

Abstract
The pathophysiology of microhemorrhages in the context of cerebral amyloid angiopathy (CAA) remains poorly understood. Here we used in vivo two-photon microscopy in aged APP/PS1 mice with mild-to-moderate CAA to assess the formation of microhemorrhages and their spatial relationship with vascular Aβ depositions in the surrounding microvascular network. Mice with chronic cranial windows were intravenously injected with fluorescent dextran to visualize the vessels and a fluorescently labeled anti-fibrin antibody to visualize microhemorrhages. Focal vessel irradiations resulted in extravascular fibrin-positive clots at individual rupture sites that remained visible for weeks. Spontaneous extravascular fibrin-positive clots were more often observed in 19-month-old transgenic APP/PS1 mice compared to their wild-type littermate controls (p = 0.039), after heparin administration. In the transgenic mice, these spontaneous leakage sites frequently occurred at arteriolar segments without CAA at bifurcations or vessel bends. These findings suggest that the presence of vascular Aβ per se does not directly predispose vessels to leak, but that complex flow dynamics within CAA-affected vascular networks likely play a role. Our in vivo approach for the detection of individual spontaneous leakage sites may be used in longitudinal studies aimed to assess structural and functional alterations at the single-vessel level leading up to microhemorrhage formation.
Keywords: Blood–brain barrier, cerebral amyloid angiopathy, fibrin, in vivo microscopy, microbleeds
Introduction
Cerebral small vessel disease (SVD) refers to pathological alterations of the small arteries and venules of the brain and is a major contributor to age-related cognitive decline and dementia.1 Whereas the small vessels escape detection by conventional magnetic resonance imaging (MRI), manifestations of SVD can be readily detected during life and include white matter hyperintensities, infarcts, and microhemorrhages.2 Advancements in the development and optimization of blood-sensitive MRI sequences have resulted in increased detection of microhemorrhages,3,4 which have been linked to a higher risk of stroke, mortality, and dementia.5 Regarding their pathophysiology, microhemorrhages in deep locations of the brain have been associated with hypertensive arteriopathy, whereas cortical microhemorrhages are indicative of cerebral amyloid angiopathy (CAA).6 CAA is characterized by the accumulation of amyloid β (Aβ) in the walls of small cortical arterioles and leptomeningeal vessels and increases the risk of large bleeds that are often fatal in patients with the disease.7 Although neuropathological examinations have confirmed a positive association between cortical microhemorrhages and severe CAA,8,9 the underlying mechanisms and sequence of events that lead to microhemorrhage formation are poorly understood. This is mainly due to a lack of relevant experimental animal models and the fact that microhemorrhages easily escape detection upon neuropathological examination. Moreover, microscopic observations in the brain post-mortem are cross-sectional in nature and do not allow to assess directly causes and consequences of bleeding. Furthermore, histopathology is often limited by the assessment of single thin brain sections, which does not allow studying lesions in relation to the surrounding microvascular network. Therefore, we utilized in vivo two-photon microscopy in a mouse model of Aβ overproduction with mild-to-moderate CAA and developed a novel approach to assess the formation of microhemorrhages as well as their exact sites of leakage in real-time in 3D and their spatial relationship with vascular Aβ depositions in the surrounding microvascular network.
Material and methods
Animals
Seven-to-nine months old and 18-to-20 months old male and female transgenic (Tg) APPswe/PSEN1dE9 (APP/PS1) mice and their wild-type (WT) littermates were included in this study. APP/PS1 mice express the human mutant amyloid precursor protein (APP) gene containing the Swedish K594N/M595L mutation and the human presenilin 1 gene lacking exon 9, under control of the prion protein promotor.10 Mice were purchased from The Jackson Laboratory (stock nr. 34829) and bred in our animal facility on a C57BL/6J background. While aging, mice were housed up to four individuals in one cage on a 12-h light/dark cycle with food and water provided ad libitum. After surgery, mice were housed individually. A subset of a separate set of naïve mice (10/12) used for ex vivo histopathology carried a Gad2-Cre transgene, unrelated to the current study.
All animal procedures were performed with the approval of the Massachusetts General Hospital Animal Care and Use Committee and experiments were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The ARRIVE guidelines were followed for reporting animal experiments.
Ex vivo histopathology
Naïve 20-month-old mice were euthanized by CO2 asphyxiation and transcardially perfused with 20 mL phosphate-buffered saline (PBS). Brains were extracted and stored at 4°C in a fixation solution containing 4% paraformaldehyde and 15% glycerol, in PBS. Next, the intact brains were embedded in paraffin in coronal orientation and cut in 6 µm-thick sections, as follows: throughout the brain, four adjacent sections were cut skipping 100 µm between each set of four sections. The olfactory bulbs and cerebellum were excluded. The first section at each level of the brain was stained with hematoxylin and eosin (H&E) to assess general morphology, and the second section with Perls’ Prussian blue to detect Iron-positive deposits, both following standard protocols. Aβ immunohistochemistry was performed on a single section per brain to confirm the genotype of each mouse.
Surgical procedures
Cranial window surgeries were performed as described previously, with minor modifications.11,12 Mice were anesthetized with 5% isoflurane (mixed with pure O2) and placed in a stereotactic frame. Throughout the procedure, isoflurane levels were maintained at ∼2% and adjusted if necessary. After shaving and partial removal of the scalp, a craniotomy (measuring 5 mm in diameter) was performed exposing both hemispheres at the level of the somatosensory cortices. A cover glass (measuring 8 mm in diameter) with sterile PBS was placed over the brain and secured in place with a mixture of dental cement and Krazy Glue. Post-surgically, mice received a single subcutaneous injection of (0.05 mg/kg) buprenorphine and (300 mg/mL) Tylenol was added to their drinking water for three days. Mice could recover from the surgery for at least three weeks before they were imaged.
In vivo two-photon microscopy
Imaging was performed using the commercially available FluoView FV1000MPE two-photon laser-scanning system mounted on a BX61WI microscope, equipped with a 25× (numerical aperture 1.05) dipping water immersion objective (Olympus). A Mai Tai® DeepSee™ Ti:Sapphire mode-locked laser (Spectra-Physics) generated two-photon excitation at 800 nm. External detectors containing three photomultiplier tubes (PMTs, Hamamatsu) collected emitted light in the range of 420–460 nm (blue), 495–540 nm (green), and 575–630 (red). PMT settings were kept constant for each imaging session, but laser power was adjusted as needed. The day prior to imaging mice were intraperitoneally injected with 300 µL (5 mg/kg dissolved in 3% Cremophor® and PBS) Methoxy-X04 (Glixx lab), a blue fluorescent Congo red derivative that crosses the blood–brain barrier and labels Aβ depositions (both parenchymal plaques and CAA) for in vivo detection.13 Prior to imaging, mice were anesthetized with isoflurane (5% inhalation, maintained at 1.5–2% throughout the imaging session) and intravenously injected with 200 µL (12.5 mg/mL) 70 kDa fluorescein dextran (Invitrogen) to provide an angiogram. To identify extravascular clots at leakage sites, mice also received an intravenous injection of 150 µL (0.5 mg/mL) anti-fibrin antibody (BioMedica Diagnostics) conjugated with DyLight® 594 (Thermo Fisher Scientific).14
Laser irradiation experiments
To visualize microhemorrhage dynamics in real-time in 19-month-old APP/PS1 mice, arterioles and venules were irradiated to allow circulating fluorescent dextran and anti-fibrin to extravasate. Vessel irradiation was achieved by focusing the laser at maximum power in bidirectional scanning mode on a targeted arteriole or venule at 6× magnification for 20 s per attempt. Irradiation was immediately stopped when dextran was observed to extravasate from the irradiated site. If no extravasation was observed after six attempts, the vessel segment was excluded from further analysis. A Z-stack was acquired prior to laser irradiation at a speed of 4 µs/pixel, resolution of 1 µm2 per pixel, matrix of 512 × 512, and a step size of 5 µm. Immediately following a successful irradiation, serial Z-stacks were acquired for a period of 21 min with 3-min intervals at 1× magnification. All mice were re-imaged again the next day and after three weeks to evaluate irradiation sites. All Tg mice received an additional intraperitoneal injection of Methoxy-X04 the day prior to the imaging session at three weeks.
Heparin experiments
The formation of spontaneous microhemorrhages was assessed in two separate cohorts of APP/PS1 mice, at 8 months and 19 months of age. First, several baseline Z-stacks were acquired in systematically sampled areas throughout the window, at a speed of 4 µs/pixel, resolution of 1 µm2 per pixel, matrix of 512 × 512, a step size of 5 µm, and a magnification of 1×. Moreover, the window was screened visually with epifluorescence to capture any additional sites of leakage. Next, to lower the threshold for spontaneous microhemorrhages to occur, each mouse received an intravenous as well as subcutaneous injection containing (0.16 U/g) heparin in PBS.15 The same areas were re-imaged after 15 min and the window was screened visually again to capture any additional sites of leakage. During the imaging sessions, the investigator was not blinded for genotype.
Image analysis
Acquired two-photon images were visually assessed and analyzed in FIJI.16 Arteriolar CAA burden was measured on maximum intensity projections of the Z-stacks taken prior to laser irradiation, in the (blue) Methoxy-X04 channel. For each irradiation site, the corresponding region of interest (ROI) was first outlined on the (green) dextran channel, and then copied to the (blue) Methoxy-X04 channel. After transforming the maximum intensity projections into a binary image, CAA burden was quantified for each ROI as the number of positive pixels divided by the total number of pixels within each ROI. Vessel diameters were calculated based on maximum intensity projections of the (green) dextran channel. In the heparin experiments, for each fibrin clot-containing image, if visible, the parent artery, first-order, second-order, and third-order arteriolar branches were identified at 1× magnification. Pial surface vessels were defined as parent arteries, penetrating arterioles as first-order branches, followed by higher order branches. A segment was considered CAA-positive if more than two distinct Methoxy-X04-positive CAA deposits were present. In addition, the distance between the edge of each fibrin clot to the nearest CAA deposit was measured.
Statistics
Graphs were created and statistical analyses performed in Graphpad Prism (version 5.03). Differences between genotype and conditions were compared using a χ2 test for proportions and a Mann–Whitney U test or t-test for continuous variables. Linear correlations were calculated using a Spearman’s rank correlation coefficient. For visualization of correlations, linear regression analysis was performed to generate fitting lines.
Results
Microhemorrhages on histopathology
To assess evidence of red blood cell extravasation in APP/PS1 mice that exhibit moderate CAA at older age, we first screened for iron-positive deposits with ex vivo histopathology. The brains of 12 20.0 ± 0.6 months old naïve APP/PS1 mice (7 Tg (4 F) and 5 WT (4 F) littermates) were extracted and cut in serial sections to assess presence and number of areas with focal parenchymal iron-positive deposits. On average, 40 sections per brain were visually screened on a brightfield microscope, which revealed that focal areas with clusters of Iron-positive deposits were more frequently present in Tg APP/PS1 mice (mean ± standard deviation (SD) 21 ± 8) compared to WT mice (mean ± SD 11 ± 3), p = 0.029) (Figure 1). The majority of these areas consisted of only a few iron-positive deposits, suggestive of subtle blood–brain barrier leakage rather than frank microhemorrhages. Areas with >20 deposits and evidence of hemosiderin-containing macrophages on the adjacent H&E-stained section – indicative of old microhemorrhages – were rare and only observed in Tg APP/PS1 mice (six old microhemorrhages in n = 4 Tg mice, p = 0.065) (Figure 1).
Figure 1.

Histopathological evidence of leakage in the brains of 20-month-old APP/PS1 mice. Representative examples of areas with focal iron-positive deposits on a Prussian blue-stained section of the post-mortem brain of a 20-month-old Tg APP/PS1 mouse (a) and an age-matched WT mouse (b). A higher number of areas with clusters of focal iron-positive deposits was observed in Tg compared to WT mice (c, t-test p = 0.029). Areas with clusters of >20 focal iron-positive deposits (d, e) with evidence of hemosiderin-containing macrophages on the adjacent H&E-stained section (insets) were rare and only observed in Tg mice (f, Mann–Whitney U test p = 0.065). Lines and error bars in C indicate mean and standard deviations. Lines in F indicate median. Note that each dot in C and F represents a mouse. *p < 0.05.
In vivo visualization of focal microhemorrhages in APP/PS1 mice
To assess microhemorrhages in real-time in relation to their surrounding microvasculature, we next utilized in vivo two-photon microscopy in 20-month-old Tg APP/PS1 mice and their WT littermates with chronic cranial windows. Angiograms were visualized in vivo by injecting fluorescein dextran in the bloodstream prior to imaging. Mice were co-injected with fluorescently labeled anti-fibrin. Focal laser irradiations elicited dextran extravasation and rapid extravascular fibrin clot formation at the site of leakage in both arterioles (21/33 targeted) and venules (8/11 targeted) in n = 6 mice (3 Tg (2 F), 3 WT (3 F)) (Figure 2). Notably, over the course of 20 min, fibrin clots appeared to ‘wrap around’ arterioles, whereas this was not observed for venules. The next day, 20/21 clots on arterioles were still visible, whereas only 3/8 clots on venules (p = 0.022). After three weeks, 10/16 clots on arterioles were still visible, compared to 1/6 clots on venules (p = 0.056) (Figure 2). Notably, arteriole diameter was not associated with time to rupture (Spearman’s ρ 0.03, p = 0.88), but more laser irradiation attempts were needed to achieve dextran extravasation from arterioles in Tg APP/PS1 mice compared to their WT littermates (p = 0.0044). No significant correlation was observed between CAA burden at the irradiation site and time to rupture (Spearman’s ρ 0.36, p = 0.27) (Figure 2).
Figure 2.

Visualizing microhemorrhage formation in vivo in real-time with two-photon microscopy in 20-month-old APP/PS1 mice. Representative example of the temporal progression of extravascular fibrin clot formation with in vivo two-photon microscopy in a 20-month-old WT mouse, depicted here immediately (a, aʹ, c, cʹ), 9 min (a″, c″), and 18 min (a‴, c‴) after focal irradiation of an arteriole and a venule. Note the increasing circumferential appearance of the extravascular fibrin clot over time on the arteriole, but not the venule. Extravascular fibrin clots surrounding arterioles were more often still (partly) visible after one day (not shown) and after three weeks (b), compared to venules (d). In 20-month-old Tg APP/PS1 mice, a similar temporal progression of extravascular fibrin clot formation after focal irradiation was observed, in venules, and in arterioles with low (eʹ) and high (e″) CAA burden. Notably, more laser irradiation attempts were needed to achieve dextran extravasation from arterioles in Tg APP/PS1 mice compared to their WT littermate controls (f, Mann–Whitney U test p = 0.0044). However, no significant correlation was observed between CAA burden at the irradiation site and time to rupture (g, Spearman’s ρ 0.36, p = 0.27). Note the additional extravascular fibrin clot on an arteriole from a previous irradiation in panel C (indicated with *). Lines in F indicate median. **p < 0.01.
Spontaneous and heparin-induced microhemorrhages in old APP/PS1 mice
Next, the prevalence of spontaneous microhemorrhages was assessed in real-time in two separate cohorts of Tg APP/PS1 mice and their WT littermates with chronic cranial windows at 8 months (n = 9; 4 Tg (2 F), 5 WT (3 F)) and 19 months of age (n = 16; 8 Tg (1 F), 8 WT (1 F)), after dextran and anti-fibrin were injected intravenously. Only sites of local dextran extravasation that showed evidence of focal extravascular clot formation were counted. Clots on newly formed vessels that occasionally grow over the pial surface after chronic window implantation were excluded from the analysis. Mice were administered a high dose of heparin with the aim to lower the threshold for microhemorrhage formation. At eight months of age, no extravasations were observed at baseline in either the Tg APP/PS1 mice or their WT littermates, but heparin administration triggered a few sites to leak (Figure 3). At 19 months of age, baseline extravasations were observed in both Tg APP/PS1 mice (6/8) and their WT littermates (4/8, p = 0.30), whereas heparin administration resulted in a higher occurrence of leakage sites in Tg (7/8) compared to WT mice (3/8, p = 0.039) (Figure 3).
Figure 3.

Spontaneous microhemorrhage formation after heparin administration in 19-month-old APP/PS1 mice. Spontaneous extravasations were not observed in 8-month-old Tg APP/PS1 mice (b) or their WT littermate controls (a), and only occasionally after heparin administration (c). At 19 months of age, both Tg APP/PS1 mice (e) and their WT littermate controls (d) had evidence of spontaneous extravasation (f, χ2 test p = 0.30). Subsequent heparin administration resulted more often in extravasations in Tg compared to WT mice (f, χ2 test p = 0.039). Lines in C and F indicate median. Note that each dot or triangle in C and F represents a mouse. *p < 0.05.
Characterization of leakage sites in old APP/PS1 mice
The formation of extravascular fibrin clots at the sites of dextran extravasation allowed us to determine the exact site of leakage and the characteristics of the vessels at the leakage sites in relationship to the surrounding microvasculature and local CAA burden. Notably, leakage sites only occurred on arterioles, but not venules. The arteriolar diameters at the site of leakage were on average greater in 19-month-old Tg APP/PS1 mice (mean ± SD 19 ± 7 μm), compared to their age-matched WT littermates (mean ± SD 13 ± 5 μm, p = 0.0054) (Figure 4). In Tg APP/PS1 mice, approximately half of all fibrin clots (15/27, 56%) were found on first-order arteriolar branches, as compared to parent arterioles (1/27, 4%), second-order (9/27, 33%), and third-order arteriolar branches (2/27, 7%). One additional clot was found at the capillary level. Notably, CAA-negative first-order arteriolar branches (13/67, 19%) were more likely to bleed compared to CAA-positive first-order branches (2/67, 3%) (p < 0.0001) (Table 1). Ten fibrin clots either directly colocalized with or were found close to (<20 µm) CAA deposits, whereas 18 where located remotely from CAA (on average 83 ± 60 µm away, range 21–216 µm). In terms of vessel morphology, most clots were observed at vessel branchpoints or bends (20/28, 71%) (Figure 4, Supplemental Figure 1).
Figure 4.
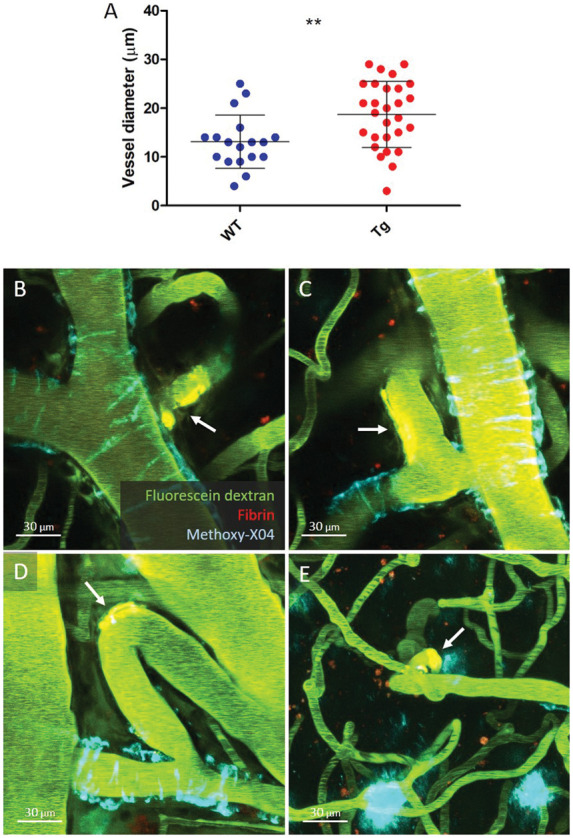
Characterization of spontaneous leakage sites in 19-month-old Tg APP/PS1 mice. Spontaneous extravasations in Tg APP/PS1 mice were on average found on larger arterioles (mean diameter at leakage sites 19 ± 7 µm) compared to their WT littermate controls (13 ± 5 µm) (a, t-test p = 0.0054). In Tg APP/PS1 mice, the majority of extravascular fibrin clots (arrows) were found on CAA-negative first order arteriolar branches (b, c, d), as compared to higher order arteriolar segments (e). Notably, most clots (71%) were observed at vessel branchpoints (b, c) or bends (d, e) and remote from CAA deposits. Lines and error bars in A indicate mean and standard deviations. Note that the vessel in B is the same as depicted in Figure 3(e). Note that the Methoxy-X04 deposit directly adjacent to the clot in panel E is a parenchymal plaque and not CAA. **p < 0.01.
Table 1.
Characterization of vessel types affected by fibrin clots in Tg APP/PS1 mice.
| Total number | Without clots | With clots | χ2 (p-value) | ||
|---|---|---|---|---|---|
| CAA+ parent artery | 30 | 29 | 1 | ||
| CAA− parent arterya | 0 | 0 | 0 | n/a | |
| CAA+ 1st order arteriole | 42 | 40 | 2 | ||
| CAA− 1st order arteriolea | 25 | 12 | 13 | 0.0001 | |
| CAA+ 2nd order arteriole | 11 | 10 | 1 | ||
| CAA− 2nd order arteriolea | 44 | 36 | 8 | 0.47 | |
| CAA+ 3rd order arteriole | 2 | 2 | 0 | ||
| CAA− 3rd order arteriolea | 25 | 23 | 2 | 0.68 | |
aVessels that were classified as parent arteries had an average diameter of 40 ± 11 µm, first-order arterioles 22 ± 4 µm, second-order arterioles 16 ± 5 µm, and third-order arterioles 13 ± 4 µm.
Discussion
In this study, we demonstrated the feasibility of visualizing individual focal sites of microhemorrhage formation in relation to their surrounding microvasculature in real-time with in vivo two-photon microscopy in Tg mice. Our findings suggest that the vessels in old mice with CAA are more prone to leakage compared to non-Tg age-matched control mice after heparin administration. Leakage does not immediately occur from CAA-laden vessels, but rather from vulnerable vessel segments at bends or bifurcations close to or remote from vascular Aβ depositions.
Focal laser irradiations coupled with optical imaging have previously been used to assess microhemorrhage dynamics in real-time in vivo17 to determine effects on the surrounding tissue18 or the influence of drug treatment on microhemorrhage size.15,19 Here we utilized this technique in combination with the detection of extravasated fluorescently labeled fibrin to identify the exact sites of microhemorrhages in vivo as a detection method for screening spontaneously occurring microhemorrhages in subsequent experiments. Using the same fluorescently labeled antibody, a previous study observed increased and more rapid fibrin extravasation after treatment with matrix metalloproteinase-9 (MMP9) in Tg2576 mice compared to age-matched WT littermate controls.14 MMP9 was topically applied on the surface of the mouse brain, resulting in widespread extravasation of fibrin. Therefore, individual leakage sites could not be determined, or whether leakage originated from vessel segments with or without vascular Aβ depositions. In our study, we observed focal extravascular clot formation after targeted laser irradiations that were comparable between 20-month-old APP/PS1 mice and their WT littermate controls. No notable differences were observed in terms of clot morphology between Tg and WT mice, whereas clots had a different appearance on arterioles compared to veins. The circumferential appearance of the fibrin clots on arterioles (and not veins) resembles the spiral-like deposition of CAA between vascular smooth muscle cells and could potentially reflect cellular uptake of fibrin in the perivascular space, an interesting topic for future studies.20,21 Unexpectedly, in mice with CAA, it took significantly longer for laser irradiation to rupture arterioles compared to arterioles in control mice, with a positive trend towards more attempts needed to rupture vessels with increased CAA burden. This may indicate a relative protective effect of mild-to-moderate CAA locally or the possibility that Methoxy-X04, the fluorescent dye that binds Aβ depositions, absorbed some of the laser power.
Using fluorescent fibrin as a marker to detect individual microhemorrhages in vivo, we observed an age-related increase of heparin-induced leakage sites in APP/PS1 mice. Previous studies have observed an increase in size of separately induced microhemorrhages in heparin-treated mice compared to vehicle-treated mice,19,22 indicating prolonged bleeding times of existing bleeds after heparin administration. Using a more sensitive detection method to assess small individual leakage sites, we observed heparin-induced microhemorrhages more often in 20-month-old APP/PS1 mice compared to their WT littermate controls. Although we took care to exclude leakage from newly formed vessels at the surface of the brain or the edge of the cranial window, it cannot be excluded that some microhemorrhages indeed originated from these permeable vessels and therefore may have resulted in false positive findings. That said, the increased number of microhemorrhages observed in Tg mice was driven by microhemorrhages on larger diameter lower order arteriolar branches (the segments of interest), and not on the smaller newly formed vessels. Interestingly, microhemorrhages were only observed on arterioles and in particular on arteriolar segments without CAA. Although laser-induced fibrin clots were visible longer on arterioles compared to venules, the increased observation of spontaneous microhemorrhages on arterioles is unlikely due to this difference in detection. This is because the formation of the heparin-induced microhemorrhages was observed in real-time (<1 h after administration) and therefore clots on veins would have been detectable within that timeframe. A more likely explanation for the observed arteriolar predilection for microhemorrhages – which is in line with neuropathological observations in humans23 – is that Aβ affects arterioles more often than veins in the context of CAA, as arterioles are believed to be the main routes for perivascular Aβ clearance.24 Also, Aβ deposition is associated with smooth muscle cell degeneration, which reduces vessel wall integrity and increases the risk for microhemorrhages.25 Even in the absence of vascular Aβ depositions, the presence of increased soluble Aβ may induce smooth muscle cell degeneration in vessel segments remote from the visible Aβ depositions.26,27 The frequent observation of microhemorrhages on bends and bifurcations in 20-month-old mice suggests a role for flow network dynamics in combination with focal damage. It may indicate that these anatomically vulnerable segments are progressively weakened due to sustained turbulence in those areas and thus likely to bleed in the context of vessel wall degeneration and overall increased Aβ levels.25 Notably, microhemorrhages in brain tissue of CAA cases also seem to originate from penetrating arterioles at the sites where leptomeningeal vessels branch into the cortex, similarly to our observations of clots on first-order arteriolar branches in mice.23 Future studies in mice are warranted to unravel potential functional alterations, such as blood flow or pulsatility changes, within affected vascular networks at those vulnerable locations.
Although 20 months represents an old age for the APP/PS1 mice, the observed CAA appears substantially less advanced compared to humans with severe CAA, suggesting that the spontaneous microhemorrhages in the mice may not be directly comparable to those in post-mortem human brain tissue of CAA patients. First, the mouse microhemorrhages are much smaller in size and perhaps better resemble subtle blood–brain barrier leakage than frank hemorrhages, as in most cases no extravasation of red blood cells was observed in vivo. Our ex vivo histopathological examination also confirmed that old microhemorrhages are only rarely observed in the brains of Tg APP/PS1 mice post-mortem. Second, microhemorrhages in post-mortem human brains are only observed in the context of moderate-to-severe CAA.8,9,28 In these brains, Aβ depositions are not only found on leptomeningeal vessels, which is the case in APP/PS1 mice, but cover penetrating cortical arterioles and sometimes capillaries as well, resulting in vasculopathies such as fibrinoid necrosis and microaneurysms in the most severe cases. We have previously reported absence of vascular Aβ at the bleeding site in vessels involved in microhemorrhages in autopsy cases with severe CAA,29 but serial sectioning reveals extensive vessel remodeling prior to bleeding, including thickening of the wall and degeneration of vascular smooth muscle cells,23 indicating severe vessel wall degeneration not observed in the mice. Therefore, the mechanisms underlying the microhemorrhages (or leakage sites) observed in mice with CAA may not necessarily resemble what happens in patients with severe CAA. Alternatively, they could represent blood–brain barrier leakage that may happen in an earlier stage of the pathophysiology of CAA.
Observations from both post-mortem human brain tissue and our in vivo findings in mice suggest that just the presence of vascular Aβ per se is not sufficient to make vessels bleed directly but that more complex vascular network dynamics likely play a role. Future studies of spontaneous microhemorrhage formation in real-time in mice with more severe CAA and vascular remodeling, such as APP23 mice,30,31 would be informative. Furthermore, our in vivo approach allows the longitudinal assessment of the role of vascular dysfunction (e.g. impaired evoked vascular reactivity) on the formation of spontaneous lesions at the single-vessel level, which is currently not feasible in humans.
Supplemental Material
Supplemental material, JCB899377 Supplemental Figure for In vivo characterization of spontaneous microhemorrhage formation in mice with cerebral amyloid angiopathy by Susanne J van Veluw, Matthew P Frosch, Ashley A Scherlek, Daniel Lee, Steven M Greenberg and Brian J Bacskai in Journal of Cerebral Blood Flow & Metabolism
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health [R01 NS096730; S10 RR025645; R01 EB000768; K99 AG059893; RF1 NS110054] and the Netherlands Organization for Scientific Research, ZonMW [Veni 91619021].
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ORCID iD: Susanne J van Veluw https://orcid.org/0000-0002-7957-8643
Supplemental material: Supplemental material for this article is available online.
Authors’ contributions
Susanne J. van Veluw was involved in the concept and design of the study, analysis and interpretation of the data, drafting and revising of the manuscript, and obtained funding.
Matthew P. Frosch was involved in the concept and design of the study and revising of the manuscript.
Ashley A. Scherlek was involved in the analysis and interpretation of the data and revising of the manuscript.
Daniel Lee was involved in the analysis and interpretation of the data and revising of the manuscript.
Steven M. Greenberg was involved in the concept and design of the study and revising of the manuscript, and obtained funding.
Brian J. Bacskai was involved in the concept and design of the study and revising of the manuscript, and obtained funding.
References
- 1.Pantoni L.Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010; 9: 689–701. [DOI] [PubMed] [Google Scholar]
- 2.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013; 12: 822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009; 8: 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haller S, Vernooij MW, Kuijer JP, et al. Cerebral microbleeds: imaging and clinical significance. Radiology 2018; 287: 11–28. [DOI] [PubMed] [Google Scholar]
- 5.Charidimou A, Shams S, Romero JR, et al. Clinical significance of cerebral microbleeds on MRI: A comprehensive meta-analysis of risk of intracerebral hemorrhage, ischemic stroke, mortality, and dementia in cohort studies (v1). Int J Stroke 2018; 13: 454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vernooij MW, Van der Lugt A, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology 2008; 70: 1208–1214. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg SM, Charidimou A.Diagnosis of cerebral amyloid angiopathy: evolution of the Boston criteria. Stroke 2018; 49: 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vonsattel JP, Myers RH, Hedley-Whyte ET.Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991; 30: 637–649. [DOI] [PubMed] [Google Scholar]
- 9.Van Veluw SJ, Charidimou A, Van der Kouwe AJ, et al. Microbleed and microinfarct detection in amyloid angiopathy: a high-resolution MRI-histopathology study. Brain 2016; 139: 3151–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jankowsky JL, Slunt HH, Ratovitski T, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng 2001; 17: 157–165. [DOI] [PubMed] [Google Scholar]
- 11.Arbel-Ornath M, Hudry E, Eikermann-Haerter K, et al. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol 2013; 126: 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuchibhotla KV, Wegmann S, Kopeikina KJ, et al. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc Natl Acad Sci U S A 2014; 111: 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klunk WE, Bacskai BJ, Mathis CA, et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 2002; 61: 797–805. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L, Arbel-Ornath M, Wang X, et al. Matrix metalloproteinase 9-mediated intracerebral hemorrhage induced by cerebral amyloid angiopathy. Neurobiol Aging 2015; 36: 2963–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lauer A, Cianchetti FA, Van Cott EM, et al. Anticoagulation with the oral direct thrombin inhibitor dabigatran does not enlarge hematoma volume in experimental intracerebral hemorrhage. Circulation 2011; 124: 1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura N, Schaffer CB, Friedman B, et al. Targeted insult to subsurface cortical blood vessels using ultrashort laser pulses: three models of stroke. Nat Methods 2006; 3: 99–108. [DOI] [PubMed] [Google Scholar]
- 18.Rosidi NL, Zhou J, Pattanaik S, et al. Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS One 2011; 6: e26612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan S, Brophy M, Nishimura N, et al. Aspirin treatment does not increase microhemorrhage size in young or aged mice. PLoS One 2019; 14: e0204295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christie R, Yamada M, Moskowitz M, et al. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol 2001; 158: 1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weller RO, Nicoll JA.Cerebral amyloid angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res 2003; 25: 611–616. [DOI] [PubMed] [Google Scholar]
- 22.Foerch C, Rosidi NL, Schlunk F, et al. Intravenous tPA therapy does not worsen acute intracerebral hemorrhage in mice. PLoS One 2013; 8: e54203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Veluw SJ, Scherlek AA, Freeze WM, et al. Different microvascular alterations underlie microbleeds and microinfarcts. Ann Neurol 2019; 86: 279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keable A, Fenna K, Yuen HM, et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta 2016; 1862: 1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frösen J, Joutel A.Smooth muscle cells of intracranial vessels: from development to disease. Cardiovasc Res 2018; 114: 501–512. [DOI] [PubMed] [Google Scholar]
- 26.Davis J, Cribbs DH, Cotman CW, et al. Pathogenic amyloid beta-protein induces apoptosis in cultured human cerebrovascular smooth muscle cells. Amyloid 1999; 6: 157–164. [DOI] [PubMed] [Google Scholar]
- 27.Blaise R, Mateo V, Rouxel C, et al. Wild-type amyloid beta 1-40 peptide induces vascular smooth muscle cell death independently from matrix metalloprotease activity. Aging Cell 2012; 11: 384–393. [DOI] [PubMed] [Google Scholar]
- 28.Van Veluw SJ, Biessels GJ, Klijn CJ, et al. Heterogeneous histopathology of cortical microbleeds in cerebral amyloid angiopathy. Neurology 2016; 86: 867–871. [DOI] [PubMed] [Google Scholar]
- 29.Van Veluw SJ, Kuijf HJ, Charidimou A, et al. Reduced vascular amyloid burden at microhemorrhage sites in cerebral amyloid angiopathy. Acta Neuropathol 2017; 133: 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reuter B, Venus A, Heiler P, et al. Development of cerebral microbleeds in the APP23-transgenic mouse model of cerebral amyloid angiopathy – a 9.4 Tesla MRI study. Front Aging Neurosci 2016; 8: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shih AY, Hyacinth HI, Hartmann DA, et al. Rodent models of cerebral microinfarct and microhemorrhage. Stroke 2018; 49: 803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, JCB899377 Supplemental Figure for In vivo characterization of spontaneous microhemorrhage formation in mice with cerebral amyloid angiopathy by Susanne J van Veluw, Matthew P Frosch, Ashley A Scherlek, Daniel Lee, Steven M Greenberg and Brian J Bacskai in Journal of Cerebral Blood Flow & Metabolism