

Abstract
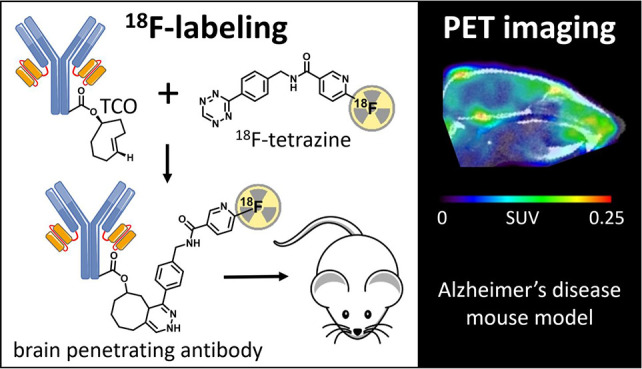
Antibodies are attractive as radioligands due to their outstanding specificity and high affinity, but their inability to cross the blood–brain barrier (BBB) limits their use for CNS targets. To enhance brain distribution, amyloid-β (Aβ) antibodies were fused to a transferrin receptor (TfR) antibody fragment, enabling receptor mediated transport across the BBB. The aim of this study was to label these bispecific antibodies with fluorine-18 and use them for Aβ PET imaging. Bispecific antibody ligands RmAb158-scFv8D3 and Tribody A2, both targeting Aβ and TfR, were functionalized with trans-cyclooctene (TCO) groups and conjugated with 18F-labeled tetrazines through an inverse electron demand Diels–Alder reaction performed at ambient temperature. 18F-labeling did not affect antibody binding in vitro, and initial brain uptake was high. Conjugates with the first tetrazine variant ([18F]T1) displayed high uptake in bone, indicating extensive defluorination, a problem that was resolved with the second and third tetrazine variants ([18F]T2 and [18F]T3). Although the antibody ligands’ half-life in blood was too long to optimally match the physical half-life of fluorine-18 (t1/2 = 110 min), [18F]T3-Tribody A2 PET seemed to discriminate transgenic mice (tg-ArcSwe) with Aβ deposits from wild-type mice 12 h after injection. This study demonstrates that 18F-labeling of bispecific, brain penetrating antibodies is feasible and, with further optimization, could be used for CNS PET imaging.
Keywords: Fluorine-18, antibody radioligand, positron emission tomography (PET), inverse electron demand Diels−Alder reaction, tetrazine, trans-cyclooctene (TCO)
Introduction
Antibody-based PET radioligands have mainly been used for applications in the field of oncology, while targets in the CNS have so far been reserved for ligands based on small molecules.1 Antibodies can display very specific and high affinity binding to conformational or defined linear epitopes of, e.g., protein targets. Such properties are more difficult to achieve with small molecular radioligands but could be of particular importance for imaging of soluble or diffuse aggregates of amyloid-β (Aβ). Current PET ligands used for diagnosis of Alzheimer’s disease (AD), e.g., [11C]PiB, can only detect insoluble, fibrillar Aβ which is a less dynamic marker of the disease process. However, one challenge with antibody-based radioligands is their size (antibodies are conventionally larger than 150 kDa), and hence, they are strongly hindered in passing the blood–brain barrier (BBB). However, by engineering an antibody to interact with the transferrin receptor (TfR), endogenously expressed on endothelial cells, it can cross the BBB via receptor mediated transcytosis.2−10
A second challenge is the long circulation time associated with antibodies. Antibodies may display biological half-lives of several weeks. For PET imaging, this is a problem, because it will increase the nonspecific signal derived from radioactivity in blood. When engineered into bispecific formats, i.e. with the addition of an antibody fragment that binds to TfR, the long half-life of IgG antibodies is reduced, although it may still be in the range of days rather than hours.2,7,11,12 The half-life in blood also appears to be correlated to size. Our previously developed RmAb158-scFv8D3, a 210 kDa bispecific antibody used for PET imaging of Aβ protofibrils,13 has a biological half-life in blood of 19 h.2 A somewhat smaller Tribody, composed of a TfR binding Fab fragment and two single chain variable fragments (scFv) targeting Aβ, has a reduced size of 100 kDa and a half-life in blood of approximately 9 h.7
Traditionally, protein-based radioligands have been labeled with radiometals, such as zirconium-89 (t1/2 = 78.4 h), with long physical half-lives and thereby unfavorable dosimetry. This is especially problematic for studying chronic diseases of the brain. The decreased biological half-life of engineered bispecific antibodies permits the use of radionuclides with shorter decay half-lives. Fluorine-18 (18F) is an attractive radionuclide for clinical PET radioligands, as it has a shorter decay half-life and higher percentage of positron decay. Hence, development of methods for 18F-radiolabeling of protein-based ligands is essential.
The aim of this study was to develop methods to label bispecific antibody ligands with 18F under mild reaction conditions and evaluate their feasibility as PET radioligands for Aβ. The antibody ligands were functionalized with trans-cyclooctene (TCO) groups which in turn were coupled to 18F-labeled tetrazines by inverse electron demand Diels–Alder reactions (IEDDA) performed in aqueous solutions. Three different 18F-labeled tetrazines were synthesized and used in the coupling reactions. Ex vivo and PET imaging studies were carried out in a mouse model of Aβ pathology (tg-ArcSwe) and in wild-type (wt) control mice.
Results and Discussion
TCO Modification of Antibody Ligands
The bispecific antibody ligands RmAb158-scFv8D3 and Tribody A2 were selected for 18F-labeling based on their brain penetrating properties and different sizes. The prospect of labeling these large molecules, 210 kDa and 100 kDa, respectively, was challenging as the antibodies needed to be employed at the microgram scale. To enable labeling under mild reaction conditions, the Tribody A2 was designed with lysine rich linkers between its TfR and Aβ binding domains to facilitate lysine targeted modifications without affecting its functionality. Previously, [18F]F-Py-TFP has been conjugated directly with lysine residues in peptides at the milligram scale.14−17 However, because our aim was to label these sizable antibodies at the microgram scale, we opted for the highly efficient IEDDA reaction where [18F]tetrazines were conjugated with TCO groups attached to the antibody. The functionalization of the antibodies with TCO prior the labeling step facilitated confirmation of their reactivity toward target proteins before and after the modification.
TCO modification of RmAb158-scFv8D3 and Tribody A2 did not affect binding to either of their target proteins, as demonstrated with TfR and Aβ ELISA binding analyses (Figure 1A,B). The ability of the antibody ligands to click with a tetrazine was studied in vitro by incubation with tetrazine functionalized BSA, followed by SDS–PAGE analysis to visualize the result of this reaction (Figure 1C,D). Both antibody ligands appeared as a single band on the gel, confirming that TCO modification did not induce aggregation or degradation (Figure 1C,D, lane 2). These bands largely disappeared as stable antibody–BSA conjugates of different sizes were formed (Figure 1C,D, lane 3), suggesting that all antibody molecules were TCO modified and fully reacted with the tetrazine functionalized BSA.
Figure 1.

ELISA analysis of RmAb158-scFv8D3 (A) and Tribody A2 (B) binding to TfR and Aβ before and after TCO modification revealed no change in reactivity to either of the target proteins after modification. Unreduced SDS–PAGE of the in vitro click reaction between TCO-modified RmAb158-scFv8D3 (C) or Tribody A2 (D) and tetrazine–BSA added in excess. Both ligands reacted almost completely, resulting in the formation of high molecular weight complexes of various sizes, as indicated by arrows in parts C and D. In addition, the analysis demonstrated that both antibodies appeared as a single distinct band (middle lane), without signs of aggregation or degradation.
The band appearing around 125 kDa in the BSA–Tribody A2 conjugate analysis (Figure 1D, lane 3) could be mistaken for nonreacted TCO–Tribody A2 but is in fact a BSA dimer band, which appeared also for BSA analyzed alone (Figure 1C,D, lane 1). In addition, the formation of high molecular weight conjugates indicated by arrows in Figure 1C,D suggests that each antibody molecule contained several TCOs that could react with multiple tetrazine–BSA molecules to form larger complexes. This also implies that several tetrazine molecules could attach to the antibody ligand, resulting in a higher molar activity of the 18F-labeled product.
Radiochemistry
The conventional 18F-fluorination, performed on the tosylate precursor 1 (Figure 2), yielded approximately 3 GBq (GBq = gigabecquerel) of [18F]T1 with >99% radiochemical purity when starting with 20 GBq [18F]fluoride. The total synthesis time was 48 min. The water/ethanol eluent for the semipreparative HPLC purification was selected to enable direct coupling with any TCO-modified substrate, without the need for reformulation. Considering that only small volumes can be injected in mice, it was important to maximize radioactivity concentration. For this reason, the product eluting from the semipreparative HPLC was fractionated, and the fraction with highest activity was used for the conjugation reaction, typically with a volume of 0.8 mL and an activity concentration of approximately 0.9 GBq/mL.
Figure 2.

Synthesis of [18F]T1 was performed according to a previously published method with minor modifications.18
The two step procedure used for the synthesis of [18F]T2 and [18F]T3 was based on the previously reported [18F]F-Py-TFP prosthetic group and its precursor (Figure 3) that combines a remarkably high reactivity toward nucleophilic aromatic substitution with an activated ester functionality for swift coupling, for example, with amines.17 The high reactivity has been exemplified in the unusual and efficient 18F-fluorination on an anion exchange cartridge which omits conventional drying of the trapped [18F]fluoride.14 In our hands, [18F]F-Py-TFP was obtained in 59 ± 3% radiochemical yield when performing the 18F-fluorination on solid support. To simplify the previously described method, precursor 2 was removed online by eluting the formed [18F]F-Py-TFP in acetonitrile through a MCX cation exchange cartridge, omitting intermediate C18 SepPak purification and allowing the product to be eluted straight into the reaction vial where the coupling with the aminotetrazine compound was performed. The recovery of activity from the MCX cartridge was 63 ± 5%, and the subsequent coupling with the CH3- and H-tetrazines gave [18F]T2 and [18F]T3 in 71–80 and 62–64% radiochemical yields and with a radiochemical purities of 97.5 ± 3.5 and 98.5 ± 1.8%, respectively. The synthesis time for both [18F]T2 and [18F]T3 was approximately 50 min. Efficient removal of tetrazine precursors (3a and 3b) was important to avoid the possibility that they react with TCO in the IEDDA reaction, which would reduce radiochemical yield and molar activity of the labeled antibody ligand. This was accomplished by semipreparative purification with an acidic eluent (0.01% TFA) to protonate the tetrazine amine precursor, resulting in a very short retention time on the C18 HPLC column and complete separation from the 18F-labeled tetrazines. The overall radiochemical yields of the isolated tetrazines [18F]T2 and [18F]T3 were 18 ± 6 and 16 ± 3%, respectively, based on the starting activity of [18F]fluoride (typically 16–20 GBq).
Figure 3.

[18F]Fluoride was trapped on the Chromabond cartridge (a), washed with acetonitrile and eluted by incorporation into precursor 2 passed over the cartridge in acetonitrile. Unreacted precursor was removed by Oasis MCX Plus cartridge connected in series. [18F]T2 and [18F]T3 were obtained by direct amidation of the activated [18F]F-Py-TFP ester (b).
All three 18F-labeled tetrazines were successfully coupled to the TCO-modified antibody ligands. The molar activities obtained were 116 ± 53 GBq/μmol (554 ± 253 MBq/mg) for [18F]T1-RmAb158-scFv8D3 (n = 4), 39 ± 10 GBq/μmol (161 ± 101 MBq/mg) for [18F]T2-RmAb158-scFv8D3 (n = 5), 29 ± 7 GBq/μmol (137 ± 33 MBq/mg) for [18F]T3-RmAb158-scFv8D3 (n = 10) and 79 ± 13 GBq/μmol (706 ± 116 MBq/mg) for [18F]T3-Tribody A2 (n = 2).
The purification of the labeled antibodies was performed using gel filtration which removed small molecule impurities, i.e. unreacted [18F]tetrazine was removed but unreacted antibody remained. Hence, the molar activities obtained for the labeled antibodies relied on efficient incorporation of 18F, a requirement less important when labeling smaller molecules where the precursor can be separated from the 18F-labeled product after the reaction. Therefore, to achieve a high molar activity of the 18F-labeled product, an excess of [18F]tetrazine was added to the reaction, thereby minimizing the fraction of unreacted antibody. However, this also resulted in a higher fraction of unreacted [18F]tetrazine, and hence, a lower radiochemical yield. Thus, for 18F-labeling of the tetrazine, a high radiochemical yield is desired, while in the IEDDA reaction, complete conjugation of the TCO-functionalized antibody is more important than a high radiochemical yield. The labeled antibodies were obtained in 46 ± 30% radiochemical yield (5–93%, n = 11) based on [18F]tetrazine when purified by NAP-5 and 15 ± 11% when purified by Zeba 7K (4–39%, n = 12).
The IEDDA reaction was performed with an approximate ratio of 1 MBq [18F]tetrazine to 1 μg of antibody, and the amount of antibody in the syntheses varied between 7 and 190 μg. This mass corresponded to 30–860 pmol of TCO-modified RmAb158-scFv8D3; hence, satisfactory radiochemical yields were obtained despite reducing the reaction scale far below what is normally used in 18F-labeling (μmol).
In Vitro Evaluation of Radiolabeled RmAb158-scFv8D3
The impact of 18F-labeling on RmAb158-scFv8D3 function was studied in vitro with TfR and Aβ ELISA, demonstrating that the 18F-tetrazine click reaction did not affect the binding of the antibody ligand, as compared with nonmodified antibody (Figure 4A). Autoradiography was performed on brain sections from tg-ArcSwe and wt mice to assess antibody binding in tissue, where Aβ deposits are formed in vivo in complex biological processes, closely resembling the AD brain. As expected, [18F]RmAb158-scFv8D3 displayed a binding pattern closely resembling the distribution of Aβ deposits in this mouse model,13 with a high intensity signal in the cortex, hippocampus, and thalamus. A fainter background signal was also seen in the wt mouse brain, potentially from antibody ligand binding to TfR expressed on neurons (Figure 4B).
Figure 4.

(A) ELISA evaluation of TfR and Aβ binding before and after 18F-labeling of RmAb158-scFv8D3. (B) Autoradiography of [18F]T1-RmAb158-scFv8D3 on cryosections from tg-ArcSwe and wt mouse brain, demonstrating specific binding to areas with high Aβ burden.
An antibody applied to a tissue section will be exposed to the whole surface of Aβ deposits in the brain. It will thereby have more available binding sites than an antibody administered through the blood, reaching its target via transcytosis through the brain endothelium and thereby encountering only a fraction of Aβ deposits. This distinction is important to consider when comparing autoradiography with PET, especially for antibodies which display an extremely slow rate of diffusion in the brain parenchyma compared with that of small molecules.
Ex Vivo Studies
The 18F-labeled antibody ligands were injected in wt mice whereafter brain and blood were isolated at 2 h post administration to investigate transport across the BBB. In general, 18F-labeled RmAb158-scFv8D3, regardless of tetrazine used for the radiolabeling, was distributed into the brain in concentrations substantially higher than expected for proteins that are not engineered to penetrate the BBB.2,4,5,11 The smaller antibody ligand Tribody A2, conjugated with [18F]T3, displayed brain concentrations somewhat higher than those of the RmAb158-scFv8D3 conjugates (Figure 5A). Interestingly, brain uptake of Tribody A2 was higher when radiolabeled with 18F compared to iodine-125,7 likely because 18F-labeling mainly occurred at lysine residues within the linkers of Tribody A2. These linkers are not involved in target binding and were designed with a high density of lysine residues to attract the TCO groups. 125I-labeling, on the other hand, occurs at tyrosine residues, which could be present anywhere in the amino acid sequence. At 12 h, i.e., at a time point when unbound ligand had been partly eliminated from the brain, there was still no significant difference in brain concentrations of [18F]T3-RmAb158-scFv8D3 and [18F]T3-Tribody A2 in tg-ArcSwe compared wt mice (Figure 5B).
Figure 5.

(A) Ex vivo brain concentrations at 2 h post administration of RmAb158-scFv8D3 conjugated with [18F]T1, [18F]T2 or [18F]T3 and Tribody A2, conjugated with [18F]T3. The red line represents the level of unmodified IgG concentrations in the brain at the same time after injection (approximately SUV of 0.01). (B) Ex vivo brain concentrations of [18F]T3-RmAb158-scFv8D3 and [18F]T3-Tribody A2 in tg-ArcSwe and wt mice 12 h postadministration. No significant genotype related differences were observed. (C) Biodistribution of activity at 10–12 h postadministration of the 18F-labeled ligands. (D) Radioactivity concentration in skull at different time points after administration. Radiolabeling with [18F]T1 lead to a time dependent increase in radioactivity in the skull, while low activity concentrations were observed at all time points after radiolabeling with [18F]T2 and [18F]T3.
Major organs were isolated from mice at 10–12 h after administration to investigate the peripheral distribution of the two ligands. There were no differences in organ distribution between wt and tg-ArcSwe mice. However, [18F]T3-Tribody A2 displayed higher concentrations in the spleen, which may be a result of higher TfR affinity and thereby tighter binding to TfR on the surface of red blood cells which are abundant in the spleen (Figure 5C). After coupling with [18F]T1, radioactivity in the femoral bone and skull was high (Figure 5C). Therefore, activity uptake in the skull over time was studied in more detail. Coupling with [18F]T1 lead to an increase in activity concentrations with time in skull, indicating defluorination and accumulation of 18F in bone structures. Indications of defluorination were not observed with [18F]T2 and [18F]T3 (Figure 5D). These results demonstrate that the choice of tetrazine is central, especially for protein-based radioligands that require longer residence time in the circulation before imaging is possible. According to the literature, T3 should be associated with faster reaction kinetics,19 but as used in the present study, where the IEDDA reaction took place in vitro, there were no essential differences between [18F]T2 and [18F]T3 with regards to yield of the subsequent RmAb158-scFv8D3 radiolabeling or in vivo performance.
PET Imaging
PET imaging was performed with [18F]T1-RmAb158-scFv8D3, [18F]T3-RmAb158-scFv8D3, and [18F]T3-Tribody A2 in tg-ArcSwe mice. Extensive defluorination was observed when the [18F]T1 tetrazine was used to radiolabel RmAb158-scFv8D3 (Figure 6A), whereas no defluorination was seen for [18F]T3-labeled antibody ligands, confirming the ex vivo results. PET imaging with [18F]T3-RmAb158-scFv8D3 (Figure 6B) and [18F]T3-Tribody A2 (Figure 6C) both resulted in PET images where a signal could be detected in the brain, but no significant differences in SUV were observed between wt and tg-ArcSwe mice. However, when expressed as SUV ratio (SUVR) of the frontal cortex with cerebellum as reference region, the smaller ligand [18F]T3-Tribody A2 clearly tended to discriminate between wt and tg-ArcSwe mice (Figure 6D). This difference was not apparent in the ex vivo studies where activity was measured in the whole brain (Figure 5B). Such measurement is likely to “dilute” the specific signal originating from the frontal cortex, which constitutes only a relatively small part of the whole brain. In addition, the use of a reference region, which serves as an internal control within each individual brain, will likely reduce confounding factors related to individual differences in blood flow or other physiological parameters that could affect distribution of the radioligand.
Figure 6.

Summed PET images obtained after administration of 18F-labeled antibody ligands in old tg-ArcSwe and wt mice. (A) [18F]T1-RmAb158-scFv8D3 PET in tg-ArcSwe (n = 1) and wt (n = 1) mice 7–8 h postinjection. Both genotypes had a high uptake in bone, likely due to defluorination. [18F]T3-RmAb158-scFv8D3 (B) and [18F]T3-Tribody A2 (C) PET in old tg-ArcSwe and wt mice 11–12 h postinjection (n = 2 per group). Note the difference in scale between part A (0–2.5 SUV) and part B (0–0.25 SUV). (D) Quantification of PET images shown in parts B and C expressed as SUVR (SUV ratio of frontal cortex over cerebellum). Dashed line represents SUVR at unity, which is expected in wt animals lacking a specific signal in the cortex.
The small differences between tg-ArcSwe and wt mice in this study are likely due to the time of scanning, i.e., at 12 h postinjection. Previous studies have shown that Tribody A2 displays a somewhat faster systemic clearance than that of the larger RmAb158-scFv8D3.2,6,7 Thus, at 12 h, less unbound antibody ligand will be present in the blood and brain, and therefore, the specific-to-nonspecific signal ratio is more favorable with [18F]T3-Tribody A2 compared to [18F]T3-RmAb158-scFv8D3. Still, 12 h postinjection is a very “early” time point for scanning with these radioligands; previous studies using iodine-124 radiolabeled variants have reported that PET scanning can at the earliest differentiate between tg-ArcSwe and wt animals at 1–2 days postinjection.2,7 However, with the decay half-life of 110 min for 18F, scanning at time points beyond 12 h was not possible. This is also the rationale for not including more animals in the present study, as it was assumed that neither of the two antibodies would be eliminated fast enough for PET scanning at 12 h postinjection. Instead, antibody ligands with shorter circulation time must be developed or alternative labeling strategies, e.g., involving copper-64 (half-life12.7 h) could be explored.
In addition to the PET imaging results discussed above, the study showed that 18F radiolabeling of antibody ligands can be performed with a novel method associated with short reaction times, high radiolabeling yields, and biologically functional products. The conjugates with [18F]T2 and [18F]T3 were characterized by extremely high stability against defluorination in vivo, enabling scanning several hours after injection without observed accumulation in bone. It should be noted that the bispecific nature of the antibody ligands used in this study probably makes them more sensitive than most antibodies. With the current method, they were transported into the brain via TfR mediated transcytosis to a higher or similar extent as described in previous studies.2,6,7,11
Antibody- and protein-based drugs (biologicals) are, today, the fastest growing class of therapeutic drugs. Therefore, it is also important to develop in vivo biomarkers to assess target engagement and therapeutic efficacy. For many of the novel biologicals studied in clinical trials there are no such biomarkers. Thus, engineering a biological drug candidate into a version with faster pharmacokinetics, e.g., by size reduction or functionalized with a BBB transport enhancing moiety when the primary target is localized in the brain, may enable its use as a PET biomarker to directly assess the function and effect of the drug candidate. However, and analogous to the development of radioligands based on small molecule drug candidates, this is only feasible if the radioligand candidate can be radiolabeled with a clinically relevant radionuclide.
In conclusion, we have presented a novel and general method for 18F radiolabeling of proteins and demonstrated that antibody radioligands produced with the method were stable in vivo with regards to biological functionality and defluorination.
Methods
Expression of Bispecific Antibody Ligands RmAb158-scFv8D3 and Tribody A2
Bispecific RmAb158-scFv8D3,2 based on the Aβ protofibril selective monoclonal antibody mAb15820 and TfR-antibody 8D3,21,22 was expressed as described previously.23 Briefly, Expi293f mammalian cells were transiently transfected with pcDNA3.4 vectors, carrying the sequence of the heavy chain and the light chain of RmAb158-scFv8D3. Cell medium containing expressed RmAb158-scFv8D3 was purified using a protein G column on a ÄKTA system (5 mL HiTrap Protein G HP, 17-0405-01, GE Healthcare) and then dialyzed with phosphate buffered saline (PBS, pH 7.4).
Tribody A2,7 consisting of an 8D3 Fab fragment recombinantly fused to two scFv fragments of mAb158 with lysine rich linkers, was expressed and supplied by the company that is behind the Tribody technology (Biotecnol Ltd., London, UK).
TCO Modification of Antibody Ligands
Lysine residues in RmAb158-scFv8D3 and Tribody A2 were functionalized with an NHS-activated trans-cyclooctene (TCO) tag to facilitate conjugation with 18F-labeled tetrazines. Sodium carbonate buffer (1 M, 3 μL, pH 8.0) was added to PBS (96 μL) containing RmAb158-scFv8D3 or Tribody A2 (1.0–1.5 mg/mL), followed by addition of axial TCO–NHS (product number (prod. nr.) CP-6016, Conju-Probe, San Diego, USA) in DMSO (10 mM, 0.8–1.2 μL) at a 10–20x TCO–antibody ligand molar ratio. Following a 2.5 h incubation in darkness while shaking at 600 rpm, the solution was purified from remaining unreacted TCO with Zeba spin desalting columns (7K MWCO, 0.5 mL, 89882, Thermofisher) and eluted in PBS. For the purpose of in vitro evaluation of the modified antibody ligands, bovine serum albumin (BSA; Sigma-Aldrich, Stockholm, Sweden) (2 mg/mL in PBS) was functionalized with tetrazine-PEG5-NHS (prod. nr. 900913, Sigma-Aldrich) in DMSO (10 mM) at a 20x tetrazine–BSA molar ratio, using the same protocol as above.
In Vitro Analysis of TCO Modification
To verify that the TCO modification was functional, TCO-modified RmAb158-scFv8D3 or Tribody A2 was mixed with 5x molar excess of tetrazine–BSA (2.0 mg/mL) and incubated for 15 min while shaking at 600 rpm. Bolt 4x LDS sample buffer (B0007, Thermofisher) was added to the sample and then loaded on a 4–12% 12 well Bolt Bis-Tris SDS–PAGE gel (NW04122BOX, Thermofisher; it was run at 200 V for 23 min, and the gel was stained for protein with PageBlue staining solution (24620, Thermofisher).
Synthesis of [18F]tetrazines
Three 18F-labeled tetrazines were synthesized: 3-(3-[18F]fluoropropyl)-6-methyl-1,2,4,5-tetrazine ([18F]T1), 6-[18F]fluoro-N-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)nicotinamide ([18F]T2), and N-(4-(1,2,4,5-tetrazin-3-yl)benzyl)-6-[18F]fluoronicotinamide ([18F]T3).
[18F]T1 was obtained by direct 18F-fluorination on a tosylate precursor 1 (Figure 2) using a previously reported procedure with slight modifications.18 Cyclotron produced [18F]fluoride (typically 20 GBq) was concentrated on a Sep-Pak Accell Plus QMA Carbonate Plus Light Cartridge (Waters) and then eluted with a solution of kryptofix 2.2.2 (10 mg) and K2CO3 (1.4 mg) in acetonitrile/water (8:2 v/v, 1 mL) into a septum equipped vial. The water was removed by heating at 120 °C under a helium stream for 13 min. A solution of (3-(6-methyl-1,2,4,5-tetrazin-3-yl)propyl) p-tolylsulfonate (8 mg, Pharmasynth AS) in acetonitrile (0.8 mL) was added, and the reaction mixture was heated at 90 °C for 10 min before it was diluted with water (1.5 mL), passed over a Sep-Pak Alumina N Plus Light Cartridge (Waters) to remove any unreacted [18F]fluoride, and then purified by semipreparative HPLC. Column = ACE HL C18 5 μm 250 × 10 mm, eluent = water/ethanol 18% B, flow 5 mL/min. Column effluent was monitored using a UV detector (254 nm) and radiodetector. A fraction representing 70% of the product peak was collected between 7.1 and 7.5 min, discarding the beginning and end of the peak to maximize the activity concentration in the collected fraction. The radiochemical purity was assessed by analytical HPLC. Column = Chromolith Performance RP-18e 4.6 × 100 mm (Merck), eluent = water/acetonitrile 2% B, flow 4 mL/min. Retention time = 6.6 min. The identity of the product was confirmed by coinjection with 3-(3-fluoropropyl)-6-methyl-1,2,4,5-tetrazine (Pharmasynth AS).
Tetrazines [18F]T2 and [18F]T3 were labeled by a two-step synthesis procedure (Figure 3). Cyclotron produced [18F]fluoride (16–20 GBq) in aqueous solution was trapped and concentrated on Chromabond PS-HCO3 Shorty/45 mg (Macherey-Nagel). The water was removed by rinsing with acetonitrile (5 mL), and the cartridge was connected to an Oasis MCX Plus cartridge (Waters) preconditioned with acetonitrile (3 mL), by switching a valve. The trapped [18F]fluoride was reacted on the column with a solution of N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium trifluoromethanesulfonate 2 (15 mg) in acetonitrile (0.8 mL) which was passed over the Chromabond cartridge followed by neat acetonitrile (0.7 mL) at a flow of 0.4 mL/min. The effluent containing the formed [18F]F-Py-TFP continued over a MCX Plus cartridge, which removed 2, and into a septum equipped vial (5 mL) containing either (4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)methanamine hydrochloride 3a (1.0 mg) for the synthesis of [18F]T2 or (4-(1,2,4,5-tetrazin-3-yl)phenyl)methanamine hydrochloride 3b (1 mg) for [18F]T3. The amines were predissolved in DMSO (0.1 mL) to enhance their solubility. The two cartridges were then purged with a stream of nitrogen gas to rinse the residual solution into the vial. Triethylamine (5 μL) was added, and the mixture was heated for 15 min at 55 °C. After dilution with aqueous trifluoroacetic acid (3 mL, 0.1%), the labeled tetrazine was isolated by semipreparative HPLC. Column = Kinetex C18 5 μm (150 × 10 mm), eluent = water/ethanol/TFA 70:30:0.01%, flow 5 mL/min. Column effluent was monitored using a UV detector (254 nm) and a radiodetector. To maximize activity concentration, the product peak was collected in fractions of 10 s corresponding to 0.8 mL volumes. Retention times = [18F]T2 8.5 min, [18F]T3 6.5 min. The fraction with highest activity was used for antibody labeling. The radiochemical purity was assessed by analytical HPLC. Column = Chromolith Performance RP-18e column 4.6 × 100 mm (Merck), eluent = water/acetonitrile 2% B, flow 4 mL/min. Retention times = [18F]T2 6.6 min, [18F]T3 5.4 min. The identities of the products were confirmed by coinjection with authentic reference standards.
18F-Labeling of Antibody Ligands
TCO–RmAb158-scFv8D3 and TCO–Tribody A2 were reacted for 15 min at room temperature with 18F-labeled tetrazines [18F]T1, [18F]T2, or [18F]T3 in an aqueous solution containing 18–30% ethanol, supplemented with 10% 10x PBS, at an approximate ratio of 1 MBq [18F]fluorotetrazine per 1 μg of antibody. The 18F-labeled antibody was purified from free [18F]fluorotetrazine with either an NAP-5 size exclusion column (GE Healthcare AB, Uppsala, Sweden) or, for small volumes, a Zeba 7K spin desalting column and eluted in PBS.
ELISA Evaluation of Radiolabeled Antibody Ligands
To verify that TCO modification or 18F-labeling of the antibody ligands did not affect their functionality, TfR and Aβ ELISA binding assays were performed, as previously described.4 In brief, 96-well half area plates (Corning) were coated with TfR (1 μg/mL; Sinobiological, Beijing, China) or Aβ (100 nM; Innovagen, Lund, Sweden) and blocked with BSA (1% in PBS), followed by addition of serial dilutions of the antibody ligands. Bound ligand was detected with HRP-conjugated antimouse-IgG-F(ab′)2 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), and signals were developed with K Blue Aqueous TMB substrate (Neogen Corp., Lexington, KY, USA) and read with a spectrophotometer at 450 nm. All dilutions were made in ELISA incubation buffer (PBS, 0.1% BSA, 0.05% Tween-20).
Autoradiography
Autoradiography was performed as previously with minor modifications.24 Sagittal cryosections (20 μm) from an 18-month-old tg-ArcSwe mouse and an age-matched wt control were equilibrated to room temperature and preincubated 15 min in autoradiography buffer (PBS, pH 7.4; 0.1% BSA; 0.05% Tween-20) before addition of [18F]T1-RmAB158-scFv8D3 (0.3 MBq/ml). Sections were incubated 120 min, washed three times (for 5 min each time) in PBS, dried for 10 min at 37 °C, and then exposed to phosphor imaging plates (MS, MultiSensitive, PerkinElmer, Downers Grove, IL, USA) overnight and scanned in a Cyclone Plus phosphor imager (PerkinElmer) at 600 dpi.
Animals
The tg-ArcSwe mouse model used in this study is maintained on a C57/Bl6 background and expresses human AβPP with the Arctic (E693G)25 and the Swedish (KM670/671NL)26 mutations, which increase Aβ aggregation and production, respectively. As a result, the tg-ArcSwe model has elevated soluble Aβ protofibril levels and increased intraneuronal Aβ accumulation already detectable at four months of age and visible plaque pathology from six months of age onward.27,28 Both male and female mice were used in this study, and wt littermates were used as controls. Animals were housed in an animal facility at Uppsala University and given access to food and water ad libitum. All procedures were approved by the Uppsala County Animal Ethics board (C17/14; 5.8.18-13350/17), following the rules and regulations of the Swedish Animal Welfare Agency, and were in compliance with the European Communities Council Directive of 22 September 2010 (2010/63/EU). All efforts were made to minimize animal suffering and to reduce the number of animals used.
Ex Vivo Analyses
The antibody radioligands were administered intravenously to tg-ArcSwe and wt mice. Administered activity and number of studied mice is given in Table 1.
Table 1. Number of Animals and Injected Radioactivity.
|
ex
vivo biodistribution at 2 h |
PET
and ex vivo biodistribution at 10–12 h |
|||
|---|---|---|---|---|
| radioligand | animal (wt) | injected radioactivity (MBq) | animal (wt/tg) | injected radioactivity (MBq) |
| [18F]T1-RmAb158-scFv8D3 | 3 | 0.35 ± 0.02 | 1/1 | 16.8 ± 3.5 |
| [18F]T2-RmAb158-scFv8D3 | 2 | 0.78 ± 0.11 | 0/0 | naa |
| [18F]T3-RmAb158-scFv8D3 | 3 | 0.55 ± 0.03 | 2/2 | 22.5 ± 6.0 |
| [18F]T3-Tribody A2 | 3 | 4.1 ± 0.26 | 2/2 | 19.1 ± 4.4 |
na = not applicable.
A terminal blood sample was obtained in all mice from the heart prior to intracardial perfusion with 50 mL of saline (0.9% NaCl) during a time period of 2 min. For biodistribution purposes, lung, liver, kidney, heart, pancreas, spleen, femoral and skull bone, muscle, and urine samples were isolated. The brain was divided into hemispheres; the left hemisphere was further divided into the cerebral and cerebellar part. The radioactivity in the blood samples and the organs was measured with a well counter (GE Healthcare, Uppsala, Sweden). The concentrations, quantified as standardized uptake value (SUV), were calculated as the following:
PET
PET imaging was performed in 5 tg-ArcSwe and 5 wt mice at an age of 18–20 months, when tg-ArcSwe mice have developed advanced Aβ pathology. Prior to the start of the PET scan, the animal was placed in a prone position on a preheated bed in the gantry of the PET/CT scanner (intrinsic resolution 1.35 mm, Triumph Trimodality System, TriFoil Imaging, Inc., Northridge, CA, USA). Anaesthesia was maintained throughout the study using 1.5–2.0% isoflurane in a 0.5 L/min flow of 50% oxygen and 50% medical air. The acquisition time in the PET scanner was 60 min, followed by a CT examination for 3 min with a field of view (FOV) of 8 cm. The PET data were reconstructed using an OSEM 3D algorithm (20 iterations). The CT raw files were reconstructed using filtered back projection (FBP). All subsequent processing of the PET and CT images was performed in imaging software Amide 1.0.4 (Loening and Gambhir, 2003).
Acknowledgments
We would like to acknowledge Prof. Lars Nilsson for developing the transgenic mouse model and Bioarctic AB for providing the RmAb158 sequence. We are also grateful to Raffaella Rossin and Marc S. Robillard, Tagworks Pharmaceuticals, for technical advice on TCO modification of antibodies. The molecular imaging work in this study was performed at the SciLifeLab Pilot Facility for Preclinical PET–MRI, a Swedish nationally available imaging platform at Uppsala University, Sweden, financed by the Knut and Alice Wallenberg Foundation. This work was financially supported by grants from the Swedish Research Council (2017-02413 and 2018-02715), Alzheimerfonden, Hjärnfonden, Torsten Söderbergs stiftelse, Hedlunds stiftelse, Åke Wibergs stiftelse, Konung Gustaf V:s och Drottning Victorias frimurarestiftelse, Åhlén-stiftelsen, Magnus Bergalls stiftelse, Stohnes stiftelse, and Stiftelsen för Gamla tjänarinnor. BioArctic AB contributed financially with the salary of one researcher (J.R.).
Author Contributions
S.S. and X.T.F. contributed equally to the study. S.S., X.T.F., J.E., and D.S. designed the study; L.L. contributed to the study design. J.E. and J.R. performed radiochemical work with input from D.E.O. S.S., X.T.F., R.F., and D.S. performed in vitro and in vivo experiments and analyzed results together with J.E. and J.R. S.S., X.T.F., J.E., and D.S. wrote the manuscript with feedback from all authors.
This work was financially supported by grants from the Swedish Research Council (2017-02413 and 2018-02715), Alzheimerfonden, Hjärnfonden, Torsten Söderbergs stiftelse, Hedlunds stiftelse, Åke Wibergs stiftelse, Konung Gustaf V:s och Drottning Victorias frimurarestiftelse, Åhlén-stiftelsen, Magnus Bergvalls stiftelse, Stohnes stiftelse, and Stiftelsen för Gamla tjänarinnor. BioArctic AB contributed financially with the salary of one researcher (J.R.).
The authors declare the following competing financial interest(s): L.L. is cofounder and shareholder of BioArctic AB.
References
- van Dongen G. A.; Poot A. J.; Vugts D. J. (2012) PET imaging with radiolabeled antibodies and tyrosine kinase inhibitors: immuno-PET and TKI-PET. Tumor Biol. 33 (3), 607–15. 10.1007/s13277-012-0316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultqvist G.; Syvanen S.; Fang X. T.; Lannfelt L.; Sehlin D. (2017) Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 7 (2), 308–31.8. 10.7150/thno.17155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M.; Buciak J. L.; Friden P. M. (1991) Selective Transport of an Antitransferrin Receptor Antibody through the Blood-Brain-Barrier Invivo. J. Pharmacol Exp Ther 259 (1), 66–70. [PubMed] [Google Scholar]
- Sehlin D.; Fang X. T.; Cato L.; Antoni G.; Lannfelt L.; Syvanen S. (2016) Antibody-based PET imaging of amyloid beta in mouse models of Alzheimer’s disease. Nat. Commun. 7, 10759. 10.1038/ncomms10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehlin D.; Stocki P.; Gustavsson T.; Hultqvist G.; Walsh F. S.; Rutkowski J. L.; Syvanen S. (2020) Brain delivery of biologics using a cross-species reactive transferrin receptor 1 VNAR shuttle. FASEB J. 34, 13272–13283. 10.1096/fj.202000610RR. [DOI] [PubMed] [Google Scholar]
- Sehlin D.; Syvanen S.; et al. (2019) Engineered antibodies: new possibilities for brain PET?. Eur. J. Nucl. Med. Mol. Imaging 46 (13), 2848–2858. 10.1007/s00259-019-04426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen S.; Fang X. T.; Hultqvist G.; Meier S. R.; Lannfelt L.; Sehlin D. (2017) A bispecific Tribody PET radioligand for visualization of amyloid-beta protofibrils - a new concept for neuroimaging. NeuroImage 148, 55–63. 10.1016/j.neuroimage.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Niewoehner J.; Bohrmann B.; Collin L.; Urich E.; Sade H.; Maier P.; Rueger P.; Stracke J. O.; Lau W.; Tissot A. C.; Loetscher H.; Ghosh A.; Freskgard P. O. (2014) Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81 (1), 49–60. 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- Yu Y. J.; Zhang Y.; Kenrick M.; Hoyte K.; Luk W.; Lu Y.; Atwal J.; Elliott J. M.; Prabhu S.; Watts R. J.; Dennis M. S. (2011) Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 3 (84), 84ra44. 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
- Sehlin D.; Fang X. T.; Meier S. R.; Jansson M.; Syvanen S. (2017) Pharmacokinetics, biodistribution and brain retention of a bispecific antibody-based PET radioligand for imaging of amyloid-beta. Sci. Rep. 7 (1), 17254. 10.1038/s41598-017-17358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson T.; Syvanen S.; O’Callaghan P.; Sehlin D. (2020) SPECT imaging of distribution and retention of a brain-penetrating bispecific amyloid-beta antibody in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 9 (1), 37. 10.1186/s40035-020-00214-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen S.; Hultqvist G.; Gustavsson T.; Gumucio A.; Laudon H.; Soderberg L.; Ingelsson M.; Lannfelt L.; Sehlin D. (2018) Efficient clearance of Abeta protofibrils in AbetaPP-transgenic mice treated with a brain-penetrating bifunctional antibody. Alzheimer's Res. Ther. 10 (1), 49. 10.1186/s13195-018-0377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S. R.; Syvanen S.; Hultqvist G.; Fang X. T.; Roshanbin S.; Lannfelt L.; Neumann U.; Sehlin D. (2018) Antibody-Based In Vivo PET Imaging Detects Amyloid-beta Reduction in Alzheimer Transgenic Mice After BACE-1 Inhibition. J. Nucl. Med. 59 (12), 1885–1891. 10.2967/jnumed.118.213140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuli F.; Zhang X.; Jagoda E. M.; Choyke P. L.; Swenson R. E. (2016) Facile room temperature synthesis of fluorine-18 labeled fluoronicotinic acid-2,3,5,6-tetrafluorophenyl ester without azeotropic drying of fluorine-18. Nucl. Med. Biol. 43 (12), 770–772. 10.1016/j.nucmedbio.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuli F.; Zhang X.; Woodroofe C. C.; Jagoda E. M.; Choyke P. L.; Swenson R. E. (2017) Fast indirect fluorine-18 labeling of protein/peptide using the useful 6-fluoronicotinic acid-2,3,5,6-tetrafluorophenyl prosthetic group: A method comparable to direct fluorination. J. Labelled Compd. Radiopharm. 60 (3), 168–175. 10.1002/jlcr.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N.; Machulla H. J.; Solbach C.; Winter G.; Reske S. N.; Zlatopolskiy B. (2011) Radiosynthesis of a new PSMA targeting ligand ([18F]FPy-DUPA-Pep). Appl. Radiat. Isot. 69 (7), 1014–8. 10.1016/j.apradiso.2011.03.041. [DOI] [PubMed] [Google Scholar]
- Olberg D. E.; Arukwe J. M.; Grace D.; Hjelstuen O. K.; Solbakken M.; Kindberg G. M.; Cuthbertson A. (2010) One step radiosynthesis of 6-[(18)F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([(18)F]F-Py-TFP): a new prosthetic group for efficient labeling of biomolecules with fluorine-18. J. Med. Chem. 53 (4), 1732–40. 10.1021/jm9015813. [DOI] [PubMed] [Google Scholar]
- Denk C.; Svatunek D.; Filip T.; Wanek T.; Lumpi D.; Frohlich J.; Kuntner C.; Mikula H. (2014) Development of a (18) F-labeled tetrazine with favorable pharmacokinetics for bioorthogonal PET imaging. Angew. Chem., Int. Ed. 53 (36), 9655–9. 10.1002/anie.201404277. [DOI] [PubMed] [Google Scholar]
- Karver M. R.; Weissleder R.; Hilderbrand S. A. (2011) Synthesis and evaluation of a series of 1,2,4,5-tetrazines for bioorthogonal conjugation. Bioconjugate Chem. 22 (11), 2263–70. 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund H.; Sehlin D.; Johansson A. S.; Nilsson L. N.; Gellerfors P.; Paulie S.; Lannfelt L.; Pettersson F. E. (2007) Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J. Neurochem. 103 (1), 334–45. 10.1111/j.1471-4159.2007.04759.x. [DOI] [PubMed] [Google Scholar]
- Kissel K.; Hamm S.; Schulz M.; Vecchi A.; Garlanda C.; Engelhardt B. (1998) Immunohistochemical localization of the murine transferrin receptor (TfR) on blood-tissue barriers using a novel anti-TfR monoclonal antibody. Histochem. Cell Biol. 110 (1), 63–72. 10.1007/s004180050266. [DOI] [PubMed] [Google Scholar]
- Boado R. J.; Zhang Y.; Wang Y. T.; Pardridge W. M. (2009) Engineering and Expression of a Chimeric Transferrin Receptor Monoclonal Antibody for Blood-Brain Barrier Delivery in the Mouse. Biotechnol. Bioeng. 102 (4), 1251–1258. 10.1002/bit.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X. T.; Sehlin D.; Lannfelt L.; Syvanen S.; Hultqvist G. (2017) Efficient and inexpensive transient expression of multispecific multivalent antibodies in Expi293 cells. Biol. Proced. Online 19, 11. 10.1186/s12575-017-0060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X. T.; Hultqvist G.; Meier S. R.; Antoni G.; Sehlin D.; Syvanen S. (2019) High detection sensitivity with antibody-based PET radioligand for amyloid beta in brain. NeuroImage 184, 881–888. 10.1016/j.neuroimage.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Nilsberth C.; Westlind-Danielsson A.; Eckman C. B.; Condron M. M.; Axelman K.; Forsell C.; Stenh C.; Luthman J.; Teplow D. B.; Younkin S. G.; Naslund J.; Lannfelt L. (2001) The ’Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 4 (9), 887–93. 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- Mullan M.; Crawford F.; Axelman K.; Houlden H.; Lilius L.; Winblad B.; Lannfelt L. (1992) A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1 (5), 345–7. 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Lord A.; Kalimo H.; Eckman C.; Zhang X. Q.; Lannfelt L.; Nilsson L. N. (2006) The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 27 (1), 67–77. 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Lord A.; Englund H.; Soderberg L.; Tucker S.; Clausen F.; Hillered L.; Gordon M.; Morgan D.; Lannfelt L.; Pettersson F. E.; Nilsson L. N. (2009) Amyloid-beta protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS J. 276 (4), 995–1006. 10.1111/j.1742-4658.2008.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]