected starting materials 20, 22, 24, 31, 33, and 35 with aqueous HBr at 70 °C for 4–5 h, followed by dilution with acetone and then concentration (iterated three times), afforded a mixture that was suitable for vacuum filtration to provide the desired phenols 21, 23, 25, 32, 34, and 36 in high yields (80–100%) and excellent purity. The moderate yield of imidazole 23 (50%) was due to its higher aqueous solubility. All of the starting materials produced reaction mixtures as emulsions from which all of the products precipitated. The products derived from vanillin (21, 23, and 25) were so finely powdered that the use of an HPLC filter paper was crucial for their recovery, while a regular double-layered filter paper worked well for those derived from isovanillin (32, 34, and 36). The three-time iteration of dilution with acetone and concentration was imperative for the success of vacuum filtration. For the syntheses of amines 25 and 36, corresponding azides 24 and 35 were subjected to a Staudinger reduction with P(OEt)3, followed by treatment with aqueous HBr to achieve acidic hydrolysis and debenzylation simultaneously in one pot.
B. Synthesis of 7-Hydroxy-3-nitroindenoisoquinolines (Scheme 5).
Scheme 5. Synthesis of 7-Hydroxy-3-nitroindenoisoquinolines.

a
aReagents and conditions: (a) BnBr, DMF, K2CO3, 23 °C; (b) 3-bromopropylamine hydrobromide, Et3N, Na2SO4, CHCl3, 23 °C; (c) anhydride 14, CHCl3, 0–23 °C; (d) (i) SOCl2, 23 °C, (ii) AlCl3, 1,2-dichloroethane, 0–23 °C; (e) morpholine, 1,4-dioxane, 70 °C; (f) imidazole, 1,4-dioxane, 70 °C; (g) (i) NaN3, DMSO, 23 °C, (ii) P(OEt)3, benzene, reflux, (iii) HCl, MeOH, 70 °C.
Commercially available salicylaldehyde 38 was O-benzyl protected to give 39, which reacted with 3-bromopropylamine to afford Schiff base 40. Condensation of 40 with anhydride 14 in CHCl3 yielded cis acid 41, which upon treatment with SOCl2, followed by AlCl3 in 1,2-dichloroethane, provided indenoisoquinoline bromide 42 in good yield (63%). The 1H NMR spectrum of 42 indicated excellent purity without chromatographic purification, and it also indicated that AlCl3 induced debenzylation to afford the free phenol. Interestingly, the use of AlCl3 to simultaneously remove the benzyl group was tried in connection with the chemistry described in Schemes 1 and 2, but in those cases it led to complicated mixtures and no debenzylation products could be isolated. Displacement of the bromide in 42 with morpholine, imidazole, or NaN3, followed by a Staudinger reduction of the azide intermediate and acidic hydrolysis with methanolic HCl, provided the desired amines 43, 44, and 45, respectively. The pure products were isolated without chromatographic purification.
C. Synthesis of 8- and 10-Hydroxy-3-nitroindenoisoquinolines.
A similar approach was implemented to prepare 8-hydroxy-3-nitroindenoisoquinolines (Scheme 6). The commercially available 3-hydroxybenzaldehyde 46 was protected with a benzyl group to give 47, which was then converted to Schiff base 48 upon treatment with 3-bromopropylamine hydrobromide under basic conditions. Condensation of 48 with anhydride 14 provided a mixture of the desired cis acid 49 and its trans diastereomer. Boiling the mixture in CHCl3, followed by filtration, helped remove the unwanted trans acid and provide the pure cis acid 49 as a sole product. However, treatment of 49 with SOCl2 0 to 23 °C, followed by AlCl3 in refluxing 1,2-dichloroethane, did not yield the desired bromide 50. Gratifyingly, a profitable transformation was observed when cis acid 49 was heated with SOCl2 (neat) at reflux for 4 h, during which the solution turned to clear orange. Removal of SOCl2, followed by filtering the residue and washing with ether, provided an orange solid whose 1H NMR spectrum revealed that it was an approximately 2:1, inseparable mixture of 8- and 10-(benzyloxy)indenoisoquinolines 51 and 52.
Scheme 6a.

aReagents and conditions: (a) BnBr, DMF, K2CO3, 23 °C; (b) 3-bromopropylamine hydrobromide, Et3N, Na2SO4, CHCl3, 23 °C; (c) anhydride 14, CHCl3, 0 to 23 °C; (d) (i) SOCl2, 0–23 °C, (ii) AlCl3, 1,2-dichloroethane, reflux; (e) SOCl2, reflux.
Treatment of the mixture of bromides 51 and 52 with morpholine and imidazole, followed by chromatographic purification, allowed the isolation of each pure morpholinyl and imidazolyl 8- and 10-benzyloxy compounds 53, 54, 57, and 58 (Scheme 7). In all cases, the 8-substituted products eluted first (higher Rf value), and the two regioisomers were distinguished based on the differences in 1H NMR multiplicity of the three aromatic protons in the D-ring: the 8-substitiuted products have 2 doublets and 1 singlet (or 1 doublet with a small meta coupling constant), while the 10-substituted products have 2 doublets and 1 multiplet. All of the benzyl-protected materials were subjected to a 3 h debenzylation with aqueous HBr (48 wt %) to provide the desired 8- and 10-hydroxyindenoisoquinolines 55, 56, 59, and 60 in good yields and purities. The azides 61 and 62, prepared by reaction of the bromides 51 and 52 with sodium azide in DMSO at room temperature, were subjected to a Staudinger reduction, followed by simultaneous acidic hydrolysis and deprotection with aqueous HBr, to provide pure HBr salts of amines 63 and 64 in excellent purity and moderate yields (Scheme 7).
Scheme 7.

Synthesis of 8- and 10-Hydroxy-3-nitroindenoisoquinolinesa
aReagents and conditions: (a) morpholine, 1,4-dioxane, 70 °C; (b) HBr, H2O, 70 °C; (c) imidazole, 1,4-dioxane, 70 °C; (d) NaN3, DMSO, 23 °C; (e) (i) P(OEt)3, benzene, reflux, (ii) HBr, H2O, 70 °C.
D. Synthesis of 9-Hydroxy-3-nitroindenoisoquinolines (Scheme 8).
Scheme 8. Synthesis of 9-Hydroxy-3-nitroindenoisoquinolines.

a
aReagents and conditions: (a) 3-bromopropylamine hydrobromide, Et3N, Na2SO4, CHCl3, 23 °C; (b) anhydride 14, CHCl3, 0–23 °C; (c) (i) SOCl2, 23 °C, (ii) AlCl3, 1,2-dichloroethane, 0–23 °C; (d) (i) morpholine, THF, 70 °C, (ii) HBr, MeOH, 23 °C; (e) (i) imidazole, THF, 70 °C, (ii) HCl, MeOH, 23 °C; (f) (i) NaN3, DMSO, 23 °C, (ii) PPh3, THF, 70 °C, (iii) HBr, MeOH, 70 °C.
4-Benzyloxybenzaldehyde (65) is commercially available or can be prepared easily by the methodology shown in Scheme 5 for 2-benzyloxybenzaldehyde. Treatment of 65 with 3-bromopropylamine hydrobromide and Et3N provided Schiff base 66, which upon condensation with anhydride 14 produced cis acid 67 in good yield and excellent purity. Treatment of acid 67 with SOCl2 for 16 h at room temperature yielded a yellow syrup upon removal of SOCl2. The yellow syrup was then treated with AlCl3 in 1,2-dichloroethane to provide the benzyl-free indenoisoquinoline phenol 68 after aqueous workup. Treatment of 68 with morpholine or imidazole in THF provided the corresponding displacement products, which were then stirred in freshly made methanolic HBr or HCl to afford the HBr and HCl salts 69 and 70, respectively. This extra step was meant to facilitate later biological testing because the salts were more soluble in DMSO than the neutral compounds. The synthesis of amine 71 by the previous methodology involving reduction of the azide intermediate with P(OEt)3 in benzene (as shown Scheme 5) was not successful due to complications in purification and isolation of the compound in solid form. The Staudinger reaction was therefore reattempted by treating the azide intermediate, obtained from 68, with PPh3 in THF (instead of P(OEt)3 in benzene), followed by 4 h acidic hydrolysis with methanolic HBr. This modification provided the desired amine 71 in 32% yield with excellent purity.
BIOLOGICAL RESULTS AND DISCUSSION
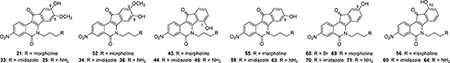
All of the proposed compounds (collectively shown in Figure 5) were subjected to determinations of Top1 and TDP1 inhibitory potencies and antiproliferative activities. Top1 inhibition was recorded as the ability of a drug to poison Top1 and induce enzyme-linked DNA breaks in the Top1-mediated DNA cleavage assay.43 The anti-Top1 potency was graded on the basis of visual inspection of the number and intensities of the bands corresponding to DNA fragments and was recorded on a semiquantitative scale relative to 1 μM camptothecin (CPT, 1, Figure 1): 0, no inhibitory activity; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity; ++++, equipotent; +++++, more potent. Ambiguous scores between two values are designated with parentheses (e.g., ++(+) would be between ++ and +++). The loss of potency at 100 μM of drug concentration, as evidenced by the pale intensity or disappearance of the DNA cleavage bands (Figure 6), results from the drug intercalating into free DNA, rendering it a poorer substrate for the Top1-catalyzed cleavage reaction. Some of the indenoisoquinolines act as Top1 poisons (i.e., inhibiting the DNA religation reaction) at low drug concentrations and as Top1 suppressors (i.e., inhibiting the DNA cleavage reaction) at high drug concentrations. The steric bulk of the substituent on the lactam side chain seems to play a role in determining whether a given indenoisoquinoline poisons or suppresses Top1, with larger substituents showing a preference for DNA intercalation into the binary Top1–DNA complex instead of free DNA.44 In the present case, the 3-nitro group seems to facilitate intercalation into free DNA so that 55, 56, 59, and 60, with large substituents on the side chain, all act as Top1 suppressors at high drug concentrations (Figure 6).
Figure 6.

Top1-mediated DNA cleavage induced by indenoisoquinolines 55, 56, 59, and 60. From left to right: lane 1, DNA alone; lane 2, DNA + Top1; lane 3, CPT (l), 1 μM; lane 4, MJ-III-65, 1 μM; lanes 5–20, 55, 56, 59, and 60 at 0.1, 1.0, 10, and 100 μM, respectively. Numbers and arrows on the left indicate arbitrary cleavage site positions. MJ-III-65 is the positive indenoisoquinoline control.45
TDP1 inhibition was measured as the ability of a drug to inhibit the enzyme-catalyzed hydrolysis of the phosphodiester linkage between tyrosine and the 3′-end of a DNA substrate (N14Y), thus preventing the generation of an oligonucleotide with a free 3′-phosphate (N14P).21 The anti-TDP1 potency, as indicated by the disappearance of the gel band for N14P, was recorded as IC50 based on the following semiquantitative scale: 0, IC50 >111 μM; +, IC50 between 37 and 111 μM; ++, IC50 between 12 and 37 μM; +++, IC50 between 1 and 12 μM; +++ +, IC50 < 1 μM. Representative gels demonstrating dose-dependent Top1 and TDP1 inhibitory activities of some target indenoisoquinolines are depicted in Figures 6 and 7, respectively.
Figure 7.

Inhibition of recombinant TDP1 hydrolysis of N14Y to N14P by indenoisoquinolines 21, 23, 25, 36, and 72. From left to right: lane 1, DNA alone; lane 2, DNA + TDP1; lanes 3–22, compounds 21, 23, 25, 36, and 72 (each at 1.4, 4.1, 12.3, 37, and 111 μM). Bis(indenoisoquinoline) 72 (TDP1: +++) was used as the standard.
The antiproliferative activities of all of the target compounds were evaluated in the National Cancer Institute’s Developmental Therapeutics Program screen (NCI-DTP) against 60 common human cancer cell lines (the “NCI-60”). AH compounds were initially tested in a one-dose prescreening assay at moderately high concentration (10 μM), and those that passed a cutoff threshold were tested at five concentrations ranging from 100 μM to 10 nM. The 50% growth-inhibitory concentration (GI50) in each cell line was determined by interpolation of data points received from the five-dose screen. The cytotoxicity of a compound was reported as a mean-graph midpoint (MGM) of the average GI50 value across the entire panel of 60 cell lines, where GI50 values that fell outside the tested range (100 μM to 10 nM) were assigned as the maximal (100 μM) and minimal (10 nM) drug concentrations, respectively, used in the screening. Top1 and TDP1 inhibitory potencies, and the antiproliferative activities of all target compounds are collectively shown in Table 1. For comparison, activities of compounds 1 (Figure 1), 4–5 and 8–11 (Figure 2) are included.
Table 1.
Top1 and TDP1 Inhibitory Potencies and Antiproliferative Activities of Nitrated Hydroxyindenoisoquinolines
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cytotoxicity (GI50 in μM)a |
||||||||||||
| compd | Top1b cleavage | TDP1c inhibition | MGMd (μM) | lung HOP-62 | colon HCT-116 | CNS SF-539 | melanoma UACC-62 | ovarian OVACAR-3 | renal SN12C | prostate DU-145 | breast MCF7 | |
| camptothecin | 134 | ++++ | –e | 0.040 ± 19f | <0.010 | 0.030 | <0.010 | <0.010 | 0.22 | 0.020 | <0.010 | <0.010 |
| 7-methoxy | 434,37 | ++++ | +++ | 1.4 | 1.1 | 0.72 | 1.4 | 2.3 | 2.3 | 7.1 | 1.1 | 1.6 |
| 9-methoxy | 534,37 | +++++ | +++ | 0.027 ± 8 | <0.010 | <0.010 | <0.010 | <0.010 | 2.8 | <0.010 | – | 3.3 |
| 8-hydroxy-9-methoxy | 833 | +++ | + | 3.1 ± 0.3 | 0.18 | 2.3 | 0.28 | 0.070 | 8.3 | 0.19 | 0.20 | 0.030 |
| 933 | ++(+) | 0 | 42 ± 7 | >100 | >100 | – | 0.33 | >100 | – | >100 | 0.050 | |
| 9 -hydroxy-8-methoxy | 1033 | +++++ | 0 | 0.087 ± 63 | 0.020 | 0.030 | 0.020 | 0.020 | 0.030 | 0.020 | 0.020 | 0.020 |
| 1133 | ++++(+) | 0 | 0.055 ± 3 | <0.010 | <0.010 | <0.010 | <0.010 | 0.020 | <0.010 | <0.010 | <0.010 | |
| 9-hydroxy-8-methoxy | 21 | ++ | 0 | 0.021 ± 2 | <0.010 | <0.010 | <0.010 | <0.010 | – | <0.010 | <0.010 | <0.010 |
| 23 | + | 0 | 0.016 ± 2 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | |
| 25 | ++ | ++ | 0.017 ± 2 | <0.010 | <0.010 | <0.010 | <0.010 | 0.027 | <0.010 | <0.010 | <0.010 | |
| 8-hydroxy-9-methoxy | 32 | ++ | 0 | 0.20 ± 1 | 0.079 | 0.13 | 0.13 | 0.039 | 0.72 | 0.063 | 0.11 | 0.035 |
| 34 | ++(+) | ++ | 0.74 ± 5 | 0.085 | 0.071 | 0.049 | 0.049 | 0.32 | 0.051 | 0.051 | 0.020 | |
| 36 | ++ | ++ | 0.066 ± 2 | 0.47 | 0.15 | 0.66 | 0.95 | 1.7 | 0.45 | 0.43 | 0.17 | |
| 7-hydroxy | 43 | ++ | 0 | – | – | – | – | – | – | – | – | – |
| 44 | +++ | 0 | – | – | – | – | – | – | – | – | – | |
| 45 | ++ | ++ | – | – | – | – | – | – | – | – | – | |
| 8-hydroxy | 55 | ++++ | 0 | 0.056 ± 6 | 0.025 | 0.026 | 0.021 | 0.020 | 0.041 | 0.036 | 0.034 | <0.010 |
| 59 | +++ | 0 | 0.072 ± 6 | – | <0.010 | <0.010 | <0.010 | 0.023 | <0.010 | <0.010 | <0.010 | |
| 63 | +++(+) | +++ | 0.41 ± 1 | – | 0.22 | 0.21 | 0.21 | 0.27 | 0.27 | 0.35 | 0.12 | |
| 9-hydroxy | 68 | +++++ | + | 0.022 ± 2 | – | <0.010 | <0.010 | <0.010 | 0.023 | <0.010 | <0.010 | <0.010 |
| 69 | +++++ | 0 | 0.014 ± 1 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | <0.010 | |
| 70 | +++(+) | 0 | 0.023 ± 2 | <0.010 | <0.010 | <0.010 | <0.010 | 0.019 | <0.010 | <0.010 | <0.010 | |
| 71 | ++++ | + | 0.12 ± 1 | 0.056 | 0.078 | 0.030 | 0.041 | 0.22 | 0.046 | 0.052 | 0.026 | |
| 10-hydroxy | 56 | ++ | 0 | 3.5 | 12 | 0.55 | 0.20 | 0.39 | 29 | – | – | 0.064 |
| 60 | +++ | 0 | 0.23 | 0.081 | 0.13 | 0.15 | 0.18 | 0.23 | 0.17 | 0.13 | 0.056 | |
| 64 | +++ | ++ | 2.0 | 1.9 | 0.47 | 1.7 | 2.5 | 2.9 | 3.4 | 1.2 | 0.25 | |
The listed cytotoxicity GI50 values are concentrations that induce 50% growth inhibition and are the results of single determination.
Top1 inhibitory potency in the Top1-mediated DNA cleavage assay is graded relative to 1 μM camptothecin (1): 0, no inhibitory activity; +, 20–50% activity; ++, 50–75% activity; +++, 75–95% activity; ++++, equipotent; +++++, more potent. Ambiguous scores between two values are designated with parentheses (e.g., ++(+) would be between ++ and +++).
TDP1 inhibition is reported as IC50 based on the following scale (μM): 0, IC50 > 111; +, IC50 37–111; ++, IC50 12–37; +++, IC50 1–12; ++++, IC50 < 1.
Mean-graph midpoint (MGM) is an approximate average of GI50 values across the entire panel of 60 human cancer cell lines successfully tested, calculated by assigning MGM GI50 values of 0.01 and 100 μM to compounds whose MGM GI50 values fall outside the testing range of 0.01–100 μM.
–, not tested; for TDP1, the values were not available from previous reports; for MGM, the compound had a low activity in the initial one-dose testing at 10 μM that did not pass the predetermined cutoff value to warrant the five-dose testing required for the GI50 determination; for GI50 of individual cell lines, no values available in the NCI-60 reports, probably as a result of unsuccessful tests.
MGM GI50 values with a standard deviation have their individual GI50 value for each cell line as an average of two determinations, otherwise one determination was done.
As observed in Table 1, all of the compounds exhibited at least some Top1 inhibitory activity. The nitrated 9-hydroxy-8-methoxy series 21, 23, and 25 displayed only weak to moderate anti-Top1 potencies (+ or ++) even though their 2,3-dimethoxy analogues 10 and 11 were excellent Top1 inhibitors (+++++). Furthermore, the nitrated 8-hydroxy-9-methoxy series 32, 34, and 36 showed a similar extent of Top1 inhibition as observed in their 2,3-dimethoxy analogues 8 and 9 (++ or ++ +). In all cases, the nitrated compounds displayed a significant improvement in terms of cytotoxicity when compared to their corresponding dimethoxy analogues, with the 9-hydroxy-8-methoxy series 21, 23, and 25 possessing low nanomolar antiproliferative potencies (MGM values of 16–21 nM). Although the apparent enhancement of cytotoxicity brought about by the presence of the 3-nitro group was in agreement with our initial hypothesis, the stark contrasts in Top1 inhibition among these very similar analogues was rather surprising for several reasons.
First, the electron-withdrawing nature of the 3-nitro group is proposed to confer biological activity to the indenoisoquinolines because it facilitates the π–π stacking by enhancing charge-transfer interactions between the aromatic indenoisoquinoline ring system and the flanking DNA base pairs, and it also provides a favorable hydrogen bond to the Asn722 residue of Top1 in the spacious area provided by the cleaved DNA strand, thus moving the inhibitor away from the crowded environment of the uncleaved DNA strand. As a result, favorable A-ring electronics may offset the deleterious steric effect of bulky substituents at the 9-position that clash with the unbroken DNA strand.34–36 As noted above, Morrell et al. reported that 3-nitroindenoisoquinolines bearing bulky groups like methoxycarbonyl, ethoxy, or halogen (bromine, iodine) at the 9-position exhibited excellent Top1 inhibitory potencies (+ ++ to ++++).34
Second, even though this effect did not enhance Top1 inhibition, it was expected to at least retain, instead of drastically attenuate the activity, as shown by the drastic drop in the nitrated 9-hydroxy-8-methoxy series 21, 23, and 25 (+ or ++) compared to their dimethoxy analogues 10 and 11 (++++ +). Such a decrease in Top1 inhibition is even more striking if one takes into account that for the 2,3-dimethoxy series, the 9-hydroxy-8-methoxy compounds 10 and 11 are much more potent than their 8-hydroxy-9-methoxy regioisomers 8 and 9. Rationalizing this observation through docking studies led to the hypothesis that for the 2,3-dimethoxy series, the 9-methoxy group protrudes near the phosphodiester backbone of the uncleaved DNA strand, thus representing a less favorable fit as compared to the 8-methoxy group.33 However, for the 3-nitro series, the results were completely opposite because the 9-hydroxy-8-methoxy compounds 21, 23, and 25 were not better but were even somewhat less active than their 8-hydroxy-9-methoxy regioisomers 32, 34, and 36.
Third, the anti-Top1 potency and antiproliferative activity g enerally correlate well, with better Top1 inhibitors also displaying higher cytotoxicity. However, this was not the case for the nitrated 9-hydroxy-8-methoxy series 21, 23, and 25 because these relatively weak Top1 inhibitors (+ or ++) exhibited cytotoxicities (16–21 nM) 4-fold higher than their strong Top1 inhibitor (+++++) dimethoxy analogues 10 and 11 (55–87 nM). The reason for the lack of a strong correlation between Top1 inhibition and antiproliferative activity observed for the nitrated series remains unknown, but it is probably a complicated consequence of several factors, including pharmacokinetics (cellular permeability, solubility, and metabolism) and possible off-target effects.
All of the nitrated hydroxyindenoisoquinolines that do not possess a D-ring methoxy group displayed similar or better Top-1 inhibitory potency than their analogues with a methoxy group. In contrast to the 9-hydroxy-8-methoxy series, a strong correlation between Top1 inhibition and antiproliferative activity was observed for these compounds. Indeed, the order of Top1 inhibition and cytotoxicity went from the 9-hydroxyl series 68–71 as the most active and cytotoxic (Top1 inhibition ++++ or more, MGM 14–117 nM) to the 8-hydroxyl series 55, 59, and 63 (+++ to ++++, 56–407 nM), and finally to the equipotent 7-hydroxyl series 43–45 and the 10-hydroxyl series 56, 60, and 64 (++ to +++ for both series, 234 to 3550 nM for the 10-hydroxyl) as the least active and least cytotoxic. The 7-hydroxyl series 43–45 had very low cytotoxicities in the initial one-dose testing at 10 μM that did not pass the predetermined cutoff value to warrant the subsequent five-dose testing required for the GI50 determination in the NCI-60. Interestingly, the 9-hydroxyl series 68−71 displayed potencies and cytotoxicities comparable to or even greater than camptothecin (1, ++++, 40 nM) and the 3-nitro-9-methoxy standard 5 (+++++, 27 nM). Therefore, the 9-hydroxyl group fulfilled our initial mission of replacing the 9-methoxy group in compounds 5−7 with a reactive functional group that could be derivatized to produce prodrugs or linked to targeting moieties or antibodies for improvement of selectivity for cancer cells vs normal cells. The better overall activities of the 8- and 9-hydroxyl series as compared to the 7- and 10-hydroxyl series suggest that the 8- and 9-positions are optimal for forming favorable interactions between the D-ring hydroxyl group and their surroundings in the ternary cleavage complex. In agreement with the docking results reported by Morrell (Figure 4),36 the 8- and 9-hydroxyl groups are within a reasonable distance to form a water-mediated hydrogen bond from the oxygen atom to the phosphodiester DNA backbone, and the electron-rich oxygen atom also provides an electrostatic potential that is complementary to the outer edges of the nonscissile DNA strand. Furthermore, because the 9-hydroxyl group can be considered to be directly across from and in resonance with the 3-nitro group, the slightly higher activities observed for the 9-hydroxyl series might result from resonance contributors involving donation of electron density from the phenol to the nitro group. Within each series, the position of the hydroxyl group in the D-ring is a more important determinant of Top1 inhibitory activity than the identity of the amine group in the side chain.
Interestingly, the indenoisoquinoline bromide 68, a member of the 9-hydroxyl series, was a more potent Top1 poison (++++ +) than CPT (1, ++++). Indeed, the following two points are apparent from the inspection of the gels in Figure 8: (1) 68 trapped Top1 cleavage complexes at a concentration as low as 5 nM, whereas CPT had lesser activity at that concentration, and (2) in contrast to the other indenoisoquinolines reported in this study, the bromide 68 did not intercalate into free DNA to suppress the cleavage reaction at a concentration as high as 100 μM. It might be reasonable to pose the question of whether the superior potency of 68 came about as a result of its DNA alkylation via displacement of the bromide group in the inhibitor. To answer this question, the kinetics of ternary cleavage complex reversal for 68 were studied, and the results showed that it formed drug−Top1−DNA cleavage complexes that did not reverse as quickly as CPT at their common cleavage sites (70, 92, 97, and 119) (Figure 9). The ternary cleavage complex complexes induced by 68 were quite persistent but did reverse over time, while those induced by CPT reversed much faster. Obviously 68 forms more stable cleavage complexes than CPT. The decrease in intensity of the cleavage bands induced by 68 over time also suggest that the drug did not alkylate or form an irreversible ternary cleavage complex.
Figure 8.

Comparison of Top1 inhibitory potencies between camptothecin (CPT, 1) and indenoisoquinoline bromide 68 in a dose-dependent Top1-mediated DNA cleavage. From left to right: lane 1, DNA alone; lane 2, DNA + Top1; lanes 3−16, CPT (1) and 68 at 0.005, 0.01, 0.05, 0.1, 1.0, 10, and 100 μM, respectively.
Figure 9.

Kinetics of reversal assay of Top1-mediated DNA cleavage complexes induced by CPT vs bromide 68. The forward reaction was terminated with 0.35 M NaCl.
Table 1 reveals that most nitrated hydroxyindenoisoquinolines were either inactive or weakly active against TDP1 with 0 or + activity. However, there were also compounds that exhibited good anti-TDP1 potencies (++ to +++), with the 8-hydroxyindenoisoquinoline amine 63 displaying the highest activity (+++). With an excellent anti-Top1 potency of ++++, amine 63 is the most potent dual Top1−TDP1 inhibitor reported in the present study. A thorough inspection of TDP1 inhibition within each series in Table 1 revealed an apparent trend: compounds with a primary amine side chain consistently exhibited higher potencies than their morpholine and imidazole analogues, with one single exception of the imidazole 34 possessing an anti-TDP1 potency (++) comparable to its amino analogue 36. This trend also consistently applied across the whole series of hydroxyindenoisoquinolines, with or without a methoxy. This observation is in agreement with the hypothesis that the positively charged ammonium cation is of importance in orienting the ammonium group toward the hydrophilic region in the TDP1 active site so that the aromatic indenoisoquinoline ring system could reside in close proximity and form favorable contacts with some key residues in the catalytic region (H263, K265, N283, H493, K495, and N516), thus conferring TDP1 inhibitory activities to the indenoisoquinolines.3 Moreover, it is also apparent from Table 1 that most indenoisoquinolines with a morpholine or imidazole side chain are inactive against TDP1 (0 activity), with the imidazole 34 as the single exception (++ activity). Similar observations regarding indenoisoquinoline TDP1 inhibitors were reported by Conda-Sherian37 and Lv,46 in which compounds bearing a bulky group on the lactam side chain (e.g., morpholine, imidazole, Boc-protected amino, N-methylpiperidine) showed no TDP1 inhibition.
When compared to the lead compounds 4−5, the target compounds were, in general, somewhat weaker TDP1 inhibitors. The primary amines 25, 36, 45, 63, 64, and 71 were all TDP1 inhibitors. The most advantageous position for a single hydroxyl group in the D-ring for the highest TDP1 inhibitory activity is at C-8, followed by C-7 and C-10, which confer equal activity. Placing the hydroxyl group at C-9 is the least effective.
CONCLUSION
A series of 18 nitrated 7-, 8-, 9-, and hydroxyindenoisoquinolines were designed as dual Top1−TDP1 inhibitors based on the ideas that (1) they are the demethylated analogues of the lead compounds 4 and 5, both of which are potent dual Top1−TDP1 inhibitors, (2) the hydroxyl group might serve as a replacement of the methoxy group in the lead compounds 4 and 5 and provide a point of attachment for prodrug modules, targeting moieties, or antibodies, and (3) both the 3-nitro group in the A-ring and the hydroxyl group in the D-ring of indenoisoquinolines were previously shown separately to enhance both Top1 inhibitory and antiproliferative activities of Top1 poisons. All of the target compounds were synthesized using the condensation of homophthalic anhydride and Schiff bases bearing appropriate substituents, and were evaluated for dual Top1−TDP1 inhibition and antiproliferative activities. The 9-hydroxyl series comprised of compounds 68−71 was the most active Top1 inhibitors in the series. Among them, bromide 68 exhibited many unique features: (1) its anti-Top1 potency and cytotoxicity surpassed those of CPT, (2) it acted as a Top1 poison and was not a Top1 suppressor at a concentration as high as 100 μM, whereas many other indenoisoquinolines start to suppress DNA cleavage at this concentration, (3) it induced a drug−DNA−Top1 ternary cleavage complex that reversed slower than that of camptothecin, and (4) there was no evidence of DNA alkylation. All the indenoisoquinolines bearing a terminal amine on their lactam side chains had TDP1 inhibitory activities, while their morpholine and imidazole analogues were generally inactive. The relative TDP1 inhibitory potencies of amines 45 (++), 63 (+++), 64 (++), and 71 (+) indicate that the most advantageous location for installation of a single hydroxyl group in the D-ring is at C-8. The 8-hydroxyindenoisoquinoline amine 63 is the most potent dual Top1−TDP1 inhibitor reported in the present studies (++++ for Top1 and + ++ for TDP1). Its activities are also comparable to the bis(indenoisoquinoline) 72 (Figure 7), which is the most potent dual Top1−TDP1 inhibitor. These results detail successful potency optimization of phenolic indenoisoquinolines as Top1−TDP1 inhibitors.
EXPERIMENTAL SECTION
General.
Solvents and reagents were purchased from commercial vendors and were used without further purification. Melting points were determined using capillary tubes with a Mel-Temp apparatus and were uncorrected. Infrared spectra were obtained as films on KBr pellets with CHCl3 as the solvent, using a PerkinElmer 1600 series or Spectrum One FTIR spectrometer, and were baseline-corrected. 1H NMR spectra were recorded at 300 or 500 MHz, using Bruker ARX300 or Bruker Avance 500 spectrometers with a QNP probe or TXI 5 mm/BBO probe, respectively. Mass spectral analyses were performed at the Purdue University Campus-Wide Mass Spectrometry Center. ESI-MS studies were performed using a FinniganMAT XL95 (FinniganMAT Corp., Bremen, Germany) mass spectrometer. The instrument was calibrated to a resolution of 10000 with a 10% valley between peaks using the appropriate polypropylene glycol standards. EI/CI-MS studies were performed using a Hewlett-Packard Engine or GCQ FinniganMAT mass spectrometer. APCI-MS studies were performed using an Agilent 6320 ion trap mass spectrometer. Combustion microanalyses were performed at Midwest Microlab, LLC (Indianapolis, IN). All reported values were within 0.4% of calculated values. Analytical thin layer chromatography was carried out on Baker-flex silica gel IB2-F plastic-backed TLC plates. Compounds were visualized with both short and long wavelength UV light and ninhydrin staining unless otherwise specified. Silica gel flash column chromatography was performed using 40−63 μM flash silica gel. Analytical HPLC studies were performed using a Waters 1525 binary HPLC pump with a Waters 2487 dual wavelength absorbance detector and an injection volume of 10 μL. A Sunrise C18 5 μM 100 Å reverse-phase column with dimensions of 15 cm × 4.6 mm (ES Industries) was used for all analytical HPLC experiments. For purities estimated by HPLC, the major peak accounted for ≥95% of the combined total peak area when monitored by a UV detector at 254 nm unless otherwise specified. All yields refer to isolated compounds.
4-Nitrohomophthalic Acid.47
Homophthalic acid (12, 10.0 g, 55.5 mmol) was slowly added to fuming HNO3 (35 mL) at 0 °C. After the addition was complete, the clear-yellow solution was warmed to room temperature, and stirring was continued for 5 h, during which a white precipitate was formed. Ice was slowly added to the cloudy mixture until the volume was doubled. The mixture was sonicated until the ice melted, and then it was filtered. The residue was washed with cold H2O to afford the product 13 as an amorphous white solid (8.83 g, 71%): mp 219−220 °C (lit.47 217 °C). 1H NMR (300 MHz, DMSO-d6) δ 8.61 (d, J = 2.6 Hz, 1 H), 8.37 (dd, J = 2.6 and 5.6 Hz, 1 H), 7.67 (d, J = 8.4 Hz, 1 H), 4.10 (s, 2 H).
4-Nitrohomophthalic Anhydride (14).48
Diacid 13 (8.83 g, 39.2 mmol) was diluted in acetyl chloride (30 mL), and the mixture was stirred and heated at reflux for 4 h. The yellow solution was then evaporated to dryness. The resulting residue was filtered and washed with 50% CHCl3 in hexane to provide the product 14 as pale-yellow solid (5.75 g, 71%); mp 147−148 °C (lit.48 154−155 °C). 1H NMR (300 MHz, DMSO-d6) δ 8.66 (s, 1 H), 8.55 (d, J = 8.1 Hz, 1 H), 7.73 (d, J = 7.9 Hz, 1 H), 4.41 (s, 2 H).
Benzylvanillin (16).49
Vanillin (15, 5.00 g, 32.9 mmol), benzyl bromide (5.90 g, 34.5 mmol), and K2CO3 (9.08 g, 65.7 mmol) were diluted in DMF (50 mL). The yellow mixture was stirred at room temperature for 2 h, poured into a solution of Et2O−H2O (200 mL, 1:1), and stirred for 10 min. The ethereal layer was separated. The aqueous layer was extracted with Et2O (50 mL × 2). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The crude residue was washed with hexane to provide the pure product 16 as a white solid (7.91 g, 99%); mp 49−51 °C (lit.49 61 °C). 1H NMR (300 MHz, CDCl3) δ 9.84 (s, 1 H), 7.44−7.36 (m, 7 H), 7.00 (d, J = 8.2 Hz, 1 H), 5.25 (s, 2 H), 3.95 (s, 3 H).
N-[4′-(Benzyloxy)-3′-methoxybenzylidene]-3-bromopropyl-1-amine (17).33
Benzylvanillin 16 (3.00 g, 12.4 mmol), 3-bromopropylamine hydrobromide (3.12 g, 14.2 mmol), Et3N (1.39 g, 13.6 mmol), and Na2SO4 (3.52 g, 24.8 mmol) were diluted in CHCl3 (100 mL). The mixture was stirred at room temperature for 16 h and then washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to provide the product 17 as a yellow syrup (4.49 g, 100% + residual solvent). IR (film) 2937, 2841, 1646, 1602, 1587, 1512, 1456, 1415, 1270, 1233, 743 cm−1. 1H NMR (300 MHz, CDCl3) δ 8.22 (s, 1 H), 7.45−7.30 (m, 6 H), 7.10 (dd, J = 1.7 and 6.4 Hz, 1 H), 6.90 (d, J = 8.2 Hz, 1 H), 5.20 (s, 2 H), 3.95 (s, 3 H), 3.74 (t, J = 6.1 Hz, 2 H), 3.51 (t, J = 6.5 Hz, 2 H), 2.27 (m, 2 H).
cis-3-[4-(Benzyloxy)-3-methoxyphenyl]-N-(3-bromopropyl)-4-carboxy-3,4-dihydro-7-nitro-1(2H)-isoquinolone (18).
Schiff base 17 (7.48 g, 20.7 mmol) was diluted in CHCl3 (50 mL) and cooled to 0 °C, and anhydride 14 (4.28 g, 20.7 mmol) was added. The red mixture was stirred at 0 °C for 2 h and then at room temperature for 3 h. The mixture was filtered, and the residue was washed with 50% CHCl3 in hexane to afford the product 18 as a white solid (7.55 g, 64%); mp 145−146 °C. IR (film) 3079, 1748, 1621, 1520, 1493, 1418, 1349, 1177, 755 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.06 (d, J = 2.4 Hz, 1 H), 8.36 (dd, J = 2.5 and 6.0 Hz, 1 H), 7.87 (d, J = 8.8 Hz), 7.35−7.28 (m, 5 H), 6.71 (d, J = 8.9 Hz, 1 H), 6.50 (m, 2 H), 5.13 (d, J = 6.4 Hz, 1 H), 5.04 (s, 2 H), 4.80 (d, J = 6.4 Hz, 1 H), 4.04 (m, 1 H), 3.63 (s, 3 H), 3.48 (m, 2 H), 3.28 (m, 1 H), 2.31 (m, 1 H), 2.18 (m, 1 H). ESI-MS m/z (rel intensity) 569/571 ([MH]+, 27/28). HRMS (+ESI) calcd for C27H25BrN2O7 MH+, 569.0923; found. 569.0932.
9-(Benzyloxy)-6-(3-bromopropyl)-8-methoxy-3-nitro-5H-indeno-[1,2-c]isoquinoline-5,11(6H)-dione (19).
Cis acid 18 (1.50 g, 2.63 mmol) was diluted in SOCl2 (50 mL) and the mixture was stirred at room temperature for 16 h. The red solution was evaporated to dryness. The resulting residue was diluted with CHCl3 (50 mL) and treated slowly with saturated aqueous NaHCO3 (100 mL). The mixture was stirred at room temperature for 10 min, and the two layers were separated. The aqueous layer was extracted with CHCl3 (100 mL × 2). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with CHCl3 to provide the product 19 as a reddish-brown solid (722 mg, 50%); mp 218−220 °C (dec). IR (film) 1677, 1611, 1504, 1427, 1336, 1300, 746 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.15 (d, J = 2.4 Hz, 1 H), 8.78 (d, J = 9.0 Hz, 1 H), 8.47 (dd, J = 2.5 and 6.7 Hz, 1 H), 7.44−7.36 (m, 6 H), 7.31 (d, J = 1.8 Hz, 1 H), 5.26 (s, 2 H), 4.71 (t, J = 7.0 Hz, 2 H), 4.06 (s, 3 H), 3.74 (t, J = 5.7 Hz, 2 H), 2.51 (m, 2 H). ESI-MS m/z (rel intensity) 549/551 ([MH]+, 42/53). HRMS (+ESI) calcd for C27H21BrN2O6 MH+, 549.0661; found, 549.0672.
9-(Benzyloxy)-8-methoxy-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11[6H]-dione (20).
Bromide 19 (320 mg, 0.58 mmol) and morpholine (304 mg, 3.49 mmol) were diluted in 1,4-dioxane (30 mL). The mixture was stirred at room temperature for 16 h and then evaporated to dryness. The resulting residue was diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with a gradient of MeOH in CHCl3 (2% to 4%) to provide the product 20 as a brown solid (140 mg, 44%); mp 233−234 °C (dec). IR (film) 1673, 1612, 1557, 1507, 1428, 1333, 1300, 667 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.15 (d, J = 2.5 Hz, 1 H), 8.75 (d, J = 9.0 Hz, 1 H), 8.45 (dd, J = 2.4 and 6.5 Hz, 1 H), 7.47−7.35 (m, 5 H), 7.31 (s, 1 H), 7.21 (s, 1 H), 5.25 (s, 2 H), 4.63 (t, J = 7.3 Hz, 2 H), 4.01 (s, 3 H), 3.66 (m, 4 H), 2.60 (t, J = 6.7 Hz, 2 H), 2.46 (m, 4 H), 2.14 (m, 2 H). ESI-MS m/z (rel intensity) 556 ([MH]+, 100). HRMS (+ESI) calcd for C31H29N3O7 MH+, 556.2084; found, 556.2076.
9-Hydroxy-8-methoxy-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (21).
Compound 20 (50 mg, 0.090 mmol) was diluted in aqueous HBr (48 wt %, 35 mL), and the mixture was heated at 70 °C for 5 h, during which it turned to a black emulsion. The cooled mixture was concentrated to remove HBr. The concentrate was then diluted with acetone (10 mL) and concentrated again. This procedure was done three times. The final mixture was filtered through an HPLC filter paper, and the residue was washed with acetone and CHCl3 to provide the desired product 21 as a black solid (47.5 mg, 97%); mp >400 °C. IR (film) 3206, 1697, 1641, 1614, 1558, 1506, 1427, 1335, 792 cm−1. 1H NMR (300 MHz, CDCl3) δ 10.45 (s, 1 H), 9.49 (s, 1 H), 8.87 (s, 1 H), 8.65 (d, J = 8.8 Hz, 1 H), 8.55 (d, J = 8.2 Hz, 1 H), 7.23 (s, 1 H), 7.08 (s, 1 H), 4.63 (m, 2 H), 4.00−3.95 (m, 7 H), 3.61 (m, 4 H), 3.10 (m, 2 H), 2.28 (m, 2 H). ESI-MS m/z (rel intensity) 447 (MH+, 89). HRMS (+ESI) calcd C24H23N3O7 for MH+, 466.1614; found, 466.1618; HPLC purity: 100% (MeOH, 100%), 97.8% (MeOH−H2O, 90:10). Anal. Calcd for C24H24BrN3O7·0.1H2O: C, 52.59; H, 4.45; N, 7.67. Found: C, 52.28; H, 4.24; N, 7.30.
6-(3-(1H-Imidazol-1-yl)propyl)-9-(benzyloxy)-8-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (22).
Bromide 19 (100 mg, 0.182 mmol) and imidazole (124 mg, 1.82 mmol) were diluted in 1,4-dioxane (30 mL). The mixture was heated at 70 °C for 16 h and then evaporated to dryness. The resulting residue was diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with 4% MeOH in CHCl3 to provide the product 22 as a brown solid (36.2 mg, 37%); mp 235−236 °C (dec). IR (film) 1662, 1612, 1555, 1424, 1291, 746 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.17 (d, J = 2.3 Hz, 1 H), 8.77 (d, J = 9.0 Hz, 1 H), 8.48 (dd, J = 2.4 and 6.6 Hz, 1 H), 7.61 (s, 1 H), 7.46−7.30 (m, 5 H), 7.12 (s, 1 H), 7.05 (s, 1 H), 6.85 (s, 1 H), 5.24 (s, 2 H), 4.61 (t, J = 6.8 Hz, 2 H), 4.27 (t, J = 6.5 Hz, 2 H), 3.86 (s, 3 H), 2.42 (m, 2 H). ESI-MS m/z (rel intensity) 537 (MH+, 100). HRMS (+ESI) calcd for C30H24N4O6 MH+, 537.1774; found, 537.1784.
6-(3-(1H-Imidazol-1-yl)propyl)-9-hydroxy-8-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (23).
Compound 22 (50 mg, 0.093 mmol) was diluted in aqueous HBr (48 wt %, 35 mL) and the mixture was heated at 70 °C for 5 h, during which it turned to a brown emulsion. The cooled mixture was concentrated to remove HBr. The concentrate was then diluted with acetone (10 mL) and concentrated again. This procedure was done three times. The final mixture was filtered through an HPLC filter paper, and the residue was washed with acetone and CHCl3 to provide the desired product 23 as a pale-brown solid (24.6 mg, 50%); mp >400 °C. IR (film) 3398, 1680, 1609, 1557, 1492, 1429, 1385, 1338, 859 cm−1. 1H NMR (300 MHz, CDCl3) δ 10.43 (s, 1 H), 9.11 (s, 1 H), 8.86 (s, 1 H), 8.65 (d, J = 9.2 Hz, 1 H), 8.54 (d, J = 6.5 Hz, 1 H), 7.83 (s, 1 H), 7.68 (s, 1 H), 7.23 (s, 1 H), 7.08 (s, 1 H), 4.60 (m, 2 H), 4.37 (m, 2 H), 3.97 (s, 3 H), 2.50 (m, 2 H, under the water peak). ESI-MS m/z rel intensity) 466 (MH+, 100). HRMS (+ESI) calcd for C23H18N4O6 MH+, 447.1305; found, 447.1303. HPLC purity: 100% (MeOH, 100%), 96.7% (MeOH−H2O, 90:10). Anal. Calcd for C23H19BrN4O6·0.5H2O: C, 51.51; H, 3.76; N, 10.45. Found: C, 51.33; H, 3.46; N, 10.30.
6-(3-Azidopropyl)-9-(benzyloxy)-8-methoxy-3-nitro-5H-indeno-[1,2-c]isoquinoline-5,11(6H)-dione (24).
Bromide 19 (150 mg, 0.273 mmol) and NaN3 (178 mg, 2.73 mmol) were diluted in DMSO (50 mL), and the mixture was stirred at room temperature for 16 h. The deep-red solution was diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with CHCl3, to provide the product 24 as a deep-red solid (36.2 mg, 26%); mp 205−207 °C (dec). IR (film) 2090, 1673, 1610, 1579, 1502, 1427, 847 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.14 (d, J = 2.3 Hz, 1 H), 8.75 (d, J = 9.0 Hz, 1 H), 8.45 (dd, J = 2.3 and 6.7 Hz, 1 H), 7.48−7.35 (m, 5 H), 7.28 (s, 1 H), 5.26 (s, 2 H), 4.61 (t, J = 6.9 Hz, 2 H), 4.07 (s, 3 H), 3.79 (t, J = 5.7 Hz, 2 H), 2.16 (m, 2 H). ESI-MS m/z (rel intensity) 512 (MH+, 100). HRMS (+ESI) calcd for C27H21N5O6 MH+, 512.1570; found, 512.1576.
6-(3-Aminopropyl)-9-hydroxy-8-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (25).
Azide 24 (30 mg, 0.059 mmol) was diluted in benzene (50 mL), and triethyl phosphite (29.2 mg, 0.176 mmol) was added. The mixture was heated at reflux for 16 h and then allowed to cool to room temperature. Aqueous HBr (48 wt %, 30 mL) was added, and the mixture was heated at 70 °C for 5 h, during which it turned to a brown/red emulsion. The cooled mixture was concentrated to remove benzene and HBr. The concentrate was then diluted with acetone (10 mL) and concentrated again. This procedure was done three times. The final mixture was filtered through an HPLC filter paper, and the residue was washed with acetone and CHCl3 to provide the desired product 25 as a brown solid (26.0 mg, 93%); mp 285−287 °C (dec). IR (film) 3243, 2848, 1705, 1641, 1614, 1562, 1488, 1336, 1207, 1133, 868 cm−1. 1H NMR (300 MHz, CDCl3) δ 10.41 (s, 1 H), 8.83 (d, J = 2.3 Hz, 1 H), 8.60 (d, J = 9.0 Hz, 1 H), 8.51 (dd, J = 2.5 and 6.5 Hz, 1 H), 7.74 (br s, 3 H), 7.19 (s, 1 H), 7.03 (s, 1 H), 4.58 (m, 2 H), 3.98 (s, 3 H), 3.01 (m, 2 H), 2.14 (m, 2 H). ESI-MS m/z (rel intensity) 396 (MH+, 100). HRMS (+ESI) calcd for C20H19N3O6 MH+, 396.1196; found, 396.1199. HPLC purity: 100% (MeOH, 100%), 98.6% (MeOH−H2O, 90:10). Anal. Calcd for C20H18BrN3O6: C, 50.44; H, 3.81; N, 8.82. Found: C, 50.13; H, 3.75; N, 8.59.
Benzylisovanillin (27).50
Isovanillin 26 (5.00 g, 32.9 mmol), benzyl bromide (5.90 g, 34.5 mmol), and K2CO3 (9.08 g, 65.7 mmol) were diluted in DMF (50 mL). The yellow mixture was stirred at room temperature for 2 h and then poured into a solution of Et2O−H2O (200 mL, 1:1) and stirred for 5 min. The ethereal layer was separated. The aqueous layer was extracted with Et2O (50 mL × 2). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The crude residue was washed with hexane to provide the pure product 27 as a white solid (7.34 g, 92%); mp 48−49 °C (lit.50 61−62 °C). 1H NMR (300 MHz, CDCl3) δ 9.82 (s, 1 H), 7.48−7.46 (m, 4 H), 7.39−7.36 (m, 3 H), 7.01 (d, J = 8.5 Hz, 1 H), 5.20 (s, 2 H), 3.97 (s, 3 H).
N-[3′-(Benzyloxy)-4′-methoxybenzylidene]-3-bromo-1-propylamine (28).33
Benzylisovanillin 27 (3.00 g, 12.4 mmol), 3-bromopropylamine hydrobromide (3.12 g, 14.2 mmol), Et3N (1.39 g, 13.6 mmol), and Na2SO4 (3.52 g, 24.8 mmol) were diluted in CHCl3 (100 mL). The mixture was stirred at room temperature for 16 h and then washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to provide the product 28 as a yellow syrup (4.49 g, 100% + residual solvent). IR (film) 2935, 2837, 1642, 1602, 1583, 1512, 1437, 1265, 1137, 741 cm−1. 1H NMR (300 MHz, CDCl3) δ 8.21 (s 1 H), 7.49−7.44 (m, 3 H), 7.40−7.33 (m, 3 H), 7.21 (dd, J = 6.4 and 1.8 Hz, 1 H), 6.92 (d, J = 8.2 Hz, 1 H), 5.20 (s, 2 H), 3.93 (s, 3 H), 3.73 (t, J = 5.4 Hz, 2 H), 3.51 (t, J = 6.5 Hz, 2 H), 2.26 (m, 2 H).
cis-3-[3-(Benzyloxy)-4-methoxyphenyl]-N-(3-bromopropyl)-4-carboxy-3,4-dihydro-7-nitro-1(2H)-isoquinolone (29).
Schiff base 28 (4.486 g, 12.38 mmol) was diluted in CHCl3 (50 mL), and the mixture was cooled to 0 °C for 10 min. Anhydride 14 (2.56 g, 12.38 mmol) was then added. The red mixture was stirred at 0 °C for 2 h and then at room temperature for 3 h. The mixture was filtered, and the residue was washed with 50% CHCl3 in hexane to provide the product 29 as a white solid (4.867 g, 69%); mp 165−167 °C. IR (film) 3078, 1754, 1630, 1519, 1352, 1259, 1272, 1142, 701 cm−1. 1H NMR (300 MHz, CDCl3) δ 8.99 (d, J = 2.3 Hz, 1 H), 8.28 (dd, J = 8.4 and 2.5 Hz, 1 H), 7.74 (d, J = 8.4 Hz, 1 H), 7.41−7.35 (m, 6 H), 6.70 (d, J = 8.4 Hz, 1 H), 6.48 (d, J = 8.5 Hz, 1 H), 6.38 (d, J = 2.0 Hz, 1 H), 5.23 (d, J = 14 Hz, 1 H), 5.04 (s, 2 H), 4.64 (d, J = 6.5 Hz, 1 H), 3.94 (m, 1 H), 3.83 (s, 3 H), 3.43 (m, 2 H), 3.22 (m, 1 H), 2.20 (m, 1 H), 2.10 (s, 1 H). ESI-MS m/z (rel intensity) 445 ([MH − COOH − Br]+, 100). HRMS (+ESI) calcd for C27H25BrN2O7 [MH − COOH − Br]+, 445.1763; found, 445.1771.
8-(Benzyloxy)-6-(3-bromopropyl)-9-methoxy-3-nitro-5H-indeno-[1,2-c]isoquinoline-5,11[6H]-dione (30).
Cis acid 29 (1.50 g, 2.63 mmol) was diluted in SOCl2 (50 mL) at 0 °C and stirred for 16 h, during which the mixture warmed to room temperature. The red solution was evaporated to dryness, and the residue was diluted with CHCl3 (50 mL) and treated slowly with saturated NaHCO3 (100 mL). The mixture was stirred at room temperature for 10 min, and the two layers were separated. The aqueous layer was extracted with CHCl3 (100 mL × 2). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with CHCl3 to provide the product 30 as a greenish-brown solid (578 mg, 40%); mp 239−240 °C. IR (film) 1671, 1613, 1557, 1507, 1488, 1335, 1297, 746 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.15 (d, J = 3.3 Hz, 1 H), 8.78 (d, J = 9.0 Hz, 1 H), 8.47 (dd, J = 6.7 and 2.4 Hz, 1 H), 7.44−7.31 (m, 5 H), 6.91−6.88 (m, 2 H), 5.30 (s, 2 H), 4.59 (t, J = 7.9 Hz, 2 H), 4.02 (s, 3 H), 3.62 (t, J = 6.0 Hz, 2 H), 2.35 (m, 2 H). APCI-MS m/z (rel intensity) 549/550 ([MH]+, 100). HRMS (+EI/CI) calcd for C27H21BrN2O6 MH+, 549.0661; found, 549.0655.
8-(Benzyloxy)-9-methoxy-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11[6H]-dione (31).
Bromide 30 (87 mg, 0.16 mmol) and morpholine (83 mg, 5.2 mmol) were diluted in 1,4-dioxane (30 mL). The mixture was stirred at room temperature for 16 h and then evaporated to dryness. The resulting residue was diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with a gradient of MeOH in CHCl3 (0% to 2%), to provide the product 31 as a deep-purple solid (46 mg, 52%) after being triturated with ether; mp 229−230 °C. IR (film) 3399, 1698, 1672, 1614, 1556, 1330, 1304 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.14 (d, J = 2.3 Hz, 1 H), 8.76 (d, J = 9.0 Hz, 1 H), 8.45 (dd, J = 6.7 and 2.4 Hz, 1 H), 7.42−7.35 (m, 5 H), 7.31 (s, 1 H), 7.12 (s, 1 H), 5.30 (s, 2 H), 4.47 (t, J = 7.3 Hz, 2 H), 4.03 (s, 3 H), 3.55 (m, 4 H), 2.41 (m, 6 H), 1.88 (m, 2 H). ESI-MS m/z (rel intensity) 556 ([MH]+, 100). HRMS (+ESI) calcd for C31H29N3O7 MH+, 556.2084; found, 556.2082. HPLC purity: 99.2% (MeOH, 100%), 99.0% (MeOH−H2O, 90:10).
8-Hydroxy-9-methoxy-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (32).
Compound 31 (25 mg, 0.045 mmol) was diluted in aqueous HBr (48 wt %, 30 mL), and the mixture was heated at 70 °C for 5 h, during which it turned to a brown emulsion. The cooled mixture was concentrated to remove HBr. The concentrate was then diluted with acetone (10 mL) and concentrated again. This procedure was done three times. The final mixture was filtered under vacuum, and the residue was washed with acetone and CHCl3 to provide the desired product 32 as a pale plum-colored solid (24.6 mg, 100%); mp 288−289 °C. IR (film) 3259, 1682, 1608, 1556, 1506, 1390, 1334, 1275, 1209, 861 cm−1. 1H NMR (300 MHz, CDCl3) δ 10.28 (s, 1 H), 9.58 (s, 1 H), 8.84 (d, J = 2.4 Hz, 1 H), 8.65 (d, J = 8.9 Hz, 1 H), 8.55 (dd, J = 2.5 and 6.5 Hz, 1 H), 7.30 (s, 1 H), 7.26 (s, 1 H), 4.50 (m, 2 H), 3.92 (s, 3 H), 3.63 (m, 4 H), 3.45 (m, 4 H), 3.10 (m, 2 H), 2.24 (m, 2 H). ESI-MS m/z (rel intensity) 466 (MH+, 100). HRMS (+ESI) calcd for C24H23N3O7 MH+, 466.1614; found, 466.1606. HPLC purity: 98.0% (MeOH, 100%), 97.2% (MeOH−H2O, 90:10). Anal. Calcd for C24H24BrN3O7: C, 52.76; H, 4.43; N, 7.69. Found: C, 52.51; H, 4.39; N, 7.35.
6-(3-(1H-Imidazol-1-yl)propyl)-8-(benzyloxy)-9-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (33).
Bromide 30 (100 mg, 0.182 mmol) and imidazole (124 mg, 1.82 mmol) were diluted in 1,4-dioxane (30 mL). The mixture was heated at 70 °C for 16 h and then evaporated to dryness. The resulting residue was diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with 4% MeOH in CHCl3, to provide the product 33 as a brown solid (35.0 mg, 36%); mp 236−237 °C (dec). IR (film) 1671, 1612, 1557, 1507, 1334, 1299 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.14 (d, J = 2.3 Hz, 1 H), 8.76 (d, J = 9.0 Hz, 1 H), 8.47 (dd, J = 2.4 and 6.6 Hz, 1 H), 7.52 (s, 1 H), 7.38−7.36 (m, 4 H), 7.32 (m, 2 H), 7.08 (s, 1 H), 6.94 (s, 1 H), 6.83 (s, 1 H), 5.25 (s, 2 H), 4.40 (t, J = 6.5 Hz, 2 H), 4.03 (s, 3 H), 4.01 (t, J = 7.1 Hz, 2 H), 2.00 (m, 2 H). ESI-MS m/z (rel intensity) 537 (MH+, 100). HRMS (+ESI) calcd for C30H24N4O6 MH+, 537.1774; found, 537.1780.
6-(3-(1H-Imidazol-1-yl)propyl)-8-hydroxy-9-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (34).
Compound 33 (30 mg, 0.056 mmol) was diluted in aqueous HBr (48 wt %, 30 mL) and the mixture was stirred at 70 °C for 5 h, during which it turned to a brown emulsion. The cooled mixture was concentrated to remove HBr. The concentrate was then diluted with acetone (10 mL) and concentrated again. This procedure was done three times. The final mixture was filtered under vacuum, and the residue was washed with acetone and CHCl3 to provide the desired product 34 as a pale plum-colored solid (23.6 mg, 80%); mp 285−286 °C. IR (film) 3301, 1669, 1616, 1562, 1334, 1265, 1165, 854 cm−1. 1H NMR (300 MHz, CDCl3) δ 10.27 (s, 1 H), 9.12 (s, 1 H), 8.80 (d, J = 2.2 Hz, 1 H), 8.62 (d, J = 8.9 Hz, 1 H), 8.52 (dd, J = 2.3 and 6.7 Hz, 1 H), 7.83 (s, 1 H), 7.68 (s, 1 H), 7.22 (s, 2 H), 4.48 (m, 2 H), 4.42 (t, J = 6.7 Hz, 2 H), 3.91 (s, 3 H), 2.41 (m, 2 H). ESI-MS m/z (rel intensity) 447 (MH+, 100). HRMS (+ESI) calcd for C23H18N4O6 MH+, 447.1305; found, 447.1308. HPLC purity: 97.8% (MeOH, 100%), 97.3% (MeOH−H2O, 90:10). Anal. Calcd for C23H19BrN4O6· 0.1H2O: C, 52.21; H, 3.66; N, 10.59. Found: C, 51.91; H, 3.55; N, 10.32.
6-(3-Azidopropyl)-8-(benzyloxy)-9-methoxy-3-nitro-5H-indeno-[1,2-c]isoquinoline-5,11(6H)-dione (35).
Bromide 30 (150 mg, 0.273 mmol) and NaN3 (178 mg, 2.73 mmol) were diluted in DMSO (30 mL), and the mixture was stirred at room temperature for 16 h. The deep-red solution was diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with CHCl3 to provide the product 35 as a brown solid (40.5, 29%); mp 222−224 °C (dec). IR (film) 2099, 1713, 1666, 1614, 1508, 1340, 751 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.14 (d, J = 2.2 Hz, 1 H), 8.77 (d, J = 9.0 Hz, 1 H), 8.46 (dd, J = 2.4 and 6.5 Hz, 1 H), 7.44−7.37 (m, 5 H), 7.31 (s, 2 H), 5.33 (s, 2 H), 4.52 (t, J = 7.1 Hz, 2 H), 4.02 (s, 3 H), 3.67 (t, J = 5.9 Hz, 2 H), 2.01 (m, 2 H). ESI-MS m/z (rel intensity) 512 (MH+, 28).
6-(3-Aminopropyl)-8-hydroxy-9-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (36).
Azide 35 (30 mg, 0.059 mmol) was diluted in benzene (50 mL), and triethyl phosphite (29.2 mg, 0.176 mmol) was added. The mixture was heated at reflux for 16 h and then allowed to cool to room temperature. Aqueous HBr (48 wt %, 30 mL) was added, and the reaction mixture was heated at 70 °C for 5 h, during which it turned to a brown/red emulsion. The cooled mixture was concentrated to remove benzene and HBr. The concentrate was then diluted with acetone (10 mL) and concentrated again. This procedure was done three times. The final mixture was filtered under vacuum, and the residue was washed with acetone and CHCl3 to provide the desired product 36 as a brown solid (24.6 mg, 88%); mp 338−340 °C (dec). IR (film) 3218, 2853, 1678, 1609, 1555, 1472, 1336, 1178, 855 cm−1. 1H NMR (300 MHz, CDCl3) δ 10.34 (s, 1 H), 8.85 (d, J = 2.5 Hz, 1 H), 8.66 (d, J = 9.0 Hz, 1 H), 8.55 (dd, J = 2.5 and 6.4 Hz, 1 H), 7.73 (br s, 3 H), 7.28 (s, 2 H), 4.49 (m, 2 H), 3.92 (s, 3 H), 2.99 (m, 2 H), 2.08 (m, 2 H). ESI-MS m/z (rel intensity) 396 (MH+, 44). HRMS (+ESI) calcd for C20H17N3O6 MH+, 396.1196; found, 396.1191. HPLC purity: 100% (MeOH, 100%), 98.6% (MeOH−H2O, 90:10). Anal. Calcd for C20H18BrN3O6·0.5H2O: C, 49.50; H, 3.95; N, 8.66. Found: C, 49.39; H, 3.77; N, 8.48.
2-(Benzyloxy)benzaldehyde (39).51
Compound 38 (5.00 g, 0.041 mol) was diluted in DMF (30 mL), followed by addition of benzyl bromide (7.70 g, 0.045 mol) and K2CO3 (11.3 g, 0.082 mmol). The yellow mixture was stirred at room temperature for 3 h until it turned cloudy white. The mixture was then diluted in H2O (100 mL) and extracted with ether (50 mL × 3). The combined extract was washed with H2O (50 mL × 3) and brine (50 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to yield the product 39 as a colorless liquid (8.69 g, 100%). 1H NMR (300 MHz, CDCl3) δ 10.57 (s, 1 H), 7.88 (d, J = 2.0 Hz, 1 H), 7.86 (d, J = 1.6 Hz, 1 H), 7.54−7.38 (m, 5 H), 7.07−7.05 (m, 2 H), 5.20 (s, 2 H).
N-[2-(Benzyloxy)benzylidene]-3-bromo-1-propylamine (40).
3-Bromopropylamine hydrobromide (3.56 g, 16.2 mmol) was diluted in CHCl3 (30 mL) and Et3N (1.64 g, 16.2 mmol). The mixture was stirred until the salt dissolved completely, and then compound 39 (3.00 g, 14.1 mmol) and Na2SO4 (4.02 g, 28.3 mmol) were added. The mixture was stirred at room temperature for 16 h, diluted with CHCl3 (100 mL), and then washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to yield the product 40 as a yellow syrup (4.69 g, 100%). IR (film) 2894, 1637, 1599, 1452, 1245, 754 cm−1. 1H NMR (300 MHz, CDCl3) δ 8.82 (s, 1 H), 7.96 (d, J = 7.7 Hz, 1 H), 7.43−7.34 (m, 6 H), 7.00 (m, 2 H), 5.14 (s, 2 H), 3.78 (dt, J = 0.9 and 5.4 Hz, 2 H), 3.52 (t, J = 6.6 Hz, 2 H), 2.28 (m, 2 H). ESI-MS m/z (rel intensity) 354/356 (MNa+, 99/100).
cis-4-Carboxy-3,4-dihydro-N-(3-bromopropyl)-3-[2-(benzyloxy)phenyl]-7-nitro-1(2H)-isoquinolone (41).
Schiff base 40 (4.69 g, 0.014 mmol) was diluted in CHCl3 (50 mL) at 0 °C, and anhydride 14 (2.92 g, 0.014 mmol) was added. The red mixture was stirred at 0 °C for 1 h and then at room temperature for 3 h. The cloudy orange mixture was filtered, and the residue was washed with 50% CHCl3 in hexane to provide the product 41 as a pale-yellow solid (3.48 g, 46%); mp 145−147 °C. IR (film) 3075, 1749, 1621, 1579, 1525, 1486, 1353, 1162, 760 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.65 (d, J = 2.6 Hz, 1 H), 8.38 (dd, J = 2.6 and 5.9 Hz, 1 H), 7.73 (dd, J = 0.7 and 8.0 Hz, 1 H), 7.46−7.31 (m, 5 H), 7.22 (m, 1 H), 7.05 (d, J = 8.3 Hz, 1 H), 6.74 (d, J = 3.9 Hz, 2 H), 5.74 (d, J = 6.9 Hz, 1 H), 5.05 (s, 2 H), 3.88 (m, 1 H), 3.51 (m, 3 H), 3.01 (m, 1 H), 2.11 (m, 1 H), 1.96 (m, 1 H); ESI-MS m/z (rel intensity) 415 ([MH − COOH − Br]+, 100). HRMS (+ESI) calcd for C26H23BrN2O6 (MH − COOH − Br)+, 415.1658; found, 415.1654.
6-(3-Bromopropyl)-7-hydroxy-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione (42).
Cis acid 41 (0.50 g, 0.93 mmol) was diluted in SOCl2 (30 mL), and the mixture was stirred at room temperature for 5 h. The resulting yellow solution was evaporated to dryness. The yellow syrup was diluted in 1,2-dichloroethane (30 mL) at 0 °C, followed an addition of AlCl3 (0.37 g, 2.78 mmol), and the mixture was stirred for 15 min. Stirring was continued at room temperature for 7 h. The black mixture was then diluted with CHCl3 (50 mL) and washed with cold 6 N HCl (100 mL). The aqueous layer was extracted with CHCl3 (50 mL × 2). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The yellow organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue was triturated with ether, filtered, and washed with excess ether to provide the product 42 in high purity as a yellow amorphous solid (253 mg, 63%); mp 172–175 °C. IR (film) 1719, 1674, 1606, 1516, 1342 cm−1. 1H NMR (300 MHz, CDCl3) δ 9.58 (d, J = 9.3 Hz, 1 H), 9.29 (d, J = 2.6 Hz, 1 H), 8.61 (dd, J = 2.6 and 6.7 Hz, 1 H), 8.01 (dd, J = 1.2 and 7.1 Hz, 1 H), 7.70 (dt, J = 1.3 and 7.2 Hz, 1 H), 7.53–7.42 (m, 2 H), 4.72 (t, J = 7.2 Hz, 2 H), 3.50 (t, J = 5.9 Hz, 1 H), 2.69 (m, 2 H). ESI-MS m/z (rel intensity) 351 ([MH – HBr]+, 100).
7-Hydroxy-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione (43).
Bromide 42 (70 mg, 0.16 mmol) and morpholine (71 mg, 0.81 mmol) were diluted in 1,4-dioxane (30 mL). The yellow mixture was heated at 70 °C for 16 h. The solution was then diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue was triturated with acetone, filtered, and washed with ether to provide the product 43 as a brown solid (68.7 mg, 97%); mp 216–218 °C. IR (film) 3413, 1719, 1675, 1606, 1515, 1342, 1117 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 9.39 (d, J = 9.3 Hz, 1 H), 8.99 (d, J = 2.4 Hz, 1 H), 8.68 (dd, J = 2.6 and 6.5 Hz, 1 H), 8.12 (d, J = 8.3 Hz, 1 H), 7.79 (t, J = 7.5 Hz, 1 H), 7.59 (d, J = 8.3 Hz, 1 H), 7.50 (t, J = 7.5 Hz, 1 H), 4.60 (m, 2 H), 3.17 (m, 4 H), 2.04–1.98 (m, 8 H). ESI-MS m/z (rel intensity) 436 (MH+, 100). HRMS (+ESI) calcd for C23H21N3O6 MH+, 436.1509; found, 436.1505. HPLC purity: 95.0% (MeOH, 100%), 96.7% (MeOH–H2O, 90:10).
6-(3-(1H-Imidazol-1-yl)propyl)-7-hydroxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (44).
Bromide 42 (100 mg, 0.23 mmol) and imidazole (79 mg, 1.16 mmol) were diluted in 1,4-dioxane (30 mL). The yellow mixture was heated at 70 °C for 16 h. The solution was then diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue was triturated with acetone, filtered, and washed with ether to provide the product 44 as a yellow solid (77.2 mg, 79%); mp 173–175 °C. IR (film) 1717, 1675, 1606, 1515, 1342, 1119, 756 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 9.38 (d, J = 9.3 Hz, 1 H), 8.97 (d, J = 2.5 Hz, 1 H), 8.67 (dd, J = 2.5 and 6.7 Hz, 1 H), 7.85 (d, J = 7.7 Hz, 1 H), 7.76 (t, J = 7.4 Hz, 1 H), 7.56 (d, J = 8.0 Hz, 1 H), 7.38 (t, J = 8.5 Hz, 1 H), 4.37 (m, 2 H), 4.14 (m, 2 H). ESI-MS m/z (rel intensity) 417 (MH+, 100). HRMS (+ESI) calcd for C22H16N4O5 MH+, 417.1199; found, 417.1202.
6-(3-Aminopropyl)-7-hydroxy-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione Hydrochloride (45).
Bromide 42 (100 mg, 0.23 mmol) and NaN3 (45 mg, 0.70 mmol) were diluted in DMSO (30 mL). The yellow mixture was stirred at room temperature for 16 h. The solution was then diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue was diluted in benzene (30 mL), and triethyl phosphite (116 mg, 0.70 mmol) was added. The yellow solution was heated at reflux for 16 h and then cooled to room temperature. Methanolic HCl (3 M, 40 mL) was added. The mixture was heated at 70 °C for 3 h and then concentrated to dryness. The residue was triturated with acetone, filtered, and washed with acetone to provide the product 45 as a yellow solid (56.5 mg, 60%); mp 272–274 °C (dec). IR (film) 3283, 1712, 1671, 1603, 1509, 1484, 1341, 1284, 760 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 9.41 (d, J = 9.3 Hz, 1 H), 9.00 (d, J = 2.6 Hz, 1 H), 8.70 (dd, J = 2.6 and 6.6 Hz, 1 H), 8.03 (d, J = 8.1 Hz, 1 H), 7.86 (br s, 3 H), 7.81 (t, J = 7.4 Hz, 1 H), 7.61 (d, J = 7.3 Hz, 1 H), 7.51 (t, J = 7.3 Hz, 1 H), 4.47 (t, J = 6.5 Hz, 2 H), 2.87 (m, 2 H), 2.32 (m, 2 H). ESI-MS m/z (rel intensity) 366 (MH+, 100). HRMS (+ESI) calcd for C19H15N3O5 MH+, 366.1090; found, 366.1093. HPLC purity: 96.4% (MeOH, 100%), 98.1% (MeOH–H2O, 90:10).
3-(Benzyloxy)benzaldehyde (47).52
Compound 46 (5.00 g, 0.041 mol) was diluted in DMF (50 mL), followed by addition of benzyl bromide (7.70 g, 0.045 mol) and K2CO3 (11.3 g, 0.082 mmol). The yellow mixture was stirred at room temperature for 3 h until it turned cloudy white. The mixture was then diluted in H2O (100 mL) and extracted with ether (50 mL × 3). The combined extract was washed with H2O (50 mL × 3) and brine (50 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to yield a pale-yellow syrup, which solidified upon standing at room temperature. The solid was washed with hexane and filtered to provide the product 47 as a white solid (8.69 g, 100%); mp 43–45 °C. 1H NMR (300 MHz, CDCl3) δ 9.98 (s, 1 H), 7.49–7.35 (m, 8 H), 7.28 (d, J = 2.5 Hz, 1 H), 5.13 (s, 2 H).
N-[3-(Benzyloxy)benzylidene]-3-bromo-1-propylamine (48).
3-Bromopropylamine hydrobromide (6.45 g, 29.4 mmol) was diluted in CHCl3 (50 mL) and Et3N (2.98 g, 29.4 mmol). The mixture was stirred for 5 min, and then compound 47 (5.00 g, 23.6 mmol) and Na2SO4 (6.69 g, 4.71 mmol) were added. The mixture was stirred at room temperature for 16 h and then washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to yield the product 48 as a pale-yellow syrup (7.57 g, 97%). IR (film) 2847, 1646, 1580, 1445, 1263, 689 cm−1. 1H NMR (300 MHz, CDCl3) δ 8.31 (s, 1 H), 7.46–7.28 (m, 8 H), 7.08 (dd, J = 1.0 and 1.4 Hz, 1 H), 5.11 (s, 2 H), 3.78 (dt, J = 0.9 and 5.3 Hz, 2 H), 3.52 (t, J = 6.5 Hz, 2 H), 2.29 (m, 2 H). ESI-MS m/z (rel intensity) 332/334 (MH+, 100/99).
cis-3-[3-(Benzyloxy)phenyl]-N-(3-bromopropyl)-4-carboxy-3,4-dihydro-7-nitro-1(2H)-isoquinolone (49).
Schiff base 48 (4.57 g, 13.8 mmol) was diluted in CHCl3 (50 mL) at 0 °C, and anhydride 14 (2.85 g, 13.8 mmol) was added. The red mixture was stirred at 0 °C for 1 h and then at room temperature for 2 h. The cloudy orange mixture was filtered, and the residue was diluted in CHCl3 (50 mL) and heated at reflux for 20 min. The cloudy mixture was then filtered and washed with hot CHCl3 to provide the product 49 as an off-white solid (5.08 g, 68%); mp 166–167 °C. IR (film) 3082, 1749, 1628, 1586, 1524, 1268, 1181, 698 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.59 (d, J = 2.4 Hz, 1 H), 8.25 (dd, J = 2.4 and 5.9 Hz, 1 H), 7.46 (d, J = 8.4 Hz, 1 H), 7.36–7.26 (m, 5 H), 7.20 (t, J = 7.9 Hz, 1 H), 6.87 (dd, J = 2.2 and 5.9 Hz, 1 H), 6.71 (s, 1 H), 6.63 (d, J = 7.8 Hz, 1 H), 5.10 (d, J = 5.4 Hz, 1 H), 5.00 (s, 2 H), 4.14 (m, 1 H), 3.81 (q, J = 6.7 H, 1 H), 3.59 (t, J = 7.1 Hz, 2 H), 2.96 (m, 1 H), 2.21 (m, 2 H). ESI-MS m/z (rel intensity) 495/497 ([MH – COOH]+, 91/97). HRMS (+ESI) calcd for C26H23BrN2O6 (MH – COOH)+, 495.0919; found, 495.0911.
Mixture of 8- and 10-(Benzyloxy)-6-(3-bromopropyl)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (51 and 52).
Cis acid 49 (1.48 g, 2.86 mmol) was diluted in SOCl2 (50 mL), and the mixture was heated at reflux for 4 h. The resulting red solution was evaporated to dryness. The residue was triturated with ether and filtered to provide an approximately 2:1 (by 1H NMR) mixture of 8- and 10-(benzyloxy)indenoisoquinolines (51 and 52, respectively) as an orange solid (0.32 g, 22%). Identities of the products were determined by 1H NMR. The product mixture was subjected to the next SN2 reaction without further purification.
8-(Benzyloxy)-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione (53) and 10-(Benzyloxy)-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (54).
A mixture of bromides 51 and 52 (150 mg, 0.29 mmol) and morpholine (151 mg, 1.73 mmol) was diluted in 1,4-dioxane (30 mL), and the mixture was heated at 70 °C for 16 h. The mixture was then evaporated to dryness. The resulting residue was diluted with CHCl3 (100 mL) and washed with H2O (100 mL × 4) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with a gradient of MeOH in CHCl3 (0% to 1%), to provide the 8-benzyloxy product (53) as an orange solid (60.3 mg, 40%) and the 10-benxyloxy product (54) as a red solid (48.6 mg, 32%).
8-Benzyloxy (53): mp 250–252 °C. IR (film) 1669, 1615, 1560, 1508, 1337, 1114, 841 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 9.17 (d, J = 2.4 Hz, 1 H), 8.87 (d, J = 8.9 Hz, 1 H), 8.48 (dd, J = 2.4 and 6.5 Hz, 1 H), 7.65 (d, J = 8.0 Hz, 1 H), 7.45–7.41 (m, 6 H), 6.94 (dd, J = 1.7 and 6.4 Hz, 1 H), 5.16 (s, 2 H), 4.62 (t, J = 7.2 Hz, 2 H), 3.58 (t, J = 4.4 Hz, 4 H), 2.56 (t, J = 6.1 Hz, 2 H), 2.44 (m, 4 H), 2.06 (m, 2 H). ESI-MS m/z (rel intensity) 526 (MH+, 100). HRMS (+ESI) calcd for C30H27N3O6 MH+, 526.1978; found, 526.1984.
10-Benzyloxy (54): mp 254–256 °C. IR (film) 1678, 1610, 1558, 1332, 1117, 746 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 9.18 (d, J = 2.4 Hz, 1 H), 8.95 (d, J = 9.0 Hz, 1 H), 8.49 (dd, J = 2.5 and 6.5 Hz, 1 H), 7.54–7.48 (m, 3 H), 7.45–7.34 (m, 4 H), 7.10 (d, J = 8.5 Hz, 1 H), 5.35 (s, 2 H), 4.65 (t, J = 7.9 Hz, 2 H), 3.71 (t, J = 4.4 Hz, 4 H), 2.60 (t, J = 6.2 Hz, 2 H), 2.49 (m, 4 H), 2.08 (m, 2 H). ESI-MS m/z (rel intensity) 526 (MH+, 100). HRMS (+ESI) calcd C30H27N3O6 for MH+, 526.1978; found, 526.1974.
8-(Hydroxy)-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione (55).
Compound 53 (55 mg, 0.10 mmol) was diluted with aqueous HBr (48 wt %, 30 mL), and the mixture was heated at 70 °C for 3 h. The red mixture was diluted with CHCl3 (10 mL) and acetone (10 mL) and then concentrated in vacuo. This procedure was done three times. The final concentrate was filtered through an HPLC filter paper, and the residue was washed with acetone and CHCl3 to provide the desired product 55 as an orange solid (51.6 mg, 96%); mp >400 °C. IR (film) 3353, 1668, 1615, 1562, 1506, 1435, 1338, 1258, 847 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.87 (s, 1 H), 9.48 (br s, 1 H), 8.90 (d, J = 2.4 Hz, 1 H), 8.77 (d, J = 9.0 Hz, 1 H), 8.60 (dd, J = 2.4 and 6.6 Hz, 1 H), 7.56 (d, J = 8.1 Hz, 1 H), 7.25 (s, 1 H), 6.90 (d, J = 8.0 Hz, 1 H), 4.56 (m, 2 H), 3.98 (m, 2 H), 3.64 (m, 2 H), 3.44 (m, 4 H), 3.09 (m, 2 H), 2.25 (m, 2 H). ESI-MS m/z (rel intensity) 436 (MH+, 100). HRMS (+ESI) calcd for C23H21N3O6 MH+, 436.1509; found, 436.1514. HPLC purity: 98.0% (MeOH, 100%), 97.4% (MeOH–H2O, 90:10). Anal. Calcd for C23H22BrN3O6·0.75H2O: C, 52.14; H, 4.47; N, 7.93. Found: C, 51.79; H, 4.19; N, 7.64.
10-(Hydroxy)-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione (56).
Compound 54 (45 mg, 0.086 mmol) was diluted with aqueous HBr (48 wt %, 30 mL), and the mixture was heated at 70 °C for 3 h. The red mixture was diluted with CHCl3 (10 mL) and acetone (10 mL) and then concentrated in vacuo. This procedure was done 3 times. The final concentrate was filtered through an HPLC filter paper, and the residue was washed with acetone and CHCl3 to provide the desired product 56 as a brown solid (19 mg, 43%); mp 314–316 °C (dec). IR (film) 2917, 1688, 1611, 1557, 1506, 1430, 1344, 1172, 802 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.72 (s, 1 H), 9.48 (br s, 1 H), 8.91 (d, J = 2.4 Hz, 1 H), 8.81 (d, J = 8.9 Hz, 1 H), 8.61 (dd, J = 2.5 and 6.5 Hz, 1 H), 7.50 (t, J = 7.8 Hz, 1 H), 7.39 (d, J = 7.7 Hz, 1 H), 7.09 (d, J = 8.4 Hz, 1 H), 4.56 (m, 2 H), 3.98 (m, 2 H), 3.63 (m, 2 H), 3.43 (m, 4 H), 3.09 (m, 2 H), 2.26 (m, 2 H). ESI-MS m/z (rel intensity) 436 (MH+, 100). HRMS (+ESI) calcd for C23H21N3O6 MH+, 436.1509; found, 436.1500. HPLC purity: 98.8% (MeOH, 100%), 98.3% (MeOH–H2O, 90:10). Anal. Calcd for C23H22BrN3O6: C, 53.50; H, 4.29; N, 8.14. Found: C, 53.24; H, 4.29; N, 7.91.
6-(3-(1H-Imidazol-1-yl)propyl)-8-(benzyloxy)-3-nitro-5H-indeno-[1,2-c]isoquinoline-5,11(6H)-dione (57) and 6-(3-(1H-Imidazol-1-yl)propyl)-10-(benzyloxy)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (58).
A mixture of bromides 51 and 52 (100 mg, 0.19 mmol) and imidazole (131 mg, 1.9 mmol) was diluted in 1,4-dioxane (30 mL), and the mixture was heated at 70 °C for 16 h. The mixture was then concentrated to dryness. The resulting residue was diluted in CHCl3 (100 mL) and washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated, adsorbed onto SiO2, and purified by flash column chromatography (SiO2), eluting with a gradient of MeOH in CHCl3 (0–2%) to provide the 8-benzyloxy product (57) as an orange solid (35.6 mg, 37%) and the 10-benxyloxy product (58) as an orange solid (17.8 mg, 18%).
8-Benzyloxy (57): mp 265–267 °C. IR (film) 1670, 1613, 1560, 1505, 1339, 1229, 842 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.89 (d, J = 2.4 Hz, 1 H), 8.76 (d, J = 9.0 Hz, 1 H), 8.59 (d, J = 9.1 Hz, 1 H), 7.78 (s, 1 H), 7.64 (d, J = 8.2 Hz, 1 H), 7.52 (d, J = 7.0 Hz, 2 H), 7.44–7.35 (m, 3 H), 7.26 (m, 2 H), 7.17 (d, J = 8.0 Hz, 1 H), 6.90 (m, 1 H), 5.28 (s, 2 H), 4.51 (m, 2 H), 4.16 (m, 2 H), 2.26 (m, 2 H). ESI-MS m/z (rel intensity) 507 (MH+, 100). HRMS (+ESI) calcd for MH+, 507.1668; found, 507.1662.
10-Benzyloxy (58): mp 260–261 °C. IR (film) 1671, 1612, 1562, 1502, 1454, 1331, 1264, 840 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.88 (m, 1 H), 8.78 (d, J = 9.1 Hz, 1 H), 8.54 (dd, J = 2.4 and 6.6 Hz, 1 H), 7.80 (br s, 1 H), 7.55 (d, J = 7.6 Hz, 2 H), 7.47–7.31 (m, 6 H), 6.94 (m, 2 H), 5.33 (s, 2 H), 4.48 (t, J = 7.6 Hz, 2 H), 4.26 (t, J = 6.6 Hz, 2 H), 2.24 (m, 2 H). ESI-MS m/z (rel intensity) 507 (MH+, 100). HRMS (+ESI) calcd for C29H22N4O5 MH+, 507.1668; found, 507.1673.
6-(3-(1H-Imidazol-1-yl)propyl)-8-hydroxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (59).
Compound 57 (41 mg, 0.081 mmol) was diluted in aqueous HBr (30 mL, 48 wt %), and the mixture was heated at 70 °C for 3 h. The red mixture was diluted in CHCl3 (10 mL) and acetone (10 mL) and then concentrated in vacuo. This procedure was done 3 times. The final concentrate was filtered through an HPLC filter paper, and the residue was washed with acetone and CHCl3 to provide the product 59 as a bright-orange solid (36.3 mg, 90%); mp 329–331 °C (dec). IR (film) 3064, 1669, 1615, 1562, 1506, 1340, 1255, 835 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.84 (s, 1 H), 9.04 (s, 1 H), 8.89 (d, J = 2.4 Hz, 1 H), 8.76 (d, J = 9.0 Hz, 1 H), 8.60 (dd, J = 2.5 and 6.5 Hz, 1 H), 7.80 (s, 1 H), 7.63 (s, 1 H), 7.55 (d, J = 8.0 Hz, 1 H), 7.18 (s, 1 H), 6.89 (d, J = 9.5 Hz, 1 H), 4.54 (m, 2 H), 4.40 (t, J = 6.6 Hz, 2 H), 2.42 (m, 2 H). ESI-MS m/z (rel intensity) 417 (MH+, 100). HRMS (+ESI) calcd for C22H16N4O5 MH+, 417.1199; found, 417.1195. Anal. Calcd for C22H17BrN4O5·0.5H2O: C, 52.19; H, 3.58; N, 11.07. Found: C, 51.82; H, 3.44; N, 10.67.
6-(3-(1H-Imidazol-1-yl)propyl)-10-hydroxy-3-nitro-5H-indeno-[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (60).
Compound 58 (27 mg, 0.053 mmol) was diluted in aqueous HBr (30 mL, 48 wt %), and the mixture was heated at 70 °C for 3 h. The red mixture was diluted in CHCl3 (10 mL) and acetone (10 mL) and then concentrated in vacuo. This procedure was done 3 times. The final concentrate was filtered through an HPLC filter paper, and the residue was washed with acetone and CHCl3 to provide the product 60 as a bright-orange solid (25.3 mg, 95%); mp 337–339 °C (dec). IR (film) 3347, 2946, 1694, 1666, 1615, 1563, 1503, 1428, 1336, 1287, 852 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.70 (s, 1 H), 9.11 (s, 1 H), 8.90 (d, J = 2.4 Hz, 1 H), 8.79 (d, J = 8.9 Hz, 1 H), 8.60 (dd, J = 2.5 and 6.5 Hz, 1 H), 7.83 (s, 1 H), 7.68 (s, 1 H), 7.46 (t, J = 7.9 Hz, 1 H), 7.28 (d, J = 7.4 Hz, 1 H), 7.08 (d, J = 8.5 Hz, 1 H), 4.54 (m, 2 H), 4.42 (t, J = 7.1 Hz, 2 H), 2.39 (m, 2 H). ESI-MS m/z (rel intensity) 417 (MH+, 100). HRMS (+ESI) calcd for C22H16N4O5 MH+, 417.1199; found, 417.1203. Anal. Calcd for C22H17BrN4O5·0.25H2O: C, 52.66; H, 3.52; N, 11.16. Found: C, 52.81; H, 3.49; N, 10.79.
6-(3-Aminopropyl)-8-hydroxy-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione Hydrobromide (63) and 6-(3-Amino-propyl)-10-hydroxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione Hydrobromide (64).
A mixture of bromides 51 and 52 (100 mg, 0.19 mmol) and NaN3 (63 mg, 0.96 mmol) was diluted in DMSO (30 mL), and the mixture was stirred at room temperature for 16 h. The mixture was then diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, concentrated, and purified by flash column chromatography (SiO2), eluting with CHCl3 to provide azides 61 and 62 as orange solids. Both compounds were diluted separately in benzene (20 mL) and triethyl phosphite (36 mg, 0.22 mmol) and heated at reflux for 16 h. Aqueous HBr (48 wt %, 20 mL) was added to the cooled solutions, and stirring was continued at reflux for 4 h, during which orange precipitates formed. The two mixtures were concentrated, washed with acetone (10 mL) and CHCl3 (10 mL), and concentrated again. The mixtures were then filtered and the residues were washed thoroughly with acetone and CHCl3 to provide the amines 63 (32 mg, 37% in two steps) and 64 (28 mg, 33% in two steps) as orange solids.
Amine 63: mp 353–355 °C (dec). IR (film) 3248, 1666, 1613, 1595, 1507, 1480, 1388, 1341, 1295, 864 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.86 (s, 1 H), 8.87 (d, J = 2.4 Hz, 1 H), 8.74 (d, J = 9.0 Hz, 1 H), 8.58 (dd, J = 2.6 and 6.4 Hz, 1 H), 7.73 (br s, 3 H), 7.53 (d, J = 8.0 Hz, 1 H), 7.24 (d, J = 1.5 Hz, 1 H), 6.88 (dd, J = 1.5 and 6.5 Hz, 1 H), 4.55 (t, J = 6.7 Hz, 2 H), 3.00 (m, 2 H), 2.11 (m, 2 H). ESI-MS m/z (rel intensity) 366 (MH+, 14); HRMS (+ESI) calcd for C19H15N3O5 MH+, 366.1090; found, 366.1087. HPLC purity: 97.1% (MeOH, 100%).
Amine 64: mp 337–339 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ 10.69 (s, 1 H), 8.90 (d, J = 2.4 Hz, 1 H), 8.79 (d, J = 8.9 Hz, 1 H), 8.60 (dd, J = 2.6 and 6.4 Hz, 1 H), 7.71 (br s, 3 H), 7.49–7.44 (m, 1 H), 7.39–7.35 (m, 1 H), 7.08 (d, J = 8.3 Hz, 1 H), 4.55 (t, J = 6.8 Hz, 2 H), 3.01 (m, 2 H), 2.10 (m, 2 H). MALDI-MS m/z (rel intensity) 366 (MH+, 100). HRMS (+ESI) calcd for C19H15N3O5 MH+, 366.1090; found, 366.1109. HPLC purity: 96.5% (MeOH, 100%).
N-[4-(Benzyloxy)benzylidene]-3-bromo-1-propylamine (66).
3-Bromopropylamine hydrobromide (3.56 g, 16.2 mmol) was diluted in CHCl3 (50 mL) and Et3N (1.64 g, 16.2 mmol). The mixture was stirred for 5 min, and then compound 65 (3.00 g, 14.1 mmol) and Na2SO4 (4.02 g, 28.3 mmol) were added. The mixture was stirred at room temperature for 16 h and then washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to yield the product 66 as a pale-yellow syrup (4.69 g, 100% + residual solvent). IR (film) 2839, 1645, 1605, 1509, 1246, 1166, 830 cm−1. 1H NMR (300 MHz, CDCl3) δ 8.26 (s, 1 H), 7.68 (dd, J = 1.8 and 6.9 Hz, 2 H), 7.45–7.33 (m, 5 H), 7.02 (dd, J = 1.9 and 6.9 Hz, 2 H), 5.11 (s, 2 H), 3.74 (dt, J = 0.9 and 6.2 Hz, 2 H), 3.51 (t, J = 6.6 Hz, 2 H), 2.27 (m, 2 H). ESI-MS m/z (rel intensity) 332/334 (MH+, 100/97).
cis-4-Carboxy-3,4-dihydro-N-(3-bromopropyl)-3-[4-(benzyloxy)-phenyl]-7-nitro-1(2H)-isoquinolone (67).
Schiff base 66 (4.69 g, 14.1 mmol) was diluted in CHCl3 (50 mL) at 0 °C, and anhydride 14 (2.80 g, 13.5 mmol) was added. The red mixture was stirred at 0 °C for 2 h, and then at room temperature for 2 h. The cloudy orange mixture was filtered, and the residue was washed with 50% CHCl3 in hexane to provide the product 67 as an off-white solid (5.18 g, 71%); mp 140–141 °C. IR (film) 3076, 1727, 1630, 1525, 1347, 1187, 738 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 8.71 (d, J = 2.6 Hz, 1 H), 8.39 (dd, J = 2.6 and 6.0 Hz, 1 H), 7.92 (d, J = 8.2 Hz, 1 H), 7.40–7.30 (m, 5 H), 6.92–6.83 (m, 4 H), 5.19 (d, J = 6.4 Hz, 1 H), 5.03 (d, J = 6.3 Hz, 1 H), 4.98 (s, 2 H), 3.90 (m, 1 H), 3.59 (m, 2 H), 3.03 (m, 1 H), 2.16 (m, 1 H), 2.04 (m, 1 H). ESI-MS m/z (rel intensity) 415 ([MH – COOH – Br]+, 100). HRMS (+ESI) calcd for C26H23BrN2O6 MH+, 539.0818; found, 539.0812.
6-(3-Bromopropyl)-9-hydroxy-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione (68).
Cis acid 67 (1.00 g, 1.85 mmol) was diluted in SOCl2 (50 mL), and the mixture was stirred at room temperature for 5 h. The resulting yellow solution was evaporated to dryness. The yellow syrup was diluted in 1,2-dichloroethane (40 mL) at 0 °C, followed an addition of AlCl3 (0.75 g, 5.56 mmol), and the mixture was stirred for 15 min. Stirring was continued at room temperature for 8 h. The black mixture was then diluted with CHCl3 (50 mL) and THF (30 mL) and washed with cold 6 N HCl (100 mL). The aqueous layer was extracted with CHCl3 (50 mL × 2). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The red organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue was triturated with CHCl3, filtered, and washed with excess CHCl3 to provide the product 68 in high purity as a deep-red solid (262 mg, 33%); mp 281–283 °C (dec). IR (film) 3273, 1659, 1613, 1531, 1345, 1270, 755 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.82 (s, 1 H), 8.83 (d, J = 2.1 Hz, 1 H), 8.61 (d, J = 9.0 Hz, 1 H), 8.51 (d, J = 9.2 Hz, 1 H), 7.74 (d, J = 8.6 Hz, 1 H), 6.99 (s, 1 H), 6.89 (d, J = 8.6 Hz, 1 H), 4.54 (m, 2 H), 3.78 (t, J = 6.3 Hz, 2 H), 2.33 (m, 2 H). ESI-MS m/z (rel intensity) 428/430 (M+, 99/100). HRMS (+ESI) calcd for C19H13BrN2O5 M+, 428.0008; found, 428.0000.
9-Hydroxy-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione Hydrobromide (69).
Phenol 68 (50 mg, 0.12 mmol) and morpholine (51 mg, 0.58 mmol) were diluted in THF (20 mL). The red mixture was heated at 70 °C for 16 h. The cooled solution was diluted with 3 M methanolic HBr (20 mL) and stirred at room temperature for 4 h. The deep-red solution was concentrated and filtered through an HPLC filter paper. The residue was washed with CHCl3 to provide the product 69 as a deep-brown solid (49 mg, 83%); mp 295–297 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ 10.87 (s, 1 H), 9.53 (br s, 1 H), 8.86 (d, J = 2.4 Hz, 1 H), 8.66 (d, J = 9.0 Hz, 1 H), 8.56 (dd, J = 2.4 and 6.5 Hz, 1 H), 7.70 (d, J = 8.4 Hz, 1 H), 7.04 (d, J = 2.4 Hz, 1 H), 6.93 (dd, J = 2.3 and 6.0 Hz, 1 H), 4.52 (m, 2 H), 3.98 (m, 2 H), 3.64 (m, 2 H), 3.38 (m, 4 H), 3.08 (m, 2 H), 2.21 (m, 2 H). ESI-MS m/z (rel intensity) 436 (MH+, 100). HRMS (+ESI) calcd for C23H21N3O6 MH+, 436.1509; found, 436.1508. Anal. Calcd for C23H22BrN3O6: C, 53.50; H, 4.29; N, 8.14. Found: C, 53.51; H, 4.23; N, 7.98.
6-(3-(1H-Imidazol-1-yl)propyl)-7-hydroxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione hydrochloride (70).
Bromide 68 (70 mg, 0.16 mmol) and imidazole (67 mg, 0.98 mmol) were diluted in THF (20 mL). The red mixture was heated at 70 °C for 16 h. The cooled solution was diluted with 3 M methanolic HCl (20 mL) and stirred at room temperature for 4 h. The deep-red mixture was concentrated and filtered through an HPLC filter paper. The residue was washed with CHCl3 to afford the product 70 as a brown solid (42.6 mg, 58%); mp 323–325 °C (dec). IR (film) 1668, 1611, 1503, 1428, 1334, 1263 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 9.38 (d, J = 9.3 Hz, 1 H), 8.97 (d, J = 2.5 Hz, 1 H), 8.67 (dd, J = 2.5 and 6.7 Hz, 1 H), 7.85 (d, J = 7.7 Hz, 1 H), 7.76 (t, J = 7.4 Hz, 1 H), 7.56 (d, J = 8.0 Hz, 1 H), 7.38 (t, J = 8.5 Hz, 1 H), 4.37 (m, 2 H), 4.14 (m, 2 H). ESI-MS m/z (rel intensity) 417 (MH+, 100). HRMS (+ESI) calcd for C22H16N4O5 MH+, 417.1199; found, 417.1202. HPLC purity: 100% (MeOH, 100%), 100% (MeOH–H2O, 70:30).
6-(3-Aminopropyl)-9-hydroxy-3-nitro-5H-indeno[1,2-c]-isoquinoline-5,11(6H)-dione Hydrobromide (71).
Bromide 68 (100 mg, 0.23 mmol) and NaN3 (75 mg, 1.15 mmol) were diluted in DMSO (30 mL) and stirred at room temperature for 16 h. The solution was then diluted with H2O (100 mL) and extracted with CHCl3 (50 mL × 3). The combined extract was washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to provide the crude intermediate azide. The compound was then diluted in THF (20 mL), followed by addition of PPh3 (181 mg, 0.69 mmol), and the mixture was heated at reflux for 16 h. The cooled deep-brown solution was diluted with 3 M methanolic HBr (20 mL), and stirring was continued at 70 °C for 4 h. The bright-red solution was evaporated, rediluted in CHCl3 (10 mL), and let sit for 16 h at 0 °C, during which a precipitate formed. The solution was then filtered through an HPLC filter paper, and the residue was washed thoroughly with CHCl3 to provide the product 71 (33.1 mg, 32%) as a deep-red solid; mp 360–362 °C (dec). 1H NMR (300 MHz, DMSO-d6) δ 10.83 (s, 1 H), 8.83 (d, J = 2.3 Hz, 1 H), 8.61 (d, J = 9.0 Hz, 1 H), 8.52 (dd, J = 2.5 and 6.4 Hz, 1 H), 7.74–7.67 (m, 4 H), 6.99 (d, J = 2.3 Hz, 1 H), 6.91 (dd, J = 2.2 and 6.1 Hz, 1 H), 4.52 (t, J = 7.0 Hz, 2 H), 2.99 (m, 2 H), 2.08 (m, 2 H). APCI-MS m/z (rel intensity) 366 ([MH – NH3]+, 100). HRMS (+ESI) calcd for C19H15N3O5 MH+, 366.1090; found, 366.1082; HPLC purity: 99.6% (MeOH, 100%).
Topoisomerase I-Mediated DNA Cleavage Reactions.
Human recombinant Top1 was purified from baculovirus as previously described.53 DNA cleavage reactions were performed as previously reported with the exception of the DNA substrate.31 Briefly, a 117-bp DNA oligonucleotide (Integrated DNA Technologies) encompassing the previously identified Top1 cleavage sites in the 161-bp fragment from pBluescript SK(−) phagemid DNA was employed. This 117-bp oligonucleotide contains a single 5′-cytosine overhang, which was 3′-end-labeled by fill-in reaction with [α−32P]dGTP in React 2 buffer [50 mM Tris-HCl (pH 8.0), 100 mM MgCl2, and 50 mM NaCl] with 0.5 unit of DNA polymerase I (Klenow fragment, New England BioLabs). Unincorporated [32P]dGTP was removed using mini Quick Spin DNA columns (Roche, Indianapolis, IN), and the eluate containing the 3′-end-labeled DNA substrate was collected. Approximately 2 nM radiolabeled DNA substrate was incubated with recombinant Top1 in 20 μL of reaction buffer [10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at room temperature for 20 min in the presence of various concentrations of compounds. The reactions were terminated by adding SDS (0.5% final concentration), followed by addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of each reaction mixture were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a phosphorimager and ImageQuant software (Molecular Dynamics). For simplicity, cleavage sites were numbered as previously described in the 161-bp fragment.53
Gel-Based Assay Measuring the Inhibition of Recombinant TDP1.
A 5′-[32P]-labeled single-stranded DNA oligonucleotide containing a 3′-phosphotyrosine (N14Y) was generated as described by Dexheimer et al.21 The DNA substrate was then incubated with 5 pM recombinant TDP1 in the absence or presence of inhibitor for 15 min at room temperature in reaction buffer [50 mM Tris-HCl (pH 7.5), 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween-20]. Reactions were terminated by an addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to 16% denaturing PAGE, and gels were exposed after drying to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare), and densitometric analyses were performed using the ImageQuant software (GE Healthcare).
Supplementary Material
ACKNOWLEDGMENTS
This work was made possible by the National Institutes of Health (NIH) through support with research grants U01CA089566 and P30CA023168 and by a Purdue Research Foundation Grant. This research was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (Z01-BC006161). This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
ABBREVIATIONS USED
- APCI-MS
atmospheric-pressure chemical ionization mass spectrometry
- CPT
camptothecin
- DMSO-d6
dimethyl-d6 sulfoxide
- EI/CI-MS
electron impact/chemical ionization mass spectrometry
- ESI-MS
electrospray ionization mass spectrometry
- HRMS
high resolution mass spectrometry
- MALDI-MS
matrix-assisted laser desorption ionization mass spectrometry
- PMB-Cl
p-methoxybenzyl chloride
- TDP1
tyrosyl-DNA phosphodiesterase I
- Top1
human topoisomerase type IB
Footnotes
The authors declare the following competing financial interest(s): Mark Cushman is on the Board of Directors and is an investor in Linus Oncology, Inc., which has licensed indenoisoquinoline intellectual property owned by Purdue University. Neither Linus Oncology, Inc., nor any other commercial company sponsored or provided other direct financial support to the author or his laboratory for the research reported in this article. The remaining authors have no competing and/or relevant financial interest(s) to disclose.
ASSOCIATED CONTENT
Supporting Information
SMILES molecular formula strings. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Pommier Y Topoisomerase I Inhibitors: Camptothecins and Beyond. Nature Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- (2).Wang JC DNA Topoisomerases. Annu. Rev. Biochem 1996, 65, 635–692. [DOI] [PubMed] [Google Scholar]
- (3).Nguyen TX; Morrell A; Conda-Sheridan M; Marchand C; Agama K; Bermingam A; Stephen AG; Chergui A; Naumova A; Fisher R; O’Keefe BR; Pommier Y; Cushman M Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)–Topoisomerase I (Top1) Inhibitors. J. Med. Chem 2012, 55, 4457–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pommier Y; Leo E; Zhang HL; Marchand C DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol 2010, 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yang SW; Burgin AB; Huizenga BN; Robertson CA; Yao KC; Nash HA A Eukaryotic Enzyme that Can Disjoin Dead-End Covalent Complexes between DNA and Type I Topoisomerases. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Interthal H; Pouliott JJ; Champoux JJ The Tyrosyl-DNA phosphodiesterase Tdp1 Is a Member of the Phospholipase D Superfamily. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pouliot JJ; Robertson CA; Nash HA Pathways for Repair of Topoisomerase I Covalent Complexes in Saccharomyces cerevisiae. Genes to Cells 2001, 6, 677–687. [DOI] [PubMed] [Google Scholar]
- (8).Debethune L; Kohlhagen G; Grandas A; Pommier Y Processing of Nucleopeptides Mimicking the Topoisomerase I–DNA Covalent Complex by Tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2002, 30, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dexheimer TS; Antony S; Marchand C; Pommier Y Tyrosyl-DNA Phosphodiesterase as a Target for Anticancer Therapy. Anti-Cancer Agents Med. Chem 2008, 8, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Thomas CJ; Rahier NJ; Hecht SM Camptothecin: Current Perspectives. Bioorg. Med. Chem 2004, 12, 1585–1604. [DOI] [PubMed] [Google Scholar]
- (11).Husain I; Mohler JL; Seigler HF; Besterman JM Elevation of Topoisomerase-I Messenger-RNA, Protein, and Catalytic Activity in Human Tumors—Demonstration of Tumor-Type Specificity and Implications for Cancer Chemotherapy. Cancer Res. 1994, 54, 539–546. [PubMed] [Google Scholar]
- (12).Pfister TD; Reinhold WC; Agama K; Gupta S; Khin SA; Kinders RJ; Parchment RE; Tomaszewski JE; Doroshow JH; Pommier Y Topoisomerase I Levels in the NCI-60 Cancer Cell Line Panel Determined by Validated ELISA and Microarray Analysis and Correlation with Indenoisoquinoline Sensitivity. Mol. Cancer Ther 2009, 8, 1878–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Takashima H; Boerkoel CF; John J; Saifi GM; Salih MAM; Armstrong D; Mao YX; Quiocho FA; Roa BB; Nakagawa M; Stockton DW; Lupski JR Mutation of TDP1, Encoding a Topoisomerase I-Dependent DNA Damage Repair Enzyme, in Spinocerebellar Ataxia with Axonal Neuropathy. Nature Genet. 2002, 32, 267–272. [DOI] [PubMed] [Google Scholar]
- (14).Murai J; Huang SYN; Das BB; Dexheimer TS; Takeda S; Pommier Y Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Repairs DNA Damage Induced by Topoisomerases I and II and Base Alkylation in Vertebrate Cells. J. Biol. Chem 2012, 287, 12848–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Marchand C; Lea WA; Jadhav A; Dexheimer TS; Austin CP; Inglese J; Pommier Y; Simeonov A Identification of Phosphotyrosine Mimetic Inhibitors of Human Tyrosyl-DNA Phosphodiesterase I by a Novel AlphaScreen High-throughput Assay. Mol. Cancer Ther 2009, 8, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou T; Lee JW; Tatavarthi H; Lupski JR; Valerie K; Povirk LF Deficiency in 3′-Phosphoglycolate Processing in Human Cells with a Hereditary Mutation in Tyrosyl-DNA Phosphodiesterase (TDP1). Nucleic Acids Res. 2005, 33, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Barthelmes HU; Habermeyer M; Christensen MO; Mielke C; Interthal H; Pouliot JJ; Boege F; Marko D TDP1 Overexpression in Human Cells Counteracts DNA Damage Mediated by Topoisomerases I and II. J. Biol. Chem 2004, 279, 55618–55625. [DOI] [PubMed] [Google Scholar]
- (18).Davies DR; Interthal H; Champoux JJ; Hol WGJ Insights into Substrate Binding and Catalytic Mechanism of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) from Vanadate and Tung-state-Inhibited Structures. J. Mol. Biol 2002, 324, 917–932. [DOI] [PubMed] [Google Scholar]
- (19).Antony S; Marchand C; Stephen AG; Thibaut L; Agama KK; Fisher RJ; Pommier Y Novel High-Throughput Electro-chemiluminescent Assay for Identification of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) Inhibitors and Characterization of Furamidine (NSC 305831) as an Inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liao ZY; Thibaut L; Jobson A; Pommier Y Inhibition of Human Tyrosyl-DNA Phosphodiesterase by Aminoglycoside Anti-biotics and Ribosome Inhibitors. Mol. Pharmacol 2006, 70, 366–372. [DOI] [PubMed] [Google Scholar]
- (21).Dexheimer TS; Gediya LK; Stephen AG; Weidlich I; Antony S; Marchand C; Interthal H; Nicklaus M; Fisher RJ; Njar VC; Pommier Y 4-Pregnen-21-ol-3,20-dione-21-(4-bromo-benzenesulfonate) (NSC 88915) and Related Novel Steroid Derivatives as Tyrosyl-DNA Phosphodiesterase (Tdp1) Inhibitors. J. Med. Chem 2009, 52, 7122–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Huang S.-y. N.; Pommier Y; Marchand C Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibitors. Expert Opin. Ther. Pat 2011, 21, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Takagi M; Ueda J.-y.; Hwang J-H; Hashimoto J; Izumikawa M; Murakami H; Sekido Y; Shin-ya, K. Tyrosyl-DNA Phosphodiesterase 1 Inhibitor from an Anamorphic Fungus. J. Nat. Prod. 2012, 75, 764–767. [DOI] [PubMed] [Google Scholar]
- (24).Weidlich IE; Dexheimer T; Marchand C; Antony S; Pommier Y; Nicklaus MC Inhibitors of Human Tyrosyl-DNA Phospodiesterase (hTdp1) Developed by Virtual Screening Using Ligand-Based Pharmacophores. Bioorg. Med. Chem 2010, 18, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Weidlich IE; Dexheimer T; Marchand C; Antony S; Pommier Y; Nicklaus MC Virtual Screening Using Ligand-Based Pharmacophores for Inhibitors of Human Tyrosyl-DNA Phosphodiesterase (hTdp1). Bioorg. Med. Chem 2010, 18, 2347–2355. [DOI] [PubMed] [Google Scholar]
- (26).Sirivolu VR; Vernekar SKV; Marchand C; Naumova A; Chergui A; Renaud A; Stephen AG; Chen F; Sham YY; Pommier Y; Wang Z 5-Arylidenethioxothiazolidinones as Inhibitors of Tyrosyl-DNA Phosphodiesterase I. J. Med. Chem 2012, 55, 8671–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Takemura H; Rao VA; Sordet O; Furuta T; Miao Z-H; Meng L; Zhang H; Pommier Y Defective Mre11-Dependent Activation of Chk2 by Ataxia Telangiectasia Mutated in Colorectal Carcinoma Cells in Response to Replication-Dependent DNA Double-Strand Breaks. J. Biol. Chem 2006, 281, 30814–30823. [DOI] [PubMed] [Google Scholar]
- (28).Dasika GK; Lin SCJ; Zhao S; Sung P; Tomkinson A; Lee E DNA Damage-Induced Cell Cycle Checkpoints and DNA Strand Break Repair in Development and Tumorigenesis. Oncogene 1999, 18, 7883–7899. [DOI] [PubMed] [Google Scholar]
- (29).Reinhold WC; Sunshine M; Liu H; Varma S; Kohn KW; Morris J; Doroshow J; Pommier Y CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012, 72, 3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pommier Y; Cushman M The Indenoisoquinoline Non-camptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol. Cancer Ther. 2009, 8, 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Antony S; Agama KK; Miao ZH; Takagi K; Wright MH; Robles AI; Varticovski L; Nagarajan M; Morrell A; Cushman M; Pommier Y Novel Indenoisoquinolines NSC 725776 and NSC 724998 Produce Persistent Topoisomerase I Cleavage Complexes and Overcome Multidrug Resistance. Cancer Res. 2007, 67, 10397–10405. [DOI] [PubMed] [Google Scholar]
- (32).Doroshow JH; Ji JJ; Chen A; Allen D; Zhang Y; Lawrence SM; Pfister TD; Wang L; Redon E, C. E.; Bonner W; Speranza G; Weil MK; Eiseman J; Holleran JL; Kinders RL; Beumer JH; Parchment RE; Pommier Y; Tomaszewski JE; Kummar S Proof of Mechanism (POM) in the First-in-Human Trial of Two Novel Indenoisoquinoline, Non-camptothecin Topoisomerase I (TOP1) Inhibitors. In 48th Annual Meeting of the American Society of Clinical Oncology (ASCO), Chicago, IL, June 1–5, 2012; American Society of Clinical Oncology: 2318 Mill Road, Ste 800, Alexandra, VA 22314, 2012; Vol. 30. [Google Scholar]
- (33).Cinelli MA; Reddy PVN; Lv PC; Liang JH; Chen L; Agama K; Pommier Y; van Breemen RB; Cushman M Identification, Synthesis, and Biological Evaluation of Metabolites of the Experimental Cancer Treatment Drugs Indotecan (LMP400) and Indimitecan (LMP776) and Investigation of Isomerically Hydroxylated Indenoisoquinoline Analogues as Topoisomerase I Poisons. J. Med. Chem 2012, 55, 10844–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morrell A; Placzek M; Parmley S; Grella B; Antony S; Pommier Y; Cushman M Optimization of the Indenone Ring of Indenoisoquinoline Topoisomerase I Inhibitors. J. Med. Chem 2007, 50, 4388–4404. [DOI] [PubMed] [Google Scholar]
- (35).Morrell A; Placzek M; Parmley S; Antony S; Dexheimer TS; Pommier Y; Cushman M Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors: A Systematic Study and Optimization. J. Med. Chem 2007, 50, 4419–4430. [DOI] [PubMed] [Google Scholar]
- (36).Morrell A; Antony S; Kohlhagen G; Pommier Y; Cushman M A Systematic Study of Nitrated Indenoisoquinolines Reveals a Potent Topoisomerase I Inhibitor. J. Med. Chem 2006, 49, 7740–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Conda-Sheridan M; Reddy PVN; Morrell A; Cobb BT; Marchand C; Agama K; Chergui A; Renaud A; Stephen AG; Bindu LK; Pommier Y; Cushman M Synthesis and Biological Evaluation of Indenoisoquinolines That Inhibit Both Tyrosyl-DNA Phosphodiesterase I (Tdp1) and Topoisomerase I (Top1). J. Med. Chem 2013, 56, 182–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Morrell A; Placzek MS; Steffen JD; Antony S; Agama K; Pommier Y; Cushman M Investigation of the Lactam Side Chain Length Necessary for Optimal Indenoisoquinoline Topoisomerase I Inhibition and Cytotoxicity in Human Cancer Cell Cultures. J. Med. Chem 2007, 50, 2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lebrun S; Couture A; Deniau E; Grandclaudon P Suzuki–Miyaura Cross-Coupling and Ring-Closing Metathesis: A Strategic Combination to the Synthesis of Indeno[1,2-c]isoquinolin-5,11-diones. Tetrahedron Lett. 2011, 52, 1481–1484. [Google Scholar]
- (40).Cushman M; Cheng L Total Synthesis of Nitidine Chloride. J. Org. Chem 1978, 43, 286–288. [Google Scholar]
- (41).Cushman M; Cheng L Stereoselective Oxidation by Thionyl Chloride Leading to the Indeno[1,2-c]isoquinoline System. J. Org. Chem 1978, 43, 3781–3783. [Google Scholar]
- (42).Felix AM; Heimer EP; Lambros TJ; Tzougraki C; Meienhofer J Rapid Removal of Protecting Groups from Peptides by Catalytic Transfer Hydrogenation with 1,4-Cyclohexadiene. J. Org. Chem 1978, 43, 4194–4196. [Google Scholar]
- (43).Dexheimer TS; Pommier Y DNA Cleavage Assay for the Identification of Topoisomerase I Inhibitors. Nature Protoc. 2008, 3, 1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kiselev E; DeGuire S; Morrell A; Agama K; Dexheimer TS; Pommier Y; Cushman M 7-Azaindenoisoquinolines as Topoisomerase I Inhibitors and Potential Anticancer Agents. J. Med. Chem 2011, 54, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Antony S; Kohlhagen G; Agama K; Jayaraman M; Cao S; Durrani FA; Rustum YM; Cushman M; Pommier Y Cellular Topoisomerase I Inhibition and Antiproliferative Activity by MJ-III-65 (NSC 706744), an Indenoisoquinoline Topoisomerase I Poison. Mol. Pharmacol 2005, 67, 523–530. [DOI] [PubMed] [Google Scholar]
- (46).Lv PC; Agama K; Marchand C; Pommier Y; Cushman M Design, Synthesis, and Biological Evaluation of O-2-Modified Indenoisoquinolines as Dual Topoisomerase I–Tyrosyl-DNA Phosphodiesterase I Inhibitors. J. Med. Chem 2014, 57, 4324–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bihel F; Quelever G; Lelouard H; Petit AS; da Costa CA; Pourquie O; Checler F; Thellend A; Pierre P; Kraus JL Synthesis of New 3-Alkoxy-7-amino-4-chloro-isocoumarin Derivatives as New Beta-Amyloid Peptide Production Inhibitors and Their Activities on Various Classes of Protease. Bioorg. Med. Chem 2003, 11, 3141–3152. [DOI] [PubMed] [Google Scholar]
- (48).Whitmore WF; Cooney RC Preparation of Homophthalic Cyclic Hydrazide and 4-Aminohomophthalyl Cyclic Hydrazide. J. Am. Chem. Soc 1944, 66, 1237–1240. [Google Scholar]
- (49).Guthrie DA; Frank AW; Purves CB Studies in the Polyoxyphenol Series. XI. Synthesis of Papaverine and Papaveraldine by the Pomeranz–Fritsch Method. Can. J. Chem 1955, 33, 729–742. [Google Scholar]
- (50).Duclos RI; Tung JS; Rapoport H A High-Yield Modification of the Pschorr Phenanthrine Synthesis. J. Org. Chem. 1984, 49, 5243–5246. [Google Scholar]
- (51).Patel BA; Ashby CR; Hardej D; Talele TT The Synthesis and SAR Study of Phenylalanine-Serived (Z)-5-Arylmethylidene Rhodanines as Anti-methicillin-resistant Staphylococcus aureus (MRSA) Compounds. Bioorg. Med. Chem. Lett 2013, 23, 5523–5527. [DOI] [PubMed] [Google Scholar]
- (52).Patel BA; Krishnan R; Khadtare N; Gurukumar KR; Basu A; Arora P; Bhatt A; Patel MR; Dana D; Kumar S; Kaushik-Basu N; Talele TT Design and Synthesis of L- and D-Phenylalanine Derived Rhodanines with Novel C5-Arylidenes as Inhibitors of HCV NS5B Polymerase. Bioorg. Med. Chem 2013, 21, 3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Pourquier P; Ueng L-M; Fertala J; Wang D; Park H-J; Essigmann JM; Bjornsti M-A; Pommier Y Induction of Reversible Complexes Between Eukaryotic DNA Topoisomerase I and DNA-containing Oxidative Base Damages. 7,8-Dihydro-8-oxoguanine and 5-Hydroxycytosine. J. Biol. Chem 1999, 274, 8516–8523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.