

Abstract
The mutation rate of mitochondrial DNA (mtDNA) is 17 times higher than nuclear DNA, and these mutations can cause mitochondrial disease in 1 of 10.000 people. The T10609C mutation was identified in type 2 diabetes mellitus (T2DM) patients and the C10676G mutation in cataract patients, with both mutations occurring in the ND4L gene of mtDNA that encodes ND4L protein. ND4L protein, a subunit of complex I in the respiratory complex, has been shown to play a role in the proton translocation process. The purpose of this study was to investigate the effect of both mutations on the proton translocation mechanism. Mutation mapping showed changes in amino acids M47T (T10609C) and C69W (C10676G). The 100 ns molecular dynamics (MD) simulations performed on native and mutants of ND4L-ND6 subunits. It is revealed that the native model had a similar proton translocation pathway to that of complex I from other organisms. Interestingly, the mutant M47T and C69W showed the interruption of the translocation pathway by a hydrogen bond formation between Glu34 and Tyr157. It is observed that the mutations were restricting the passage of water molecules through the transmembrane region. These results could help to develop the computational assay for the validation of a specific genetic biomarker for T2DM and cataracts.
Keywords: ND4L, diabetes mellitus, cataract, proton translocation, T10609C, C10676G
Introduction
Sequence variations of mitochondrial DNA (mtDNA) have pivotal implications associated with clinical outcomes of various diseases, with most hypervariable regions in the mtDNA genome consisting of 16.569-long nucleotide sequences.1 MtDNA is maternally inherited in the mitochondrion and not entirely influenced by recombination events. Moreover, owing to the absence of DNA repair mechanisms, the genome causes an increased mutation rate of 10- to 17-fold higher than that observed in nuclear DNA.2 Furthermore, several of these variants may contribute to mitochondrial functional defects leading to the susceptibility to various diseases.1 Indeed, mtDNA mutations are responsible for ~80% of mitochondrial diseases in adults, whereas they are only found in 20% to 25% of childhood-onset diseases. Childhood-onset mitochondrial diseases have a prevalence of 5 to 15 cases, with adult-onset disease varying between 2.9 and 9.6 per 100.000 individuals. The prevalence can vary in specific populations, such as those with genetic founder mutations or those with high consanguinity.3
Mutations in mtDNA can alter the function of respiratory chain complex proteins that play a role in the electron transfer and proton translocation process.4 It is leading to the deficiency of ATP production, which affects the energetics of cells in transduction mechanisms and metabolic pathways, as well as the regulation of enzyme, channel, and receptor activities, resulting in mitochondrial dysfunction.5,6 Clinical presentation of mitochondrial diseases can be in the form of neurological or nonneurological disorders, but most mitochondrial diseases related to mtDNA mutations have neurological characteristics, including MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes), ataxia, CPEO (chronic progressive external ophthalmoplegia), LHON (Leber hereditary optic neuropathy), and MIDD (maternally inherited diabetes and deafness).7 The A3243G mutation in the tRNALeu gene (UUR) is the most common pathogenic mtDNA mutation and is correlated with highly variable and heterogeneous disease phenotypes.8,9
Intriguingly, the A3243G was also found in patients with pure cataract, a nonneuromuscular disease,10 and hence maybe a potential biomarker specifically for cataracts as a result of the metabolic disruption of ATP metabolism due to the mutations in the respiratory complex. Other secondary mutation that also occur in the genome carrying A3243G is T10609C, which is found in type-2 diabetes mellitus (T2DM) patients but also has been reported to be related to LHON disease in a Kuwait family,11 and C10676G found in cataract patients. Both mutations occur in the gene encoding ND4L protein, one of the respiratory complex I (NADH dehydrogenase) subunits. In Escherichia coli and Thermus thermophilus, ND4L protein is associated with the proton translocation pathway.12-14 The previous study showed that the fourth proton channel at the interface of Nqo10 and Nqo11 was homologous with ND4L-ND6 subunits.12,14-17 This study aims to investigate the effect of T10609C and C10676G mutations on the fourth proton translocation pathway of respiratory complex I using molecular dynamics. The simulation of subunit ND4L-ND6 was performed using Amber18. Furthermore, the simulation data were analyzed with the visualization program VMD.
Methods
Mutation mapping
Mutation mapping was performed on the sequence of mitochondrial ND4L gene from NCBI with access number NC_012920.1 using the Genome Data Viewer. The amino acid changes that occurred due to the mutations (T10609C and C10676G) were also determined.18
Homology modeling
The structure of the human respiratory complex I (transmembrane arm) was obtained from the Protein Data Bank (PDB ID: 5XTC). This structure has the highest level of homology to ND4L and ND6 gene sequences from MITOMAP (https://www.mitomap.org/MITOMAP), with 98% of identity, and hence is the most suitable template for homology modeling. The modeling was performed using MODELLER 9.21, and the lowest DOPE score of 50 models for native and each mutant was used for further analysis.
Model evaluation
The evaluation of the protein models was done using PROCHECK19 and QMEANBrane,20 as well as the DOPE profile comparison of the model and template.21 These methods were used to rule out impossible protein structures with inappropriate stereochemical properties such as steric hindrance, improper hydrogen bonds, and distorted bond angles.
Transmembrane system building
The ND4L-ND6 subunits were placed in a hydrophobic environment in the form of a lipid bilayer membrane entirely composed of 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), the largest constituent of the inner membrane of mitochondria, which is ~ 40% as found from the experimental study.22 The lipid bilayer was built by Membrane Builder in the CHARMM-GUI (http://www.charmm-gui.org). Through a generalized and automated building process, including system size determination, as well as generation of pore water, bulk water, and ions, a realistic membrane system can be generated in 5 minutes to 2 hours depending on the system size.23,24 The explicit solvents were also used in the form of TIP3P water molecules,25 as well as K+ and Cl− ions with a concentration of 150 mM as a physiological salt solution.26
Molecular dynamics simulation
A 100 ns MD simulation was conducted using Amber18 through several minimization steps to reach the lowest energy. Then, heating was performed in 3 stages from 0 K and increasing per 100 K up to 310 K to resemble the human body temperature, which is around 37°C. The equilibration processes were adapted according to the protocol of Ng et al,27 employing a timestep of 2 fs. The long-range electrostatics were treated with the use of particle mesh Ewald technique (PME), together with the force field for protein, lipids, water, and ions. The results of MD simulation are visualized using Visual Molecular Dynamics (VMD) to observe the alleged fourth proton translocation pathway in the interface of the ND4L-ND6 subunit and the effect of both mutations on those pathways. The trajectories were analyzed by RMSD and RMSF in the cpptraj program of Amber18.
Hydrogen bond calculations
A further observation showed the possible pathway involved the Glu34 residue from ND4L and Tyr157 residue from ND6. The hydrogen bond was calculated by hbond command in the cpptraj program. The results showed the number of hydrogen bonds formed between those 2 residues with a short-range cutoff of 3.0 Å in 10 000 frames of 100 ns MD simulation.
Results
T10609C (M47T) mutation analysis
Based on the results of mutation mapping, it was found that the transition from thymine to cytosine in the position of 10609 ND4L gene causes a codon to change from ATA for methionine to ACA for threonine (M47T) (Figure 1). The respiratory complex I from Thermus thermophilus (PDB ID: 5XTC) with 98% of identity was used for homology modeling, resulting in 50 models for each native and mutants, with one model with the lowest DOPE value selected and evaluated. The Ramachandran plot analysis of all 3 models showed more than 90% of residues are plotted in the most favorable regions, which also had transmembrane energy characteristics and a well-defined structure according to the template.
Figure 1.

The T10609C mutation causes the conversion of (A) methionine (hydrophobic) to (B) threonine (hydrophilic) at residue 47 of the ND4L subunit.
In the native model, oxygen atoms from the Met47 amino acid backbone can form 3 hydrogen bonds with 2 amine groups, each on Thr51 and Asn50, and one with a hydroxyl group on the side chain of Thr51. The conformation of Thr51 can also form 2 other hydrogen bonds with Ser53; these bonds form a loop structure and turn to the next helix (Figure 2A). The M47T mutant model shows the loss of one hydrogen bond and the conformational change of Thr51 so that it no longer forms another hydrogen bond with Ser53 (Figure 2B). Amino acid changes from methionine to threonine and causes a significant alteration via the loss of hydrophobic interactions that occur between Met47 (ND4L) and Met79 of ND2 subunits (Figure 3).
Figure 2.

Alteration of interaction in the M47T (B) mutant model compared with (A) native. The green dashed line shows the hydrogen bond.
Figure 3.
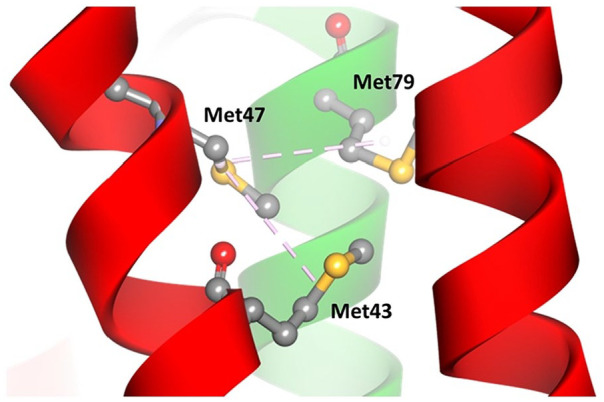
Alteration of the intramolecular interaction in the M47T mutant model. The pink dashed line shows hydrophobic interactions, the red helix is the ND4L subunit, and green is the ND2 subunit.
M47T mutations occur at the end of the helical region adjacent to the loop, causing conformational changes in the loop. The polar amino acid, threonine, forms a new hydrogen bond with Thr51 and makes the previously hydrogen bond between Thr51 and Ser53 disappear (Figure 4A). These changes make the loop structure of the mutant model longer than the native model (Figure 4B).
Figure 4.

Loop conformation changes due to the new hydrogen bond caused by the M47T mutation. The main chain of ND4L-ND6 subunit is shown by the gray spiral, green: charged amino acids, and white: uncharged or nonpolar amino acids; the blue dashed line shows the hydrogen bond.
C10676G (C69W) mutation analysis
The transversion from cytosine to guanine in the 10676 of the ND4L gene also causes changes in the codon from TGC that encodes cysteine to TGG, which encodes tryptophan (C69W) (Figure 5). The native model has 2 hydrogen bonds, one from the sulfur atoms of Cys69 residue with the hydrogen atom of the hydroxyl group on the Thr257 side chain and the other one from the backbone of Val65 residue. There is also a hydrophobic interaction between Cys69 and Leu258 because they are nonpolar residues (Figure 6A). However, the change of cysteine to tryptophan resulted in a stronger interaction between the amino acid residues. Tryptophan forms a strong hydrophobic interaction with Val73 and Ile264 (Figure 6B) so the conformation of the ND4L-ND6 subunit tends to be more stable when compared with the native model. There were also slight changes in the RMSD and RMSF values (Supplemental Figures S1 and S2), with the 3 hydrophobic interactions in the C69W mutant model changing the conformation of the helix to become more organized compared with its native model (Figure 7A-B).
Figure 5.

The C10676G mutation causes the conversion of (A) cysteine to (B) tryptophan, which is a bulky amino acid with a larger molecule size. Cysteine is more hydrophobic than tryptophan.
Figure 6.

Alteration of the interaction in the (B) mutant C69W model compared with the (A) native model. The green dashed line shows the hydrogen bond; pink shows the hydrophobic interaction. ND4L subunit: red helix, ND6 subunit: yellow helix.
Figure 7.

Helix conformations become more organized due to the strong hydrophobic interaction caused by the C69W mutation. The main chain of the ND4L-ND6 subunit is shown by the gray spiral, green: charged amino acids, and white: uncharged or nonpolar amino acids, blue dashed line: hydrogen bond, pink: hydrophobic interaction.
Proton translocation interferences
The conformational changes in Glu34 occur in both mutant models, which reverses the direction toward the top. Hence, the acidic Glu34, which has negatively charged oxygen atoms, can be stabilized by the polar Tyr157, which has hydroxyl groups, through hydrogen bonds. The interaction between these 2 residues blocks the passage of water molecules (Figure 8B-C). The difference between the M47T mutant and native is also considered by the hydration area, and it is assumed that the change in the length of the loop near the point mutation will affect the stability of the helix itself and contributes to the conformational change of the loop at the bottom side, which limits the recruitment of water molecules. However, the structure of the C69W mutant is considered more stable compared with the M47T mutant and its native model, but the conformational changes still affect the proton translocation pathway. It was observed in the hydrogen bonds analysis between the Glu34 and Tyr157 residues that the M47T mutant has 85.49%, and the C69W mutant was considered stable with 28.99% of the total frame, which is very high compared with 0.16% in the native model (Figure 9).
Figure 8.

Proton translocation pathway of the native and mutant ND4L-ND6 subunit models involving polar and charged amino acid residues. In the (A) native model, water molecules are recruited by Glu34 to pass through the membrane, whereas in the (B-C) mutant model, the conformation of Glu 34 changes results in a new hydrogen bond between Glu34 and Tyr157, thereby limiting the passage of the water molecules.
Figure 9.

Hydrogen bond analysis showing the probability of a hydrogen bond between Glu34 and Tyr157 calculated by hbond in cpptraj Amber18.
Discussion
The T10609C mutation was found in patients with T2DM carrying the primary mutation A3243G.28,29 In 2014, Behbehani et al. also reported that this mutation was related to the LHON of a family in Kuwait, based on the sequencing of several mtDNA lineages. The T10609C variant was also present in 12.4% of the sampled population of Han Chinese.30 LHON has 3 primary mutations as the main cause of about 90% of cases, G11778A, G3460A, and T14484TC, which cause abnormalities in the structure of respiratory complex I proteins that play a role in the respiration chain in mitochondria. This change in protein function is likely to cause a decrease in ATP synthesis, but the most significant is free radical production that leads to oxidative stress.31 The C10676G mutations have not been associated with any disease, but in this study, it was found as a secondary mutation in cataract patients who also carry A3243G.28,29 Both mutations occur in the ND4L gene, which encodes one of the subunits of respiratory complex I. Until now, mtDNA mutations related to nonneuromuscular diseases, such as cataracts and T2DM, are still rarely reported so this finding is of interest for further research.
As in silico study of the proton translocation pathway in the human respiratory complex I has not been reported, therefore, an approach using the model of E. coli, Y. lipolytica, and T. thermophilus is essential due to the similar structure and functions to those found in humans.14,16,17 Besides, based on the endosymbiosis theory, mitochondria in eukaryotes originate from aerobic bacteria that have evolved.32 The role of the ND4L subunit in human respiratory complex I has not been further investigated, but it is thought that there is a fourth proton translocation pathway in the interface of the ND4L-ND6 subunit of respiratory complex I.12,14,15 Proton translocation in respiratory complex I has been studied using various experimental techniques, and according to the current consensus, proton translocation occurs with a 4H+/2e− stoichiometry.33,34 Each of the 3 respiratory complex I subunits, ND2, ND4, and ND5, translocates one proton as all those 3 subunits are homologous with the Na+/H+ antiporter and have several charged amino acid residues that are conserved and located in the middle of the subunit.35-37 The 3 antiporter-type subunits (ND2, ND4, ND5) are represented for the 3H+/2e− stoichiometry. However, the consensus has stated that respiratory complex I can pump 4 protons; hence, it is thought that there is a fourth proton translocation path. Haapanen and Sharma14 reported that 2 amino acid residues have a crucial role in the fourth proton translocation pathway, namely, Glu32 and Tyr59, both of which are conserved in T. thermophilus and E. coli.16 The observations on the human ND4L-ND6 subunits also indicate the presence of these 2 amino acids.
The proton transfer process occurs by using water molecules as mediums of hopping under the Grotthuss mechanism38,39 so the movement of the water molecules will represent the process of proton translocation in respiratory complex I. In the native model, a large number of water molecules are present around the Glu34 residue of the ND4L subunit and Tyr157 of the ND6 subunit (Figure 8A) due to the downward conformation of the Glu34 residue that allows water molecules to pass through and mediate the proton translocation process. This phenomenon is similar to what was reported to occur in respiratory complex I of T. thermophilus.14
The M47T (T10609C) mutant model shows a change in the characteristics or properties of the amino acids that can change the interactions that occur around it. These changes affect the loop length near the mutation point, thereby altering the stability of the helix itself (Figure 4B). It contributes to the conformational change of the bottom side loop, which limits the entry of water molecules. Apart from affecting the concentration of ATP produced in the mitochondria, the mutation of mtDNA also induces an increase in ROS due to hyperpolarization or dysfunction of complex proteins in the respiratory chain.40 The previous experimental study using human cybrid cells revealed that the T0609C mutation increases the ROS production under hypoxic conditions and conveys susceptibility to high altitude polycythemia. The wild-type cells (ND4L:47Met) produced approximately 1.5-fold more H2O2 (DCFDA fluorescence) at 3% oxygen levels than the mutant.41 On the other phenotype which is Friedreich Ataxia (FRDA), the downregulation of the expression of ND4L gene was observed and affected the function of respiratory complex I.42
In contrast to the native model, the C69W (C10676G) mutant shows strong hydrophobic interactions between Trp69, Val73, and Ile264, which can directly stabilize the 2 adjacent helices (ND4L-ND6 subunits). This interaction constructs the helices in C69W mutant to be more organized than the native model (Figure 7B), but the position of C69W mutation is in the helix, which is next to the helix backbone of Glu34. The change from cysteine to tryptophan, which is a much larger amino acid that affects the helix, causes the Glu34 conformation to change so that water molecules cannot pass through. There is no experimental evidence yet for the C10676G mutation.
In most cases, the manifestation of the phenotype from the mutations occurs only when the threshold level is exceeded, and this phenomenon has been termed the “phenotypic threshold effect.” It is observed at the single-cell level when wild-type mtDNA can no longer “balance out” the mutated effects. For example, a heteroplasmic mutation in mtDNA will result in the co-existence of a mutated mRNA and a dysfunctional respiratory chain subunit along with its wild-type homologs. At the time this wild-type level falls below the threshold, it will cause impaired mitochondrial function.43
Both mutations that occur are thought to disrupt the structure and function of the respiratory complex I, interfere the ATP synthesis, increase ROS production, and affect calcium metabolism that leads to ATP deficiency. In cataracts, ATP deficiency affects the failure of the crystalline protein renaturation, inducing protein aggregates to form in the lens of the eye that can scatter light.44,45 Likewise, in pancreatic beta cells that lack ATP, insulin secretion is inhibited, causing T2DM.32 A previous in silico study on the relationship of mtDNA mutations with the phenotype of T2DM and cataracts was conducted by Mutia et al46 via mutation modeling in respiratory complex I using ab initio. Also, other studies regarding the G9053A mutation in the ATP6 gene,47 the T8414G mutation in the ATP8 gene,48 and the T15458C and T15663C mutations in the CYB gene49 have been reported to cause structure changes, interfering the respiration process. The methods in this study can be developed as a computational assay for validation of mutations that serve as specific biomarkers of disease. In this case, the computational assay is an analytical procedure in molecular biology to qualitatively assess or quantitatively measure the presence, amount, or functional activity of the analyte using computational methods.50,51 Currently, the MD simulation is often to be used as a computational assay for biological activity51-54 so the analysis of the proton translocation pathway as in this study also can be used as a test parameter.
Conclusions
The T10609C and C10676G mutations affect proton translocation in the mitochondrial respiratory complex I by disrupting the fourth proton translocation path in the ND4L-ND6 subunits, as evidenced by conformational changes in the Glu34 residue. This alteration leads to the formation of a hydrogen bond between Glu34 and Tyr157, which limits the passage of water molecules. Therefore, T10609C and C10676G mutations are thought to be related to the phenotype of T2DM and cataracts. These results could help to develop the computational assay for the validation of a specific genetic biomarker for T2DM and cataracts.
Supplemental Material
Supplemental material, sj-docx-1-bbi-10.1177_1177932220978672 for Molecular Dynamics Simulation of T10609C and C10676G Mutations of Mitochondrial ND4L Gene Associated With Proton Translocation in Type 2 Diabetes Mellitus and Cataract Patients by Wanda Destiarani, Rahmaniar Mulyani, Muhammad Yusuf and Iman Permana Maksum in Bioinformatics and Biology Insights
Acknowledgments
The authors wish to acknowledge the Research Center for Molecular Biotechnology and Bioinformatics – Universitas Padjadjaran for generous computational support. We are thankful to all members of the Laboratory of Computational Chemistry – Universitas Padjadjaran for helpful discussions.
Footnotes
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Master’s Thesis Grant from the Ministry of Research, Technology, & Higher Education (1827/UN6.3.1/LT/2020) and Academic Leadership Grant (ALG) from Universitas Padjadjaran (1427/UN6.3.1/LT/2020).
Author Contributions: WD, MY, and IPM conceived and designed the experiments. WD, RM, and MY analyzed the data. WD wrote the first draft of the manuscript. WD, MY, and IPM contributed to the writing of the manuscript. WD, RM, MY, and IPM agree with manuscript results and conclusions. WD and MY jointly developed the structure and arguments for the paper. WD, RM, MY, and IPM made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
ORCID iDs: Muhammad Yusuf  https://orcid.org/0000-0003-1627-1553
https://orcid.org/0000-0003-1627-1553
Iman Permana Maksum
https://orcid.org/0000-0001-8166-8421
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Wang J, Schmitt ES, Landsverk ML, et al. An integrated approach for classifying mitochondrial DNA variants: one clinical diagnostic laboratory’s experience. Genet Med. 2012;14:620-626. [DOI] [PubMed] [Google Scholar]
- 2. Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial tRNA mutations and disease. Wiley Interdiscip Rev RNA. 2010;1:304-324. doi: 10.1002/wrna.27. [DOI] [PubMed] [Google Scholar]
- 3. Nature. Mitochondrial diseases. Nat Rev Dis Prim. 20162;2:16081. doi: 10.1038/nrdp.2016.81 [DOI] [PubMed] [Google Scholar]
- 4. Koh H, Park GS, Shin SM, et al. Mitochondrial mutations in cholestatic liver disease with biliary atresia. Sci Rep. 2018;8:1-14. doi: 10.1038/s41598-017-18958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johns DR. Mitochondrial DNA and disease. N Engl J Med. 1995;333:638-644. doi: 10.1056/NEJM199509073331007. [DOI] [PubMed] [Google Scholar]
- 6. Bagshaw CR. ATP analogues at a glance. J Cell Sci. 2001;114:459-460. [DOI] [PubMed] [Google Scholar]
- 7. Gorman GS, Chinnery PF, DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Prim. 2016;2:1-22. doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- 8. Pierron D, Rocher C, Amati-Bonneau P, et al. New evidence of a mitochondrial genetic background paradox: impact of the J haplogroup on the A3243G mutation. BMC Med Genet. 2008;9:1-13. doi: 10.1186/1471-2350-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J, Guo J, Fang W, Jun Q, Shi K. Clinical features of MELAS and its relation with A3243G gene point mutation. Int J Clin Exp Pathol. 2015;8:13411-13415. [PMC free article] [PubMed] [Google Scholar]
- 10. Maksum I, Natradisastra G, Nuswantara S, Ngili Y. The effect of A3243G mutation of mitochondrial DNA to the clinical features of type-2 diabetes mellitus and cataract. Eur J Sci Res. 2013;96:591-599. [Google Scholar]
- 11. Behbehani R, Melhem M, Alghanim G, Behbehani K, Alsmadi O. ND4L gene concurrent 10609T>C and 10663T>C mutations are associated with Leber’s hereditary optic neuropathy in a large pedigree from Kuwait. Br J Ophthalmol. 2014;98:826-831. doi: 10.1136/bjophthalmol-2013-304140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Torres-Bacete J, Sinha PK, Sato M, et al. Roles of subunit NuoK (ND4L) in the energy-transducing mechanism of Escherichia coli NDH-1 (NADH:quinone oxidoreductase). J Biol Chem. 2012;287:42763-42772. doi: 10.1074/jbc.M112.422824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sazanov LA. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat Rev Mol Cell Biol. 2015;16:375-388. [DOI] [PubMed] [Google Scholar]
- 14. Haapanen O, Sharma V. Role of water and protein dynamics in proton pumping by respiratory complex I. Sci Rep. 2017;7:1-12. doi: 10.1038/s41598-017-07930-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443-448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaila VRI, Wikstrom M, Hummer G. Electrostatics, hydration, and proton transfer dynamics in the membrane domain of respiratory complex I. Proc Natl Acad Sci. 2014;111:6988-6993. doi: 10.1073/pnas.1319156111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wirth C, Zickermann V, Siegmund K, et al. Mechanistic insight from the crystal structure of mitochondrial complex I. Science. 2015;347:44-49. doi: 10.1126/science.1259859. [DOI] [PubMed] [Google Scholar]
- 18. Agarwala R, Barrett T, Beck J, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018;46:D8-D13. doi: 10.1093/nar/gkx1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laskowski RA, Macarthur MW, Thornton JM. Chapter 21.4. PROCHECK: validation of protein-structure coordinates. Int Table Crystallograph. 2012;21:684-687. [Google Scholar]
- 20. Studer G, Biasini M, Schwede T. Assessing the local structural quality of transmembrane protein models using statistical potentials (QMEANBrane). Bioinformatics. 2014;30:505-511. doi: 10.1093/bioinformatics/btu457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinforma. 2016;2016:561-5637. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horvath SE, Daum G. Lipids of mitochondria. Prog Lipid Res. 2013;52:590-614. doi: 10.1016/j.plipres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 23. Jo S, Kim T, Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qi Y, Cheng X, Lee J, et al. Computational tool mobile membrane-mimetic model. Biophysj. 2015;109:2012-2022. doi: 10.1016/j.bpj.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Florova P, Sklenovsky P, Bana P. Explicit water models affect the specific solvation and dynamics of unfolded peptides while the conformational behavior and flexibility of folded peptides remain intact. J Chem Theory Comput. 2010;6:3569-3579. [DOI] [PubMed] [Google Scholar]
- 26. Froschauer E, Nowikovsky K, Schweyen RJ. Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim Biophys Acta—Biomembr. 2005;1711:41-48. doi: 10.1016/j.bbamem.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 27. Ng HW, Laughton CA, Doughty SW. Molecular dynamics simulations of the adenosine A2a receptor in. J Chem Inf Model. 2014;54:573-581. [DOI] [PubMed] [Google Scholar]
- 28. Maksum IP, Farhani A, Rachman SD, Ngili Y. Making of the A3243g mutant template through site directed mutagenesis as positive control in PASA-Mismatch three bases. Int J Pharmtech Res. 2013;5:441-450. [Google Scholar]
- 29. Hartati YW, Nur Topkaya S, Maksum IP, Ozsoz M. Sensitive detection of mitochondrial DNA A3243G tRNALeu mutation via an electrochemical biosensor using Meldola’s blue as a hybridization indicator. Adv Anal Chem. 2013;2013:20-27. doi: 10.5923/s.aac.201307.04. [DOI] [Google Scholar]
- 30. Yao YG, Kong QP, Bandelt HJ, Kivisild T, Zhang YP. Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am J Hum Genet. 2002;70:635-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paragallo JH, Newman NJ. HHS Public Access. 2015;26:450-457. doi: 10.1016/j.coviro.2015.09.001.Human. [DOI] [Google Scholar]
- 32. Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 6th ed. New York, NY: W.H. Freeman and Company; 2013. [Google Scholar]
- 33. Ohnishi T, Nakamaru-Ogiso E, Ohnishi ST. A new hypothesis on the simultaneous direct and indirect proton pump mechanisms in NADH-quinone oxidoreductase (complex I). FEBS Lett. 2010;584:4131-4137. doi: 10.1016/j.febslet.2010.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haapanen O. Proton translocation channels in respiratory complex I probed by molecular dynamics simulations, 2016. https://trepo.tuni.fi/handle/123456789/24527.
- 35. Torres-Bacete J, Nakamaru-Ogiso E, Matsuno-Yagi A, Yagi T. Characterization of the NuoM (ND4) subunit in Escherichia coli NDH-1: conserved charged residues essential for energy-coupled activities. J Biol Chem. 2007;282:36914-36922. doi: 10.1074/jbc.M707855200. [DOI] [PubMed] [Google Scholar]
- 36. Nakamaru-Ogiso E, Kao MC, Chen H, Sinha SC, Yagi T, Ohnishi T. The membrane subunit NuoL(ND5) is involved in the indirect proton pumping mechanism of Escherichia coli complex I. J Biol Chem. 2010;285:39070-39078. doi: 10.1074/jbc.M110.157826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sato M, Sinha PK, Torres-Bacete J, Matsuno-Yagi A, Yagi T. Energy transducing roles of antiporter-like subunits in Escherichia coli NDH-1 with main focus on subunit NuoN (ND2). J Biol Chem. 2013;288:24705-24716. doi: 10.1074/jbc.M113.482968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de Grotthuss CJ. Sur la décomposition de l’eau et des corps qu’elle tient en dissolution à l’aide de l’électricité galvanique. Ann Chim. 1806; LVIII:54-73. [Google Scholar]
- 39. Cukierman S. Et tu, Grotthuss! and other unfinished stories. Biochim Biophys Acta—Bioenerg. 2005;1757:876-885. [DOI] [PubMed] [Google Scholar]
- 40. Szczepanowska J, Malinska D, Wieckowski MR, Duszynski J. Effect of mtDNA point mutations on cellular bioenergetics. Biochim Biophys Acta. 2012;1817:1740-1746. [DOI] [PubMed] [Google Scholar]
- 41. Jiang C, Cui J, Liu F, Gao L, Luo Y. Mitochondrial DNA 10609T promotes hypoxia-induced increase of intracellular ROS and is a risk factor of high altitude polycythemia. PLoS ONE. 2014;9:e87775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Salehi MH, Kamalidehghan B, Houshmand M, et al. Gene expression profiling of mitochondrial oxidative phosphorylation (OXPHOS) complex I in Friedreich ataxia (FRDA) patients. PLoS ONE. 2014;9: e94069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370:751-762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moreau KL, King JA. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med. 2012;18:273-282.22520268 [Google Scholar]
- 45. Lam D, Rao SK, Ratra V, et al. Cataract. Nat Rev Dis Prim. 2015;1:1-15. doi: 10.1038/nrdp.2015.14. [DOI] [PubMed] [Google Scholar]
- 46. Mutia RD, Maksum IP, Yusuf M, Sciences N, Padjadjaran U, Java W. Ab initio modeling of complex I human mitochondrial DNA using I-Tasser. 2010:312-325. [Google Scholar]
- 47. Maksum IP, Saputra SR, Indrayati N, Yusuf M, Subroto T. Bioinformatics study of m.9053G>a mutation at the ATP6 gene in relation to type 2 diabetes mellitus and cataract diseases. Bioinform Biol Insights. 2017;11:1-5. doi: 10.1177/1177932217728515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang W, Li R, Zhang Y, et al. Mitochondrial DNA mutations associated with type 2 diabetes mellitus in Chinese Uyghur population. Sci Rep. 2017;7:1-9. doi: 10.1038/s41598-017-17086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maksum IP, Safitri S, Yusuf M, Natradisastra Nuswantara S, Suprijana O. Structure and function of CYB in type-2 diabetes mellitus and cataract patients which associated with mitochondrial DNA mutation. 1St Int Conf Comput Sci Technol. 2010:103-110. [Google Scholar]
- 50. The American Heritage Dictionary of the English Language. Boston, MA: Houghton Mifflin; 2006. [Google Scholar]
- 51. Huggins DJ, Biggin PC, Dämgen MA, et al. Biomolecular simulations: from dynamics and mechanisms to computational assays of biological activity. Wiley Interdiscip Rev Comput Mol Sci. 2019;9:1-23. doi: 10.1002/wcms.1393. [DOI] [Google Scholar]
- 52. Thai K, Le D, Tran N, Nguyen T. Computational assay of Zanamivir binding affinity with original and mutant influenza neuraminidase 9 using molecular docking. J Theor Biol. 2015;385:31-39. doi: 10.1016/j.jtbi.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 53. Pavlin M, Spinello A, Pennati M, et al. A computational assay of estrogen receptor α antagonists reveals the key common structural traits of drugs effectively fighting refractory breast cancers. Sci Rep. 2018;8:1-11. doi: 10.1038/s41598-017-17364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hirvonen VHA, Hammond K, Chudyk EI, et al. An efficient computational assay for β-lactam antibiotic breakdown by class a β-lactamases. J Chem Inf Model. 2019;59:3365-3369. doi: 10.1021/acs.jcim.9b00442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-bbi-10.1177_1177932220978672 for Molecular Dynamics Simulation of T10609C and C10676G Mutations of Mitochondrial ND4L Gene Associated With Proton Translocation in Type 2 Diabetes Mellitus and Cataract Patients by Wanda Destiarani, Rahmaniar Mulyani, Muhammad Yusuf and Iman Permana Maksum in Bioinformatics and Biology Insights