

SUMMARY
A role for cancer cell epithelial-to-mesenchymal transition (EMT) in cancer is well established. Here, we show that, in addition to cancer cell EMT, ovarian cancer cell metastasis relies on an epigenomic mesenchymal-to-epithelial transition (MET) in host mesenchymal stem cells (MSCs). These reprogrammed MSCs, termed carcinoma-associated MSCs (CA-MSCs), acquire pro-tumorigenic functions and directly bind cancer cells to serve as a metastatic driver/chaperone. Cancer cells induce this epigenomic MET characterized by enhancer-enriched DNA hypermethylation, altered chromatin accessibility, and differential histone modifications. This phenomenon appears clinically relevant, as CA-MSC MET is highly correlated with patient survival. Mechanistically, mirroring MET observed in development, MET in CA-MSCs is mediated by WT1 and EZH2. Importantly, EZH2 inhibitors, which are clinically available, significantly inhibited CA-MSC-mediated metastasis in mouse models of ovarian cancer.
Graphical Abstract
In Brief
Fan et al. demonstrate that ovarian cancer reprograms the epigenome of stromal cells, inducing a mesenchymal-to-epithelial transition (MET) to form carcinoma-associated mesenchymal stem cells (CA-MSCs). This MET, mediated by WT1 and EZH2, enables CA-MSC:cancer cell co-metastasis. EZH2 inhibition decreases CA-MSC-mediated metastasis, presenting a potential therapeutic opportunity in ovarian cancer.
INTRODUCTION
Ovarian cancer, the deadliest gynecologic cancer, kills roughly 14,000 U.S. women yearly and 70% of all women with the disease. This high mortality is due to early, diffuse intra-abdominal metastatic spread. Ovarian cancer quickly colonizes the peritoneal cavity, building a complex tumor microenvironment (TME) that supports ovarian cancer survival, growth, and spread. This TME is a complex bionetwork of tumor, immune, and stromal cells (Tothill et al., 2008; Verhaak et al., 2013; Konecny et al., 2014; Karlan et al., 2014; Zhang et al., 2003).
Efforts to determine what drives the formation and function of the ovarian TME led to the identification of a critical stromal stem cell, the carcinoma-associated mesenchymal stem/stromal cell (CA-MSC) (McLean et al., 2011). CA-MSCs are benign stromal cells that meet all the criteria for mesenchymal stem/stromal cells (MSCs) as defined by the International Society for Cellular Therapy (ISCT) (Dominici et al., 2006). Karyotype analysis, high-resolution genotyping, and in vivo tumor initiation assays demonstrated that CA-MSCs have normal genomes and lack tumor-related mutations and that isolated CA-MSCs lack malignant potential (McLean et al., 2011; Verardo et al., 2014). CA-MSCs are distinct from carcinoma-associated fibroblasts (CAFs) and other more terminally differentiated stromal cells with long-term proliferative capacity, lineage differentiation capacity, and a unique gene expression pattern (Augsten, 2014; Coffman et al., 2019). CA-MSCs enhance ovarian cancer growth and chemotherapy resistance, increase the cancer stem cell-like (CSC) pool, and dramatically alter the stromal TME, increasing tumor-associated fibrosis and inducing angio-genesis (Coffman et al., 2016b; Spaeth et al., 2009). CA-MSCs arise from resident tissue MSCs (Coffman et al., 2019). However, the mechanism driving the formation of a CA-MSC—and, thus, the ability to target these critical stromal cells—remains unknown. CA-MSCs have widespread, durable expression changes without gain of genomic mutations. These “mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” are evocative of the exact definition of “epigenetics” by Riggs and colleagues (Martienssen et al., 1996). Therefore, we hypothesized that epigenetic reprogramming drives this conversion of a normal MSC into a CA-MSC. While cancer-related epigenetic changes are well documented in tumor cells, reciprocal changes in the stroma are less recognized (Sharma et al., 2010; Shen and Laird, 2013).
Here, we demonstrate that CA-MSCs, compared to normal MSCs, have a unique epigenetic landscape characterized by enhancer-enriched DNA hypermethylation, enrichment in repressive histone modifications, and altered chromatin accessibility. We demonstrate that normal MSCs undergo cancer-mediated epigenetic reprogramming to form a cancer-supportive CA-MSC. Unexpectedly, this reprogramming induces a partial mesenchymal-to-epithelial transition (MET), a process not previously recognized in cancer-associated stroma. This MET leads to increases in cell contact pathways and functional demonstration of increased binding of CA-MSCs to tumor cells. Direct binding of CA-MSCs to tumor cells increases ovarian cancer metastasis through a process of co-metastasis where CA-MSCs and tumor cells travel in complex to effectively colonize metastatic sites. We identify EZH2 and WT1 as potential mediators of CA-MSC epigenetic mesenchymal-to-epithelial reprogramming. Interrupting this reprogramming with an EZH2 inhibitor decreases the ability of ovarian cancer cells to metastasize in an orthotopic mouse model. Thus, in contrast to tumor cell epithelial-to-mesenchymal transition (EMT), stromal MET is critical to ovarian cancer metastasis. Importantly, in patients, this stromal MET phenotype can be altered and may present a novel therapeutic target.
RESULTS
CA-MSCs Have a Unique DNA Methylation Profile Compared to Normal Tissue MSCs
We hypothesized that CA-MSCs undergo epigenetic alterations, which drives their mitotically stable pro-tumorigenic phenotype. We used the Infinium MethylationEPIC (850K/EPIC/HM850) array to profile 13 CA-MSCs derived from high-grade serous ovarian cancer (HGSC) samples, 20 omental/adipose/fallopian tube (FT)-derived normal MSCs, and six patient tumor cell samples (Table S1). We performed data quality and sample quality controls, including purity determination and potential sample swaps. The promoter of MIR200C/141 is unmethylated in epithelial cells and methylated in mesenchymal cells (Vrba et al., 2010), and the methylation level (beta value) of MIR200C/141 directly corresponds to the fraction of non-epithelial cells (George et al., 2019; Cherniack et al., 2017). As expected, our MSCs had close to 100% methylation at the MIR200C/141 promoter site, consistent with a non-epithelial origin and high sample purity. In contrast, primary human tumor cell samples demonstrated lack of methylation at this locus (Figure 1A). One CA-MSC sample had low MIR200C/141 promoter methylation, consistent with tumor cell contamination, and this sample also clustered with sorted tumor cells (Figure S1) and was removed from all further analysis.
Figure 1. CA-MSCs Have Unique DNA Methylation Profiles compared to Normal-Tissue MSCs.
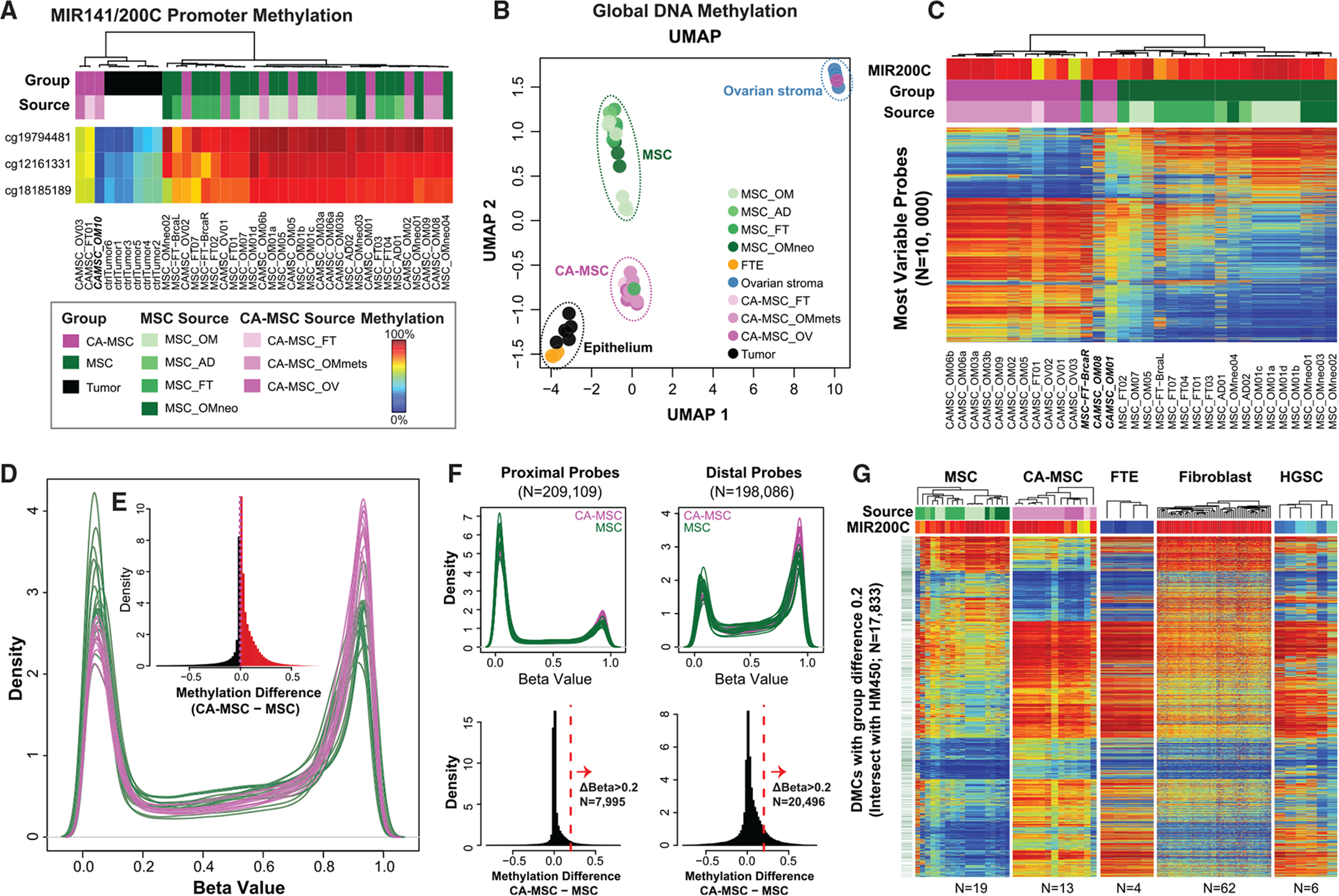
(A) DNA methylation at the MIR141/MIR200C promoter separates epithelial and mesenchymal samples. Rows indicate MIR141/MIR200C promoter region probes. Columns indicate cell type. Column-side color bars indicate sample group or tissue source. A blue-to-red gradient indicates a beta value of 0–1 (DNA methylation level of 0% to 100%) in all heatmaps. FT, fallopian tube; AD, adipose; OM, omentum; OV, ovary; OMmets, omentum metastasis.
(B) UMAP of DNA methylation. Each dot represents an individual sample. The two dimensions are the intersect set of 438,064 CpG loci present on both the EPIC and HM450 platforms.
(C) CA-MSCs have differential DNA methylation versus MSCs. Top variable CpG probes (rows) (N = 10,000; filter with standard deviation) of 13 CA-MSCs and 19 MSCs (columns). MIR141/200C promoter methylation—average of the three probes in (A)—indicates the mesenchymal component proportion per sample.
(D) Overall distribution (smoothed probability density distribution) per sample of DNA methylation beta values (x axis) for CA-MSCs (purple lines) and MSCs (green lines).
(E) Histogram of DNA methylation distribution (beta value) group differences between CA-MSCs and normal MSCs. Red indicates methylation difference > 0 (hypermethylation in CA-MSCs); black indicates methylation difference < 0 (hypomethylation in CA-MSCs).
(F) Distal regions show more frequent DNA methylation gain in CA-MSCs versus MSCs. Density and histogram plots for proximal and distal CpG probe set (vertical in order). Proximal probes, overlapping with transcript promoters (±500 bp around the transcription start site; TSS). Distal probes, located near TFBSs (±100 bp) but not within ±2 bp from the TSS. Distal elements are almost three times more likely to acquire a DNA methylation gain greater than 0.2 in CA-MSCs.
(G) DNA methylation heatmap showing differential methylation cytosine sites (DMCs; N = 17,883) between CA-MSCs and MSCs.
We also downloaded and reprocessed external DNA methylation data on normal cell types from prior publications (Patch et al., 2015; Klinkebiel et al., 2016), including normal FT epithelium (FTE) and ovary stroma, profiled on the earlier generation Infinium HumanMethylation450 (450K) array. The intersect between the two platforms (450,161 CpGs) were used in joint analysis with these external normal controls. Uniform Manifold Approximation and Projection (UMAP) reveals clustering coinciding with sample type (Figure 1B), with a few exceptions. (1) One CA-MSC (CA-MSC-OM08) grouped with the normal ovary stroma. After final pathology review, this sample was found to be derived from an ovarian carcinosarcoma, not HGSC. (2) One sample, CA-MSC-OM01, grouped with CA-MSCs on the UMAP but clustered with the normal MSCs on the unsupervised hierarchical clustering (Figure 1C) and, overall, demonstrated an intermediate profile between normal MSCs and CA-MSCs. This sample was derived from tumor-adjacent omentum without clear malignant involvement and may represent a mixture of normal MSCs and CA-MSCs. (3) MSC-FT-BrcaR, derived from the right FT of a patient with a germline BRCA2 mutation undergoing risk reduction surgery, grouped with CA-MSCs. In contrast, MSCs derived from the left FT of the same patient grouped with normal MSCs as expected. The expression-based CA-MSC classifier score, which accurately distinguishes normal MSCs from CA-MSCs (Coffman et al., 2019) for the left FT MSCs, was 0.01 (consistent with a normal MSC) and for the right FT MSCs, it was 0.95 (approaching the CA-MSC threshold of 0.96), in line with the methylation grouping and indicating that certain stromal cells derived from a high-risk patient more closely align with cancer-associated stroma. As expected, flow-sorted tumor cells were the closest to the normal FTE, the presumed cell of origin for HGSC.
MSCs Derived from Patients with Complete Response to Chemotherapy Resemble Normal MSCs
Interestingly, MSCs derived from the omentum of patients treated with neoadjuvant chemotherapy who achieved a pathologic complete response in the omental tissue clustered with the normal MSCs (n = 4; MSC-OMneo1–4). Likewise, the expression classifier scores of these MSCs ranged from 0.01 to 0.15 (within the normal MSC range). Radiographic evidence of omental involvement prior to neoadjuvant chemotherapy was documented in all cases.
CA-MSCs Demonstrate Epigenetic Alteration Characterized by Hypermethylation at Distal Enhancers
The top 10,000 most variably methylated probes across all MSC samples demonstrated a clear delineation between CA-MSCs and normal MSCs (Figure 1C). CA-MSCs demonstrated distinct gains in DNA methylation versus normal MSCs (Figures 1D and 1E). Greater than 128,000 (32% out of ~401,000 filtered probes) differentially methylated cytosines (DMCs), mapping to 22,991 differentially methylated regions (DMRs), were identified based on a threshold of a false discovery rate (FDR) < 0.05. Such difference is more pronounced at distal rather than proximal sites (Figures 1F, S1B, and S1C), with distal regions three times more likely to acquire hypermethylation of >0.2 beta value difference compared to proximal regions. In other words, the observed DNA methylation changes preferentially occurred in enhancer regions.
CA-MSC DNA Methylation Profile Demonstrates Similarity to that of Epithelium
To understand the implications of such DNA methylation alterations, we examined sites with absolute beta value difference of greater than 0.2 in CA-MSCs versus normal MSCs (n = 36,606 in HM850 loci; n = 17,904 in HM850/HM450 intersect). Unexpectedly, at these differential methylation loci, CA-MSCs closely resembled FTE samples (Figure 1G), while normal MSCs were more similar to dermal fibroblasts. Similarly, CA-MSCs bore more resemblance to sorted HGSC cells, which are also epithelial in origin.
Intrigued by this result, we further examined all sites (instead of just CA-MSC versus MSC differential sites) in the HM850/HM450 intersect. It was clear that, although globally, CA-MSCs exhibit increased similarity to FTE samples (Figure S1D), they were distinct. We continued to investigate sites shared between, or unique to, MSCs, CA-MSCs, and FTE samples. CA-MSCs had a DNA methylation profile that combined MSC and FTE features, with a limited set of CA-MSC-specific hyper- and hypo-methylated sites, compared to shared sites with either MSC or FTE (Figure 2A). Therefore, CA-MSCs likely demonstrated an intermediate state between MSC and FTE. Importantly, CA-MSCs remain distinct from cancer cells. CA-MSCs do not acquire tumor cell-related mutations, as demonstrated by our group and others previously (McLean et al., 2011; Verardo et al., 2014). We further confirmed the lack of p53 hotspot mutations in CA-MSCs by searching for the top 50 p53 mutations curated in the Catalogue of Somatic Mutations in Cancer database (COSMIC v90; GRCh37) (Tate et al., 2019) and verified that these hotspot mutations were not present in CA-MSC RNA-sequencing (RNA-seq) data (Coffman et al., 2019). CA-MSCs are also distinct from other epithelial and mesenchymal malignancies, including uterine carcinosarcoma and soft tissue sarcomas (Figure 2A).
Figure 2. Verification of Differential DNA Methylation Loci Demonstrating that Methylation Differences Are Acquired during CA-MSC Reprogramming.
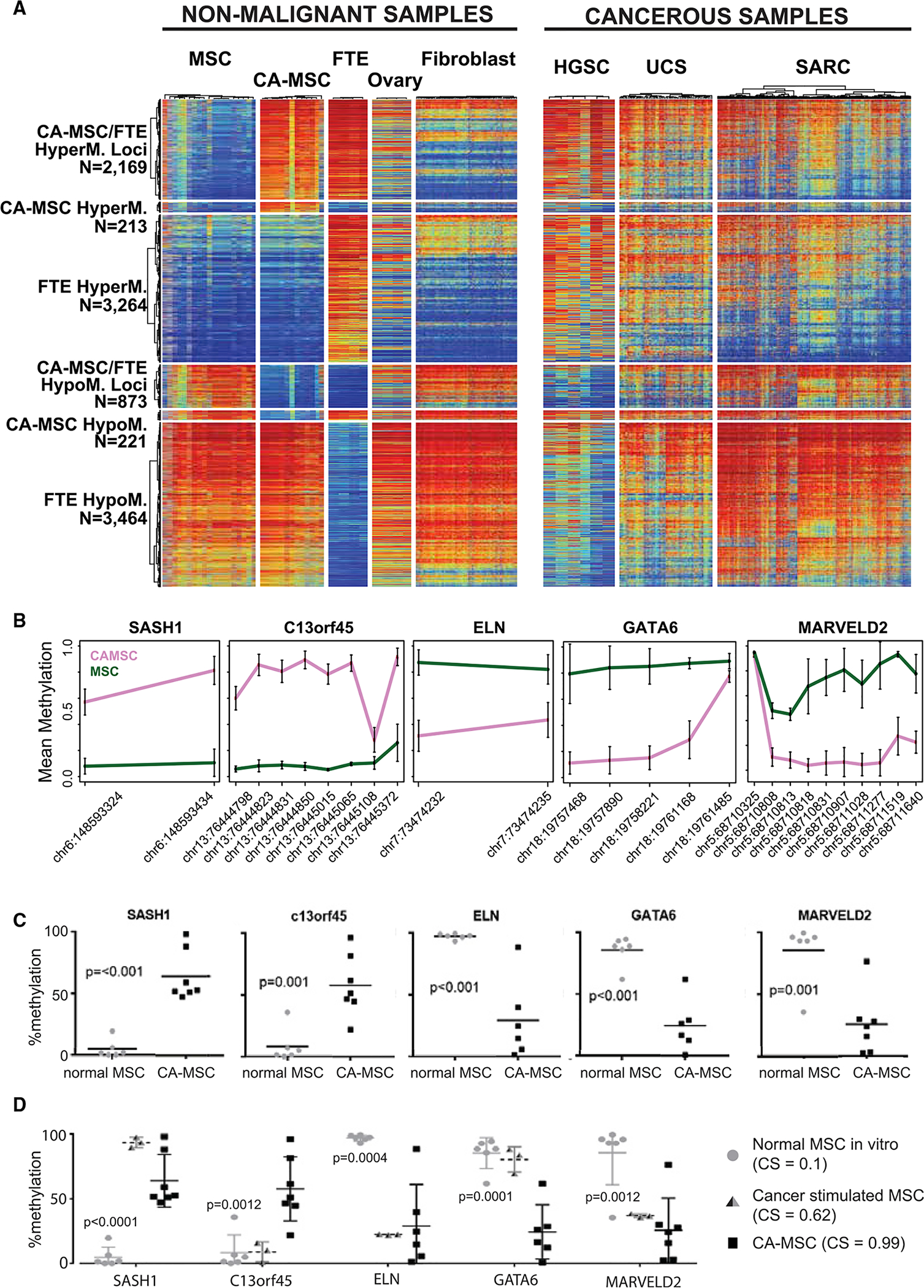
(A) DNA methylation heatmap of CpG probes hyper- or hypomethylated in CA-MSC, FTE, and MSC compared to normal ovarian stroma (ovary), fibroblast, HGSC, sarcoma (SARC), or uterine carcinosarcoma (UCS). A blue-to-red gradient indicates a beta value of 0–1.
(B) Top DMRs within five different genes chosen for quantitative methylation-specific PCR (qMSP) validation. The y axis indicates mean beta values; the x axis indicates probe location. Error bars represent SEM.
(C and D) qMSP of top DMRs validates differential methylation in 9 independent CA-MSCs and 6 MSCs. Cancer stimulation of normal MSCs in vitro induces a partial CA-MSC methylation pattern. Means and SEM of 3 independent samples are represented. The p values in (D) compare CS MSCs with normal or CA-MSCs.
Quantitative Methylation-Specific PCR (qMSP) Validates DMRs
To confirm the DNA methylation differences noted in the EPIC/HM850 array, we selected a panel of five top DMRs (CA-MSCs versus MSCs): two regions of hypermethylation (SASH1 nd C13orf45), and three regions of hypomethylation (ELN, GATA6, and MARVELD2). Average beta values for each CpG as determined by the microarray are plotted in Figure 2B. The qMSP assays were designed for each locus. qMSP was performed on an independent set of 6 additional normal MSCs and 9 CA-MSCs. Across the five loci tested, the pattern of methylation in CA-MSCs and normal MSCs was confirmed (Figure 2C).
CA-MSC-Associated DMRs Persist throughout Lineage Differentiation and Are Acquired during Cancer-Mediated Reprogramming of MSCs
To determine whether the observed differential DNA methylation is carried through differentiation and possibly impacts the phenotype of stromal fibroblast and adipocyte cells, we used the qMSP panel to characterize normal MSC and CA-MSC in vitro lineage-directed differentiation into fibroblasts or adipocytes (Figures S2A and S2B). These cells maintained their methylation status at these loci even after differentiation, indicating that CA-MSC reprogramming is independent of lineage-specific differentiation.
Given our previous work demonstrating that normal MSCs are induced by tumor cells to become CA-MSCs, we analyzed in vitro indirect cancer stimulation of normal MSCs (which yields a partial conversion to a CA-MSC) for the development of CA-MSC-specific DMRs with the qMSP assays discussed earlier. Three of the five loci tested demonstrated CA-MSC-like alterations, with acquisition of DNA methylation at SASH1 and loss of methylation at ELN and MARVELD2. C13orf45 and GATA6 loci remained unchanged (Figure 2D). This finding suggests a partial conversion to a CA-MSC DNA methylation profile with in vitro cancer stimulation. This is consistent with a change in the CA-MSC classifier score from 0.1 in normal MSCs (consistent with a normal phenotype) to 0.62 after indirect cancer stimulation (the threshold for CA-MSC is 0.96).
CA-MSCs Have Altered Chromatin Accessibility
We next analyzed the chromatin accessibility of CA-MSCs versus normal MSCs using the Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq). Clustering based on ATAC-seq peaks accurately separated normal MSCs from CA-MSCs (Figure 3A). DESeq2 identified 5,129 statistically differential peaks with an adjusted p < 0.05. Power was limited in this analysis, partly due to the small sample size after removing samples that failed quality control. However, even in the majority of sites that did not reach statistical significance, CA-MSCs in general demonstrated a decrease in accessibility compared to normal MSCs after normalizing for library size (Figure 3B), consistent with the observed widespread gain of DNA methylation in CA-MSCs. The differential peaks between CA-MSCs and MSCs were enriched for intergenic regions and depleted of CpG islands (Figures S3A and S3B). Overall, sites with reduced accessibility in CA-MSCs tended to have higher DNA methylation (Figure 3C).
Figure 3. CA-MSCs Have Altered Chromatin Accessibility compared to Normal MSCs, which Correspond to DNA Methylation and RNA Expression Changes.
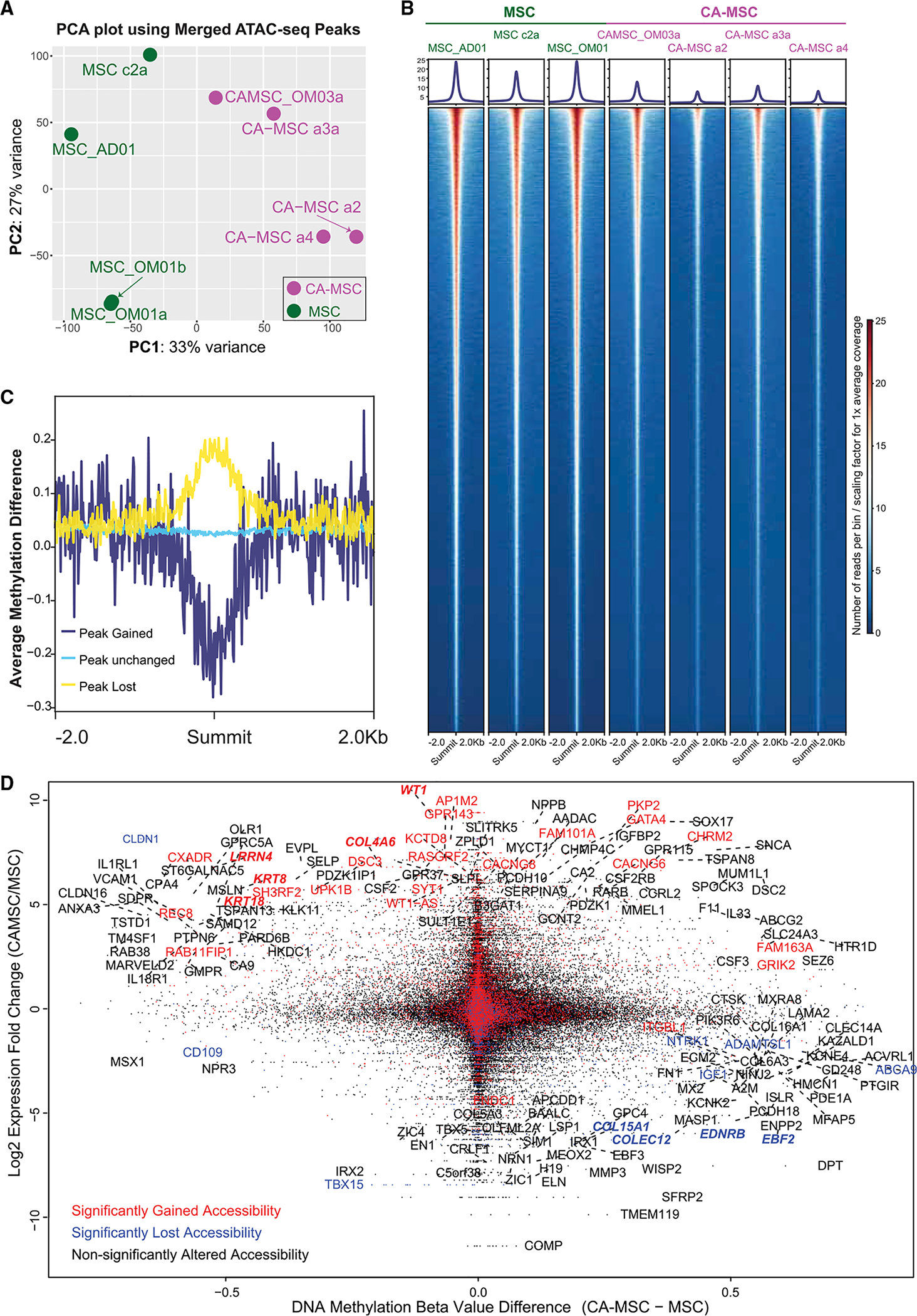
(A) Principal-component analysis (PCA) on the merged peak regions from ATAC-seq.
(B) ATAC-seq heatmap per sample centered at the summits of all the merged peaks. The heatmap is based on a 10-bp-binned ATAC-seq signal, normalized against sequencing library size per sample. Signal profile is indicated on top of each heatmap. The y axis indicates signal strength at base resolution surrounding the peak summits.
(C) DNA methylation patterns around the summits for peak group gained, unchanged, and lost calculated from (B). Line plot centered on the summits of all the peaks within each group is plotted as the x axis. The y axis indicates the average of overlapping methylation difference.
(D) Integration of DNA methylation, gene expression, and ATAC-seq calls. Probe-level DNA methylation difference (CA-MSC − MSC) for promoter probes is plotted as the x axis. Expression fold changes (log2-scaled) for the corresponding genes are plotted as the y axis. Dots/gene labels are colored based on gene-overlapping ATAC-seq peak orientations: red, open; blue, closed; and black, unaltered peaks in CA-MSCs versus MSCs. Genes with the largest expression alterations (50 gene fold change rank), and combined methylation and gene expression differences (absolute beta value difference > 0.5 and fold change > 2) are labeled with gene names.
DNA Methylation and Chromatin Accessibility Correspond to Transcriptomic Differences in CA-MSCs versus MSCs
We incorporated our DNA methylation and chromatin accessibility data (Figure 3D) with previously reported RNA-seq data (Coffman et al., 2019). In general, genes with gained peaks or lost peaks segregated based on gene expression. Genes exemplified by WT1, LRRN4, KRT8, KRT18, and COL4A6 were significantly overexpressed in CA-MSCs and demonstrated hypomethylation and open ATAC peaks. In contrast, genes such as COL15A1, EBF2, EDNRB, and COLEC12, which have decreased expression in CA-MSCs, demonstrated hypermethylation and reduced ATAC peaks.
Gene Expression Pathway Analysis Demonstrates that CA-MSCs Have Altered Collagen Synthesis and Undergo a Partial MET
Pathway analysis of the most differentially expressed genes demonstrated an upregulation of multiple pathways associated with alterations in extracellular matrix and cell:cell and cell:matrix adhesion (Figures S3C and S3D). We validated alterations in collagen with increased expression of collagen IV (COL4A6) and decreased expression of collagen XV (COL15A1) in CA-MSCs versus normal MSCs (Figure S4A). We also demonstrated that CA-MSCs deposit significant extracellular collagen IV during in vitro culture, while normal MSCs deposit extracellular collagen XV (Figure S4B). Collagen IV and XV are basement membrane proteins with apparently opposing roles in cancer; collagen IV is associated with invasion and metastasis, while collagen XV is found largely in normal tissues and decreases with cancer initiation (Clementz and Harris, 2013; Burnier et al., 2011). We also demonstrate that collagen IV is prevalent in ovarian cancer omental metastasis, while collagen XV is more abundant in normal omental tissue (Figure S4C).
We next focused on cell:cell and cell:matrix adhesion pathways, which were altered in CA-MSCs versus normal MSCs. CA-MSCs demonstrate increases in cell adhesion pathways, a largely epithelial feature, and downregulation of mesenchymal genes (Figures S3C and S3D). We took the top 25 epithelial and mesenchymal signature genes (E-genes and M-genes, respectively) from a previous study (Creighton et al., 2013), which were ranked based on the Pearson’s correlations of gene expression with the EMT score calculated from the canonical EMT markers (Lee et al., 2006). CA-MSCs clearly exhibited higher expression of the E-genes and lower expression for the M-genes. A quantitative EMT score derived with a 16-gene signature from the same study also suggested that CA-MSCs, indeed, had a more “epithelial” signature, while normal MSCs had a more “mesenchymal” signature (Figure 4A). Gene set enrichment analysis (GSEA) showcased the downregulation of several EMT-related gene sets as well (Figure 4B). This observation coalesces with the DNA methylation results, which also suggest a partial MET in CA-MSCs.
Figure 4. CA-MSCs Undergo a Partial Mesenchymal-to-Epithelial Transition (MET), Leading to Increased Tumor Cell Binding and Increased Ovarian Cancer Metastasis.
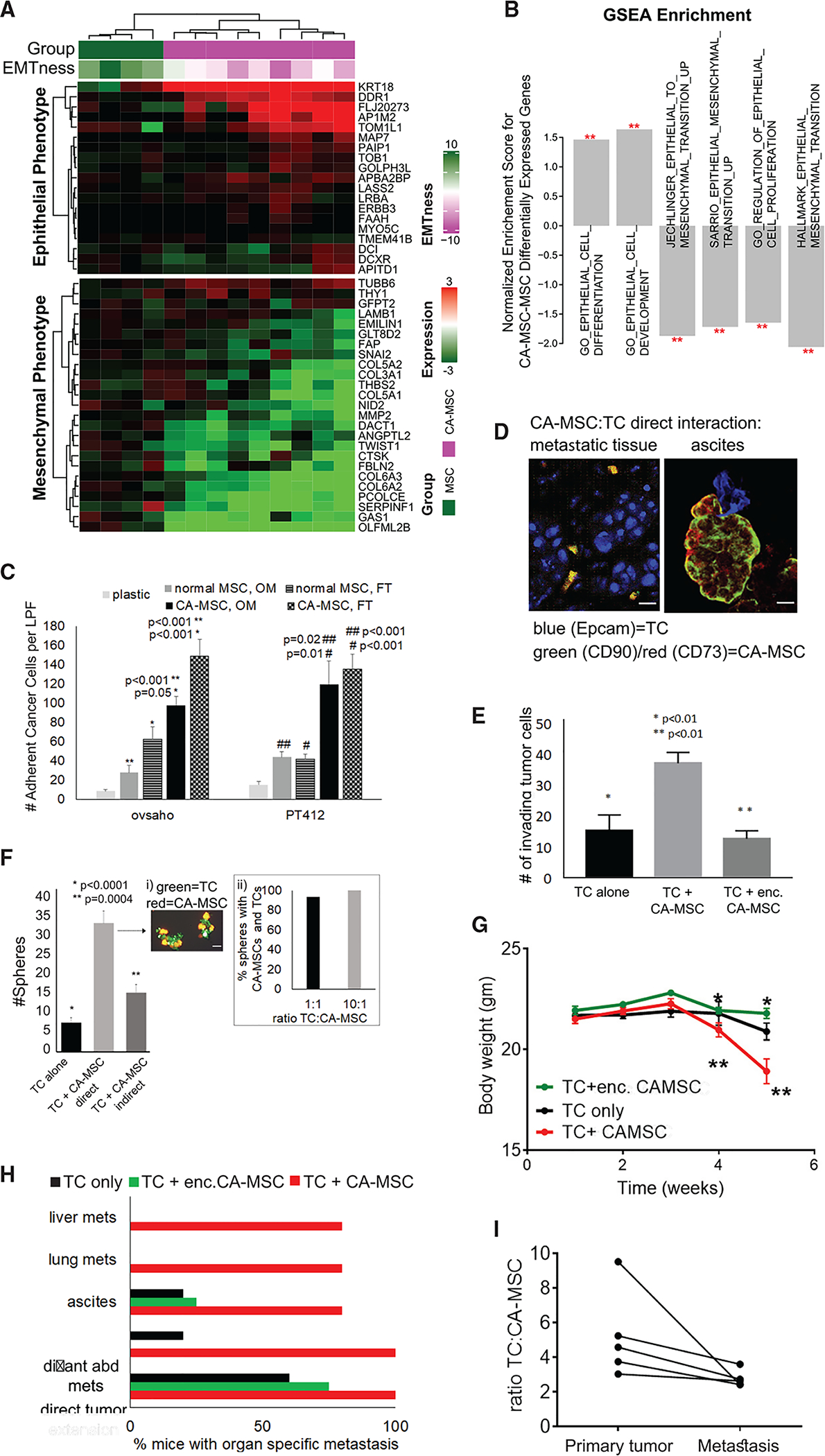
(A) Heatmap of epithelial- and mesenchymal-signature genes indicates a partial MET in CA-MSCs. EMTness score is indicated as column annotation.
(B) GSEA results support a potential MET in CA-MSCs. Genes are ranked based on CA-MSCs versus MSCs fold changes. Red stars indicate significance level. The y axis indicates normalized enrichment scores.
(C) CA-MSCs demonstrate greater cancer cell adhesion versus normal MSCs. Quantification of adherent cancer cells to plastic, normal MSCs or CA-MSCs via fluorescent microscopy, cell counts per low power field (LPF).
(D) Immunofluorescence of human HGSC metastatic omental tissue and cellular complexes isolated from malignant ascites. Confocal microscopy demonstrates tumor cells (TCs) in contact with CA-MSCs. Scale bars, 20 μm.
(E) CA-MSCs enhance the invasion of TCs through Matrigel-coated Transwells. TCs that invaded were counted per LPF. Alginate encapsulation (enc.) prevents enhanced invasion.
(F) CA-MSCs enhance TC sphere growth via direct CA-MSC:TC interactions. Spheres containing GFP-TCs were quantified via fluorescent microscopy per LPF and compared to TCs alone and TCs allowed to directly or indirectly interact with CA-MSCs. (i) Representative picture of GFP-TCs and mT-CA-MSCs within heterocellular spheres. (ii) Quantification of spheres with at least one CA-MSC per TC sphere at 1:1 and 10:1 TC:CA-MSC ratios.
(G) Orthotopic mouse model with increased metastasis when CA-MSCs are in contact with TCs. Mouse weight over time per group is plotted (*, ** indicate significant decrease in weight between time points, p < 0.05).
(H) Quantification of organs with metastasis at necropsy demonstrating increased lung, liver, and intra-abdominal metastasis in the TC + CA-MSC group, which is decreased with enc. CA-MSC. distant abd mets, distant abdominal metastasis.
(I) CA-MSCs co-localize with TCs at the primary and metastatic tumor and metastatic tumor sites have an increased proportion of CA-MSCs. Quantification of the TC-to-CA-MSC ratio in the TC + CA-MSC group.
For all pooled data, means and SEM are represented.
To test whether CA-MSCs exhibit an increase in epithelial phenotypic characteristics such as increased cell adhesion capacity, we performed in vitro cell adhesion assays of tumor cells to normal MSCs or CA-MSCs. CA-MSCs had a 3–5 fold increase in tumor cell binding capacity after 30 min of co-incubation. This pattern is true, regardless of MSC or CA-MSC origin (FT or omentum), and is consistent across two ovarian cancer cell lines and one primary patient tumor cell line tested (Figures 4C and S5A). The increased tumor cell binding confirms the pathway analysis showing increased cell:cell adhesion in CA-MSCs.
CA-MSCs Are Found in Direct Contact with Ovarian Cancer Cells in Both Patient Tissue and Ascites
To understand the physiologic relevance of a partial MET resulting in enhanced adhesion of CA-MSCs to tumor cells, we analyzed tissue from metastatic ovarian cancer and isolated cellular complexes from the ascites of ovarian cancer patients. Metastatic tissue and cytospins of ascites cellular complexes were analyzed with immunofluorescent confocal microscopy for Epcam (Pacific Blue)-positive tumor cells and CD90 (fluorescein isothiocyanate; FITC)/CD73 (red fluorescent protein [RFP])-positive CA-MSCs confirming physical interactions between CA-MSCs and tumor cells in both tissue and ascites (Figure 4D). To quantify the proportion of MSCs within malignant tissue and ascites compared to benign tissue, tissue or ascites cellular complexes were dissociated into single-cell suspensions. MSCs were identified via CD90, 73, 105+/CD45, 34, 14, 19− and the percentage of MSCs within each tissue was quantified (Figure S3E). Overall, there was an increase in MSC populations within malignant tissue versus benign tissue. To further verify that CA-MSCs are distinct from tumor cells, lack of significant PAX8 expression in CA-MSCs was confirmed within ascites complexes with immunohistochemistry (Figure S5B). PAX8 protein levels were also assessed by western blot in ascites CA-MSCs, omental metastasis CA-MSCs, normal MSCs, and tumor cells demonstrating significantly less PAX8 in MSCs versus tumor cells and no difference in low PAX8 protein levels between CA-MSCs and normal MSCs (Figure S5C). All CA-MSCs were verified for appropriate surface marker expression and differentiation capacity (Figures S5D and S5E) to meet ISCT criteria for MSCs (Dominici et al., 2006).
CA-MSCs Enhance Ovarian Cancer Metastasis
We next assessed the role of CA-MSCs in ovarian cancer metastasis. We tested whether CA-MSCs support critical steps in ovarian cancer metastasis, including migration/invasion and survival under non-adherent conditions. CA-MSCs allowed to directly interact with tumor cells (CAOV3, OVSAHO, and OVCAR3) significantly enhanced tumor cell migration and invasion through a Matrigel-coated Transwell culture (Figure 4E). CA-MSCs also enhanced the ability of tumor cells to grow under non-adherent conditions as spheres (Figure 4F). Fluorescent microscopy of CA-MSCs (expressing mT) and tumor cells (expressing GFP) demonstrated that CA-MSCs and tumor cells remained in physical contact during migration/invasion and sphere formation, indicating co-migration and formation of heterocellular spheres (Figure 4Fi). Increasing the ratio of tumor cells to CA-MSCs from 1:1 to 10:1 under non-adherent conditions resulted in each tumor cell sphere containing at least one CA-MSC, even at low CA-MSC concentrations (Figure 4Fii). This indicates a preferential direct binding between these cell types. This also mirrors the ratio of tumor cells to CA-MSCs (~10:1) found in patient ascites cellular complexes (Figure S5F).
Direct Tumor Cell:CA-MSC Interactions Mediate Enhancement of Ovarian Cancer Metastasis
To confirm that the enhancement of metastatic steps is mediated through direct CA-MSC:tumor cell interactions, we repeated the aforementioned experiments but prevented the direct interaction between CA-MSCs and tumor cells by encapsulating CA-MSCs in alginate or separating the cells by a permeable membrane (allowing paracrine signaling without direct contact). Luciferase-secreting CA-MSCs were used to verify the continued viability and effective protein secretion of encapsulated CA-MSCs (Figure S5G). Alginate encapsulated CA-MSCs failed to enhance tumor cell migration/invasion (Figure 4E). Similarly, CA-MSCs separated from tumor cells by a permeable membrane did not enhance tumor cell sphere formation (Figure 4F). Together, this indicates direct CA-MSC and tumor cell adhesion is critical to in vitro enhancement of metastasis.
We next assessed the impact of CA-MSCs in a murine model of metastasis. Ovarian cancer cells (OVCAR3) were implanted orthotopically in the ovarian bursa alone, with CA-MSCs or with alginate encapsulated CA-MSCs. Mice with tumor cells plus CA-MSCs demonstrated significant increases in metastasis and systemic illness, as measured by decreased weight over time, compared to mice with tumor cells alone or encapsulated CA-MSCs (Figure 4G). Mice with tumor cells with direct CA-MSC contact demonstrated significantly more intra-abdominal metastasis with co-localization of tumor cells and CA-MSCs at metastatic implants consistent with tumor cell:CA-MSC co-migration (Figures 4H and 4I). Mice with tumor cells alone or with encapsulated CA-MSCs demonstrated large localized ovarian tumors but did not develop ascites, distant abdominal metastasis, or lung or parenchymal liver metastasis (Figure 4H). CA-MSCs within the alginate capsule remained viable and were re-isolated at the time of necropsy. The ratio of tumor cells to CA-MSCs was calculated between primary tumor and metastatic tissues. A trend toward increased CA-MSCs within metastatic tissues was observed (p = 0.06, two-sided Wilcoxon signed-rank test; Figure 4I). Collectively, this indicates that CA-MSCs significantly enhance ovarian cancer metastasis via direct binding and co-metastasis with cancer cells.
WT1 Is a Driver of MET and Mediates CA-MSC Formation
One of the most significantly differentially regulated genes with increased RNA expression, decreased DNA methylation, and increased ATAC peak was WT1 (Figures 3D and 5A). WT1 expression is often found in cells going through METs or cells with mixed epithelial and mesenchymal properties, such as podocytes (Moore et al., 1998). WT1 overexpression has been reported to induce MET and is a regulator of MET during development (Hohenstein and Hastie, 2006; Essafi et al., 2011). While known as a tumor marker, WT1 is present in tumor-associated stroma (He et al., 2008; Hylander et al., 2006). Independent qRT-PCR validation of WT1 expression demonstrated ~100-fold increased expression in CA-MSCs versus normal MSCs with corresponding increase in protein (Figures 5B and 5C). Further, cancer-stimulated (CS) reprogramming of CA-MSCs induced WT1 expression (Figure 5C).
Figure 5. WT1 Is Associated with a Permissive Epigenetic State in CA-MSCs and Participates in the Formation of a CA-MSC.
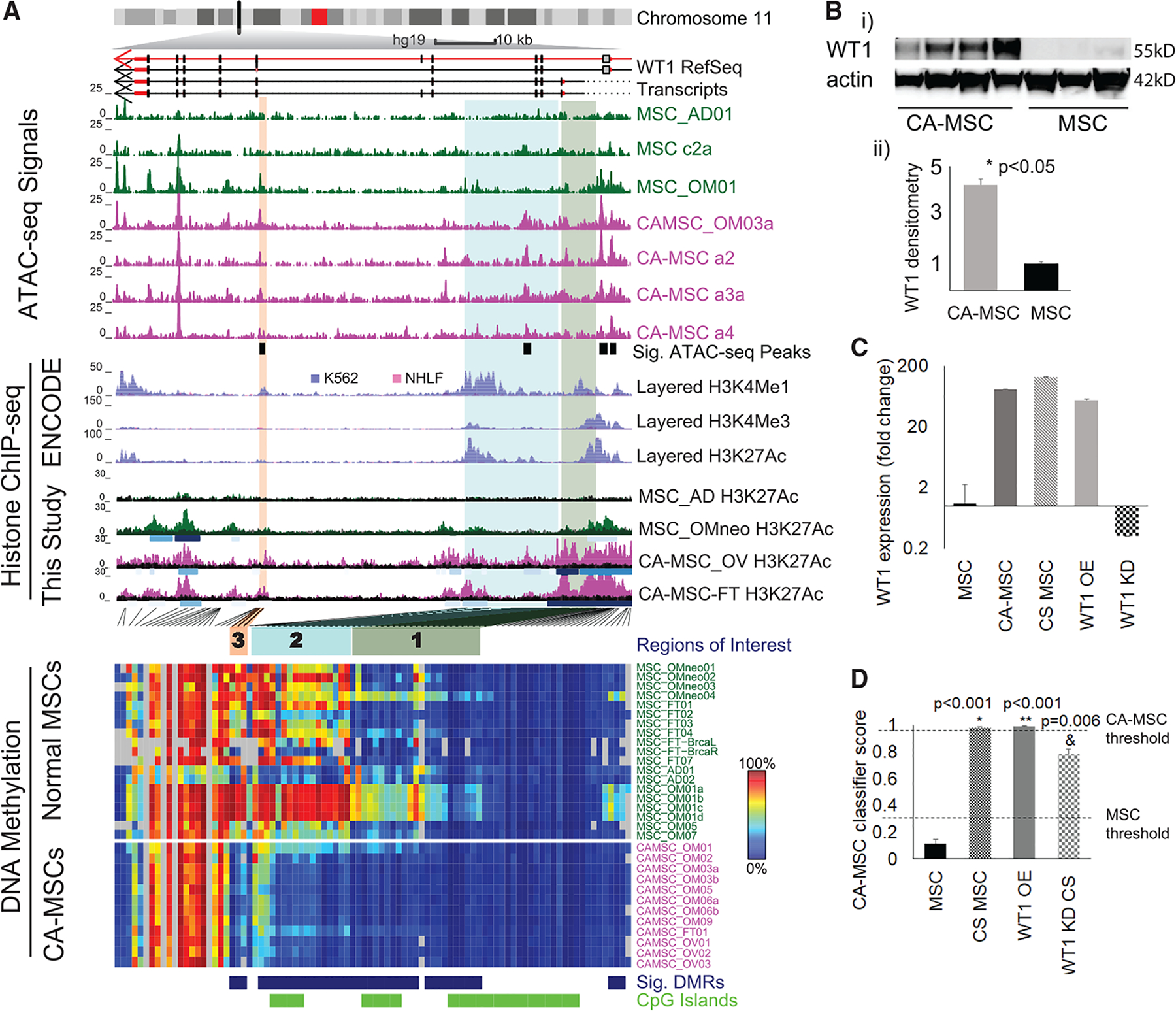
(A) Integrated view of epigenetic profiles for WT1. WT1 transcripts are plotted under the chromosome ideogram. Arrows indicate transcript direction. Normalized read count (10-bp bins) for ATAC-seq is plotted under transcripts. Significantly gained peaks (Sig. ATAC-seq Peaks) in CA-MSCs are indicated by black boxes. Layered H3K4me1 (enhancer mark), H3K4me3 (promoter mark), and H3K27ac (active chromatin mark) ChIP-seq tracks from ENCODE for two cell types, K562 (purple) and NHLF (pink), are plotted below. H3K27ac ChIP-seq signal from four MSCs (This Study) is plotted. Each track represents one sample, with called peaks as shaded boxes under each H3K27ac track and colored according to peak strength. DNA methylation level for each CpG is plotted as a heatmap. Blue, DMR probe; green, CGI probe. Three regions of interest discussed in the text are highlighted with green (region 1), cyan (region 2), and orange (region 3) shaded boxes. The locations for these regions are indicated in the heatmap with the same color.
(B) In (i): western blot of WT1 in MSCs and CA-MSCs. Actin is the loading control. In (ii): densitometry of western blot results (from a total of 10 independent MSCs and CA-MSCs).
(C) WT1 fold expression change in normal MSCs versus CA-MSCs, cancer-stimulated (CS) MSCs, WT1 OE MSCs, and WT1 siRNA knockdown (KD) MSCs.
(D) Quantification of CA-MSC conversion in CS scrambled control MSCs, WT1 OE MSCs alone, and CS WT1 KD MSCs. The y axis indicates CA-MSC classifier score, CA-MSC threshold (0.96), and MSC threshold (0.3). Means and SEM of 3 independent experiments are represented. *Group versus scrambled control without CS; **Group versus scrambled control with CS.
Interestingly, while Figure 3D showed a decrease in WT1 promoter methylation associated with WT1 overexpression, upon close examination of the entire WT1 region (Figure 5A), we discovered that, at the promoter (differentially methylated region 1, green box), DNA methylation differences were carried by only a subset of the normal MSC samples. However, within the WT1 gene body, there were two more differentially methylated regions, highlighted by cyan (region 2) and orange (region 3) boxes. These two regions were consistently methylated in normal MSCs and unmethylated in CA-MSCs. Each of these differentially methylated regions overlapped with a site with differential accessibility between CA-MSCs and MSCs as determined by ATAC-seq. These ATAC-seq differential peaks were statistically significant, even with the small sample size. In ENCODE histone modification data, both regions had enhancer marks in K562 cells, a malignant leukemia cell line, but not in normal human lung fibroblast (NHLF), a cell type that normal MSCs appear to resemble based on DNA methylation results. We further performed H3K27ac ChIP-seq with additional normal MSCs and CA-MSCs and confirmed the presence of H3K27ac marks at these putative WT1 enhancer elements discovered through differential DNA methylation in CA-MSCs. Hi-C data, DNase I-hypersensitive site (DHS) linkage data for k562 cells (Thurman et al., 2012), and regulatory interactions curated in GeneHancer database (Fishilevich et al., 2017) available through the 3D Genome Browser (Wang et al., 2018) all supported interactions between the two potential enhancers and the promoter region of gene WT1 (Figure S6). These results suggest that hypomethylation at these cryptic enhancers, associated with enhanced accessibility, likely turned this region, normally methylated in normal MSCs into an active enhancer, and greatly promotes WT1 expression. Given the global trend of hypermethylation in CA-MSCs, this specific hypomethylation is even more likely to be a driver event.
To determine whether WT1 is important in the formation of CA-MSCs, we created WT1 overexpressing (OE) normal MSCs via lentiviral transduction. Compared to lenti-GFP control MSCs, WT1 expression increased to levels approximating those of CA-MSCs (Figure 5C). Analysis of the CA-MSC classifier score in WT1 OE MSCs versus control MSCs demonstrated increase in the classifier score from 0.01 (a normal MSC score) to 0.99 (a CA-MSC score). This indicates that WT1 overexpression drives the conversion of a normal MSC to a CA-MSC. We also performed small interfering RNA (siRNA)-mediated knockdown (KD) of WT1 in normal MSCs. WT1 KD MSCs underwent cancer stimulation via hypoxic direct co-culture with ovarian cancer cells, which we previously demonstrated was the most effective in vitro condition to convert a normal MSC to a CA-MSC. CS WT1 KD MSCs, compared to CS control MSCs, demonstrated a 23% decrease in CA-MSC classifier score. We previously demonstrated a strong correlation between increasing classifier score and acquisition of tumor-promoting properties with full CA-MSC conversion occurring when classifier scores reach ≥0.96. Importantly, no WT1 KD MSCs reached the 0.96 threshold for full CA-MSC conversion (Figure 5D).
CA-MSC Expression Pattern Correlates with Altered EZH2 Binding Sites
DNA methylation alterations at enhancer regions can serve as a readout of overall activity of transcription factors, where hypermethylation often indicates an inactive transcription factor binding site (TFBS), and hypomethylation likely marks an active state (Yao et al., 2015). Enrichment analysis for annotated TFBSs identified EZH2 binding sites as the most significantly enriched category with hypermethylation in CA-MSCs (Figure 6A). Concordantly, binding sites of SUZ12, a member of the polycomb repressive complex 2 (PRC2) along with EZH2, were also enriched.
Figure 6. EZH2 and WT1 Impact CA-MSC Reprogramming and Ovarian Cancer Metastasis.
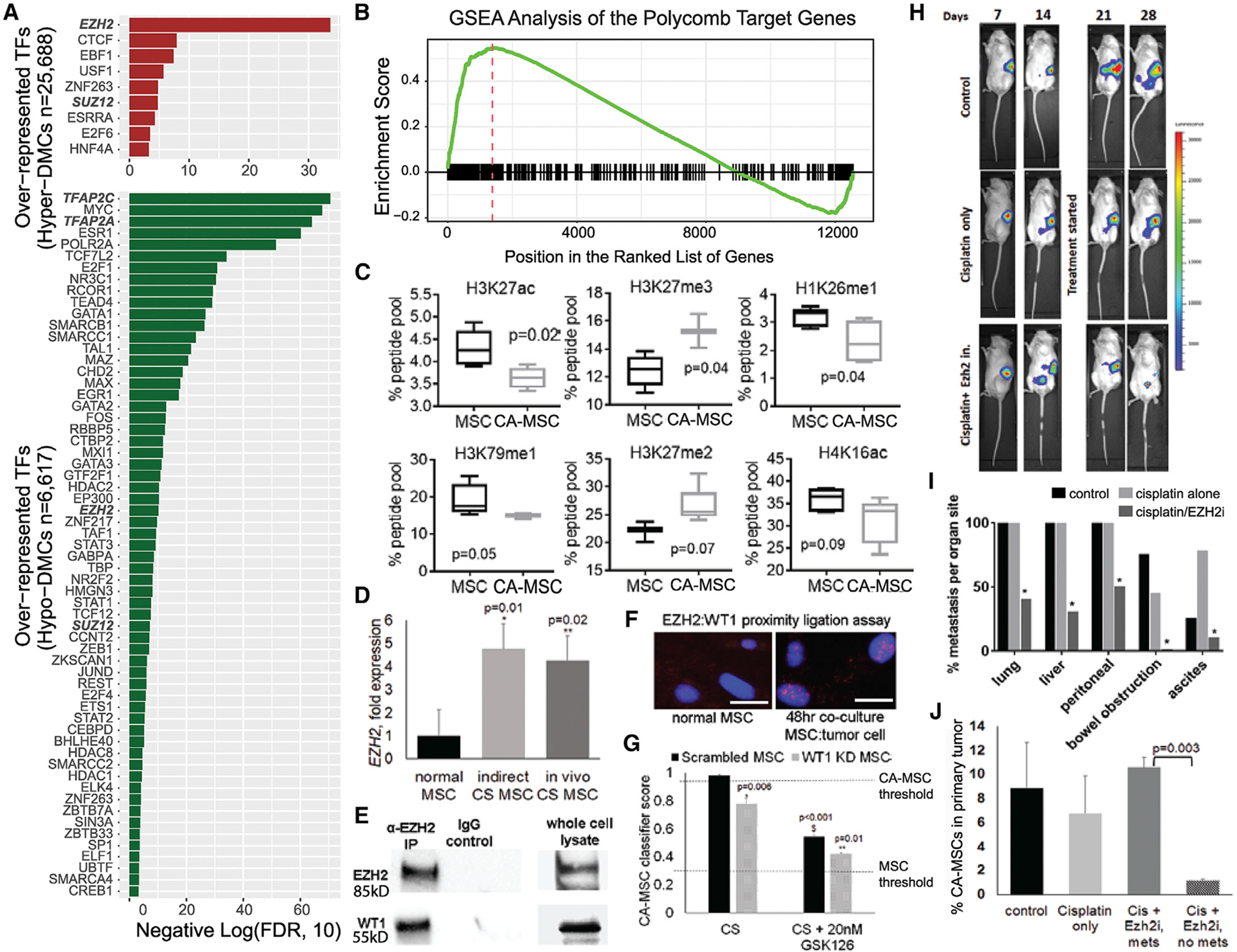
(A) Transcription factor binding site (TFBS) enrichment for distal DMCs hypermethylated (top) and hypomethylated (bottom) in CA-MSCs. The x axis indicates negative log10-transformed FDRs.
(B) GSEA plot indicates enrichment of the Polycomb group (PcG) target genes on both ends of the ranked gene list (ordered by gene fold changes in CA-MSCs versus MSCs).
(C) Boxplots of top histone modifications differentially represented in CA-MSCs versus MSCs identified by mass spectrometry.
(D) EZH2 expression is increased during in vitro and in vivo cancer stimulation with ovarian cancer cells. Fold change compared to normal MSC.
(E) EZH2 directly interacts with WT1. The coIP of EZH2 pulls down WT1.
(F) Proximity ligation assay (PLA) with EZH2 and WT1. Cancer stimulation yields nuclear signal indicating that WT1 and EZH2 are within 40 nm during CA-MSC reprogramming. Scale bars, 20 μm.
(G) WT1 knockdown (KD) with EZH2 inhibition partially blocks CA-MSC formation. Quantification of CA-MSC reprogramming in WT1 KD MSCs or scrambled control after cancer stimulation (CS) ± EZH2 inhibitor GSK126. The y axis indicates CA-MSC classifier score. CA-MSC threshold (0.96) and normal MSC threshold (0.3) depicted. *Comparison to scrambled control; **Comparison to CS scrambled control without GSK126.
(H) IVIS imaging of mice bearing luciferase-expressing cancer cells and CA-MSCs. Luciferase signal was detected within the abdomen in all mice by day 14, indicating development of metastasis.
(I) EZH2 inhibition with GSK126 significantly (p < 0.05) decreased metastatic disease at time of necropsy. Quantification of organs involved with gross metastatic disease at day 31.
(J) Quantification of viable CA-MSCs within the primary ovarian tumor at time of necropsy.
For pooled results, mean and SEM are indicated.
Interestingly, EZH2/SUZ12 binding sites were also enriched in loci with hypomethylation (Figure 6A), indicating a likely redistribution of EZH2/SUZ12 targeting. Consistent with this observation, GSEA (Figure 6B) demonstrated an enrichment in 334 EZH2 targets as marked by 1,241 common H3K27me3 peaks from MSCs derived from four different cell types (Figure S7A) in both up- and downregulated genes in CA-MSCs versus normal MSCs.
CA-MSCs Are Enriched with PRC2-Related Histone Marks
Given the role of EZH2 in histone modifications at H3K27, we investigated global changes in histone modifications between CA-MSCs and normal MSCs. We performed mass spectrometry on the most common 80 histone modifications in CA-MSCs and MSCs. Out of the 80 modifications tested, six histone marks were differentially represented in CA-MSCs versus MSCs (p < 0.1): H3K27ac, H3K27me3, H1K26me1, H3K79me1, H3K27me2, and H4K16ac (Figure 6C). Differential histone modification was verified via western blot of independent MSCs and CA-MSCs (3 MSCs and 5 CA-MSCs not used in the mass spectrometry screen). All histone differences were confirmed with the exception of H3K79me1 (Figures S7B and S7C). H3K27ac, H1K26me1, and H4K16ac are generally associated with transcriptional activation and were decreased in CA-MSCs. In contrast, H3K27me3 and H3K27me2 are associated with transcriptional repression and were increased in CA-MSCs. Three of the top six differential histone modifications are directly related to the PRC2: H3K27me2, H3K27me3, and H3K27ac. This supports a role for EZH2 differential targeting in CA-MSC formation.
Further supporting a role for differential EZH2 targeting in CA-MSCs, during CA-MSC reprogramming, EZH2 RNA expression is increased (Figure 6D). This induction occurs with paracrine tumor signaling during indirect tumor cell:MSC co-culture, conditions that, as we demonstrated earlier, induced DNA methylation changes in three of the five CA-MSC DMRs assessed in Figure 2. This indicates that EZH2 induction happens early in CA-MSC reprogramming and is not dependent on the physical interaction between CA-MSC and tumor cells necessary for the enhancement of metastasis. This induction is also seen with in vivo cancer stimulation in a xenograft mouse model as previously described (Coffman et al., 2019). This implies a role for EZH2 during the initial stages of CA-MSC formation.
EZH2 Inhibition in Combination with WT1 KD Blocks the Formation of a CA-MSC
Given our findings indicating altered EZH2 genomic targeting and known interaction between WT1 and EZH2/PRC2 targeting (Xu et al., 2011), we tested whether EZH2 and WT1 directly interact in CA-MSCs. Using co-immunoprecipitation (coIP) with an anti-EZH2 antibody in CA-MSCs, we demonstrated that WT1 is pulled down with EZH2 (Figure 6E). Further, using a proximity ligation assay (PLA), we demonstrated that WT1 and EZH2 do not interact in normal MSCs; however, during CS MSC reprogramming, WT1/EZH2 PLA signal is present in the nucleus, supporting a direct interaction during CA-MSC formation (Figure 6F). We then tested whether EZH2 inhibition, alone or in combination with WT1 KD, would further decrease the development of a CA-MSC. To verify lack of toxicity, we performed viability studies with an EZH2 inhibitor, GSK126. At doses of 20 nM (Ki = 0.59 nM, half maximal inhibitory concentration [IC50] = 9.9 nM), there was no significant decrease in cell viability of tumor cells or MSCs (Figure S8A). Next, WT1 KD and scrambled control MSCs were treated with 20 nM GSK126 during cancer stimulation. GSK126 treatment of scrambled control MSCs led to a 38% decrease in CA-MSC classifier score. GSK126 treatment of WT1 KD MSCs resulted in a 55% decrease in CA-MSC classifier score (Figure 6G). Importantly, none of the GSK126-treated MSCs reached the threshold of complete reprogramming, as defined as a classifier score ≥0.96. Similar results were seen using a different EZH2 inhibitor, Tazemetostat (Figure S8B). Collectively, this demonstrates that WT1 and EZH2 jointly mediate the formation of a CA-MSC.
EZH2 Inhibition Limits Ovarian Cancer Metastasis
We next tested whether EZH2 inhibition impacted ovarian cancer metastasis. Using the ovarian bursal orthotopic mouse model discussed earlier, OVCAR3 tumor cells and CA-MSCs were injected into the right ovarian bursa. A subset of mice bearing luciferase-expressing OVCAR3 cells underwent serial In Vivo Imaging System (IVIS) imaging to monitor tumor growth. By 7 days, localized tumor engraftment was noted, and by 14 days, intraperitoneal metastasis developed (Figure 6H). After 14 days, 10 mice were treated with cisplatin alone and 10 mice were treated with cisplatin plus GSK126, starting after the first dose of cisplatin and continued as maintenance for 3 weeks. Five additional mice were left untreated as a control. IVIS imaging demonstrated decreased luciferase signal in the primary tumors of cisplatin-treated mice versus control and decreased metastatic signal in the cisplatin + GSK126-treated mice. All mice were sacrificed on day 31 when the control group met endpoint criteria of 10% weight loss. At the time of necropsy, both the control and cisplatin-alone groups demonstrated 100% rates of gross lung, liver, and intra-peritoneal metastasis and 40%–75% rates of bowel obstruction. In contrast, mice with cisplatin and GSK126 treatment demonstrated 40% lung, 30% liver, and 50% intra-peritoneal metastasis, with no bowel obstructions, consistent with significant decrease in metastasis to all organs examined (Figure 6I). The primary tumor volume in the control group was 315 mm3 versus 97 mm3 in the cisplatin-only group and 80 mm3 in the cisplatin + GSK126 group, consistent with a significant reduction in primary tumor size with cisplatin treatment (p < 0.001) without a significant change in size with GSK126 addition (p = 0.58). This is consistent with our in vitro data demonstrating that EZH2 inhibition does not impact OVCAR3 cancer cell survival or proliferation and provides strong evidence that EZH2 inhibition targets CA-MSC-specific promotion of metastasis. We also assessed the presence of viable human CA-MSCs within the primary ovary tumor. Primary ovary tumors from the control and cisplatin-treated mice contained 9% and 7% viable CA-MSCs, respectively. In the cisplatin + GSK126 mice that developed metastasis, the primary tumor had 10.5% viable CA-MSCs compared to 1.2% viable CA-MSCs in the primary tumors of mice without metastasis (p = 0.003) (Figure 6J), demonstrating a strong correlation between viable CA-MSCs and the ability to form metastasis. This supports a potential therapeutic role for EZH2 inhibitors in treating and/or limiting CA-MSC-mediated ovarian cancer metastasis.
As ovarian cancer demonstrates both transcoelomic and hematogenous metastasis, we also used a previously described tail vein injection model of ovarian cancer, which yields robust intra-abdominal metastasis (Coffman et al., 2016a), to test the effect of EZH2 inhibition on hematogenous metastasis. Similarly, GSK126 decreased intra-abdominal metastasis by 50%–90%, adding additional evidence for a potential therapeutic role of EZH2 inhibitors in limiting ovarian cancer metastasis (Figures S8C and S8D).
Ovarian Cancer Stroma Is Marked by WT1 Expression and Stromal WT1 Loss Is Associated with Treatment Response
We next explored whether the CA-MSC phenotype correlated with disease response in patient tissue. As CA-MSCs are marked by high WT1 expression, we investigated whether WT1 overexpression could serve as a marker of cancer-supportive stroma (associated with a CA-MSC phenotype) versus normalized stroma (associated with a normal MSC phenotype). Our findings indicated that CA-MSCs derived from tissue that achieved a complete pathologic response to neoadjuvant chemotherapy undergo phenotypic normalization and resemble normal MSCs with low WT1 expression. We therefore investigated tissue obtained from interval debulking surgeries of patients treated with neoadjuvant platinum-based chemotherapy. We identified 5 patients with residual omental disease and 7 patients without residual omental disease (all HGSC histology, stage III/IV disease, with initial radiographic omental involvement). Sections were stained for WT1 and stromal WT1 levels were quantified by an H score (Figures 7A and 7B). WT1 was clearly present in stromal cells associated with residual disease (average H score = 154) but lost in tissue which achieved a pathologic complete response (average H score = 31; p < 0.0001). This indicates that the CA-MSC phenotype drives the tumor stromal microenvironment characterized by retained WT1 overexpression and also indicates that normalization of the CA-MSC phenotype is possible and associated with treatment response (Figure 7).
Figure 7. WT1 Stromal Expression Correlates with Ovarian Cancer Disease Status, and CA-MSC Normalization Is Associated with Improvement in Patient Outcomes.
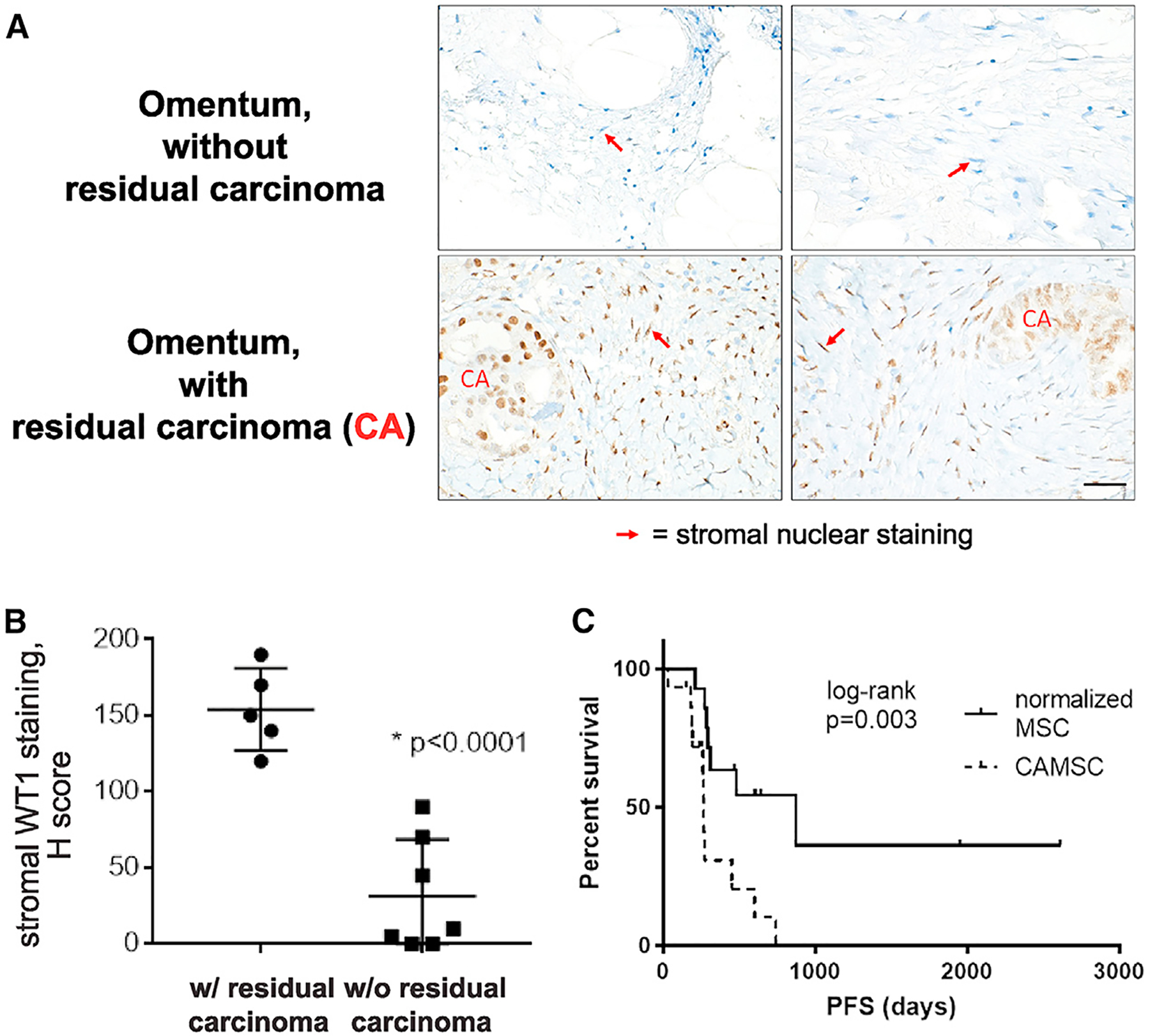
(A) WT1 immunohistochemistry of HGSC patient omentum after neoadjuvant therapy ± residual carcinoma. Stromal nuclei are indicated by red arrows. Scale bar, 20 μm.
(B) Quantification of WT1 stromal staining (H score) in tissue with residual carcinoma (n = 5) and without residual carcinoma (n = 7).
(C) Kaplan-Meier plot of progression-free survival (PFS) in women with normalized MSCs versus women with persistent CA-MSCs after neoadjuvant therapy.
Fan et al., 2020, Cell Reports 33, 108473
Normalized Epigenetic Profile of MSCs Correlates with Improved Clinical Outcome
To further determine the clinical significance of the CA-MSC phenotype, we studied MSCs from a cohort of patients (n = 29) treated with neoadjuvant therapy. Patients were treated with platinum-based chemotherapy or with metformin as part of a phase-II clinical trial (ClinicalTrials.gov: NCT01579812) (Brown et al., 2020). All patients had stage III/IV disease, and all patients had viable tumor at the time of surgical debulking. MSCs were isolated from resected tissue at time of interval surgical debulking. MSCs from 15/29 (52%) patients retained the CA-MSC phenotype, while MSCs from 14/29 (48%) had a normal-like MSC phenotype (with a normal MSC DNA methylation profile and normal classifier score). Despite small numbers, the group with normalized MSCs had significantly improved progression-free survival (PFS). Mean PFS in the CA-MSC group was 266 days versus 870 days in the normalized MSC group (log rank, p = 0.003) (Figure 7C). Distribution of age, stage, and treatment type were not different (p = 0.5 for age, and p = 0.6 for stage and treatment type) between the two groups, and in a Cox proportional hazard model adjusting for treatment type, a normalized MSC phenotype remained as a significant protective factor (p = 0.005; hazard ratio [HR] = 0.23). Overall, this indicates that the CA-MSC phenotype is clinically important, is potentially reversible, and may present a novel therapeutic avenue in ovarian cancer.
DISCUSSION
Since described about a decade ago (Polyak and Weinberg, 2009; Klymkowsky and Savagner, 2009; Thiery et al., 2009; Yilmaz and Christofori, 2009; Barrallo-Gimeno and Nieto, 2005), partial transition to a mesenchymal phenotype in the epithelial compartment (i.e., EMT) in carcinomas has been well established as a hallmark of cancer with increased metastatic potential (Hanahan and Weinberg, 2011). Here, we describe alterations in the other direction—MET—in stromal progenitors of the TME that also contribute to cancer metastasis.
As stromal progenitor cells, CA-MSCs influence the formation of the TME. We demonstrate that CA-MSCs have a unique epigenetic landscape different from that of both tumor cells and normal MSCs, characterized by DNA hypermethylation, altered chromatin accessibility, and gain of repressive histone marks. The majority of differential DNA methylation occurs in enhancer regions. It has been previously shown that enhancers are the most dynamically utilized compartment (Sur and Taipale, 2016; Calo and Wysocka, 2013; Shlyueva et al., 2014) and closely relate to cellular identity. It has become increasingly clear that DNA methylation patterns are good markers for enhancer activity (Thomassin et al., 2001; Lister et al., 2009; Stadler et al., 2011; Yao et al., 2015; Blattler and Farnham, 2013). A recent publication (Corces et al., 2018) showed that DNA methylation tracks results from ATAC-seq for distal enhancers. As an example, DNA methylation alteration at the WT1 loci helped to identify potential enhancers that are also marked by H3K27ac and ATAC-seq accessibility and/or curated as enhancers by GeneHancer (Fishilevich et al., 2017). Interestingly, at loci where CA-MSCs and MSCs differed, the DNA methylation profile of CA-MSCs resembles that of benign FTE and sorted carcinomas, while normal MSCs more closely resemble normal fibroblasts. While remaining mesenchymal in lineage, a partial MET appears to occur during CA-MSC formation, which facilitates direct binding to cancer cells, resulting in co-metastasis. This process of cancer cell and stromal cell co-metastasis is akin to the cancer “seed” traveling with its “soil” to enhance survival during metastasis and aid in distant colonization where the CA-MSC, as a stromal progenitor cell, may play a critical role in establishing the metastatic TME. This presents a new model of ovarian cancer metastasis where ovarian cancer cells reprogram normal MSCs into CA-MSCs, inducing a partial MET that enables tumor cell:CA-MSC binding, co-metastasis, and establishment of new sites of disease with the supportive stromal TME already formed.
While stromal MET has not been previously identified in cancer, MSC MET has been reported in benign conditions. De-differentiation of mesenchymal cells such as fibroblasts into induced pluripotent stem cells (iPSCs) requires MET (Li et al., 2010; Shu and Pei, 2014). During embryogenesis, stem cells often transition from EMT to MET stages, and MET in stem cell reprogramming is associated with epigenetic changes, consistent with our findings (Wu et al., 2016). Also, normal epithelial or mesothelial progenitor cells that line the female reproductive tract may contribute to CA-MSC formation through acquiring mesenchymal features and increased differentiation capacity. We are actively investigating this possibility.
WT1, one of the most significantly upregulated genes in CA-MSCs, is an important mediator of MET during development and thought to be important in maintaining the potential to transition between EMT and MET (Miller-Hodges and Hohenstein, 2012). Interestingly, WT1 impacts epigenetic regulation through directly binding the polycomb protein EZH2 and the DNA methyltransferases DNMT1 and DNMT3A (Xu et al., 2011; Szemes et al., 2013). Given the differential histone modifications (particularly related to PRC2 targets including H3K27me3) and DNA methylation changes in CA-MSCs, WT1 may be an important mediator of these epigenetic modifications. Indeed, PLA and coIP indicate that a WT1/EZH2 interaction in CA-MSCs and KD of WT1 in combination with EZH2 inhibition during cancer stimulation blocked the formation of a CA-MSC. Pharmacologic inhibition of EZH2, while not impacting ovarian cancer cell proliferation or survival, significantly decreased CA-MSC-mediated ovarian cancer metastasis in an orthotopic mouse model. Importantly, while mice had documented intra-abdominal metastasis at the time of treatment initiation, only half of the mice had residual metastatic disease after cisplatin and EZH2 treatment. This indicates that EZH2 inhibition may play a role in limiting and/or treating already established ovarian cancer metastasis as is found at the time of clinical presentation in over 70% of ovarian cancer patients.
Dichotomous roles for MSCs in cancer growth have been reported with both tumor-promoting and tumor-suppressive functions (Klymenko and Nephew, 2018). Our work implies that the CA-MSC methylation profile drives the clinical impact of MSCs in ovarian cancer with a CA-MSC profile supporting cancer growth and a normal MSC profile restricting cancer growth. This is exemplified by a 20-month increase in PFS in women with normalized MSCs versus those with CA-MSCs. Ultimately, understanding the mechanistic steps leading to CA-MSC development and the ability to reverse, replace, or create MSCs resistant to reprogramming is essential to effective therapeutic targeting of the TME and represents a novel and powerful approach to ovarian cancer treatment.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lan Coffman (coffmanl@upmc.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The DNA methylation EPIC array, ATAC-sequencing and ChIP-sequencing datasets generated in this study is available at NCBI without restriction. The accession number for the EPIC array is GSE138072, for the ATAC-seq data is SRP289005, and for the ChIP-seq data is SRP289183.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cancer cell lines
Ovarian cancer cell lines CAOV3 and OVCAR3 were purchased through ATCC. OVSAHO and the primary patient cell line, PT412, were kind gifts from Dr. R. Buckanovich. All cells were grown in DMEM (Dulbecco’s Modified Eagle Medium), 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin. Cells were incubate at 37°C in a humidified incubator equilibrated with 5% CO2. All cell lines were tested and verified negative for mycoplasma (last test 9/2019).
Human primary cell lines
Cancer associated mesenchymal stem cells (CA-MSCs) were derived from surgical resection of human ovarian cancer involving the fallopian tube, ovary and/or omental metastatic deposits. Normal mesenchymal stem cells (MSCs) were derived from surgical samples (fallopian tube or omentum) of women undergoing surgery for benign indications or risk reduction surgery. Patient’s samples were obtained in accordance with protocols approved by the University of Michigan’s IRB (HUM0009149) and University of Pittsburgh’s IRB (PRO17080326). CA-MSCs and MSCs were grown in MEBM (Mammary Epithelial cell Basal Medium) supplemented with: 1% B27, 20ng/ml epidermal growth factor (EGF), 1% penicillin-streptomycin, 20ug/ml gentamycin, 1 ng/ml hydrocortisone, 5ug/ml insulin, 100um beta-mercaptoethanol, 10ng/ml recombinant human basic- fibroblast growth factor (bFGF), and 10% fetal bovine serum (FBS). Cells were incubate at 37°C in a humidified incubator equilibrated with 5% CO2.
Mice
All experimental procedures were performed in accordance with protocol approved by the Institutional Animal Care and Use Committee at University of Pittsburgh. Female NSG (NOD scid gamma) mice (6–8 week-old) were used. All mice were group housed and kept under 12h:12h L/D conditions. Food and water were available ad libitum to all mice 24 hour period.
METHOD DETAILS
Tissue harvesting
Patients samples were obtained in accordance with protocols approved by the University of Michigan’s IRB (HUM0009149) and University of Pittsburgh’s IRB (PRO17080326). Tissue was processed for DNA, RNA and protein isolation as previously described (Coffman et al., 2016b, 2019). MSCs were isolated as previously described (McLean et al., 2011). Briefly, CA-MSCs were derived from surgical resection of human ovarian cancer involving the FT, ovary and/or omental metastatic deposits. Normal FT and omental MSCs were derived from surgical samples of women undergoing surgery for benign indications or risk reduction surgery. Cells were plated in supplemented MEBM, MSCs were selected for plastic adherence and cell surface marker expression (CD105, CD90, CD73 positive; CD45, CD34, CD14, CD19 negative). Adipocyte, osteocyte, and chondrocyte differentiation capacity was verified (following guidelines presented by the ISCT on the minimal criteria for defining multipotent mesenchymal stem cells (Dominici et al., 2006)) (Figures S8C and S8D). To ensure lack of fibroblast contamination, cells were screened for fibroblast markers fibroblast surface protein (FSP) via flow cytometry and alpha smooth muscle actin via immunohistochemistry (Figures S8C and S8D). Any cell line with a population > 5% differing from above criteria were FACs purified. Fibroblast differentiation was performed as previously described by growing MSCs with connective tissue growth factor (100 ng/ml) and ascorbic acid (50 μg/ml) for 2 weeks (Coffman et al., 2019). MSCs were maintained in culture as previously described and used at passage 5 or below (McLean et al., 2011). All cell lines were tested and verified negative for mycoplasma (last test 9/2019).
DNA methylation array
DNA was isolated from CA-MSCs derived from omental metastasis in 6 patients with high grade serous ovarian cancer (HGSC), 1 patient with ovarian carcinosarcoma, 1 patient with high grade serous carcinoma involving the FT, and 3 patients with high grade serous carcinoma involving the ovary. Samples from 4 patients with omental with initial radiographic involvement of HGSC treated with neoadjuvant chemotherapy and pathologic complete response in the omentum, 5 benign patient FT samples, 1 set of bilateral FTs from a BRCA2 germline patient, 4 benign patient normal omental MSCs and 2 adipose MSCs purchased through ATCC were also included. All normal MSCs were derived from patients undergoing surgery for benign indications. “A” and “B” indicate samples from the same patient from 2 distinct anatomic locations. A list of all samples with anatomic and histologic information is provided in Table S1. All MSCs used for DNA methylation analysis were at passage 5 or below. DNA underwent bisulphite conversion followed by analysis on the Illumina MethylationEPIC Beadchip microarray through the Genomics core at the University of Michigan and Van Andel Research Institute.
Assay for Transposase Accessible Chromatin with sequencing (ATAC-seq)
Nuclei were isolated from 4 CA-MSCs derived from omental metastasis of patients with high grade serous ovarian cancer, 2 normal omental MSCs and 2 adipose MSCs purchased from ATCC. All MSCs used for ATACseq analysis were at passage 5 or below. ATAC-seq was performed through Active Motif.
Histone mass spectrometry
CA-MSCs and MSCs used for ATAC-seq were also used for mass spectrometry to measure the relative abundance of 80 histone modifications through Active Motif. Histones were extracted from frozen cell pellets and digested with trypsin. Samples were analyzed on a triple quadrupole (QdQ) mass spectrometer directly coupled with an UltiMate 3000 Dionex nan-liquid chromatography system. Three separate mass spec runs for each sample. Each modified and unmodified form of the monitored amino acid residue was quantified as the percentage of the total pool of modifications with mean and standard deviation reported.
Quantitative real-time PCR
RNA was isolated with the RNeasy Mini Kit (QIAGEN, Hilden, Germany) and on-column DNase treatment (QIAGEN, Hilden, Germany). RNA concentration was determined with NanoDrop ND-1000 Spectrophotometer. cDNA was synthesized with the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, Grand Island, NY) as previously described [21]. SYBR green- based RT-PCR was performed using the 7900HT Sequence Detection System (Applied Biosystems, Foster City, California) and respective primers. The comparative Ct method was used for data analysis with GAPDH as the comparator gene.
Immunoblotting
Cell pellets were homogenized in RIPA buffer (Pierce, Rockford, IL) with complete protease inhibitor (Roche, Basel, Switzerland). Insoluble material was removed by centrifugation at 16,000 g at 4oC for 15mins. Protein concentrations were determined using the Bradford Protein Assay Kit (Bio-Rad, Hercules,CA). Equal amounts of protein were separated on 4%–12% NuPAGE SDS gel (Invitrogen, Grand Island, NY) and transferred onto a PVDF membrane. Anti-H3K27me3, anti-H3K27me2, anti-H4K16ac, anti-H3K27ac, anti-H3K79me1, anti-H3, anti-H4, anti-WT1 and anti-EZH2 (1:1000 dilution, Active Motif), anti-PAX8 (1:1000, Abcam) and anti-B-actin (1:10,000 dilution, Sigma-Aldrich, St. Louis, MO) were used. Bands were visualized using the ECL Kit (Pierce, Rockford, IL).
Quantitative Methylation Specific PCR (qMSP)
Bisulfite Conversion and DNA Yield
Bisulfite conversion of extracted cellular or tissue DNA was performed using the DNA Methylation-Lightning Kit (Zymo Research) according to manufacturer’s instructions and eluted into 40 μl DNA Elution buffer (Zymo Research). Post-bisulfite treatment DNA yields were quantified by MethyLight assay for the methylation-independent bisulfite converted (BSC) consensus ALU sequence, as previously described (Weisenberger et al., 2005), using primer and probe sequences: forward primer, 5′- GGT TAG GTA TAG TGG TTT ATA TTT GTA ATT TTA GTA -3′; reverse primer, 5′- ATT AAC TAA ACT AAT CTT AAA CTC CTA ACC TCA 3′, spanning a 98-bp locus, and 100nM probe, 5′- \56-FAM\ CCT ACC TTA ACC TCC C -3′ with Minor Groove Binder (MGB; ThermoFisher Scientific). PCR was performed using 10X Master Mix to yield a final volume of 25 μl and final working concentrations of 16.6mM (NH4)2SO4, 67mM Tris pH 8.8, 6.7mM MgCl2 10mM β-mercaptoethanol, 200μM of each deoxynucleotide triphosphate (dNTP) and 0.04 U/μl of Platinum Taq polymerase (ThermoFisher Scientific). Cycling conditions were 95°C for 5 minutes, followed by 50 cycles of (95°C for 5 s, 60°C for 30 s and 72°C for 30 s). Standards for quantification were created by serial dilution of pre-quantified BSC human male DNA (Promega). Amplification reactions were performed in duplicate using 96 well-plates using a CFX96 Touch Real-time PCR Detection System (Bio-Rad).
Locus-specific methylation analysis
qMSP assays (Lo et al., 1999) were developed and analytically-validated for five select loci: SASH1, c13orf45, ELN, GATA6, and MARVELD2, for which primer sequences and assay specifications are detailed in Table S1. A standard curve was created using serial dilutions of BSC Epitect unmethylated control DNA (QIAGEN) mixed with BSC CpG-Methylated HeLa Genomic DNA (New England BioLabs) (Lo et al., 1999).
Data Analysis
MSP results were analyzed using the CFX Manager v3.1 (Bio-Rad) using regression to obtain the mean quantification cycle (Cq) for each respective sample. Percent methylation was calculated for each sample using the BSC-ALU and BSC Control DNA standard curves, according to the formula % methylation = (# locus copies methylated in sample)/ (# ALU-calculated genomic copies in sample).
CA-MSC encapsulation
CA-MSCs were encapsulated into 3% alginate as previously described (Schmitt et al., 2015). Briefly, 3% w/v sodium alginate was dissolved in PBS, filtered for sterility with a 0.2um filter. Cells were mixed with the alginate solution and added dropwise into a 5mM solution of calcium chloride to allow for gelation. Alginate beads were washed 2x with PBS prior to use in assays. Cell viability over time was verified by transection of the alginate capsule after 2 weeks incubation in standard culture media (DMEM, 10% FBS, 1% pen/strep) and re-isolation and growth of previously encapsulated CA-MSCs. Also, CA-MSCs lentivirally transduced with a secreted luciferase construct were encapsulated in alginate and the media was sampled for secreted luciferase overtime using the Ready-to-glow Secreted luciferase reporter system (Takara).
Sphere assays
CA-MSCs transduced with lentiviral mT construct were mixed with lentiviral labeled GFP tumor cells (CAOV3, OVSAHO, OVCAR3) (as previously described (Coffman et al., 2019)) in a 1:1 ratio in ultra-low attachment 6 well plates grown in serum free supplemented MEBM as previously described (Coffman et al., 2019). Alternatively, CA-MSCs were added to the top of a 0.4um transwell system and tumor cells seeded in the bottom to prevent direct contact. After 5 days, the number of GFP+ spheres were counted. Experiments were repeated independently 3 times per tumor cell type.
Migration/Invasion assays
8um transwells were coated with growth factor reduced Matrigel and allowed to solidify. GFP-tumor cells (CAOV3, OVSAHO, OVCAR3) with or without mT-CA-MSCs were added to the top of the transwell. Supplemented DMEM was added to the bottom of the transwell and cell migration/invasion through the coated membrane was quantified after 24hrs via fluorescent microscopy. Alginate encapsulated CA-MSCs were added in place of control CA-MSCs to determine the impact of preventing CA-MSC:tumor cell direct interaction. Experiments were repeated independently 3 times per tumor cell type.
Adhesion assay
5×10^4 MSCs or CA-MSCs were cultured overnight in 12 well-plate to form a mono-layer. 5×10^4 fluorescently labeled tumor cells (OVSAHO, OVCAR3 or pt412) were then added. After 30 minutes, cells were washed twice with PBS and the attached tumor cells were counted using a fluorescence microscope and quantified as the number of adherent cells per low power field (10x). Experiments were repeated independently 3 times per tumor cell type.
Cancer stimulation, cancer cell:MSC co-culture
MSCs were grown in a 1:1 ratio with ovarian tumor cells allowing for direct interaction or with use of a transwell system(0.4um) to separate the two cell types to allow only indirect co-culture as previously described (Coffman et al., 2019). Cells were grown in 1:1 mixture of CA-MSC media (supplemented MEBM) and tumor cell media (supplemented DMEM) for 5 days under hypoxic conditions (1% O2). Cells were isolated via FACs separation with fluorochrome labeled MSCs or tumor cells or removed from their respective transwell chamber.
CA-MSC classifier
As previously described (Coffman et al., 2019), we created a regression model based on the expression of 6 genes which accurately distinguishes normal MSCs from CA-MSCs: Annexin A8-like protein 2 (ANXA8L2), Collagen Type XV Alpha 1 Chain (COL15A1), Cytokine Receptor Like Factor 1 (CRLF1), GATA Binding Protein 4 (GATA4), Iroquois Homeobox 2 (IRX2), and TGF-β2. Expression values are the delta CT values compared to GAPDH levels. The regression equation is:
Ascites complex isolation and flow cytometric analysis
Cellular complexes were isolated from ovarian cancer patients’ ascites as follows: Bulk ascites was obtained within 2 hours of patient paracentesis and large debris was removed via passage through a 1000um pluriStrainer (pluriSelect). Red blood cells were lysed using ACK lysis buffer (ThermoFisher Scientific). Cells were treated with DNase I to a final concentration of 0.1mg/ml with 15min incubation. Cells then underwent serial centrifugation at 400 g for 5 mins and washing with 2% FBS in PBS with visual inspection of cells after each spin. Washing and centrifugation was continued until only cellular complexes, with minimal single cells, were visualized. Cell complexes were used for down-stream applications. For flow cytometric analysis, the isolated complexes were enzymatically dissociated into a single cell suspension with trypsin (0.25%, 5–10min incubation). Cells were then stained with anti-CD90, anti-CD73, anti-CD105 (Stem-cell Technology), and anti-EpCAM (Novus) antibodies for 20 minutes at RT. The percentage of CA-MSC (CD90, 73, and 105+) and tumor cells (EpCAM+) were calculated on live cells gating.
Immunofluorescence staining
Cellular complexes were isolated from ovarian cancer patients’ ascites as above and immobilized on slides using the cytospin centrifuge as previously described (Koh, 2013). Then, the cellular complexes were fixed using methanol for 10 minutes. Immunofluorescence conjugated antibodies (anti-CD90, anti-CD73, anti-CD105 (Stem-cell Technology), and anti-EpCAM (Novus)) were added after permeabilize the cellular complexes with 0.1% Triton 100X. The stained cellular complexes were examined using Nikon A1 confocal microscope.
Orthotopic ovarian cancer mouse model
5×10^5 OVCAR3 cells were injected alone (n = 5) or with control CA-MSCs (5×10^5 cells) (n = 5) or encapsulated CA-MSCs (5×10^5 cells) (n = 5) into the ovarian bursa of NSG mice. Mouse weight and health were monitored over time and mice were sacrificed when the first group of mice met endpoint criteria of > 10% weight loss and necropsy was performed.
For experiments assessing the impact of EZH2i on metastasis, OVCAR3 and CA-MSCs were injected into the ovarian bursa as above (n = 10 mice per group in cisplatin treated arms, n = 5 mice in the control arm). Two mice per group contained OVCAR3 cells expressing luciferase. These mice underwent weekly IVIS imaging as previously described (Coffman et al., 2016b). On day 14, mice were treated with cisplatin (0.5mg/kg every other day × 3 doses) or vehicle control. Mice in the dual treatment arm started treatment on day 15 with GSK126 150mg/kg 3x weekly ongoing or vehicle control. Mice were monitored daily and all mice were sacrificed when one group met endpoint criteria of 10% weight loss.
Tail vein injection model
5×10^5 OVCAR3 cells were injected into NSG mice (n = 10 per group). Mice in the EZH2 inhibition group were treated with 300mg/kg of GSK126 (selleckchem) 3 × weekly for 1 week prior to injection and then 150mg/kg 3 × weekly for 2 weeks immediately after injection. Mice in the control group underwent parallel treatment with vehicle control and were treated with 150mg/kg 3x weekly for 2 weeks starting 2 weeks after treatment. Mice were euthanized at 4 weeks post tumor cell injection and necropsy performed. Quantitative human specific PCR was performed on whole lung isolates as previously described (Alcoser et al., 2011).
Chromatin Immunoprecipitatio
Normal and CA-MSC cells were fixed in buffer containing 1% formaldehyde, 50 mM HEPES-KOH pH 7.6, 100 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0) for 10 min at room temperature with shaking before being quenched with 125 mM glycine 5 min at room temperature. Cells were collected by scraping in cold PBS, washed 2X with cold PBS, flash frozen in liquid N2, and stored at −80°C until use. Cells were lysed in LB1 (50 mM HEPES-KOH pH 7.6, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100, Protease inhibitor cocktail (Roche cOmplete Mini tablets, EDTA-free)) for 20 min with rotation at 4°C and cleared by centrifugation at 300 × g for 5 min at 4°C. Supernatant with intact nuclei was set aside. Cell pellets were lysed again in 4x LB1 (LB1 with 2% NP-40 and 1% Triton X-100) for 20 min. Intact nuclei from this and the saved supernatant were collected by centrifugation at 1,700 × g for 5 min at 4°C. Nuclei were resuspended and washed in LB2 (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl, protease cocktail inhibitor) for 10 min with rotation at 4°C. Nuclei were collected by centrifugation at 1,700 × g for 5 min at 4°C and gently rinsed 2x with LB3 (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 0.01% NP-40, protease cocktail inhibitor) while avoiding disturbing pellet. Nuclei were resuspended in 1 mL LB3 and transferred to a 1 mL milliTUBE (Covaris). Chromatin was sheared to a range of 300–600 base-pair fragments using a Covaris E220 evolution Focused ultrasonicator with the following parameters: Peak power (140.0), Duty Factor (5.0), Cycles/Burst (200), Duration (600 s), Temperature (4°C). Sheared chromatin was quantified by Bradford Assay, and 450 mg of chromatin was brought to 500 mL in LB3 and then an additional 500 mL of ChIP Cocktail Mix (40 mM Tris-HCl pH 7.6, 150 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.5% NP-40, Protease inhibitor cocktail) was added. Prepared chromatin was pre-cleared by incubation with 20 mL of pre-washed Dynabeads Protein G magnetic beads (Invitrogen #10004D) for 3 hours at 4°C with rotation. After bead removal, 10% input (100 ml) of pre-cleared chromatin was removed and set aside. Pre-cleared chromatin was immunoprecipitated with 5 mL of H3K27me3 antibody (Cell Signaling #9733) or H3K27ac antibody (Active Motif #39133) overnight at 4°C with constant rotation. Protein G magnetic beads (35 ml/IP) were blocked in buffer containing PBS, 0.5% BSA, and 20 mg Herring Sperm DNA (Sigma #D7290) with rotation at 4°C overnight. Blocked beads were washed 3X with PBS and 0.5% BSA, then 2X with WB1 (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 5 mM EDTA pH 8.0, 0.5% NP-40, 1% Triton X-100). Immuno-chromatin complexes were incubated with blocked beads for 3 hours with rotation at 4°C. Beads were then washed 3X for 5 min with rotation at 4°C with WB1, 3X with WB2 (50 mM Tris-HCl pH 7.6, 500 mM NaCl, 5 mM EDTA pH 8.0, 0.5% NP-40, 1% Triton X-100), 2X with WB1, and 1X with Low Salt TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 50 mM NaCl). Beads were incubated in 50 mL of Elution Buffer (10 mM Tris-HCl pH 8.0, 10 mM EDTA, 150 mM NaCl, 5 mM DTT, 1% SDS) at 65°C for 15 min in 50 mL volume. The elution step was repeated, and eluates combined. Eluents and input were incubated overnight at 65°C with constant shaking to reverse crosslinks, followed by incubation at 37°C for 1 hour with DNase-free RNase A, then incubation at 37°C for 2 hours with 10 mL of Proteinase K (20 mg/ml stock). DNA was isolated with a 1.5× ratio of KAPA Pure Beads (KAPA Biosystems #KK8000) to DNA volume. After quantification with a Qubit dsDNA High Sensitivity Assay kit (ThermoFisher Scientific #Q32851), library preparation was done using a KAPA HyperPrep Kit (Kappa Biosystems #07962347001).
Transcriptome data re-processing
Raw mRNA sequencing data (FASTQ format) of 4 normal omental MSCs and 10 ovarian CA-MSCs, using Illumina TruSeq RNA Sample Preparation V2 kit (Illumina San Diego, CA, USA) and Illumina HiSeq2000 instrument 100 bp PE sequencing, were download through NCBI’s Gene Expression Omnibus (GEO) with GEO Series accession number GSE118624 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE118624) (Coffman et al., 2019). Each raw sequencing file was aligned to the human reference genome (GRCh37) using STAR version 2.7 (Dobin et al., 2013), with default settings. Estimation of gene-level abundance was carried out using RSEM version 1.3.1 (Li and Dewey, 2011). Raw reads count from RSEM were further normalized using R package edgeR (function cpm), and log-transformed for downstream analysis, with one CA-MSC sample (GSM3335697) removed due to potential tumor cell contamination.
DNA methylation data processing and QC
Raw IDATs files were processed using R package SeSAMe (Zhou et al., 2018) with noob background correction, non-linear dye bias correction, and non-detection masking (any data point not significantly different from background was replaced with NA). Probes with design issues (Zhou et al., 2017) were also masked. DNA methylation beta values, ranging from 0 to 1 (with “0” indicating fully unmethylated and “1” fully methylated), were calculated as quantitative percentage of methylated signals over both methylated and unmethylated signals. All IDATs and processed beta values have been uploaded and made available through Gene Expression Omnibus (GEO) under the accession number GSE138072.
SNP probes (‘rs’ probes) were used to examine potential sample swaps which can occur in genomic studies. No such swap was identified. DNA Methylation beta values for three MIR141/200C promoter probes (“cg12161331,” “cg18185189,” “cg19794481”) were examined to track mesenchymal content within each sample. MIR141/200C is considered to be a master regulator for epithelial/mesenchymal phenotype transition, and this process is controlled by its promoter methylation state (Vrba et al., 2010). Methylation level at these three probes used highly correlated with mesenchymal content in flow sorting results (George et al., 2019). Two samples had a failure rate of > 40% with our stringent background masking (Zhou et al., 2018) likely due to DNA degradation, but were kept in the study, using data points that passed the stringent filter.
ATAC sequencing (ATAC-seq) data processing
Quality control was carried out using the PEPATAC pipeline adopted by the TCGA ATAC-seq project (Corces et al., 2018). We followed current ENCODE QC standards and excluded four samples due to insufficient transcription start site (TSS) enrichment value (less than 6), therefore were removed from further analysis. Next, raw sequencing reads were mapped to the human reference genome (GRCh37) using Burrows-Wheeler Alignment tool (BWA) with default settings (Li and Durbin, 2009), followed by peak calling with software MACS2 (Zhang et al., 2008). Specifically, parameters of-nomodel and -f BAMPE were specified in calling open chromatin regions, for pileup of the whole fragments. Sample-based open peaks were then merged across all the samples, and fed into R package DESeq2 (Love et al., 2014) to generate a normalized reads count matrix against effective library sizes.
ChIP sequencing (ChIP-seq) data processing
Raw sequencing reads for histone marks H3K27me3 and H3K27ac were mapped to the human reference genome (GRCh37) using Burrows-Wheeler Alignment tool (BWA) with default settings (Li and Durbin, 2009). Reads for samples with multiple runs were merged ahead of mapping. Normalized signal within 10 bp bins across the genome was calculated using deeptools2 (Ramírez et al., 2016), with parameters “—normalizeUsing RPGC.” RPGC (per bin) equals the number of reads per bin divides by scaling factor for 1x average coverage. A scaling factor was computed with R package csaw (Lun and Smyth, 2016), to generate effective library size. Peak calling procedure was carried out using MACS2 (Zhang et al., 2008) with default settings.
Public available data resources
DNA methylation data (Illumina HM450 array) of ovarian stroma samples (N = 4 from the TCGA UCEC project; these samples were annotated as ‘normal ovary’ and pathologist evaluation of available slides confirms ovarian stroma histology) were downloaded from the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov), together with that of Uterine Carcinosarcoma (UCS; N = 57) and Sarcoma (SARC; N = 261). Pure, uncultured fallopian tube epithelium samples (FTE; N = 4) were downloaded through GEO database, with one sample GSM2146822 taken from Series GSE81224 (Klinkebiel et al., 2016), and the other three samples (GSM1606969, GSM1606970, and GSM1606973) from Series GSE65820 (Patch et al., 2015). Other FTE samples in the same studies either had insufficient epithelial purity or had undergone in vitro culturing and therefore not included. All DNA methylation microarray data were reprocessed from raw IDATs in the same way as described above for the MSCs. Gapped H3K27me3 chromatin peak files of MSC-derived from adipose, bone marrow, chondrocyte, and H1 stem cell were downloaded through the NIH Roadmap Epigenomics projects (http://www.roadmapepigenomics.org/data/). EZH2 target genes were then defined as genes overlapping with H3K27me3 marked regions shared by all four different cell types.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM unless otherwise stated. Details are found in the Results section and figure legends. All experiments were preformed on at least three patient derived CA-MSCs. Student’s unpaired t test or ANOVA were used to determine the statistical significance. Differences were considered significant at p < 0.05.
Identification of differential genes
Differential gene expression analysis was carried out using R package Limma (Ritchie et al., 2015). Specifically, differentially expressed genes were identified based on a p value less than 0.05, and an absolute log-transformed fold change greater than 1.5. Followed by functional enrichment analysis using R package clusterProfiler (Yu et al., 2012), with FDR cutoff at 0.05 for KEGG pathway, Gene Ontology (Biological Process), and Gene Set Enrichment Analysis (GSEA). In particular for GSEA, genes were ranked based on log-transformed fold changes by comparing sample group of CA-MSC to MSC.
Unsupervised DNA methylation analysis
Unsupervised hierarchical clustering was performed on top variable CpG probes (N = 10,000, filtered by standard deviations) across all CA-MSC and MSC samples measured on the EPIC array with the R function hclust(). Uniform Manifold Approximation and Projection (UMAP) was performed with the R package uwot.
Identification of CA-MSC/FTE/MSC-specific probe sets
For global comparison of CA-MSC/FTE/MSCs, different probe categories are defined as such: 1) CA-MSC and FTE shared hypermethylated probes: mean methylation difference > 0.4 in both CA-MSC to MSC, and FTE to MSC comparisons; 2) CA-MSC-specific hyper-methylated probes: mean methylation difference > 0.4 when comparing CA-MSC to MSC, but absolute mean methylation difference < 0.1 when comparing FTE to MSC; 3) FTE-specific hyper-methylated probes: mean methylation difference > 0.4 when comparing FTE to MSC, but absolute methylation difference < 0.1 when comparing CA-MSC to MSC; 4) CA-MSC and FTE shared hypo-methylated probes: mean methylation difference < −0.4 in both CA-MSC to MSC, and FTE to MSC comparisons; 5) CA-MSC-specific hypo-methylated probes: mean methylation difference < −0.4 when comparing CA-MSC to MSC, but absolute methylation difference < 0.1 when comparing FTE to MSC; 6) FTE-specific hypo-methylated probes: mean methylation difference < −0.4 when comparing FTE to MSC, and absolute mean methylation difference < 0.1 when comparing CA-MSC to MSC.
Identification of differential DNA methylation and functional enrichment analysis
Differentially methylated cytosines (DMCs) were calculated using R package DMRcate (Peters et al., 2015), by comparing sample group of CA-MSC to MSC, based on its default FDR cutoff of 0.05. We removed probes with any missing value (NA) in any of the samples, leaving a total of ~401,000 probes for any analyses involving DMR/DMC calling. DMCs were split into hypermethylated (hyper-) and hypomethylated (hypo-) DMCs, by comparing group of CA-MSC to MSC. Enrichment or depletion of various genomic features (annotated with R package annotatr for each probe) in hyper- and hypo DMCs were measured as odds ratios, together with significance level calculated using Chi-square test. And the probe set for DMR/DMC calling was used as the background for the enrichment or depletion analysis.
Transcription factor binding site enrichment analysis
DMCs were mapped to genes captured by mRNA sequencing, if they were located within gene promoter regions defined as ± 1.5 kb away from transcription start sites (TSSs). Annotation of CpG probes against transcription factor binding sites (TFBSs) were adopted from our previous study (Zhou et al., 2017). We then performed enrichment analysis of TFBSs over differentially methylated probes at distal regulatory elements (i.e., potential enhancers). Distal probes were defined as probes located within TFBSs, but not overlapping with ± 2 kb flanking regions surrounding TSSs. For each set of hyper- or hypo-DMCs, hypergeometric test was applied to calculate the enrichment of binding sites for each TF within the set of DMCs within all distal probes. Significance cutoff was made at p = 1e-3 after false discovery rate (FDR) correction.
Identification of differential ATAC-seq peaks, functional enrichment, and PCA analysis
Log-transformed reads count were used for further differential peak analysis, with reads counts of replicates from the same sample averaged. Enrichment or depletion of differential ATAC-seq peaks against various genomic features was carried out in a similar way as in differential methylation analysis. Peaks merged from all the samples were served as the background. Principal component analysis (PCA) was performed, based on the normalized reads count matrix. Peak summit centered heatmap per sample was generated with deepTools using normalized 10 base pair bin-based ATACseq signal (Ramírez et al., 2016).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD105 | BD PharMingen | Cat# 561443 |
| CD90 | BD PharMingen | Cat# 555596 |
| CD73 | BD PharMingen | Cat# 560847 |
| CD45 | BD PharMingen | Cat# 340664 |
| CD34 | BD PharMingen | Cat# 555824 |
| FSP | Novus | Cat# NB100–1845; RRID:AB_10000870 |
| Alpha smooth muscle actin | Abcam | Cat# AB5694; RRID:AB_2223021 |
| Anti-H3K27me3 | Active Motif | Cat# 39155; RRID:AB_2561020 |
| anti-PAX8 | Abcam | Cat# AB13611; RRID:AB_300497 |
| anti-WT1 | R&D System | Cat# IC57291V |
| anti-H3K27me2 | Active Motif | Cat# 39535; RRID:AB_2793246 |
| anti-H4K16ac | Active Motif | Cat# 61529; RRID:AB_2793671 |
| anti-H3K27ac | Active Motif | Cat# 39133; RRID:AB_2561016 |
| anti-H3 | Abcam | Cat# AB192985; RRID:AB_2650559 |
| anti-EZH2 | Abcam | Cat# AB191080; RRID:AB_304045 |
| anti-H4 | Abcam | Cat# AB177840; RRID:AB_2650469 |
| anti-EpCAM | Novus | Cat# NB600–1182AF; RRID:AB_10008035 |
| COL 4 | Abcam | Cat# AB6586; RRID:AB_305584 |
| COL 15 | Abcam | Cat# AB150463; RRID:AB_879744 |
| Bacterial and Virus Strains | ||
| LV-EF1α-tdTOMATO | SignaGen Laboratories | Cat# SL100289 |
| LV-Synapsin-GFP | SignaGen Laboratories | Cat# SL100271 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SYBR green master-Mix | Applied Biosystem | Cat# 4472908 |
| RIPA buffer | Pierce, Rockford, IL | Cat# 89900 |
| NuPAGE SDS gel | Invitrogen, Grand Island, NY | Cat# NW04122 |
| PVDF membrane | GE Healthcare Life Science | Cat# 10600001 |
| Alginate | Sigma | Cat# 71238 |
| Matrigel | Corning | Cat# 354230 |
| Red blood cells lysis buffer | ThermoFisher Scientific | Cat# 11814389001 |
| Cisplatin | Fresenius-Kabi | Cat# 4015721 |
| GSK126 | Selleckchem | Cat# S7061 |
| Dynabeads Protein G magnetic beads | Invitrogen, Grand Island, NY | Cat# 10004D |
| Herring Sperm DNA | Sigma | Cat# D7290 |
| KAPA Pure Beads | KAPA Biosystems | Cat# KK8000 |
| Critical Commercial Assays | ||
| Mycoplasma Kit | ABm | Cat# G238 |
| DNA isolation kit | QIAGEN, Hilden, Germany | Cat# 69504 |
| RNeasy Mini Kit | QIAGEN, Hilden, Germany | Cat# 28106 |
| SuperScript III First Strand Synthesis System | Invitrogen, Grand Island, NY | Cat# 18080–051 |
| Bradford Protein Assay Kit | ThermoFisher Scientific | Cat# 23227 |
| Qubit dsDNA High Sensitivity Assay kit | ThermoFisher Scientific | Cat# Q32851 |
| KAPA HyperPrep Kit | Kappa Biosystems | Cat# 07962347001 |
| Deposited Data | ||
| DNA methylation (EPIC array data) | This paper | GSE138072 |
| ATAC sequencing | This paper | SRP289005 |
| ChIP sequencing | This paper | SRP289183 |
| Mendeley dataset | This paper | https://dx.doi.org/10.17632/bdywxt6cvv.1 |
| Experimental Models: Cell Lines | ||
| OVCAR3 | ATCC | Cat# HTB-161; RRID:CVCL_0465 |
| CAOV3 | ATCC | Cat# HTB-75; RRID:CVCL_0201 |
| Experimental Models: Organisms/Strains | ||
| NSG mice | The Jackson Laboratory | Cat# 005557; RRID:IMSR_JAX:005557 |
| Software and Algorithms | ||
| PEPATAC | Corces et al., 2018 | http://pepatac.databio.org/en/latest/ |
| BWA mem algorithm | Li, 2013 | https://github.com/lh3/bwa |
| MACS2 v 2.2.7.1 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| R studio v3.4.1 and v3.4.4 | Ripley, 2001 | https://www.r-project.org |
| STAR v2.7 | Dobin et al., 2013 | https://github.com/alexdobin/STAR/releases |
| RSEM v1.3.1 | Li etal., 2010 | https://deweylab.github.io/RSEM/ |
| deepTools v2.0 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| Other | ||
| Molecular Signatures Database (MSigDB) | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| UCSC genome browser | Karolchik et al., 2003 | http://genome.ucsc.edu |
| EPIC array probe annotations | Zhou et al., 2017 | https://zwdzwd.github.io/InfiniumAnnotation |
| 4C Browser | Yue, 2018 | http://promoter.bx.psu.edu/hi-c/virtual4c.php |
| Genomic Data Commons (GDC) Data Portal | National Institutes of Health | National Cancer Institute | https://portal.gdc.cancer.gov |
| Pure, uncultured fallopian tube epithelium samples | GEO database | GSE81224; GSE65820 |
| Gapped H3K27me3 chromatin peak files | NIH Roadmap Epigenomics projects | http://www.roadmapepigenomics.org/data/ |
| Raw mRNA sequencing data (FASTQ format) | Coffman et al., 2019 | GSE118624 |
Key Resources Table references
(Corces et al., 2018; Li, 2013; Zhang et al., 2008; Ripley, 2001; Dobin et al., 2013; Li and Dewey, 2011; Ramírez et al., 2016; Subramanian et al., 2005; Karolchik et al., 2003; Zhou et al., 2017; Yue, 2018; Coffman et al., 2019)
Highlights.
Ovarian cancer induces an epigenomic MET in CA-MSCs
CA-MSCs enhance metastasis through direct binding to cancer cells and co-metastasis
MET in CA-MSCs is mediated by WT1 and EZH2
CA-MSC MET is clinically significant and correlated with patient survival
ACKNOWLEDGMENTS
We thank Marie Adams and the Van Andel Institute Genomics Core for DNA methylation microarray and ChIP-seq service and the VAI High Performance Computing Cluster supported by Zack Ramjan. L.G.C. is supported by NIH/NCI grant 7K08CA211362-02; a Tina’s Wish Rising Star grant; a Mary Kay Foundation Cancer Research grant; and a DoD Ovarian Cancer, Omics Consortium Development Award. H.S. is supported by NIH/NCI grant 1R37CA230748 and Ovarian Cancer Research Alliance’s Liz Tilberis award (373933). S.B.R. is supported by NIH grant R35GM124736. A.A.C. is supported by NIH grant F32CA225043. R.L.T. is supported by the American Cancer Society-Michigan Cancer Research Fund (PF-16-245-01-DMC).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108473.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alcoser SY, Kimmel DJ, Borgel SD, Carter JP, Dougherty KM, and Hollingshead MG (2011). Real-time PCR-based assay to quantify the relative amount of human and mouse tissue present in tumor xenografts. BMC Biotechnol 11, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augsten M (2014). Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol 4, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrallo-Gimeno A, and Nieto MA (2005). The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 132, 3151–3161. [DOI] [PubMed] [Google Scholar]
- Blattler A, and Farnham PJ (2013). Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem 288, 34287–34294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Chan DK, Shank JJ, Griffith KA, Fan H, Szulawski R, Yang K, Reynolds RK, Johnston C, McLean K, et al. (2020). Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 5, 133247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnier JV, Wang N, Michel RP, Hassanain M, Li S, Lu Y, Metrakos P, Antecka E, Burnier MN, Ponton A, et al. (2011). Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene 30, 3766–3783. [DOI] [PubMed] [Google Scholar]
- Calo E, and Wysocka J (2013). Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, Ling S, Soslow RA, Broaddus RR, et al. ; Cancer Genome Atlas Research Network (2017). Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell 31, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz AG, and Harris A (2013). Collagen XV: exploring its structure and role within the tumor microenvironment. Mol. Cancer Res 11, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman LG, Burgos-Ojeda D, Wu R, Cho K, Bai S, and Buckanovich RJ (2016a). New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res 175, 92–102.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman LG, Choi YJ, McLean K, Allen BL, di Magliano MP, and Buckanovich RJ (2016b). Human carcinoma-associated mesenchymal stem cells promote ovarian cancer chemotherapy resistance via a BMP4/HH signaling loop. Oncotarget 7, 6916–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman LG, Pearson AT, Frisbie LG, Freeman Z, Christie E, Bowtell DD, and Buckanovich RJ (2019). Ovarian Carcinoma-Associated Mesenchymal Stem Cells Arise from Tissue-Specific Normal Stroma. Stem Cells 37, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. ; Cancer Genome Atlas Analysis Network (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Gibbons DL, and Kurie JM (2013). The role of epithelial-mesenchymal transition programming in invasion and metastasis: a clinical perspective. Cancer Manag. Res 5, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop Dj., and Horwitz E (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. [DOI] [PubMed] [Google Scholar]
- Essafi A, Webb A, Berry RL, Slight J, Burn SF, Spraggon L, Velecela V, Martinez-Estrada OM, Wiltshire JH, Roberts SG, et al. (2011). A wt1-controlled chromatin switching mechanism underpins tissue-specific wnt4 activation and repression. Dev. Cell 21, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, et al. (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JW, Fan H, Johnson B, Carpenter TJ, Foy KK, Chatterjee A, Patterson AL, Koeman J, Adams M, Madaj ZB, et al. (2019). Integrated epigenome, exome and transcriptome analyses reveal molecular subtypes and homeotic transformation in uterine fibroids. Cell Rep 29, 4069–4085.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- He H, Luthringer DJ, Hui P, Lau SK, Weiss LM, and Chu PG (2008). Expression of CD56 and WT1 in ovarian stroma and ovarian stromal tumors. Am. J. Surg. Pathol 32, 884–890. [DOI] [PubMed] [Google Scholar]
- Hohenstein P, and Hastie ND (2006). The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet 15, R196–R201. [DOI] [PubMed] [Google Scholar]
- Hylander B, Repasky E, Shrikant P, Intengan M, Beck A, Driscoll D, Singhal P, Lele S, and Odunsi K (2006). Expression of Wilms tumor gene (WT1) in epithelial ovarian cancer. Gynecol. Oncol 101, 12–17. [DOI] [PubMed] [Google Scholar]
- Karlan BY, Dering J, Walsh C, Orsulic S, Lester J, Anderson LA, Ginther CL, Fejzo M, and Slamon D (2014). POSTN/TGFBI-associated stromal signature predicts poor prognosis in serous epithelial ovarian cancer. Gynecol. Oncol 132, 334–342. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. ; University of California Santa Cruz (2003). The UCSC Genome Browser Database. Nucleic Acids Res 31, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkebiel D, Zhang W, Akers SN, Odunsi K, and Karpf AR (2016). DNA Methylome Analyses Implicate Fallopian Tube Epithelia as the Origin for High-Grade Serous Ovarian Cancer. Mol. Cancer Res 14, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymenko Y, and Nephew KP (2018). Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled. Cancers (Basel) 10, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymkowsky MW, and Savagner P (2009). Epithelial-mesenchymal transition: a cancer researcher’s conceptual friend and foe. Am. J. Pathol 174, 1588–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM (2013). Preparation of cells for microscopy using cytospin. Methods Enzymol 533, 235–240. [DOI] [PubMed] [Google Scholar]
- Konecny GE, Wang C, Hamidi H, Winterhoff B, Kalli KR, Dering J, Ginther C, Chen HW, Dowdy S, Cliby W, et al. (2014). Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst 106, dju249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Dedhar S, Kalluri R, and Thompson EW (2006). The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol 172, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, arXiv:1303.3997. https://arxiv.org/abs/1303.3997. [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, et al. (2010). A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63. [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YM, Wong IH, Zhang J, Tein MS, Ng MH, and Hjelm NM (1999). Quantitative analysis of aberrant p16 methylation using real-time quantitative methylation-specific polymerase chain reaction. Cancer Res 59, 3899–3903. [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, and Smyth GK (2016). csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res 44, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen RA, Riggs AD, and Russo VEA (1996). Epigenetic Mechanisms of Gene Regulation (Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, Cabrera L, Keller E, McCauley L, Cho KR, and Buckanovich RJ (2011). Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Invest 121, 3206–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Hodges E, and Hohenstein P (2012). WT1 in disease: shifting the epithelial-mesenchymal balance. J. Pathol 226, 229–240. [DOI] [PubMed] [Google Scholar]
- Moore AW, Schedl A, McInnes L, Doyle M, Hecksher-Sorensen J, and Hastie ND (1998). YAC transgenic analysis reveals Wilms’ tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech. Dev 79, 169–184. [DOI] [PubMed] [Google Scholar]
- Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, et al. ; Australian Ovarian Cancer Study Group (2015). Whole-genome characterization of chemoresistant ovarian cancer. Nature 521, 489–494. [DOI] [PubMed] [Google Scholar]
- Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord RV, Clark SJ, and Molloy PL (2015). De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 8, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, and Weinberg RA (2009). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9, 265–273. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley BD (2001). R studio v3.4.1 (R Foundation for Statistical Computing)
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Rödel P, Anamur C, Seeliger C, Imhoff AB, Herbst E, Vogt S, van Griensven M, Winter G, and Engert J (2015). Calcium alginate gels as stem cell matrix-making paracrine stem cell activity available for enhanced healing after surgery. PLoS ONE 10, e0118937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, and Jones PA (2010). Epigenetics in cancer. Carcino-genesis 31, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, and Laird PW (2013). Interplay between the cancer genome and epigenome. Cell 153, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, and Stark A (2014). Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet 15, 272–286. [DOI] [PubMed] [Google Scholar]
- Shu X, and Pei D (2014). The function and regulation of mesenchymal-to-epithelial transition in somatic cell reprogramming. Curr. Opin. Genet. Dev 28, 32–37. [DOI] [PubMed] [Google Scholar]
- Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, and Marini F (2009). Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 4, e4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur I, and Taipale J (2016). The role of enhancers in cancer. Nat. Rev. Cancer 16, 483–493. [DOI] [PubMed] [Google Scholar]
- Szemes M, Dallosso AR, Melegh Z, Curry T, Li Y, Rivers C, Uney J, Mägdefrau AS, Schwiderski K, Park JH, et al. (2013). Control of epigenetic states by WT1 via regulation of de novo DNA methyltransferase 3A. Hum. Mol. Genet 22, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Bout-selakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47 (D1), D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, and Nieto MA (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Thomassin H, Flavin M, Espinás ML, and Grange T (2001). Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J 20, 1974–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, et al. ; Australian Ovarian Cancer Study Group (2008). Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res 14, 5198–5208. [DOI] [PubMed] [Google Scholar]
- Verardo R, Piazza S, Klaric E, Ciani Y, Bussadori G, Marzinotto S, Mariuzzi L, Cesselli D, Beltrami AP, Mano M, et al. ; Fantom Consortium (2014). Specific mesothelial signature marks the heterogeneity of mesenchymal stem cells from high-grade serous ovarian cancer. Stem Cells 32, 2998–3011. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, Fereday S, Lawrence M, Carter SL, Mermel CH, et al. ; Cancer Genome Atlas Research Network (2013). Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest 123, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrba L, Jensen TJ, Garbe JC, Heimark RL, Cress AE, Dickinson S, Stampfer MR, and Futscher BW (2010). Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS ONE 5, e8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, Li D, Choudhary MNK, Li Y, Hu M, et al. (2018). The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 19, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, Ehrlich M, and Laird PW (2005). Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res 33, 6823–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ocampo A, and Belmonte JCI (2016). Cellular Metabolism and Induced Pluripotency. Cell 166, 1371–1385. [DOI] [PubMed] [Google Scholar]
- Xu B, Zeng DQ, Wu Y, Zheng R, Gu L, Lin X, Hua X, and Jin GH (2011). Tumor suppressor menin represses paired box gene 2 expression via Wilms tumor suppressor protein-polycomb group complex. J. Biol. Chem 286, 13937–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Shen H, Laird PW, Farnham PJ, and Berman BP (2015). Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol 16, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz M, and Christofori G (2009). EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 28, 15–33. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F (2018). 4C Browser http://promoter.bx.psu.edu/hi-c/virtual4c.php.
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al. (2003). Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med 348, 203–213. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Laird PW, and Shen H (2017). Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 45, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Triche TJ Jr., Laird PW, and Shen H (2018). SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res 46, e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DNA methylation EPIC array, ATAC-sequencing and ChIP-sequencing datasets generated in this study is available at NCBI without restriction. The accession number for the EPIC array is GSE138072, for the ATAC-seq data is SRP289005, and for the ChIP-seq data is SRP289183.