

Abstract
Asthma is a heterogeneous chronic airway inflammation in which Th2 and Th17 cells are key players in its pathogenesis. We have reported that RhoA of Rho GTPases orchestrated glycolysis for Th2 cell differentiation and allergic airway inflammation by the use of a conditional RhoA-deficient mouse line. However, the role of RhoA in Th17 cells remains to be elucidated. In this study, we investigated the effects of RhoA deficiency on Th17 cells in the context of ex vivo cell culture systems and an in vivo house dust mites (HDM)-induced allergic airway inflammation. We found that RhoA deficiency inhibited Th17 differentiation and effector cytokine secretion, which was associated with the downregulations of Stat3 and Rorγt, key Th17 transcription factors. Furthermore, loss of RhoA markedly suppressed Th17 and neutrophil-involved airway inflammation induced by HDM in mice. The infiltrating inflammatory cells in the lungs and bronchoalveolar lavage (BAL) fluids were dramatically reduced in conditional RhoA-deficient mice. Th17 as well as Th2 effector cytokines were suppressed in the airways at both protein and mRNA levels. Interestingly, Y16, a specific RhoA inhibitor, was able to recapitulate the most phenotypes of RhoA genetic deletion in Th17 differentiation and allergic airway inflammation. Our data demonstrate that RhoA is a key regulator of Th17 cell differentiation and function. RhoA might serve as a potential novel therapeutic target for asthma and other inflammatory disorders.
Keywords: allergic airway inflammation, house dust mite, RhoA, Th17, Y16
1 |. INTRODUCTION
The ras homolog family member A (RhoA) of the Rho family GTPases belongs to the Ras superfamily. Like other Rho GTPases, RhoA is nucleotide-dependent proteins behaving like “molecular switches” that cycle between an inactive, GDP-bound, and an active, GTP-bound, states.1 As an intracellular signaling protein, RhoA regulates multiple pathways involved in various important biological functions such as cellular membrane transport, migration, differentiation, proliferation, and apoptosis. RhoA plays important regulatory roles in the infection, inflammation, and oncogenesis, and becomes a novel therapeutic target.2–4 In T-cells, it has been demonstrated that RhoA controls thymocyte development and migration to the periphery, as well as T-cell polarity with the use of in vitro RhoA overexpression or inactivation of RhoA by C3 transferase in transgenic mice.5–9 Recently, we have generated a conditional RhoA-deficient mouse line in which RhoA is specifically deleted in T cells. With this mouse model, we found that RhoA was required for coordinating mitochondrial function and thymocyte development.10 Moreover, RhoA orchestrated glycolysis for Th2 cell differentiation and allergic airway inflammation.11 However, the role of RhoA in Th17 cells remains to be elucidated.
Th17 cells are derived from naïve CD4+ T cells, which possess their own differentiation and regulation pathways independent from Th1 and Th2 cells. Th17 cells are preferential producers of IL-17A, IL-17F, IL-21, and IL-22.12,13 The receptors for IL-17A and IL-22 are broadly expressed on various epithelial tissues. Th17 effector cytokines, therefore, mediate crucial crosstalk between the immune system and tissues, and play indispensable roles in tissue immunity12,14 including airways.15,16 Asthma is an allergic airway inflammation affecting millions of people in the world. CD4+ Th cells, particularly Th2 cells, are traditionally believed as a key player in its pathogenesis.17–20 Recently, compelling evidence has demonstrated that Th17 cells are a major contributor to the pathogenesis of allergic airway inflammation,21–25 especially severe asthma. The latter is often characterized by mixed responses of Th2 and Th17 cells and resistant to the steroid therapy.25–29 To investigate the role of Th17 cells in the pathogenesis of asthma, we have recently established murine asthma models induced by house dust mites (HDM, Dermatophagoides sp.).25,30 HDM is one of the commonest aeroallergens naturally existed worldwide, accounting for up to 85% of asthmatics.31,32 In this model, Th17 and neutrophils are among the key responders, mimicking severe asthma.28,33
In this study, we investigated the role of RhoA in Th17 differentiation and function in the context of ex vivo cell culture systems and an in vivo murine model of HDM-trigged allergic airway inflammation with the use of T cell-specific RhoA-deficient mice and a RhoA-specific inhibitor Y16. Our data from both genetic deletion and pharmacological inhibition demonstrate that RhoA is an important regulator for Th17 differentiation and function.
2 |. MATERIALS AND METHODS
2.1 |. Mice
RhoAflox/flox mice were generated as previously described.34,35 The floxed allele contains loxP sites flanking exon 3 of the RhoA allele. To delete RhoA in vivo in T cells, RhoAflox/flox mice were mated with mice expressing Cre recombinase under the control of a CD2 proximal promoter (Jackson Laboratory, Bar Harbor, ME).10,11 In each experiment, age- (8- to 10-week old) and sex-matched RhoAflox/floxCD2Cre+ mice (hereafter, referred to as RhoA−/− or RhoA-deficient mice) and their WT littermates (RhoAflox/floxCD2Cre−, referred to as WT mice) were used. Animals were housed under specific pathogen-free conditions in the animal facility at the Cincinnati Children’s Hospital Research Foundation in compliance with the Cincinnati Children’s Hospital Medical Center Animal Care and Use Committee protocols.
2.2 |. T-cell activation and differentiation
Sorted naïve T cells (CD62LhiCD44lo) were used for T-cell activation and differentiation. Naïve T cells were activated with plate-bound anti-CD3 (clone 145-2C11, 10 μg/mL) plus soluble anti-CD28 (clone 37.51, 2 μg/mL) (BD Bioscience, San Jose, CA). For T-cell differentiation, CD4+ T cells were differentiated into Th0, Th1, Th2, Th17, or iTreg cells, as previously reported.11,25 Briefly, CD4+ T cells were stimulated by anti-CD3/CD28 for 4 days with IL-2 (20 ng/mL) in the presence of anti-IFNγ (clone 37895) and anti-IL-4 (clone 30340) (both 10 μg/mL, for Th0), anti-IL-4 and IL-12 (20 ng/mL, for Th1), anti-IFN-γ and IL-4 (20 ng/mL, for Th2). For iTreg and Th17 conditions, CD4+ T cells were stimulated by anti-CD3/CD28 for 4 days with anti-IFN-γ and anti-IL-4 (both 10 μg/mL) in the presence of TGF-β1 (5 ng/mL, for Treg),or TGF-β1 plus IL-6 (10 ng/mL, for Th17) (all from R&D Systems, Minneapolis, MN). Cells were restimulated with PMA (25 ng/mL) plus ionomycin (500 ng/mL) (Sigma, St Louis, MO) for 5 h with GolgiStop (BD Bioscience) in the last 2 h for intracellular cytokine staining; or without GolgiStop for cytokine assays in the culture supernatants by ELISA. Where indicated, RhoA inhibitor Y1636 was added to the cultures.
2.3 |. Flow cytometry
Cells were incubated with anti-CD16/32 (clone 2.4G2) (BD Bioscience) to block FcγR II/III before FACS staining. BD Cytofix/Cytoperm kit or Perm buffer III (BD Bioscience) was used for intracellular cytokine or Stat3 staining. The following conjugated monoclonal antibodies were used in this study: IL-17-PE (clone TC11-18H10), IFN-γ-FITC (XMG1.2), CD25-PerCP.Cy5.5 (clone PC61), phospho-Stat3-PE (pY705, clone 4/P-STAT3) (BD Bioscience); IL-4-PE (clone 11B11), IL-13-eFluor 660 (clone eBio13A), Foxp3-APC (clone FJK-16s), CD4-eFluor 450 (clone RM4-5) (eBioscience, San Diego, CA). Stained cells were analyzed by LSRII with FACSDiva (BD Bioscience) or FCS Express (De Novo Software, Los Angeles, CA) softwares.
2.4 |. Cytokine assay
Cytokines in the culture supernatants and BAL fluids were measured by ELISA: IL-4, IL-5, and IFNγ were measured with OptEIA kits (BD Bioscience); IL-6, IL-13, IL-17, IL-17F, IL-21, and eotaxin were measured with DuoSet ELISA kits (R&D Systems). ELISA plates were developed with TMB substrate (BD Bioscience), and read with a microplate reader. Cytokine mRNA levels were measured by real-time quantitative PCR.19,20
2.5 |. Quantitative PCR
Total RNA was extracted from lung tissues or cultured cells with the RNeasy Mini Kit (Qiagen, Valencia, CA). The cDNA was prepared by using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative real-time PCR was performed with the Platinum SYBR Green qPCR SuperMix-UDG w/RO or TaqMan Gene Expression Master Mix (Life Technologies, Carlsbad, CA) on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, Rockford, IL). The data were normalized to the 18S reference. Primers were designed with OLIGO 4.0 software as reported.20
2.6 |. HDM-induced allergic airway inflammation
The murine airway inflammation was induced by HDM according to our recent reports.25,30,37 Briefly, mice were inoculated intratracheally (i.t.) with 20 μg of house dust mite (HDM, Stallergenes Greer, Lenoir, NC) in 40 μL PBS on day 0, 3, 7, and 14, once a day. Control mice were inoculated i.t. with PBS alone. Mice were challenged i.t. with HDM or PBS 3 times from day 21 to 23, and sacrificed 24 h after the last challenge. BAL fluid was aspirated and centrifuged. Total cells in the pellet were counted by using a hemacytometer. Differential cell counts on >400 cells were performed on cytospins stained with Shandon Kwik-Diff Stain kit (Thermo Scientific). The BAL fluid from each mouse was concentrated to 0.5 mL by centrifugation with an Amicon Ultra-4 filter unit (Millipore, Billerica, MA) for determination of cytokines by ELISA. For lung histology, the lower lobe of the right lung was fixed with 4% paraformaldehyde overnight, dehydrated, embedded in paraffin, cut into 4 μm sections, and processed for H&E and Periodic acid-Schiff (PAS) staining. Lung tissue mRNAwas analyzed by real-time PCR.
To isolate pulmonary infiltrating cells, lungs were harvested, minced, and digested with Liberase DH and DNase I (both 0.5 mg/mL, Sigma) in Hanks’ balanced buffered solution at 37μC for 30 min. The tissue was forced through a 70 μm cell strainer and RBCs were lysed with ACK lysis buffer (BD Bioscience). After washing, the freshly isolated cells were restimulated with PMA (25 ng/mL) plus ionomycin (500 ng/mL) (Sigma) for 4 h with GolgiStop (BD Bioscience) in the last 3 h for intracellular cytokine FACS stainings.
2.7 |. Statistical analysis
All experimental data were analyzed and compared for statistically significant differences using 2-tailed Student’s t or Mann-Whitney U test; ANOVA was used for comparing 3 or more groups. A P value of <0.05 was considered significant.
3 |. RESULTS
3.1 |. RhoA deficiency dampens Th17 differentiation
We have previously reported that RhoA orchestrates glycolysis for Th2-cell differentiation and allergic airway inflammation without affecting Th1 cells.11 Here, we evaluated the role of RhoA in Th17 cell differentiation with conditional RhoA-deficient mice. Th17 differentiation was significantly inhibited from RhoA-deficient CD4+ T cells under Th17 polarizing conditions with TGF-β1 plus IL-6, compared to WT cells (Fig. 1A and B). Consistent with this, IL-17 (also called IL-17A) levels in the culture supernatants were also markedly lower from RhoA-deficient CD4+ T cells under neutral conditions with anti-CD3/CD28 stimulation (Fig. 1C) or under Th17 polarizing conditions (Fig. 1D). Additionally, the mRNA levels of IL-17, IL-17F, and IL-21, 3 key Th17 effector cytokines, were significantly downregulated in RhoA-deficient Th17-polarized cells and even under nonpolarizing or Treg-polarizing conditions (Fig. 1E), consistent basically with protein-level data by FACS and ELISA. IL-21 has autocrine effect to promote Th17 maturation.38 On the other hand, IL-22 was not affected by RhoA deficiency in Th17 cells (Fig. 1E). Of note, IL-22 was even not efficiently induced by TGF-β1 plus IL-6 stimulation in WT Th17 cells, supporting the notion that additional stimulation by IL-23 is needed to induce IL-22 secretion from fully committed Th17 cells.25,39
FIGURE 1. RhoA deficiency dampens Th17 differentiation.
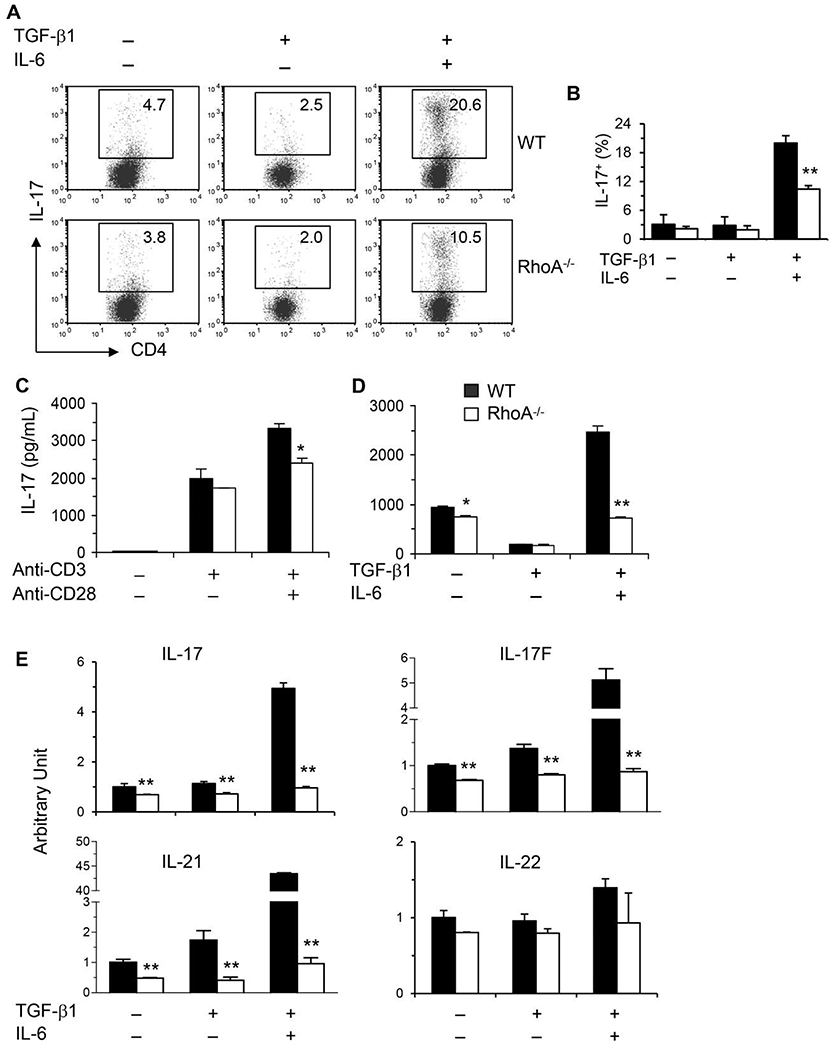
(A and B) Purified CD4+ T cells were differentiated under Th17-polarizing conditions (TGF-β1 + IL-6) for 4 days and restimulated with PMA plus ionomycin for 5 h with BD GolgiPlug in the last 2 h. Cells were collected for IL-17 intracellular staining. Percentages of IL-17+ cells are shown in representative dot plots (A) and summarized in a bar graph (B). (C and D) Supernatants were collected from CD4+ T cells stimulated with or without anti-CD3/CD28 mAbs (C) or differentiated under Th17-polarizing conditions and restimulated with PMA/ionomycin (D)for ELISA assays of IL-17. (E) Total RNA was extracted from the cultured cells for real-time PCR analysis. The mRNA levels of Th17 effector cytokines as indicated are shown (arbitrary unit). The data are normalized to an 18S reference. Results (B-E) are expressed as mean + SD of triplicates, representative of 3 independent experiments. *P < 0.05, **P < 0.01 compared to WT cells
3.2 |. RhoA deficiency downregulates key Th17 transcription factors and signaling molecules
To begin to delineate underlying molecular mechanisms whereby RhoA deficiency affects Th17 cells, several transcription factors key to Th17 differentiation were determined by quantative PCR under Th17 polarizing condition in vitro. Retinoic acid-related orphan receptor gamma t (Rorγt), a specific Th17 transcription factor, was reduced in RhoA-deficient Th17 cells (Fig. 2A).
FIGURE 2. RhoA deficiency downregulates Th17 transcription factors.
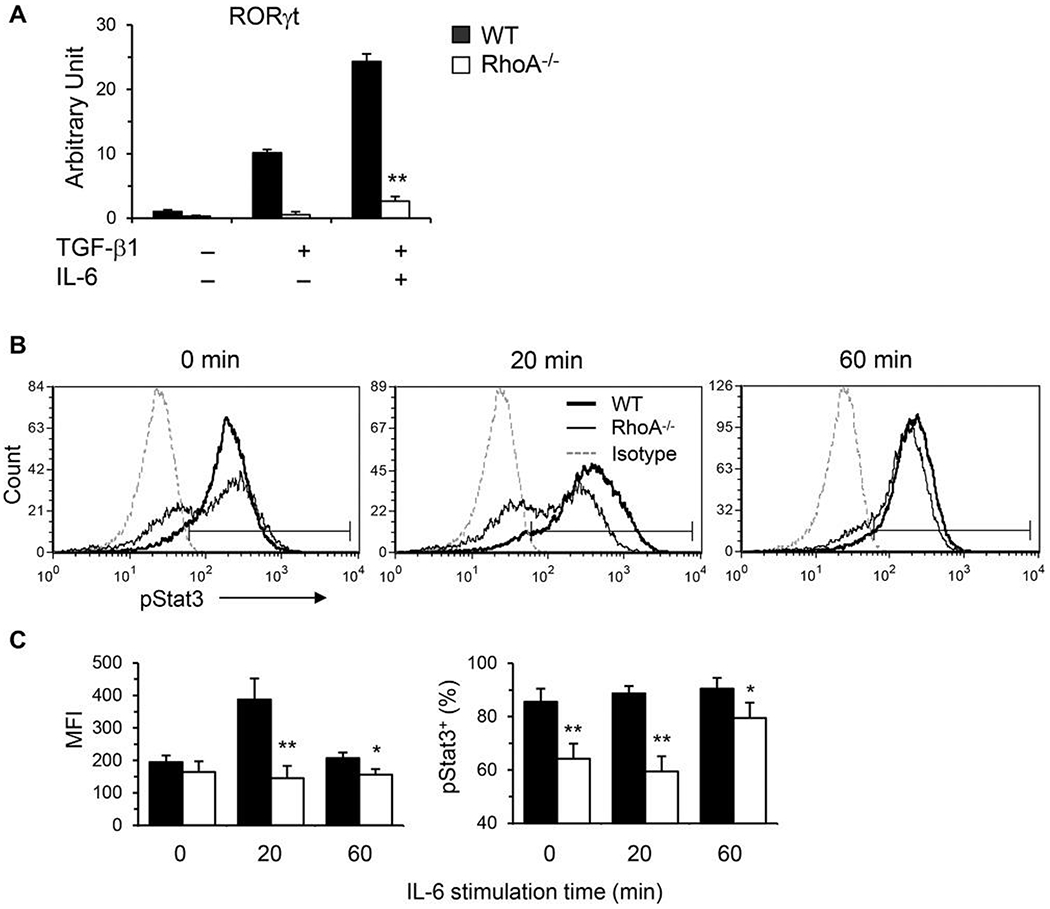
(A) Total RNA was extracted from splenic CD4+ T cells differentiated under Th17 conditions and restimulated with PMA/ionomycin for real-time PCR analysis. The mRNA levels of RORγt are shown. The data are normalized to an 18S reference. (B and C) Naïve splenic CD4+ T cells were stimulated with plate-bound anti-CD3 in the presence of free anti-CD28 for 3 days. Cells were washed and restimulated with recombinant mouse IL-6 (25 ng/mL) for the indicated time. Cells were harvested and processed for intracellular FACS staining for pStat3 (pY705) following BD Bioscience’s protocols. Representative histograms (B), the mean fluorescence intensity (MFI) and percentages of pStat3+ cells (C) are shown. Naïve CD4+ T cells were pooled from 5 to 6 mice. Results (means + SD of triplicates) are representative of 2 independent experiments. *p < 0.05, **P < 0.01 versus WT cells
IL-6 induces the phosphorylation of signal transducer and activator of transcription 3 (Stat3) to promote Th17 differentiation. The level of phospho-Stat3 (pStat3) was markedly increased from activated WT CD4+ T cells with IL-6 restimulation. This upregulation of pStat3 was significantly impaired by the loss of RhoA (Fig. 2B and C). Collectively, our data suggest that RhoA regulates Th17 cells at least in part through transcription factors Rorγt and Stat3. More in-depth investigation is clearly necessary to better define how RhoA regulates Th17 cells.
3.3 |. Effect of RhoA deficiency on Treg cells
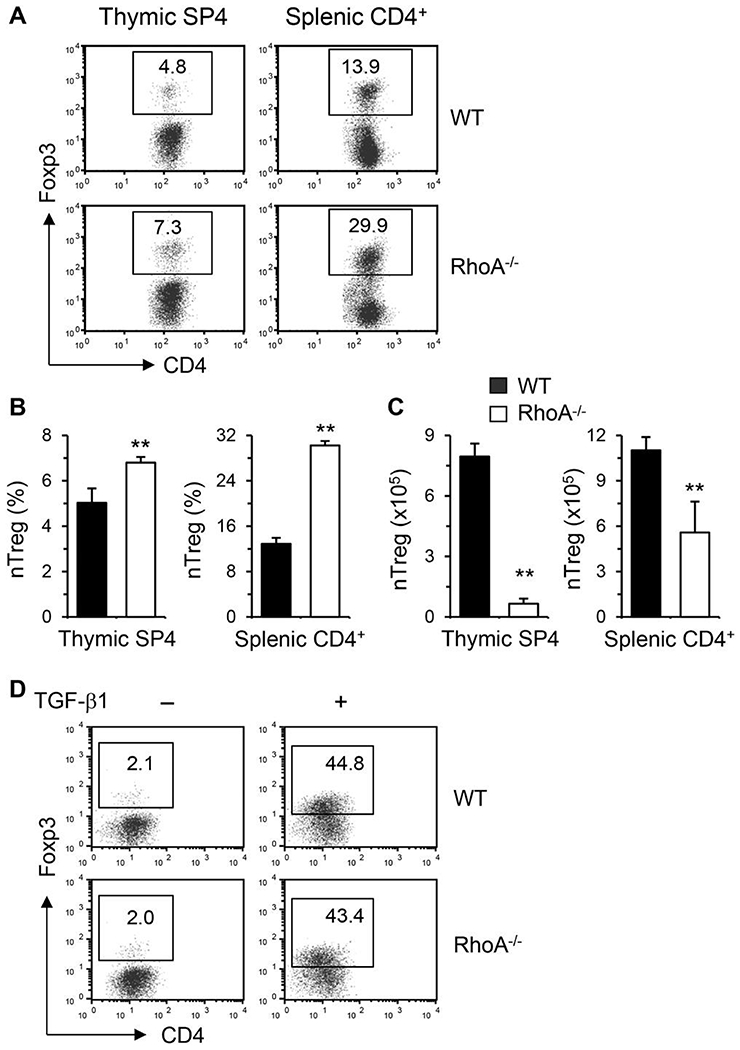
Treg cells, classified as natural Treg (nTreg) that are differentiated in the thymus and induced Treg (iTreg) that are differentiated from peripheral naïve T cells, possess immunosuppressive function. Treg and Th17 cells are reciprocally regulated and the balance of these 2 subsets plays a vital role in maintenance of immune homeostasis. We, thus, evaluated the effects of RhoA deficiency on Treg development. The percentages of nTreg were increased about 2-fold in the thymus and spleen from RhoA-deficient mice (Fig. 3A and B). However, the absolute numbers of nTreg cells in the thymus and spleen were actually markedly reduced in RhoA−/− mice (Fig. 3C), because of significantly lower total thymocytes and splenocytes.10,11 On the other hand, iTreg cells were similar in both genotypes when naïve CD4+ T cells were polarized to Treg in the presence of TGF-β1, indicating that RhoA-deficient CD4+ T cells retain the ability to differentiate into iTreg (Fig. 3D). Thus, it seems that RhoA has no direct effect on Treg induction and the upregulated proportions of nTreg in RhoA−/− mice might reflect a compensatory mechanism for their low absolute numbers.
FIGURE 3. Effect of RhoA deficiency on Treg cells.
(A-C) Total thymocytes and splenocytes were prepared from conditional RhoA-deficient mice and their WT littermates. Cells were processed for Treg staining. Percentages of natural Treg (nTreg, Foxp3+) in thymic single CD4+(SP4) and splenic CD4+ cells are shown in representative dot plots (A) and summarized in bar graphs (B). The absolute numbers of nTreg are also shown (C) (mean + SD, n = 5). (D) Naive CD4+ T cells were differentiated under Treg polarizing condition with TGF-β1 for 4 days and restimulated with PMA plus ionomycin. Percentages of Foxp3+ cells are shown in representative dot plots. Data are representative of 2 independent experiments with similar results. **P < 0.01 compared to WT mice
Taken together, RhoA deficiency significantly inhibited Th17 differentiation and effector cytokine secretion, without affecting Treg induction.
3.4 |. RhoA deficiency suppresses HDM-induced allergic airway inflammation
Since our in vitro data show that RhoA controls Th17 differentiation and cytokine production, we next evaluate if RhoA is necessary for an optimal Th17 response in vivo. We have developed a HDM-induced model for allergic airway inflammation (Fig. 4A), in which Th17 and Th2 cells, Th17-induced neutrophils and Th2-induced eosinophils are among the dominant responders.25,30 Requirement of RhoA for airway inflammation was investigated in the next series of experiments (Fig. 4B–I).
FIGURE 4. RhoA deficiency inhibits HDM-induced allergic airway inflammation.
(A) RhoA−/− mice and their WT littermates were inoculated and challenged intratracheally (i.t.) with HDM to induce allergic airway inflammation. Control groups were inoculated i.t. with PBS alone. Mice were sacrificed 24 h after the last challenge. (B and C) Quantification of total BAL cells (B), eosinophils (Eo), macrophages (Mø), neutrophils (Neu), and lymphocytes (Lym) (C) in BAL fluids. (D-G) Representative Kwik-Diff staining for BAL cytospins (E), H&E (F), and PAS staining (G) of same areas in lung tissue sections. Percentages of PAS-stained bronchi are summarized (D). (H) Cytokine and chemokine levels in BAL fluids determined by ELISA. (I) mRNA levels of cytokines and chemokines in lung tissues determined by real-time PCR. Data are normalized to an 18S reference and expressed as arbitrary units. Results (means + SE, n = 6-8) are representative of 2 independent experiments. *P < 0.05, **P < 0.01 versus HDM-challenged WT groups
The numbers of total BAL cells increased over 40-fold in WT (RhoAflox/floxCD2Cre−) mice i.t. inoculated with HDM compared to PBS-inoculated ones (Fig. 4B). However, this increase of BAL cells was markedly inhibited in RhoA-deficient (RhoAflox/floxCD2Cre+) mice with HDM inoculation. By differential counting, all the major cell types, including eosinophils, macrophages, neutrophils, and lymphocytes, were reduced in RhoA−/− mice (Fig. 4C and E). As revealed by lung histological analysis, mixed inflammatory cells were seen around blood vessels and bronchi in the lungs of HDM-inoculated WT mice. This massive infiltration of inflammatory cells was markedly suppressed in RhoA−/− mice (Fig. 4F). The mucus production from airway, which is important for the pathogenesis and severity of asthma, was also significantly reduced in RhoA−/− mice as revealed by PAS staining (Fig. 4D and G). In control groups inoculated with PBS, there was no PAS stained bronchus in both genotypes. Based on our in vitro data (Fig. 1) and previous report,11 we reasoned that the inhibitory effects of RhoA deletion on allergic airway inflammation were associated with reduced Th17 and Th2 cell function in vivo. Thus, we tested various cytokines in BAL fluids and lungs. IL-17 and Th2 effector cytokines, such as IL-4, IL-5 and IL-13, in BAL fluids were markedly downregulated from RhoA−/− mice as compared to that from WT littermates in response to HDM challenge (Fig. 4H). Consistently, the mRNA levels of these cytokines in the lungs followed similar patterns as the protein levels in BAL fluids (Fig. 4I).
Additionally, we examined several asthma-related mediators of non-T-cell origin including eotaxin, MUC-5AC, and Gob-5. Eotaxin can selectively recruit eosinophils from the airway microvessels into the lung tissue, playing a key role in the onset of asthma.40 Gob-5 gene is responsible for the synthesis of mucin glycoproteins, the main proteins in mucus, by upregulation of MUC-5AC gene expression.19,41 In support of decreased eosinophils (Fig. 4C and E), we found that eotaxin was downregulated in the lungs from HDM-inoculated RhoA−/− mice (Fig. 4I). Consistent with a reduction in mucus secretion (Fig. 4D and G), MUC-5AC and Gob-5 were decreased in RhoA−/− mice upon HDM exposure (Fig. 4I). Taken together, the above findings suggest that RhoA deficiency significantly inhibited HDM-induced allergic inflammation with attenuated Th17, as well as Th2 responses in the airways.
3.5 |. RhoA specific inhibitor Y16 effectively blocks Th17 differentiation
Our genetic data indicate that RhoA is required for Th17 differentiation and function. To substantiate these findings, Y16, a RhoA-specific inhibitor developed in our laboratory,36 was examined for its effect on Th17 differentiation. Y16 effectively blocked the production of Th17 effector cytokines, IL-17, and IL-21, in a dose-dependent manner when purified splenic CD4+ T cells were cultured under nonpolarizing conditions with anti-CD3/CD28 stimulation for 2 days (Fig. 5A). Moreover, Y16 was able to effectively inhibit Th17 differentiation. Under Th17 skewing conditions, IL-17 secreting cells were significantly reduced by Y16 treatment as evaluated with intracellular FACS staining (Fig. 5B and C). Furthermore, IL-17 and IL-21 were markedly reduced in the supernatants of cultures treated with Y16 (Fig. 5D). Y16 also effectively suppressed Th2 differentiation and effector cytokine secretion (Supporting Information Fig. S1). However, Y16 seemed not to affect Th1 differentiation (Supporting Information Fig. S1) and iTreg induction (Fig. 6A and B). Together, these data from RhoA specific inhibitor were consistent with those from RhoA genetic deficiency for the inhibition of Th17 differentiation and cytokine secretion.
FIGURE 5. RhoA specific inhibitor Y16 blocks Th17 differentiation.
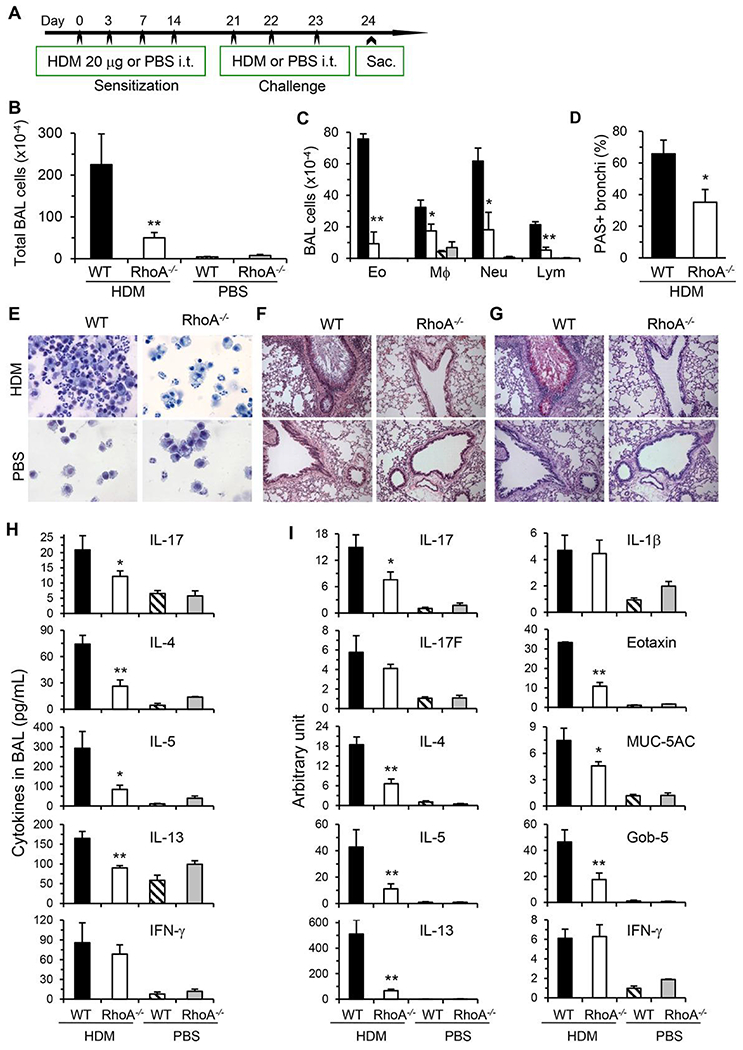
(A) Purified naïve CD4+ T cells (1 × 106/mL) pooled from 8 WT mice were stimulated with plate-bound anti-CD3 plus free anti-CD28 for 2 days without or with Y16 (0~50 μM). IL-17 and IL-21 in the culture supernatants were determined by ELISA. (B-D) Naïve CD4+ T cells were differentiated under Th17 conditions for 4 days and restimulated with PMA plus ionomycin for 5 h, in the presence of vehicle (Veh) or Y16 (30 μM) throughout the culture. Cells were collected for IL-17/IFN-γ intracellular staining. Percentages of IL-17+ or IFN-γ+ CD4+ T cells are shown in representative dot plots (B) and summarized in a bar graph (C). IL-17 and IL-21 in the culture supernatants were determined by ELISA (D). Data are representative of 2 independent experiments. **P < 0.01 compared to vehicle-treated groups
FIGURE 6. Y16 has no effect on Treg induction in vitro.
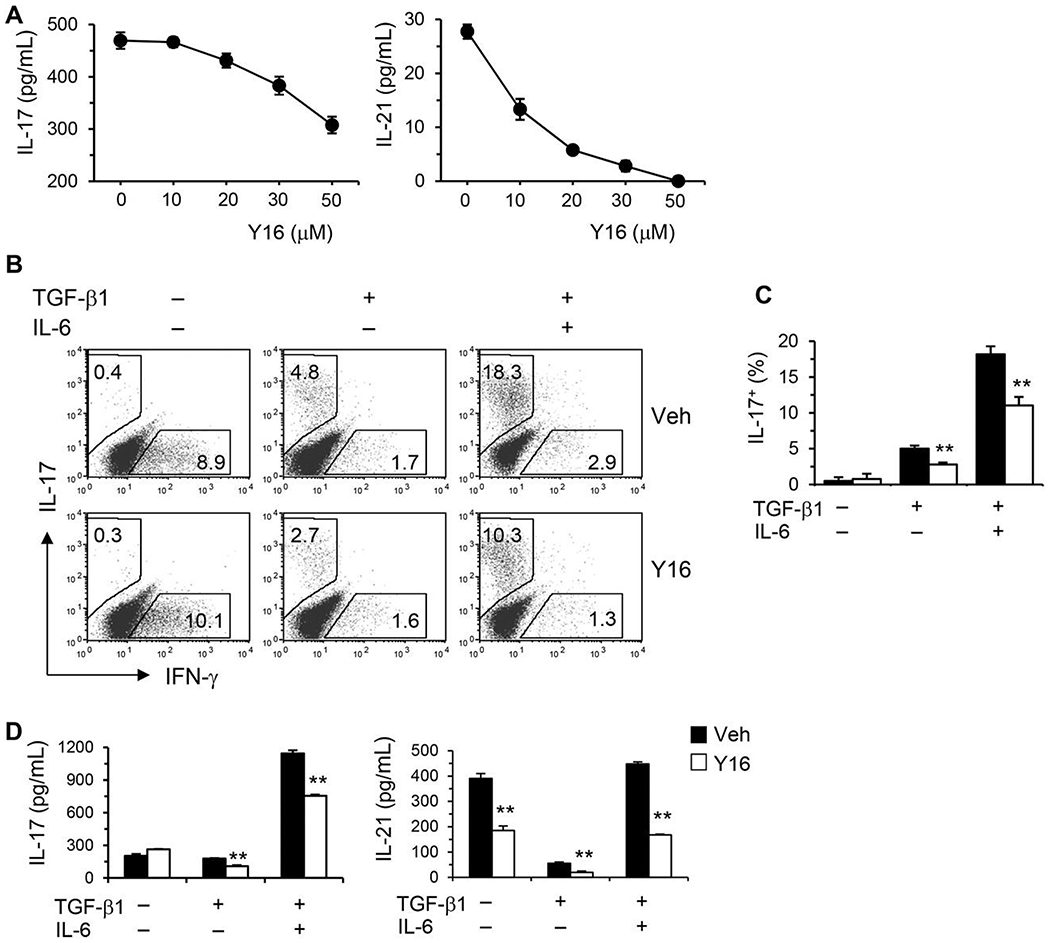
Naïve CD4+ T cells were differentiated under Treg/Th17 conditions for 4 days and restimulated with PMA plus ionomycin for 5 h, in the presence of vehicle (Veh) or Y16 (30 μM) throughout the culture. Cells were collected for Treg staining. Percentages of Foxp3+ CD4+ T cells are shown in representative dot plots (A) and summarized in a bargraph (B). Data are representative of 2 independent experiments
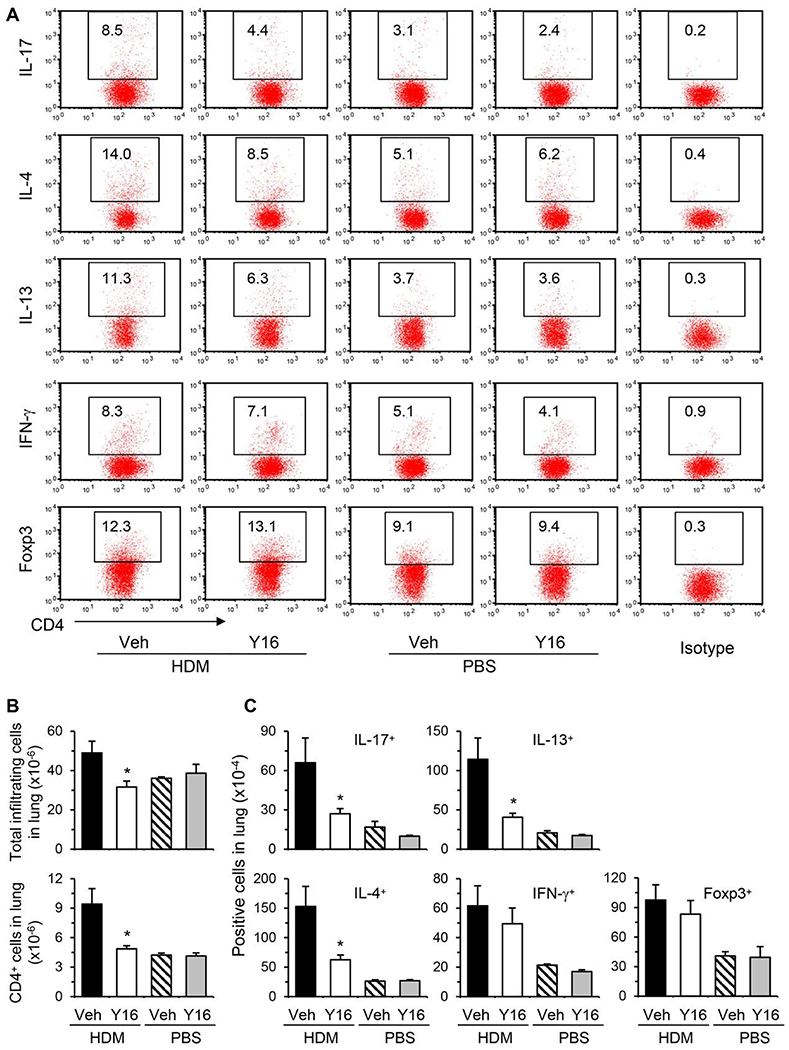
3.6 |. Y16 alleviates HDM-induced allergic airway inflammation with reduced pulmonary Th17 and Th2 cells
Finally, we investigated the effect of Y16 on allergic airway inflammation. WT mice were treated with Y16 daily starting 1 day before HDM inoculation until 1 day before sacrifice (Fig. 7A). Y16 treatment markedly inhibited airway inflammation as indicated by reduced numbers of all the major infiltrating cell types, including eosinophils, macrophages, neutrophils, and lymphocytes, in BAL fluids and the lungs (Fig. 7B–G). Consistently, inflammatory mediators, including Th17/Th2 cytokines, eotaxin, and Gob-5, in BAL fluids (Fig. 7H) and their mRNA levels in the lungs (Fig. 7I), were downregulated. However, Th1 cytokine IFN-γ was not changed by Y16 treatment. Accordingly, mucus hypersecretion was markedly alleviated in Y16 treated HDM-inoculated mice as evaluated by PAS staining (Fig. 7D and G). By FACS analysis of pulmonary infiltrating cells, Th17 and Th2 but not Th1 and Treg cells were markedly downregulated (Fig. 8A–C), which likely contributed to the significant reductions of total pulmonary infiltrating cells and CD4+ T cells (Fig. 8B) upon Y16 treatment. Collectively, these findings indicate that Y16 can recapitulate the most phenotypes of RhoA genetic deletion not only in vitro, but also in vivo in allergic airway inflammation.
FIGURE 7. Y16 alleviates HDM-induced allergic airway inflammation.
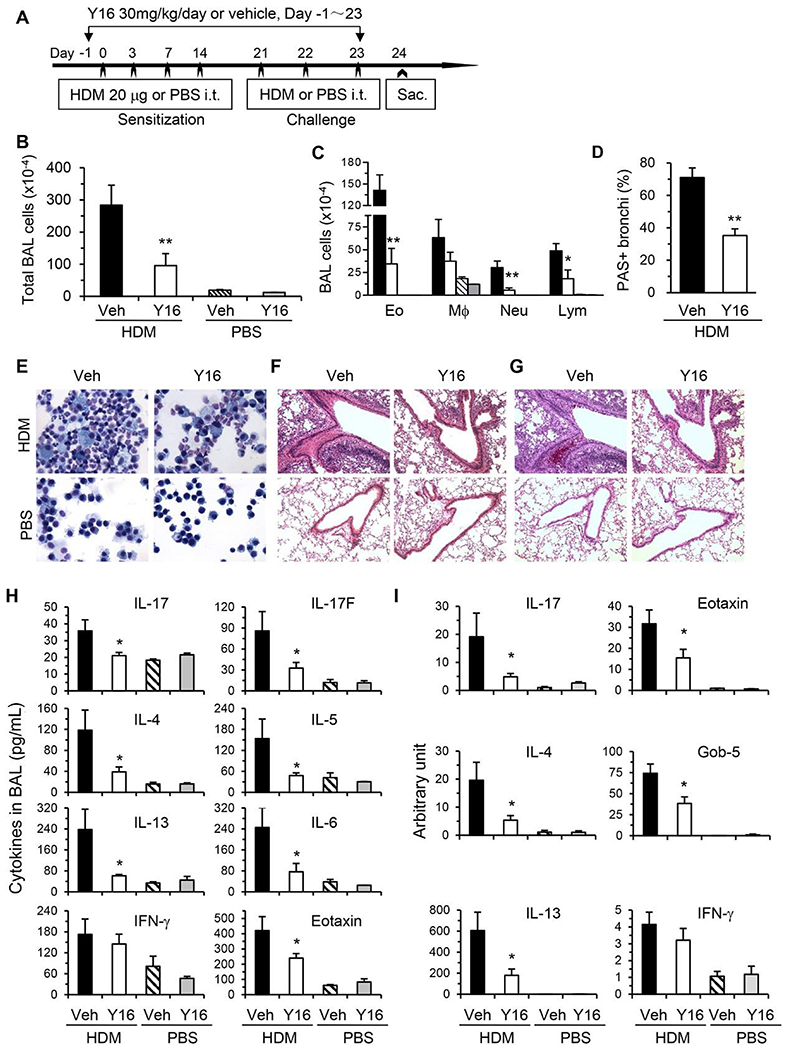
WT mice were injected intraperitoneally (i.p.) daily with Y16 (30 mg/kg) or vehicle (Veh), starting 1 day before HDM inoculation until 1 day before sacrifice (A). Total cells (B) and differential cell counts (C) in BAL fluids, representative Kwik-Diff staining for BAL cytospins (E), H&E (F), and PAS (G) staining of same areas in lung tissue sections are shown. Percentages of PAS stained bronchi are summarized (D). Cytokine and chemokine levels in BAL fluids (H), and their mRNA levels in lung tissues determined by real-time PCR (I) are shown. Results (means + SE, n = 4-8) are representative of 2 independent experiments. *P < 0.05, **P < 0.01 versus HDM-challenged vehicle-treated groups
FIGURE 8. Y16 treatment downregulates pulmonary Th17 and Th2 cells from HDM-inoculated mice.
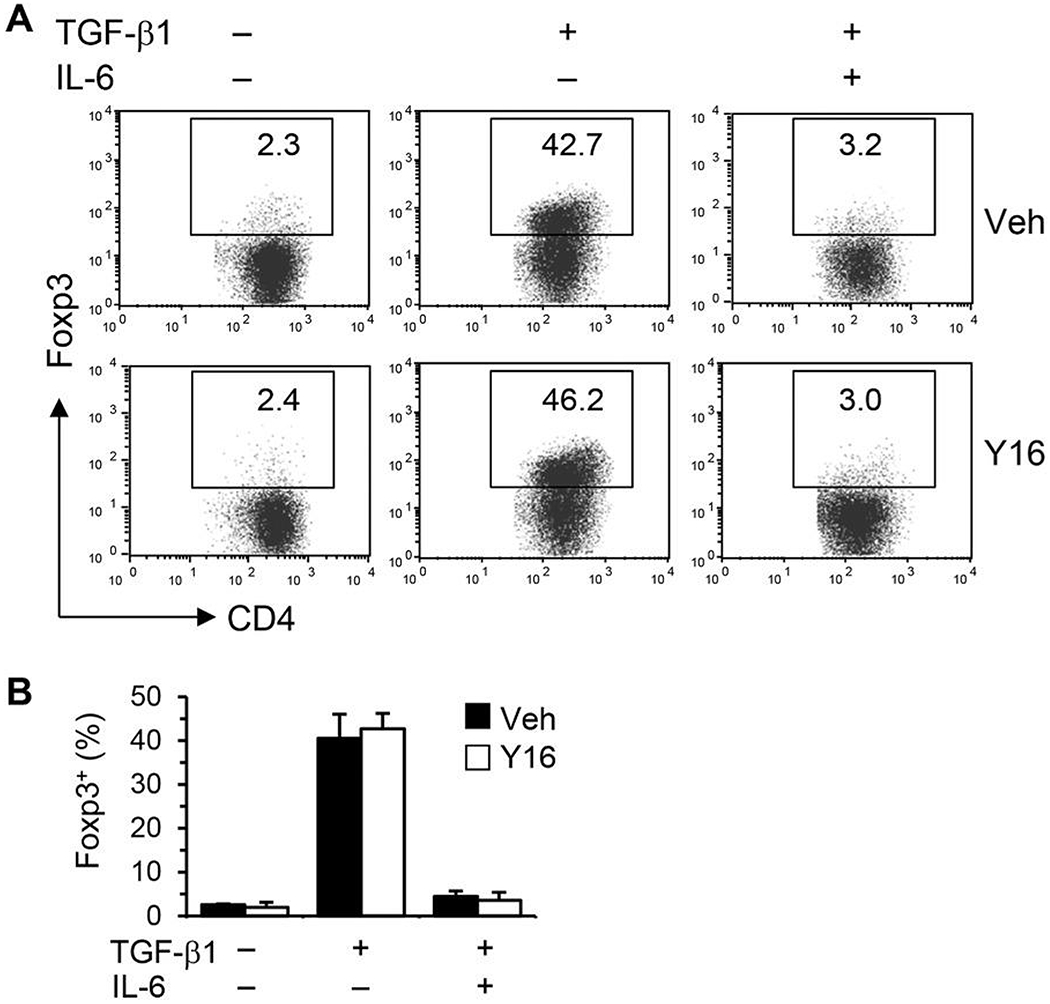
Allergic airway inflammation was induced by HDM and treated with Y16 as described in Fig. 7A. Mice were sacrificed 24 h after the last HDM challenge and the pulmonary infiltrating cells were isolated. The cells were restimulated in vitro with PMA plus ionomycin for 4 h with BD GolgiPlug in the last 3 h for FACS stainings. Percentages of cytokine or Foxp3 positive cells gated on CD4+ T cells are shown in representative dot plots (A). The isotype controls from pooled cells are also shown (A, Right). The numbers of total pulmonary infiltrating cells and CD4+ T cells (B), cytokine or Foxp3 positive cells gated on CD4+ cells (C) are summarized in bar graphs (means + SE, n = 3-6). *P < 0.05 versus HDM-challenged vehicle-treated groups
Interestingly, when Y16 was administrated post-HDM sensitization but before challenge (Supporting Information Figure S2A), there was no significant inhibition in inflammatory cell infiltration (Supporting Information Figure S2B–Figure E) and Th17/Th2 cytokine secretions in BAL fluids (Supporting Information Figure S2F). These results suggest that Y16 regulates Th17 and Th2 inflammatory immune responses by affecting T-cell activation and differentiation but not inflammatory cell recruitment into the lung. Hence, RhoA-specific inhibitor Y16 may preferentially serve as a prophylactic versus therapeutic regimen.
4 |. DISCUSSION
Unraveling the signaling pathways that promote T-cell differentiation into different lineages is of paramount importance.19 We have previously reported that RhoA is required for T-cell activation and Th2 differentiation without affecting Th1 cells. The contributions of RhoA to Th17 cells are yet to be elucidated. In this study by using conditional RhoA-deficient mice, we provided evidences that loss of RhoA downregulated Th17-differentiation and cytokine secretion in vitro and attenuated HDM-induced airway inflammation in vivo. To our knowledge, this is the first report demonstrating the effects of RhoA on Th17 cell differentiation and RhoA-Th17 axis in airway inflammation with genetic-deletion approaches.
The mechanisms whereby RhoA regulates Th17 cells remain elusive. We found that RhoA promoted Th17-related transcription factors Stat3 and Rorγt. Other signaling cascades may also be involved in RhoA-mediated Th17 differentiation. In this aspect, mammalian target of rapamycin (mTOR)-regulated glycolysis has been shown to be essential for Th17 differentiation.42 As we have shown that RhoA promotes mTOR signaling in activated T cells and RhoA promotes Th2 differentiation through glycolysis,11 it will be interesting to determine whether mTOR-regulated glycolysis contributes to RhoA-mediated Th17 differentiation in future. In addition, RhoA functions through activation of its immediate downstream effectors such as Rho-associated protein kinase (ROCK), mDia, and protein kinase N.43 Blockade of ROCK activity by its specific inhibitors Y27632 or fasudil is associated with the downregulation of Th17 function and improvement for several autoimmune and inflammatory diseases, such as systemic lupus erythematosus,44 OVA-induced murine allergic lung inflammation,45 and viral myocarditis.46 Thus, ROCK likely contributes to RhoA-regulated Th17 signaling and differentiation.
Y16 is a specific RhoA inhibitor developed in our laboratory, which fits into a surface groove of the DH-PH domain of leukemia-associated Rho GEF (LARG), a G-protein-regulated Rho GEF involved in RhoA activation.36 Y16 has been used for RhoA functional studies of a wide range, from endothelial barrier and macrophage phenotypes to contraction of intrapulmonary artery and airway smooth muscle.47–50 The main purpose for using Y16 in this study is to complement genetic deletion of RhoA to demonstrate the role of RhoA in Th17-cell differentiation in vitro and Th17-involved allergic airway inflammation in vivo. Nonetheless, our finding that Y16 alleviated HDM-induced murine allergic airway inflammation might demonstrate a proof-of-principle for pharmacological targeting of RhoA in prevention and therapy of asthma. Particularly, RhoA activity is increased in airway biopsies of asthma patients51 and HDM is a common indoor allergens that is a strong inducer of Th17 cells and neutrophils,21,25,30,52 2 types of inflammatory cells important for the pathogenesis of severe asthma.28,33 Thus, Y16 might have important clinical implications in treatment of severe asthma that is refractory to current therapies.
The recently discovered innate lymphoid cells (ILCs) have broadened our understanding of the pathogenesis of immune diseases including allergy and asthma. ILCs can be divided into 3 groups, ILC1, ILC2, and ILC3, which can also produce cytokines of type1 (such as IFN-γ and IL-12), type 2 (IL-5 and IL-13), and IL-17/IL-22, respectively.53,54 The impacts of RhoA on ILCs remain to be elucidated as a future direction of our studies.
Taken together, our data from both genetic deletion and pharmacological inhibition demonstrate that RhoA is one of the key regulators for Th17 differentiation and function. More detailed investigations are obviously required to fully clarify the underlying molecular mechanisms. RhoA might become a potential novel therapeutic target for asthma and other inflammatory disorders.
Supplementary Material
HIGHLIGHTS.
RhoA deficiency inhibits Th17 cell differentiation in vitro.
RhoA deficiency downregulates key Th17-related transcription factors Stat3 and Rorγt.
RhoA deficiency suppresses house dust mite-induced Th17-mediated allergic airway inflammation.
RhoA specific inhibitor Y16 recapitulates the most effects of RhoA genetic deletion on Th17 differentiation and function.
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health (R01 GM108661, R21 CA198358, and R56 HL141499 to F.G), Cincinnati Children’s Hospital Medical Center (GAP funding to F.G), National Natural Science Foundation of China (81871268, 81373116 to J.Q.Y), Jiangsu Provincial Department of Science and Technology (BM2018020), and Jiangsu Provincial Project of Invigorating Health Care through Science, Technology and Education, China (to J.Q.Y).
Abbreviations:
- HDM
house dust mite
- i.p.
intraperitoneally
- i.t.
intratracheally
- ILC
innate lymphoid cell
- KO
knock-out
- LARG
leukemia-associated Rho GEF
- mTOR
mammalian target of rapamycin
- MUC-5AC
mucin 5AC
- PAS
periodic acid-Schiff
- RhoA
ras homolog family member A
- ROCK
rho-associated protein kinase
- Rorγt
retinoic acid-related orphan receptor gamma t
- Stat3
signal transducer and activator of transcription 3
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Mulloy JC, Cancelas JA, Filippi MD, Kalfa TA, Guo F, Zheng Y. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115: 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou X, Florian MC, Arumugam P, et al. RhoA GTPase controls cytokinesis and programmed necrosis of hematopoietic progenitors. J Exp Med. 2013;210:2371–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loirand G Rho kinases in health and disease: from basic science to translational research. Pharmacol Rev. 2015;67:1074–1095. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Peyrollier K, Kilic G, Brakebusch C. Rho GTPases and cancer. Biofactors. 2014;40:226–235. [DOI] [PubMed] [Google Scholar]
- 5.Henning SW, Galandrini R, Hall A, Cantrell DA. The GTPase Rho has a critical regulatory role in thymus development. Embo J. 1997;16: 2397–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cleverley S, Henning S, Cantrell D. Inhibition of Rho at different stages of thymocyte development gives different perspectives on Rho function. Curr Biol. 1999;9:657–660. [DOI] [PubMed] [Google Scholar]
- 7.Cantrell DA. GTPases and T-cell activation. Immunol Rev. 2003;192: 122–130. [DOI] [PubMed] [Google Scholar]
- 8.del Pozo MA, Vicente-Manzanares M, Tejedor R, Serrador JM, Sanchez-Madrid F. Rho GTPases control migration and polarization of adhesion molecules and cytoskeletal ERM components in T lymphocytes. Eur J Immunol. 1999;29:3609–3620. [DOI] [PubMed] [Google Scholar]
- 9.Corre I, Gomez M, Vielkind S, Cantrell DA. Analysis of thymocyte development reveals that the GTPase RhoA is a positive regulator of T-cell receptor responses in vivo. J Exp Med. 2001;194:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Konstantinidis DG, Yang JQ, et al. Gene targeting RhoA reveals its essential role in coordinating mitochondrial function and thymocyte development. J Immunol. 2014;193:5973–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang JQ, Kalim KW, Li Y, et al. RhoA orchestrates glycolysis for TH2 cell differentiation and allergic airway inflammation. J Allergy Clin Immunol. 2016;137:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouyang W, Kolls JK, Zheng Y. The biological functions of T-helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T-cell lineage with regulatory T-cell ties. Immunity 2006;24:677–688. [DOI] [PubMed] [Google Scholar]
- 14.Chen K, Kolls JK. Interluekin-17A(IL17A). Gene. 2017;614:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAleer JP, Kolls JK. Directing traffic: iL-17 and IL-22 coordinate pulmonary immune defense. Immunol Rev. 2014;260:129–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rathore JS, Wang Y. Protective role of Th17 cells in pulmonary infection. Vaccine. 2016;34:1504–1514. [DOI] [PubMed] [Google Scholar]
- 17.Ling MF, Luster AD. Allergen-specific CD4(+) T cells in human asthma. Ann Am Thorac Soc. 2016;13(Suppl 1):S25–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muehling LM, Lawrence MG, Woodfolk JA. Pathogenic CD4+ T cells in patients with asthma. J Allergy Clin Immunol. 2017;140:1523–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang JQ, Leitges M, Duran A, Diaz-Meco MT, Moscat J. Loss of PKC lambda/iota impairs Th2 establishment and allergic airway inflammation in vivo. Proc Natl Acad Sci USA. 2009;106:1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang JQ, Liu H, Diaz-Meco MT, Moscat J. NBR1 is a new PB1 signalling adapter in Th2 differentiation and allergic airway inflammation in vivo. Embo J. 2010;29:3421–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGee HS, Stallworth AL, Agrawal T, Shao Z, Lorence L, Agrawal DK. Fms-like tyrosine kinase 3 ligand decreases T helper type 17 cells and suppressors of cytokine signaling proteins in the lung of house dust mite-sensitized and -challenged mice. Am J Respir Cell Mol Biol. 2010;43:520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180:720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong CK, Lun SW, Ko FW, et al. Activation of peripheral Th17 lymphocytes in patients with asthma. Immunol Invest. 2009;38:652–664. [DOI] [PubMed] [Google Scholar]
- 24.Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 cells: new players in asthma pathogenesis. Allergy. 2011;66:989–998. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Dong P, Zhao J, et al. PKClambda/iota regulates Th17 differentiation and house dust mite-induced allergic airway inflammation. Biochim Biophys Acta. 2018;1864:934–941. [DOI] [PubMed] [Google Scholar]
- 26.Chesne J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL-17 in severe asthma. Where do we stand? Am J Respir Crit Care Med. 2014;190:1094–1101. [DOI] [PubMed] [Google Scholar]
- 27.Al-Ramli W, Prefontaine D, Chouiali F, et al. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. 2009;123:1185–1187. [DOI] [PubMed] [Google Scholar]
- 28.Brandt EB, Kovacic MB, Lee GB, et al. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J Allergy Clin Immunol. 2013;132:1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irvin C, Zafar I, Good J, et al. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol. 2014;134:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu S, Fan X, Yang Y, et al. Schistosoma japonicum infection downregulates house dust mite-induced allergic airway inflammation in mice. PLoS One. 2017;12:e0179565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gregory LG, Lloyd CM. Orchestrating house dust mite-associated allergy in the lung. Trends Immunol. 2011;32:402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calderon MA, Kleine-Tebbe J, Linneberg A, et al. House dust mite respiratory allergy: an overview of current therapeutic strategies. J Allergy Clin Immunol Pract. 2015;3:843–855. [DOI] [PubMed] [Google Scholar]
- 33.Brandenberger C, Li N, Jackson-Humbles DN, Rockwell CE, Wagner JG, Harkema JR. Enhanced allergic airway disease in old mice is associated with a Th17 response. Clin Exp Allergy. 2014;44: 1282–1292. [DOI] [PubMed] [Google Scholar]
- 34.Katayama K, Melendez J, Baumann JM, et al. Loss of RhoA in neural progenitor cells causes the disruption of adherens junctions and hyperproliferation. Proc Natl Acad Sci USA. 2011;108:7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melendez J, Stengel K, Zhou X, et al. RhoA GTPase is dispensable for actomyosin regulation but is essential for mitosis in primary mouse embryonic fibroblasts. J Biol Chem. 2011;286:15132–15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shang X, Marchioni F, Evelyn CR, et al. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc Natl Acad Sci USA. 2013;110:3155–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang JQ, Kalim KW, Li Y, et al. Rational targeting Cdc42 restrains Th2 cell differentiation and prevents allergic airway inflammation. Clin Exp Allergy. 2019;49:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605–34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conroy DM, Williams TJ. Eotaxin and the attraction of eosinophils to the asthmatic lung. Respir Res. 2001;2:150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Busse PJ, Zhang TF, Srivastava K, et al. Chronic exposure to TNF-alpha increases airway mucus gene expression in vivo. J Allergy Clin Immunol. 2005;116:1256–1263. [DOI] [PubMed] [Google Scholar]
- 42.Shi LZ, Wang R, Huang G, et al. HIF1alpha-dependent glycolytic path-way orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thumkeo D, Watanabe S, Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur J Cell Biol. 2013;92:303–315. [DOI] [PubMed] [Google Scholar]
- 44.Rozo C, Chinenov Y, Maharaj RK, et al. Targeting the RhoA-ROCK pathway to reverse T-cell dysfunction in SLE. Ann Rheum Dis. 2017;76: 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dos Santos TM, Righetti RF, Camargo LDN, et al. Effect of anti-IL17 antibody treatment alone and in combination with rho-kinase inhibitor in a murine model of asthma. Front Physiol. 2018;9:1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai K, Wang Y, Tai S, et al. Fasudil exerts a cardio-protective effect on mice with coxsackievirus B3-induced acute viral myocarditis. Cardiovasc Ther. 2018;36:e12477. [DOI] [PubMed] [Google Scholar]
- 47.Zhang XE, Adderley SP, Breslin JW. Activation of RhoA, but not Rac1, mediates early stages of S1P-induced endothelial barrier enhancement. PLoS One. 2016;11:e0155490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen W, Zhao Y, Li XC, Kubiak JZ, Ghobrial RM, Kloc M. Rho-specific guanine nucleotide exchange factors (Rho-GEFs) inhibition affects macrophage phenotype and disrupts Golgi complex. Int J Biochem Cell Biol. 2017;93:12–24. [DOI] [PubMed] [Google Scholar]
- 49.MacKay CE, Shaifta Y, Snetkov VV, Francois AA, Ward JPT, Knock GA. ROS-dependent activation of RhoA/Rho-kinase in pulmonary artery: role of Src-family kinases and ARHGEF1. Free Radic Biol Med. 2017;110:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaifta Y, MacKay CE, Irechukwu N, et al. Transforming growth factor-beta enhances Rho-kinase activity and contraction in airway smooth muscle via the nucleotide exchange factor ARHGEF1. J Physiol. 2018;596:47–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhattacharya M, Sundaram A, Kudo M, et al. IQGAP1-dependent scaffold suppresses RhoA and inhibits airway smooth muscle contraction. J Clin Invest. 2014;124:4895–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reithofer M, Jahn-Schmid B. Allergens with protease activity from house dust mites. Int J Mol Sci. 2017;18:1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ardain A, Porterfield JZ, Kloverpris HN, Leslie A. Type 3 ILCs in lung disease. Front Immunol. 2019;10:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doherty TA, Broide DH. Airway innate lymphoid cells in the induction and regulation of allergy. Allergol Int. 2019;68:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.