

Abstract
Despite promising developments in computational tools, peptide-class II MHC (MHCII) binding predictors continue to lag behind their peptide-class I MHC counterparts. Consequently, peptide–MHCII binding is often evaluated experimentally using competitive binding assays, which tend to sacrifice throughput for quantitative binding detail. Here, we developed a high-throughput semiquantitative peptide–MHCII screening strategy termed microsphere-assisted peptide screening (MAPS) that aims to balance the accuracy of competitive binding assays with the throughput of computational tools. Using MAPS, we screened a peptide library from Zika virus envelope (E) protein for binding to four common MHCII alleles (DR1, DR4, DR7, DR15). Interestingly, MAPS revealed a significant overlap between peptides that promiscuously bind multiple MHCII alleles and antibody neutralization sites. This overlap was also observed for rotavirus outer capsid glycoprotein VP7, suggesting a deeper relationship between B cell and CD4+ T cell specificity which can facilitate the design of broadly protective vaccines to Zika and other viruses.
Keywords: B cell epitopes, CD4+ T cell epitopes, MHC class II, peptide binding prediction, promiscuous peptide, rotavirus, vaccine, Zika virus
1 |. INTRODUCTION
Zika virus is a type of flavivirus, which is a family of structurally similar enveloped viruses transmitted by ticks and mosquitoes. The primary antigenic target on flaviviruses including Zika virus is the envelope (E) protein, which binds to host cell receptors and mediates virus entry.1 The Zika virus E protein has three domains, EDI (residues 1–52, 132–193, and 280–296), EDII (residues 52–132 and 193–280), and EDIII (residues 296–406)2 and is highly similar to the E protein of other flaviviruses, ranging from 39.5% similarity for tick-borne encephalitis to 57.8% similarity for dengue virus 1.3
Because the Zika virus E protein is similar to the E protein of other flaviviruses, antibodies that bind E protein are often cross-reactive. However, rather than neutralizing a heterologous flavivirus infection, these cross-reactive antibodies tend to exacerbate the infection by promoting the internalization and replication of virus in Fc-receptor expressing cells.4 This phenomenon, known as antibody-dependent enhancement (ADE), can be life threatening and underscores the importance of eliciting a highly specific, neutralizing antibody response against E protein in areas where multiple flaviviruses circulate. A number of recent studies have shown that antibodies targeting the EDIII domain of the Zika virus E protein tend to be potently neutralizing2,5,6 and less cross-reactive with E proteins from other flaviviruses than antibodies targeting EDI or EDII.2 Therefore, the EDIII domain of Zika virus E protein is of special interest for Zika virus vaccine development.
Although a robust and neutralizing antibody response generally correlates with flavivirus immunity, less is known about the contribution of T cells to clearing flavivirus infection. However, it was recently shown that CD4+ T cell signaling plays a fundamental role in sustaining antibody-mediated resistance to Zika virus infection.7 In addition, it is thought that T cells play an important role in clearing flavivirus infections from the central nervous system (CNS).8,9 In a recent study, it was shown that no antibodies are present in the CNS during persistent Zika virus infection in nonhuman primates, and decreased Zika viral load in the CNS correlated with the initiation of a CD8+ T cell response.10 This observation is especially interesting considering that while most individuals infected with Zika virus are asymptomatic, serious neurological complications including microcephaly11 and Guillain-Barre syndrome12 have been observed at rates up to 1 in 100 and 1 in 5,000 infections, respectively.13 Because a targeted T cell response to Zika virus could play a role in mitigating these neurological complications as well as sustaining the production of neutralizing antibodies, the identification of both CD8+ and CD4+ T cell epitopes within Zika virus proteins is an active area of research.14
However, before a T cell can recognize a specific antigenic peptide, that peptide must first be presented by a major histocompatibility complex (MHC) molecule. Both class I and class II MHC molecules (MHCI and MHCII, respectively) present peptides through interactions between specific peptide residues (termed the peptide binding register, or PBR) and the MHC peptide-binding groove. MHC molecules are highly polymorphic15,16 and most of the genetic diversity is manifested in the peptide-binding groove of different MHC alleles. As a result, different MHC alleles generally exhibit different binding specificities.17 Because the frequency of MHC alleles varies among ethnically diverse populations, immunodominant peptides from viral18 proteins capable of promiscuously binding multiple MHC alleles are of considerable interest for broadly protective peptide-based therapuetics.19–21
Over the past 20 years, several in silico strategies have been devised to identify such promiscuous MHCII-binding peptides including matrix based methods,22,23 structure based methods,24–27 and machine learning methods using artificial neural networks.28–31 The accuracy of in silico peptide–MHC binding predictors is measured by the area under (AUC) the receiver operating characteristic (ROC) curve, with a value of 1 indicating a perfect prediction and a value of 0.5 indicating a completely random prediction. Currently, machine learning methods using artificial neural networks are among the most accurate in silico predictors, achieving an AUC of approximately 0.85–0.95 for peptide–MHCI binding predictions and 0.75–0.85 for peptide–MHCII binding predictions.32 The accuracy of peptide–MHC binding predictors can often be improved by combining the top-performing individual predictors into a consensus method,33,34 which is the strategy recommended by the Immune Epitope Database and Analysis Resource (IEDB).35 Although in silico peptide–MHCII binding predictors perform well during cross-validation with standardized datasets, they tend to underperform when applied to new datasets or datasets containing peptides of different lengths.31 In a study of 21 different peptide–MHCII binding predictors, no individual predictor was found to be suitable for the prediction of promiscuous MHCII-binding peptides.17 Moreover, these predictors were characterized by high false-positive rates and even the most accurate could only identify 50% of actual T cell epitopes from four antigenic protein libraries.17
Given the limited accuracy and high false-positive rate of in silico peptide-MHCII binding predictors, a demand exists for high-throughput systems capable of reliably identifying promiscuous MHCII-binding peptides. To this end, a wide range of experimental methods have been applied to measure peptide-MHCII binding, including ELISA,36,37 fluorescence polarization,38 gel-filtration with radiolabeled peptides,36,39 fluorescence resonance energy transfer,40,41 surface plasmon resonance (SPR),42 cell-surface display,43 and bead-based methods.44 While many of these techniques are well established and yield quantitative peptide-MHCII binding data, the vast majority tend to sacrifice throughput in favor of quantitative detail. For example, competition based assays like ELISA,36,37 fluorescence polarization,38 and some bead-based methods44 involve titrating the target peptide for competitive binding with a labeled reference peptide. Although the quantitative binding data derived from these competition assays is critical to improving in silico peptide-MHCII binding prediction algorithms, they typically involve 8–12 point titrations in triplicate for reliable data. The amount of MHCII protein required for competitive binding assays could be greatly reduced, and the study of peptide-MHCII binding made more efficient, if a preliminary screening strategy were used to identify MHCII binding peptides from a large library of nonbinders using a binary classification scheme. Once identified, these MHCII binding peptides could be further studied in detail.
Here, we developed and validated a high-throughput, semiquantitative assay for prescreening MHCII binding peptides termed microsphere-assisted peptide screening (MAPS). MAPS was designed to strike a balance between the quantitative detail offered by conventional competition-based binding assays and the throughput offered by in silico predictors by directly measuring peptide-MHCII binding using flow cytometry. Using MAPS, we identified five peptides within the Zika virus E protein that promiscuously bound four common human MHCII alleles including DR1, DR4, DR7, and DR15. Of the five promiscuous MHCII-binding peptides identified, IEDB in silico binding predictors predicted only one, suggesting that computational peptide-MHCII binding predictors alone may miss a significant number of promiscuous MHCII-binding peptides. In addition, we observed a substantial overlap between promiscuous MHCII-binding peptides and antibody neutralization sites in the Zika virus E protein. A similar overlap was also observed for the rotavirus outer capsid glycoprotein VP7. Taken together, these results demonstrate that MAPS is a reliable, high-throughput method for rapidly identifying promiscuous MHCII-binding peptides.
2 |. MATERIALS AND METHODS
2.1 |. Protein design, expression, and peptide synthesis
Human MHCII proteins were assembled by isolating the extracellular domains of the alpha chain HLA-DRA (UniProt: P01903, residues 26–216) and each beta chain allele: HLA-DRB1*01:01 (UniProt: P04229, residues 30–227), HLA-DRB1*04:01 (UniProt:P13760, residues 30–227), HLA-DRB1*07:01 (UniProt: P13761, residues 30–227), HLA-DRB1*15:01 (UniProt: P01911, residues 30–227). The leucine zipper dimerization motifs Fos and Jun were fused to the C-terminus of the DRA and DRB1 chains, respectively, as described elsewhere.45 The 15 amino acid AviTag™ (GLNDIFEAQKIEWHE) was fused to the C-terminus of the Fos dimerization motif on the DRA chain to allow for biotinylation, and a 6X-Histidine tag was fused to the C-terminus of the complete recombinant DRA chain for purification. The N-terminus of each DRB1 chain was fused to the invariant chain CLIP87–101 peptide via a thrombin-cleavable linker to allow for peptide exchange, as described elsewhere.46 The C-terminus of the Jun-dimerization motif of each DRB1 was fused to a 6X-Histidine tag for purification. Finally, the N-terminus of each dimeric chain was fused to the baculovirus gp64 signal peptide and ligated into separate baculovirus transfer vectors pAcGP67A (BaculoGold Baculovirus Expression System, Pharmingen BD Biosciences, San Jose, CA). The DNA sequence of each construct was verified by Sanger sequencing.
Spodoptera frugiperda (Sf9) insect cells were transfected with transfer vectors carrying the recombinant DRA chain and each recombinant beta chain (DR1, DR4, DR7, and DR15) with linearized Baculovirus DNA (Pharmingen BD BaculoGold Biosciences, San Jose, CA) using Cellfectin II (Invitrogen, Waltham, MA) according to the manufacturer’s protocol. Low-titer P0 viral stocks carrying the recombinant DRA and DRB1 genes were isolated from the transfection supernatant and amplified separately in Sf9 cells to create high-titer P1 viral stocks. Each human MHCII heterodimer was expressed by coinfecting High-Five cells at a density of 2.0M/mL with equal volumes of high-titer P1 DRA and DRB1 baculovirus stocks. MHCII protein was harvested 72 hr after infection and purified using affinity chromatography with Ni-NTA beads according to the manufacturer’s protocol (Qiagen). SDS-PAGE analysis was performed to evaluate protein purity.
Approximately 0.5 mg of each peptide used in this work was chemically synthesized by Sigma-Aldrich (Woodlands, TX). Each peptide was designed to be 20 amino acids long and was fused to an N-terminal dinitrophenyl (DNP) tag. Peptide libraries covering the Burkholderia pseudomallei alkyl hydroperoxide reductase (AhpC) protein (UniProt: Q63T73 residues, 1–180), the rotavirus outer capsid glycoprotein VP7 (UniProt: P11853 residues 41–320), and the Zika virus E protein (UniProt: A0A024B7W1 residues 291–794) were designed such that each 20mer peptide overlapped with the preceding peptide in the sequence by 10 amino acids. DNP-tagged variants of DR1-binding peptide the HA306–318 were also synthesized to determine how relative PBR position affects MAPS signal. Peptides in the libraries that could not be chemically synthesized were not included in the MAPS analysis, and explain any nonconsecutive sequences in Figures S3 and S4.
2.2 |. Biotinylation and peptide exchange
After purifying the human MHCII-CLIP proteins, the DRA chain of each heterodimer was biotinylated. Biotinylation reactions were performed with the AviTag™ biotinylation kit (Avidity LLC, Aurora, CO) according to the manufacturer’s protocol. The biotinylation efficiency for each allele was assessed using a streptavidin gel-shift assay. Briefly, biotinylated MHCII-CLIP proteins were incubated with excess streptavidin for 1 hr at 30°C. The complexes were then analyzed using SDS-PAGE, and biotinylation efficiency was evaluated by estimating the fraction of the biotinylated DRA chain that shifted following incubation with streptavidin.
Peptide exchange was performed similar to previously published methods,47 with some modifications. Prior to peptide exchange, the CLIP87–101 peptide fused to each DR beta chain was cleaved. CLIP87–101 cleavage was performed by incubating the MHC-CLIP protein with restriction grade thrombin (Novagen, Madison, WI) for 2 hr at 37°C at a concentration of 10 units per milligram of protein. Thrombin-induced CLIP87–101 cleavage was confirmed using SDS-PAGE (Figure S1c). Following CLIP87–101 cleavage, peptide exchange was performed by transferring the empty DR alleles into peptide exchange buffer containing 50 mM sodium citrate pH 5.2, 1% octylglucoside, and 100 mM NaCl. DR alleles were then incubated with 25M excess DNP-tagged peptides for 16 hr at 37°C. The peptide exchange reaction was neutralized by adding 1/5 volume of 1M Tris pH 8.0.
2.3 |. MAPS and flow cytometry
Following peptide exchange, 2 μg of each exchanged MHCII was incubated with 100,000 streptavidin-coated microspheres (Bangs Laboratories, Fishers, IN) in 1% bovine serum albumin (BSA) for 1 hr at room temperature. In parallel, an equivalent amount of DNP-tagged peptide used in the peptide exchange reaction was incubated with streptavidin-coated microspheres in the absence of MHCII for calculating the MAPS signal. After loading, the microspheres were washed in 1% BSA and stained with 2 ng/μL of rat anti-DNP antibody (Clone LO-DNP-2, Invitrogen) for 30 min at room temperature. Stained microspheres were washed in 1% BSA and stained with 2 ng/μL of secondary goat-anti-rat PE (Invitrogen) for 30 min at room temperature. The stained microspheres were then washed twice in 1% BSA and resuspended in 500 μL for analysis by flow cytometry. Flow cytometry was performed by gating on the population of single microspheres and analyzing the median fluorescence intensity (MFI) from the DNP fluorescent staining. The MAPS signal was determined by normalizing the peptide-MHCII DNP MFI by the MFI of the microspheres incubated with the peptide alone. This normalization accounted for nonspecific binding of peptides to the streptavidin-coated microspheres. Peptide-MHCII interactions exhibiting a MAPS signal >5 were defined as binding interactions as this threshold results in a false-positive rate of <5% (see details in the Section 3).
2.4 |. IEDB peptide-MHCII binding prediction
IEDB peptide-MHCII binding predictions were performed by entering each 20mer peptide sequence and predicting its binding to DRB1*01:01, DRB1*04:01, DRB1*07:01, and DRB1*15:01 using the IEDB recommended prediction method. The output data was broken down into six 15mers for each 20mer sequence provided. The IEDB consensus method33,48 was used to predict peptide MHCII binding, which provided a binding percentile rank for each 15mer. The percentile rank binding score was calculated by comparing the predicted peptide-MHCII binding affinity of the target peptide against 5 million random 15mers from the SWISSPROT database. Accordingly, a low percentile rank indicated a high predicted binding affinity while a high percentile rank indicated a low predicted binding affinity. The lowest percentile rank from the six 15mers derived from each single 20mer sequence was selected as the binding score for the 20mer. Peptides were said to be binders if the percentile rank was less than or equal to 20, in accordance with a previous study concerned with promiscuous MHCII-binding peptides.49
2.5 |. Structural analysis
PyMOL50 was used to make the structural images of the Zika Virus E protein (PDB:5JHM)1 and the rotavirus VP7 protein. The comparative model for the VP7 protein was generated with SWISS-MODEL51 and used Chain A from the PDB:3FMG for the template.52
3 |. RESULTS
3.1 |. MAPS strategy and validation
MAPS is performed by first expressing a diverse panel of human MHCII alleles in insect cells (Figure 1, Step 1a). The alpha chain (DRA) of each MHCII was fused to a biotinylation site and each beta chain allele (DRB1*01:01 – DR1, DRB1*04:01 – DR4, DRB1*07:01 – DR7, DRB1*15:01 – DR15) was fused to the invariant chain peptide CLIP via a cleavable thrombin linker.46,53 The cleavable linker allows the invariant CLIP peptide to be exchanged for any peptide of interest, allowing a single MHCII-CLIP construct to be used to measure thousands of unique peptide-MHCII binding events in a high throughput manner. The alpha and beta chains of each MHCII construct were also fused to the leucine zipper dimerization motifs Fos and Jun, respectively, to stabilize the heterodimer during peptide exchange.45 The four MHCII alleles chosen in this study are expected to cover approximately 34% of the U.S. population based on known MHCII allele frequencies54 and demographic data (Figure S1a). Following purification, each MHCII-CLIP heterodimer was biotinylated and the degree of biotinylation was assessed by a streptavidin (SAv) shift assay and all alleles were >98% biotinylated (Figure S1b). Thrombin-linker cleavage was evaluated similarly (Figure S1c), and the reduction in the size of the beta chain of each allele confirmed CLIP dissociation.
FIGURE 1.
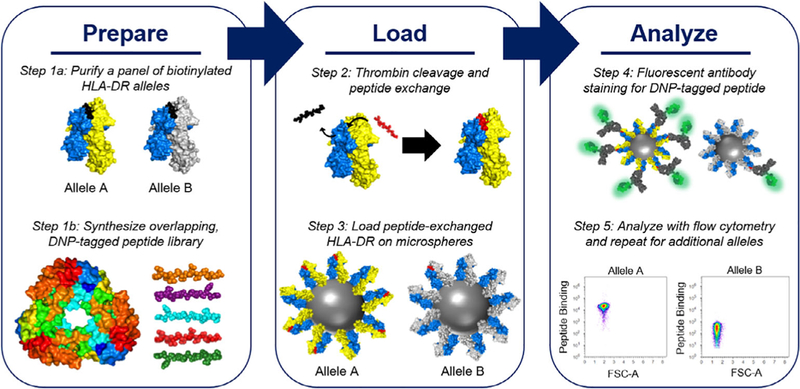
MAPS involves three phases: prepare, load, and analyze. The preparation phase involves (1a) purifying and biotinylating a panel of diverse human MHCII alleles and (1b) synthesizing an overlapping DNP-tagged peptide library. The loading phase involves (2) loading the DNP-tagged peptides from the library onto each MHCII allele via peptide exchange, and (3) loading the biotinylated, peptide-exchanged MHCII on streptavidin-coated microspheres. The analysis phase involves (4) staining the loaded microspheres for the DNP-tagged peptide and (5) analyzing resulting signal using flow cytometry. DNP, dinitrophenyl; MAPS, microsphere-assisted peptide screening
In parallel with MHCII expression and purification, an overlapping peptide library is synthesized to be screened for binding to each MHCII allele (Figure 1, Step 1b). Each peptide library consists of dinitrophenyl-(DNP) tagged 20mers with 10 amino acid overlaps. These DNP-tagged 20mers are then exchanged for the CLIP peptide (Figure 1, Step 2) and loaded onto ~4 μm diameter SAv-coated microspheres (Figure 1, Step 3). Once loaded, the microspheres are stained for the DNP-tagged peptide using fluorescently labeled anti-DNP antibody and then analyzed using flow cytometry (Figure 1, Steps 4 and 5), where the fluorescent DNP-peptide staining should correlate with binding affinity.
As a proof of concept, we first tested MAPS using the well-characterized interaction between DR1 and the influenza peptide HA306–318 tagged with DNP (DNP-PKYVKQNTLKLAT, PBR in bold). DR1-bound CLIP was exchanged with HA306–318, and the resulting peptide exchange mixture was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). It has been shown that DR1 without any peptide occupying the peptide binding groove (empty DR1) and DR1-CLIP molecules are not stable in SDS; however, DR1 binding of HA306–318 promotes SDS stability by protecting a sensitive site in the DR1 peptide-binding groove from SDS.55 As expected, both the empty DR1 (generated in this study by cleaving the thrombin linker in the DR1-CLIP without adding any peptide) and the DR1-CLIP molecules dissociated into their respective alpha- and beta-chains in the presence of SDS while DR1-HA306–318 migrated as an SDS-stable heterodimer (Figure 2a), indicating efficient peptide exchange.
FIGURE 2.
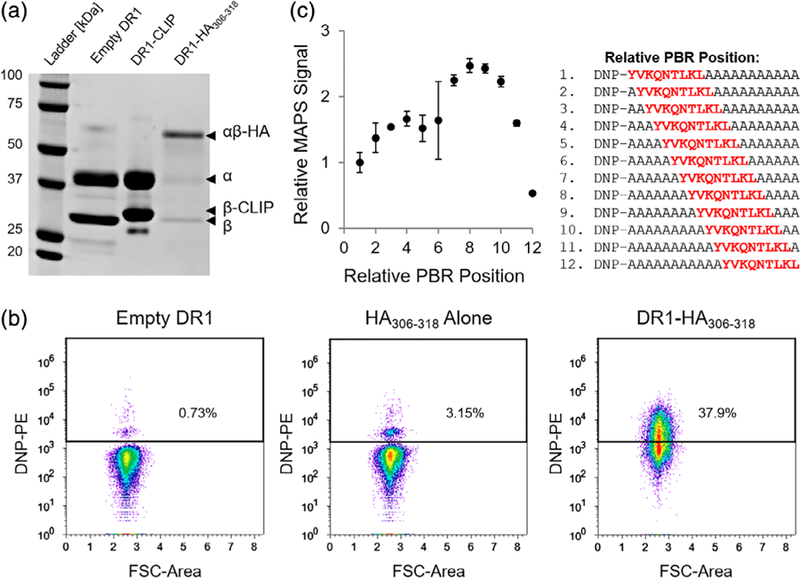
Validation of MAPS strategy with DR1-HA306–318 interaction. (a) SDS-PAGE analysis of the SDS-stability of various DR1 complexes. After thrombin cleavage of the CLIP peptide in the DR1-CLIP protein, the target HA306–318 peptide was added to allow DR1-HA306–318 complex formation through peptide exchange, or no peptide was added (empty DR1) as a control. (b) Flow cytometry dot plots of DNP-signal detected on microspheres loaded with: empty DR1 (left), HA306–318 peptide alone (middle), or DR1-HA306–318 (right). (c) Relative MAPS signal plotted with respect to the relative position of the HA306–318 PBR within a 20mer peptide. The peripheral flanking residues were mutated to alanine. Relative MAPS signal represents the MAPS signal of each peptide normalized by the MAPS signal observed for the peptide with a relative PBR position of 1. DNP, dinitrophenyl; MAPS, microsphere-assisted peptide screening; PBR, peptide binding register
After confirming the successful CLIP→HA306–318 peptide exchange in DR1, we used MAPS to measure the anti-DNP fluorescence of microspheres loaded with the DR1-HA306–318 peptide exchange mixture. When analyzed using flow cytometry, DR1-HA306–318 microspheres exhibited significantly greater DNP staining than microspheres loaded with either empty DR1 or the HA306–318 peptide alone (Figure 2b), confirming that DNP staining is specific and peptide-MHCII binding dependent. While MAPS successfully stained the DR1-HA306–318 complexes, the incomplete shift (i.e., 37.9%) of the DNP-positive beads was surprising given the close to 100% peptide exchange efficiency observed in Figure 2a and the high binding affinity between DR1 and the HA306–318 peptide (IC50 ~34 nM).36 This apparent inconsistency led us to hypothesize that the position of the 9mer PBR relative to the DNP tag might affect the accessibility of the DNP tag during antibody staining. For example, if the PBR is located at the N-terminus of a given peptide, the beta-sheet and alpha helices of the peptide-binding groove might obstruct antibody binding to the DNP tag, resulting in a lower signal. To test this hypothesis, we synthesized 12 DNP-tagged 20mers (Figure 2c, right panel), in which the relative position of the HA306–318 PBR was shifted from the N-terminus (relative PBR position of 1) to the C-terminus (relative PBR position of 12). The remaining residues were mutated to alanine to minimize complex interactions with peripheral flanking residues.
The relative MAPS signal for each PBR-variant of the HA306–318 peptide was normalized such that the signal observed equals one when the PBR is at the N-terminus (relative PBR position of 1). As expected, the relative MAPS signal was dependent on the position of the PBR within the peptide (Figure 2c, left panel). The relative MAPS signal was highest when the PBR was positioned near the middle of the 20mer peptide (between residues seven and 10), which resulted in up to 2.5 times greater signal than when the PBR was located at the N-terminus. Interestingly, the relative MAPS signal decreased approximately fivefold from its maximum when the PBR was positioned at the C-terminus of the peptide, suggesting that the accessibility of the DNP tag might also be obstructed when the N-terminus of the peptide is significantly overhanging, or that peptides with a C-terminal PBR do not bind MHC molecules strongly. While we investigated the effect of the PBR position on binding for a single peptide-MHC pair, we anticipate that other pairs would behave similarly.
Even though the MAPS signal was found to be sensitive to the relative position of the PBR within a given peptide, the actual number of missed binding peptides with unique PBRs should be minimal when screening overlapping peptides because most unique PBRs will appear twice (i.e., in consecutive, overlapping peptides). In addition, although the sensitivity of MAPS to the relative PBR position tends to reduce the overall accuracy of the approach, this sensitivity could have unanticipated advantages. For example, MAPS used in conjunction with more rigorous protein–protein binding assays could help identify unique PBRs within a particular binding peptide, which is a significant obstacle to improving the accuracy of computational peptide–MHCII binding predictions.30
3.2 |. Evaluating MAPS performance with AhpC reference peptide library
After validating the MAPS strategy with DR1-HA306–318 and observing its sensitivity to the PBR position, we next aimed to evaluate the overall accuracy of MAPS. To this end, a 16-peptide library of overlapping 20mers (Figure S2a) with known binding affinity to each MHCII allele in our panel (DR1, DR4, DR7, and DR15) was synthesized based on the alkyl hydroperoxide reductase (AhpC) protein of B. pseudomallei.56 MAPS was performed for each AhpC peptide–MHCII combination, and binding was quantified as the MAPS signal, which was defined as the ratio of the median fluorescence intensity (MFI) of the DNP-tagged peptide bound to MHCII (e.g., Figure 2b, right panel) over the MFI of the DNP-tagged peptide incubated with SAv-microspheres without MHCII (e.g., Figure 2b, middle panel). The MAPS signal of each peptide–MHCII combination was then plotted with respect to the known IC50 value56 of the peptide as shown in Figure S2b. The general inverse relationship observed between the MAPS signal and the IC50 value indicates that the MAPS signal largely correlates with peptide–MHC binding affinity. Although the MAPS strategy is generally accurate, we observed that MAPS missed some known binding peptides within the AhpC peptide library (i.e., peptides known to bind a particular MHCII allele, but exhibited a low MAPS signal). Taken together, these results demonstrate that the MAPS signal provides a semiquantitative measure of peptide-MHC binding affinity and is susceptible to false-negatives. These missed binders are likely a consequence of MAPS sensitivity to PBR position (Figure 2c), which suggests that the missed binders have either N-terminal or C-terminal PBRs.
The AhpC reference peptide library was also screened for predicted binding to each MHCII allele using the in silico bioinformatics tools provided by IEDB.35 Peptide-MHCII binding was predicted using the IEDB recommended consensus method,33 which combined the predictions provided by the stabilized matrix method SMM-align,22 the artificial neural network-based method NN-align,29 and either the Sturniolo19 method or the combinatorial library (CombLib)48 method. The binding score of the consensus prediction was given as a percentile rank, which scores each peptide’s predicted binding affinity against the binding affinities of 5 million random 15mers from the SWISSPROT database. Peptides given a low percentile rank were predicted to be strong binders while peptides given a high percentile rank were not predicted to interact strongly with that particular MHCII allele. We defined a predicted binder as a peptide with a percentile rank of less than or equal to 20, in line with previous studies of promiscuous MHCII-binding peptides.49 The accuracy of the IEDB binding predictions were then compared to the experimental results using MAPS by plotting the respective ROC curve of each (Figure 3). Based on this analysis, the AUC for the MAPS ROC curve was found to be approximately 0.851, while the AUCs for the predictor ROCs were significantly lower, ranging from 0.615 for the SMM-Align method to 0.741 for the NN-Align method. The IEDB consensus method, which combines the output of the SMM-Align method and the NN-Align method yielded an AUC of 0.694. These results indicate that while the MAPS strategy is susceptible to false-negatives depending on the relative position of the PBR within a peptide, it significantly outperforms the predictions provided by IEDB for 20mer peptide-MHCII binding. Based on the ROC analysis, peptide-MHCII interactions producing a MAPS signal >5 were classified as binding peptides in this study, as this threshold results in a false-positive rate of <5%.
FIGURE 3.
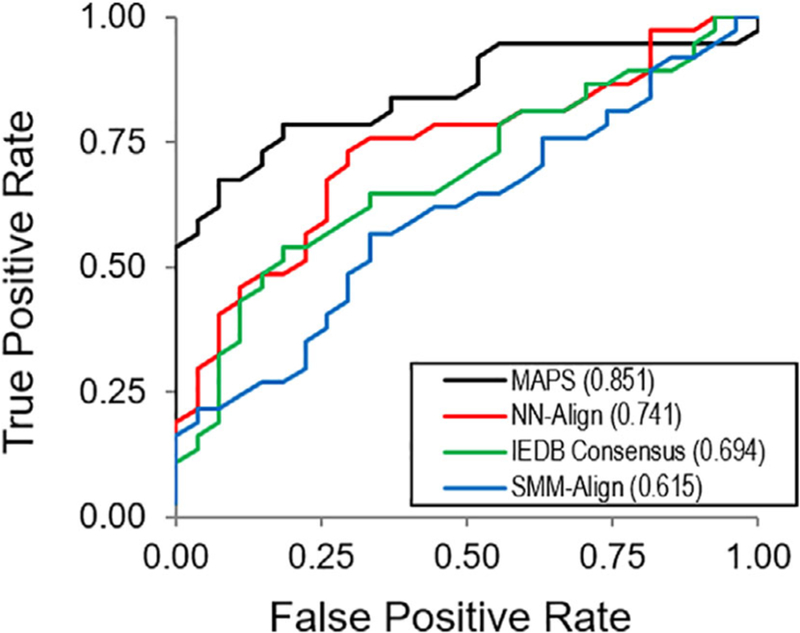
ROC analysis of MAPS strategy compared to IEDB peptide-MHCII binding predictors for the AhpC reference peptide library. The AUC of each curve is provided in the legend. AUC, area under curve; IEDB, Immune Epitope Database and Analysis Resource; MAPS, microsphere-assisted peptide screening; ROC, receiver operating characteristic
3.3 |. MAPS of Zika virus envelope protein
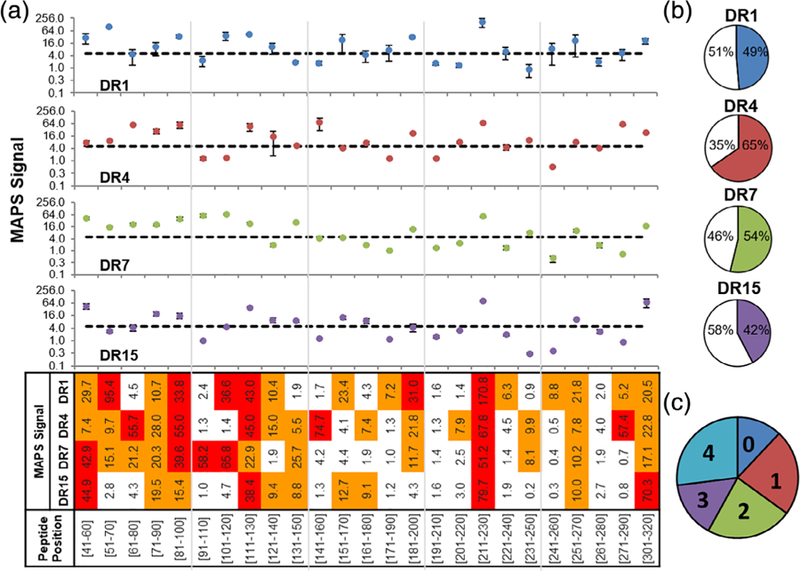
After quantifying the overall accuracy of the MAPS strategy and defining a peptide-binding threshold using the AhpC reference library, we next aimed to identify promiscuous MHCII-binding peptides within the Zika virus E protein. We synthesized a 37-peptide library of overlapping DNP-tagged 20mers from the Zika virus E protein (ZikVE) (Figure S3). Each ZikVE peptide was exchanged into the four MHCII alleles, and the resulting peptide-MHCII binding was measured by MAPS (Figure 4a). Of the four alleles screened, DR1 bound the most ZikVE peptides (49%), followed by DR4 (41%), DR7 (35%), and DR15 (30%) (Figure 4b). Based on the criterion that promiscuous MHCII-binding peptides exhibit a MAPS signal >5 for all four MHCII alleles, five (~14%) promiscuous MHCII-binding peptides within the E protein library (Figure 4c) were identified: ZikVE51–70, ZikVE131–150, ZikVE191–210, ZikVE311–330, ZikVE351–370 (Figures 4a and 5a). Of the five promiscuous MHCII-binding ZikVE peptides identified by MAPS, only two (ZikVE131–150 and ZikVE351–370) were predicted to bind all four alleles by the IEDB consensus prediction (Table S1). Interestingly, only DR4 was predicted to bind all five promiscuously binding peptides using the IEDB consensus method. Together these results provide further evidence that the IEDB consensus method for predicting promiscuous 20mer peptide-MHCII binding is generally inaccurate and that MAPS is able to identify promiscuous binders missed by computational methods.
FIGURE 4.
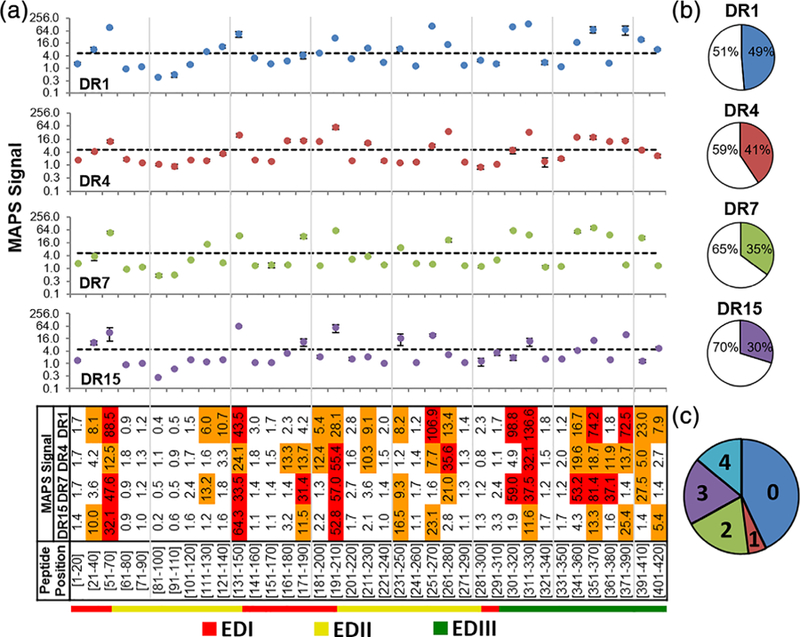
MAPS analysis of overlapping peptides from Zika virus E protein for four human MHCII alleles. (a) MAPS signal of each ZikVE peptide. Data are shown as the mean of two independent experiments. Error bars signify standard deviation. Dashed lines represent the peptide-binding threshold of MAPS signal of 5. For the heat map, orange and red indicate MAPS signals of 5–30 and >30, respectively. (b) Pie-charts representing the percentage of ZikVE peptides that bound to each MHCII allele. Colored slices represent the fraction of binders. (c) A pie-chart representing the fractions of ZikVE peptides binding 0, 1, 2, 3, or 4 MHCII alleles. MAPS, microsphere-assisted peptide screening
FIGURE 5.
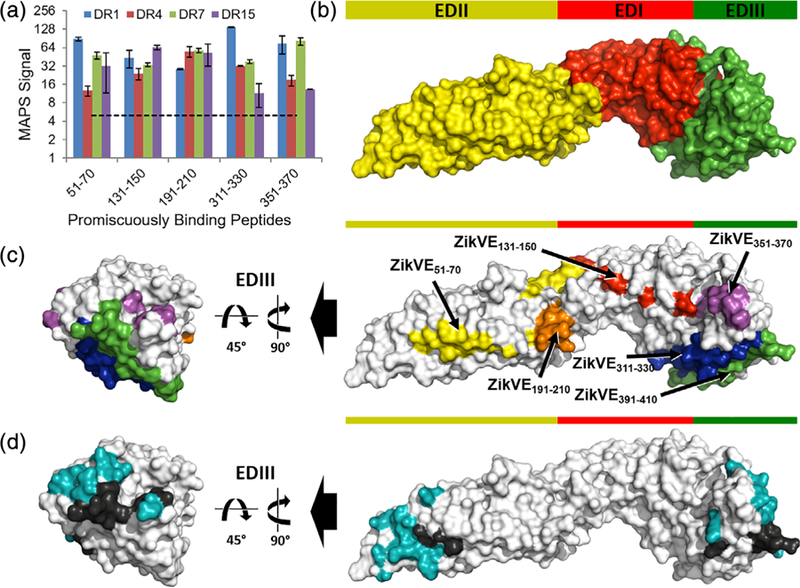
Structural analysis of MAPS-identified promiscuous MHCII-binding peptides in the Zika virus E protein. (a) MAPS signal for each promiscuous MHCII-binding E protein peptide. Dashed lines represent the peptide-binding threshold of MAPS signal of 5. E protein structural analysis of (b) the protein domains, (c) the five promiscuous MHCII-binding peptides as well as the quasi-promiscuous peptide ZikVE391–410 identified by MAPS and (d) the B cell epitopes reported previously.5,57 In (d), the residues of the B cell epitopes overlapping with the promiscuous MHCII-binding peptides are in black, and the nonoverlapping residues are in teal. In (c) and (d), only EDIII is shown in the image on the left for clarity. MAPS, microsphere-assisted peptide screening
Furthermore, all five of the MAPS-identified promiscuous MHCII-binding ZikVE peptides have been reported to be true CD4+ T cell epitopes in human-DR transgenic mouse models.14 Given their immunological relevance as T cell epitopes, we further explored the immunogenicity of these ZikVE peptides by analyzing their position within the folded E protein. In contrast to other flaviviruses, a Zika virus vaccine does not currently exist. Therefore, limited information of antibody neutralization sites on a vaccine strain is available as a point of comparison. However, it is well documented that antibodies binding the EDIII domain tend to be potently neutralizing and less prone to ADE than antibodies binding the EDI and EDII domains.2 Analyzing the position of each promiscuous MHCII-binding ZikVE peptide within the structure of the E protein revealed that one promiscuous MHCII-binding peptide is within the EDI domain (ZikVE131–150), while two are within the EDII (ZikVE51–70 and ZikVE191–210) and EDIII domains (ZikVE311–330 and ZikVE351–370) (Figure 5b,c).
While information regarding specific antibody-contacting residues on the ZikVE protein is still emerging, a 2017 study5 reported that a neutralizing antibody in Zika-infected individuals contacted residues A311, T351, and L352, among others on the E protein. These residues overlap with the EDIII promiscuous MHCII-binding peptides ZikVE311–330 and ZikVE351–370 (Figure 5d). Moreover, the same study5 also reported that mutating the lysine at residue 394 to alanine eliminated antibody binding, suggesting that antibody recognition of K394 is critical for neutralization. While the relevant peptide ZikVE391–410 did not fulfil our criterion as a promiscuous MHCII-binding peptide, it exhibited a MAPS signal >5 for three of the four alleles tested (DR1, DR4, and DR7, See Figure 4a). If we include ZikVE391–410 as a quasi-promiscuously binding peptide in our analysis, eight of 18 reported5 antibody neutralization sites on the ZikVE protein were also present in promiscuous MHCII-binding ZikVE peptides (Figure 5d). Similarly, another study57 reported that antibodies contacting residues M68, S70, V153, T315, and P354 (among others) were potently cross neutralizing to Dengue and Zika virus. In addition to the T315 and P354 residues found in the promiscuous MHCII-binding EDIII peptides ZikVE311–330 and ZikVE351–370, M68 and S70 are found in the promiscuous MHCII-binding EDII peptide ZikVE51–70 (Figure 5d). In total, six of the 16 E protein residues responsible for the polar and salt-bridge antibody contacts57 are also present in the MAPS-identified promiscuous MHCII-binding ZikVE peptides. The observed overlap between promiscuous MHCII-binding ZikVE peptides identified by MAPS and antibody neutralization sites on the Zika virus E protein (Figure 5d) was striking and could have important implications in the design of peptide-based vaccines and diagnostic tools for Zika virus.
3.4 |. MAPS of rotavirus outer capsid glycoprotein VP7
The significant overlap between promiscuous MHCII-binding peptides and antibody neutralization sites observed for the ZikVE protein was unexpected. However, in-depth studies involving the immune response to Zika virus are relatively recent, and our understanding of T cell epitopes and B cell epitopes (BCEs) within the ZikVE protein is still developing. To determine if this phenomenon is unique to the ZikVE protein, we next synthesized a 25-peptide library of overlapping 20mers derived from the well-characterized rotavirus outer capsid glycoprotein VP7 (Figure S4) and performed similar analyses.
Rotavirus is the most common cause of diarrheal disease in children worldwide,58 and remains a significant health challenge in developing countries despite being extensively characterized and the target of two approved vaccines.59,60 Each 20mer in the VP7 peptide library was exchanged into the four MHCII alleles, and peptide-MHCII binding was evaluated by MAPS (Figure 6a). DR4 bound the most VP7 peptides (65%) followed by DR1 (61%), DR7 (54%), and DR15 (42%) (Figure 6b). Interestingly, more VP7 peptides tended to bind promiscuously than the ZikVE peptides screened (See Figures 4c and 6c), potentially related to its wider spread in humans. Based on the criteria that promiscuous MHCII-binding peptides exhibit a MAPS signal >5 for all four MHCII alleles, seven promiscuous MHCII-binding peptides within the VP7 protein were identified: VP741–60, VP771–90, VP781–100, VP7111–130, VP7211–230, VP7251–270, and VP7301–320 (Figures 6a and 7a). Of these seven promiscuous MHCII-binding peptides, only four were predicted to bind all four alleles by the IEDB consensus prediction (Table S2). Further, similar to the promiscuous MHCII-binding ZikVE peptides, only DR4 was predicted to bind all seven MAPS-identified promiscuous MHCII-binding VP7 peptides using the IEDB consensus method. Taken together, these results further suggest that in silico strategies alone remain insufficient to reliably predict peptide-MHCII binding.
FIGURE 6.
MAPS analysis of overlapping peptides from VP7 protein for four human MHCII alleles. (a) MAPS signal of each VP7 peptide. Data are shown as the mean of two independent experiments. Error bars signify standard deviation. Dashed lines represent peptide-binding threshold of MAPS signal of 5. For the heat map, orange and red indicate MAPS signals of 5–30 and >30, respectively. (b) Pie-charts representing the percentage of VP7 peptides that bound to each MHCII allele. Colored slices represent the fraction of binders. (c) A pie-chart representing the fraction of VP7 peptides binding 0, 1, 2, 3, or 4 MHCII alleles. MAPS, microsphere-assisted peptide screening
FIGURE 7.

Structural analysis of MAPS-identified promiscuous MHCII-binding VP7 peptides. (a) MAPS signal for each promiscuous MHCII-binding VP7 peptide. VP7 structural analysis (b) of the promiscuous MHCII-binding peptides VP781–100, VP7111–130, and VP7211–230, and (c) the B cell epitopes (BCEs) reported previously.52 In (c), the residues of the BCEs overlapping with the promiscuous MHCII-binding peptides VP781–100, VP7111–130, or VP7211–230 are in black, and the nonoverlapping residues are in teal. (d) Amino acid sequence overlaps between the three dominant BCEs on the VP7 protein and the promiscuous MHCII-binding peptides VP781–100, VP7111–130, and VP7211–230. Overlapping residues are indicated in red. MAPS, microsphere-assisted peptide screening
To investigate any potential overlap between the promiscuous MHCII-binding VP7 peptides identified by MAPS and antibody neutralization sites on the folded VP7 protein, a structural analysis was performed. In contrast to Zika virus, for which no vaccine exists and little data regarding dominant antibody neutralization sites on the E protein is available, two vaccines for rotavirus have been licensed and the antibody neutralization sites on the VP7 protein are well characterized. VP7 protein contains three dominant antigenic epitopes: 7–1a (BCE 1), 7–1b (BCE 2), and 7–2 (BCE 3),52 all of which are located near the interface of separate VP7 trimer units. Interestingly, three of the seven promiscuous MHCII-binding VP7 peptides (VP781–100, VP7111–130, VP7211–230) identified by MAPS overlapped with these dominant antigenic epitopes (Figure 7b,c). The most striking overlap was observed for VP781–100, which included eight of the 14 residues of BCE 1. Further, VP781–100 and VP7111–130 together cover nearly all of BCE 1, overlapping with 12 of the 14 residues (Figure 7d). The VP7211–230 peptide that was associated with the greatest MAPS signal also overlapped with dominant antigenic epitopes, albeit to a lesser extent, sharing three of the six residues of BCE 2 and two of the nine residues of BCE 3 (Figure 7d). Therefore, as with the promiscuous MHCII-binding ZikVE peptides, we observed considerable overlap between the promiscuous MHCII-binding VP7 peptides and antibody neutralization sites on the folded VP7 protein (Figure 7c). These surprising results hint at a deeper relationship between acquired B cell and T cell specificity during viral infection.
4 |. DISCUSSION
Viral infections and their vaccination are often studied in the context of a neutralizing antibody response; however, CD4+ T cells also play a critical role in viral immunity.61 Therefore, identifying T cell epitopes is an important part of understanding how viral antigens are processed and presented, as well as predicting which viral protein fragments are likely to be recognized by T cells.
Early findings have suggested that, in contrast to other flaviviruses in which T cell epitopes are primarily located on nonstructural proteins (NS), T cell epitopes in Zika virus appear to be located on structural proteins including the E protein.2,8 Here, we screened an overlapping peptide library derived from the Zika virus E protein for binding to four common human MHCII alleles and identified five promiscuous MHCII-binding peptides (ZikVE51–70, ZikVE131–150, ZikVE191–210, ZikVE311–330, ZikVE351–370). Interestingly, a 2018 study14 aiming to identify T cell epitopes in the Zika virus E protein found that ZikVE51–70, ZikVE131–150, and ZikVE191–210 existed as CD4+ T cell epitopes in HLA-DR4 transgenic mice primed with 25 μg of recombinant Zika envelope protein. Similarly, ZikVE131–150 and ZikVE311–330 were also shown to be CD4+ T cell epitopes in HLA-DR1 and HLA-DR15 transgenic mice, respectively.14 Further, when mice were challenged with Zika virus, the CD4+ T cell response to ZikVE351–370 was among the strongest as measured by IFNγ ELISpot. Therefore, all five of the promiscuous MHCII-binding ZikVE peptides identified using MAPS are indeed T cell epitopes in either humans or transgenic animal models. Moreover, while the aforementioned study14 included DR1, DR4, and DR15, our results confirm that these peptides (ZikVE51–70, ZikVE131–150, ZikVE191–210, ZikVE311–330, and ZikVE351–370) also bind DR7, which provides broader coverage to Hispanic and African American populations (10.5% and 9.8%, respectively).
In addition to identifying five promiscuous MHCII-binding ZikVE peptides, we observed a striking overlap between these peptides and previously reported antibody neutralization sites on the ZikVE protein5,57 (Figure 5c,d). Interestingly, a similar overlap between regions of the Dengue E protein recognized by CD4+ T cells and those targeted by IgG molecules has also been reported elsewhere.62 However, this paired antigen specificity does not appear to be unique to flaviviruses, as it was also observed in this study for the rotavirus VP7 protein (Figure 7b–d), and has been described elsewhere for a model E. coli antigen,63 suggesting a deeper relationship between B cell receptor (BCR) binding and antigen presentation to CD4+ T cells.64 One explanation for the shared B cell/T cell antigen specificity is that BCR bound residues on the antigenic protein are protected from degradation during B-cell antigen processing.64 If these BCR-protected residues are derived from a virus structural protein, they will likely be internalized and presented on MHCII during virus infections, which would preferentially expand CD4+ T cells of the same or similar specificity.62 Regardless of the exact mechanism, shared B/T cell specificity creates the possibility of predicting CD4+ T cell epitopes from known antibody neutralization sites and vice versa, which could be relevant to the design of highly immunogenic vaccines in the future.65
While we demonstrated the MAPS strategy using pathogenic peptide libraries, high-throughput methods for identifying MHCII-binding peptides are also relevant to the development of peptide-based tools and therapeutics for treating cancer,66,67 and autoimmune conditions.68 Ultra high-throughput computational tools have come a long way in the past two decades; however, reliably predicting peptide-MHCII binding in silico remains a challenge.33 This challenge arises largely from the open-ended peptide-binding groove of MHCII molecules, which allows for the presentation of variable-length peptides with a number of possible peptide conformations and secondary interactions that are difficult to predict. In addition, MHCII are characterized by substantial allelic diversity, which is manifested in the peptide-binding grooves of different MHCII alleles.
The MAPS strategy reported here provides a more accurate, high-throughput peptide-MHCII screening strategy that seeks to balance the throughput of computational systems and the quantitative detail of more conventional peptide-binding assays. MAPS is best suited for situations where a handful of pathogenic proteins with known sequences have been identified because MAPS scales linearly with the number of pathogenic proteins screened but is much higher throughput than conventional experimental methods used to identify peptide-MHCII binders.69 While quantitative binding data is critical to improving the accuracy of computational peptide-MHCII binding predictors, this data alone provides an incomplete picture of peptide-MHCII binding. Identification of the 9mer PBR core that facilitates the principle interaction between a peptide and an MHCII molecule is also important. In fact, PBR identification is one of the greatest challenges associated with predicting peptide-MHCII binding.54 Although the MAPS strategy described here was designed to rapidly identify promiscuous MHCII-binding peptides, the method used to detect peptide binding was found to be sensitive to the relative position of the PBR core within 20mer peptides (Figure 2c). While this unforeseen sensitivity complicates the translation of the observed MAPS signal to peptide-MHC binding affinity, it does provide some insight into the location of the PBR. As a result, MAPS coupled with quantitative measurements of peptide-MHCII binding affinity might provide a basis for the informed prediction of the PBR within a 20mer peptide.
The emergence of Zika virus and the consequent epidemiological crisis underscores the need for deployable, high-throughput systems capable of rapidly characterizing the immune responses to viral infection, which includes identifying relevant peptide-MHCII interactions. The MAPS strategy introduced here addresses such a need by providing a simple method for identifying immunologically relevant, promiscuous MHCII-binding peptides. Incorporation of the MAPS strategy into the vaccine development process could accelerate T cell epitope identification and expedite the development and manufacturing of novel vaccines.70 Beyond vaccine development, our observations indicate that MAPS and similar screening strategies should provide valuable insights relevant to broadly applicable peptide-based therapies, vaccines, and diagnostic tools necessary to protect ethnically diverse populations.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (NSF) CAREER Award 1653611, grants 1511720 and 1645229, the National Institutes of Health (NIH) grant OD020053, and the MCubed program at the University of Michigan. FW was additionally supported by the Cancer Center Support Grant (NIH P30 CA046592). The authors thank all group members from Dr. Fei Wen’s lab for their constructive feedback.
Funding information
Division of Chemical, Bioengineering, Environmental, and Transport Systems, Grant/Award Numbers: 1511720, 1645229, 1653611; University of Michigan; National Institutes of Health, Grant/Award Numbers: CA046592, OD020053; National Science Foundation
Footnotes
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Dai L, Song J, Lu X, et al. Structures of the Zika virus envelope protein and its complex with a Flavivirus broadly protective antibody. Cell Host Microbe 2016;19(5):696–704. [DOI] [PubMed] [Google Scholar]
- 2.Stettler K, Beltramello M, Espinosa DA, et al. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 2016;353(6301):823–826. [DOI] [PubMed] [Google Scholar]
- 3.Xu X, Vaughan K, Weiskopf D, et al. Identifying candidate targets of immune responses in Zika virus based on homology to epitopes in other Flavivirus species. PLoS Curr 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paul LM, Carlin ER, Jenkins MM, et al. Dengue virus antibodies enhance Zika virus infection. Clin Transl Immunol 2016;5(12):e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbiani DF, Bozzacco L, Keeffe JR, et al. Recurrent potent human neutralizing antibodies to Zika virus in Brazil and Mexico. Cell 2017; 169(4):597–609.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tai W, He L, Wang Y, et al. Critical neutralizing fragment of Zika virus EDIII elicits cross-neutralization and protection against divergent Zika viruses. Emerg Microbes Infect 2018;7(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pimentel-Coelho PM, Papa MP, Vale AM, et al. Critical role of CD4+ T cells and IFNγ signaling in antibody-mediated resistance to Zika virus infection. Nat Commun 2018;9(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lima NS, Rolland M, Modjarrad K, Trautmann L. T cell immunity and Zika virus vaccine development. Trends Immunol 2017;38(8): 594–605. [DOI] [PubMed] [Google Scholar]
- 9.Zellweger RM, Miller R, Eddy WE, White LJ, Johnston RE, Shresta S. Role of humoral versus cellular responses induced by a protective dengue vaccine candidate. PLoS Pathog 2013;9(10):e1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aid M, Abbink P, Larocca RA, et al. Zika virus persistence in the central nervous system and lymph nodes of rhesus monkeys. Cell 2017; 169(4):610–620.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mlakar J, Korva M, Tul N, et al. Zika virus associated with microcephaly. N Engl J Med 2016;374(10):951–958. [DOI] [PubMed] [Google Scholar]
- 12.Petersen E, Wilson ME, Touch S, et al. Rapid spread of Zika virus in the Americas--implications for public health preparedness for mass gatherings at the 2016 Brazil Olympic games. Int J Infect Dis 2016; 44:11–15. [DOI] [PubMed] [Google Scholar]
- 13.Lessler J, Chaisson LH, Kucirka LM, et al. Assessing the global threat from Zika virus. Science 2016;353(6300):aaf8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reynolds CJ, Suleyman OM, Ortega-Prieto AM, et al. T cell immunity to Zika virus targets immunodominant epitopes that show cross-reactivity with other Flaviviruses. Sci Rep 2018;8(1):672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 2015;43(D1):D423–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen F, Rubin-pitel SB, Zhao H. Engineering of therapeutic proteins. In: Park SJ, Cochran JR, eds. Protein Engineering and Design Boca Raton, FL: CRC Press, Taylor & Francis Group; 2009:153–177. [Google Scholar]
- 17.Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinformatics 2008;9(suppl 12):S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dar H, Zaheer T, Rehman MT, et al. Prediction of promiscuous T-cell epitopes in the Zika virus polyprotein: an in silico approach. Asian Pac J Trop Med 2016;9(9):844–850. [DOI] [PubMed] [Google Scholar]
- 19.Sturniolo T, Bono E, Ding J, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol 1999;17(6):555–561. [DOI] [PubMed] [Google Scholar]
- 20.Hill BD, Zak A, Khera E, Wen F. Engineering virus-like particles for antigen and drug delivery. Curr Protein Pept Sci 2017;19(1): 112–127. [DOI] [PubMed] [Google Scholar]
- 21.Wen F, Zhao H. Construction and screening of an antigen-derived peptide library displayed on yeast cell surface for CD4+ T cell epitope identification. Methods Mol Biol 2013;1061:245–264. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics 2007;8:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reche PA, Glutting J-P, Reinherz EL. Prediction of MHC class I binding peptides using profile motifs. Hum Immunol 2002;63(9):701–709. [DOI] [PubMed] [Google Scholar]
- 24.Kumar N, Mohanty D. MODPROPEP: a program for knowledge-based modeling of protein-peptide complexes. Nucleic Acids Res 2007;35(suppl 2):549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hattotuwagama CK, Doytchinova IA, Flower DR. Toward the prediction of class I and II mouse major histocompatibility complex-peptide-binding affinity. In: Flower DR, ed. Immunoinformatics Totowa, NJ: Humana Press; 2007:227–245. [DOI] [PubMed] [Google Scholar]
- 26.Liao WWP, Arthur JW. Predicting peptide binding to major histocompatibility complex molecules. Autoimmun Rev 2011;10(8):469–473. [DOI] [PubMed] [Google Scholar]
- 27.Huang M, Huang W, Wen F, Larson RG. Efficient estimation of binding free energies between peptides and an MHC class II molecule using coarse-grained molecular dynamics simulations with a weighted histogram analysis method. J Comput Chem 2017;38(23): 2007–2019. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen M, Lundegaard C, Worning P, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 2003;12(5):1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics 2009;10:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreatta M, Karosiene E, Rasmussen M, Stryhn A, Buus S, Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015;67(11–12):641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo H, Ye H, Ng HW, et al. Machine learning methods for predicting HLA-peptide binding activity. Bioinform Biol Insights 2015;9(suppl 3): 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen M, Lund O, Buus S, Lundegaard C. MHC class II epitope predictive algorithms. Immunology 2010;130(3):319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol 2008;4(4): e1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen KK, Andreatta M, Marcatili P, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018;154(3):394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vita R, Overton JA, Greenbaum JA, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res 2015;43(Database issue):D405–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Justesen S, Harndahl M, Lamberth K, Nielsen L-LB, Buus S. Functional recombinant MHC class II molecules and high-throughput peptide-binding assays. Immunome Res 2009;5(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jensen PE, Moore JC, Lukacher AE. A europium fluoroimmunoassay for measuring peptide binding to MHC class I molecules. J Immunol Methods 1998;215(1–2):71–80. [DOI] [PubMed] [Google Scholar]
- 38.Yin L, Stern LJ. Measurement of peptide binding to MHC class II molecules by fluorescence polarization. Curr Protoc Immunol 2014;106 (1):5.10.1–5.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidney J, Southwood S, Oseroff C, del Guercio MF, Sette A, Grey HM. Measurement of MHC/peptide interactions by gel filtration. Curr Protoc Immunol 2001;31(1):18.3.1–18.3.19. [DOI] [PubMed] [Google Scholar]
- 40.Zarutskie JA, Busch R, Zavala-Ruiz Z, Rushe M, Mellins ED, Stern LJ. The kinetic basis of peptide exchange catalysis by HLA-DM. Proc Natl Acad Sci USA 2001;98(22):12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joshi RV, Zarutskie JA, Stern LJ. A three-step kinetic mechanism for peptide binding to MHC class II proteins. Biochemistry 2000;39(13): 3751–3762. [DOI] [PubMed] [Google Scholar]
- 42.Narayan K, Su KW, Chou CL, Khoruzhenko S, Sadegh-Nasseri S. HLA-DM mediates peptide exchange by interacting transiently and repeatedly with HLA-DR1. Mol Immunol 2009;46(15):3157–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang W, Boder ET. High-throughput engineering and analysis of peptide binding to class II MHC. Proc Natl Acad Sci USA 2010;107(30): 13258–13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernardeau K, Kerzhero J, Fortun A, et al. A simple competitive assay to determine peptide affinity for HLA class II molecules: a useful tool for epitope prediction. J Immunol Methods 2011;371 (1–2):97–105. [DOI] [PubMed] [Google Scholar]
- 45.Kalandadze A, Galleno M, Foncerrada L, Strominger JL, Wucherpfennig KW. Expression of recombinant HLA-DR2 molecules. Replacement of the hydrophobic transmembrane region by a leucine zipper dimerization motif allows the assembly and secretion of soluble DR alpha beta heterodimers. J Biol Chem 1996;271(33):20156–20162. [DOI] [PubMed] [Google Scholar]
- 46.Jang M-H, Seth NP, Wucherpfennig KW. Ex vivo analysis of thymic CD4 T cells in nonobese diabetic mice with tetramers generated from I-A(g7)/class II-associated invariant chain peptide precursors. J Immunol 2003;171(8):4175–4186. [DOI] [PubMed] [Google Scholar]
- 47.Day CL, Seth NP, Lucas M, et al. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest 2003;112(6):831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang P, Sidney J, Kim Y, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 2010;11:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oseroff C, Sidney J, Kotturi MF, et al. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol 2010;185(2):943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. [Google Scholar]
- 51.Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46(W1):W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aoki ST, Settembre EC, Trask SD, Greenberg HB, Harrison SC, Dormitzer PR. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009;324(5933):1444–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith MR, Tolbert SV, Wen F. Protein-scaffold directed nanoscale assembly of T cell ligands: artificial antigen presentation with defined valency, density, and ratio. ACS Synth Biol 2018;7(6):1629–1639. [DOI] [PubMed] [Google Scholar]
- 54.Maiers M, Gragert L, Klitz W. High-resolution HLA alleles and haplotypes in the United States population. Hum Immunol 2007;68(9):779–788. [DOI] [PubMed] [Google Scholar]
- 55.Natarajan SK, Stern LJ, Sadegh-Nasseri S. Sodium dodecyl sulfate stability of HLA-DR1 complexes correlates with burial of hydrophobic residues in pocket 1. J Immunol 1999;162(6):3463–3470. [PubMed] [Google Scholar]
- 56.Reynolds C, Goudet A, Jenjaroen K, et al. T cell immunity to the alkyl hydroperoxide reductase of Burkholderia pseudomallei: a correlate of disease outcome in acute melioidosis. J Immunol 2015;194(10):4814–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barba-Spaeth G, Dejnirattisai W, Rouvinski A, et al. Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 2016; 536(7614):48–53. [DOI] [PubMed] [Google Scholar]
- 58.Madhi SA, Cunliffe NA, Steele D, et al. Effect of human rotavirus vaccine on severe diarrhea in African infants. Malawi Med J 2016;28(3): 108–114. [PMC free article] [PubMed] [Google Scholar]
- 59.Ciarlet M, Schödel F. Development of a rotavirus vaccine: clinical safety, immunogenicity, and efficacy of the pentavalent rotavirus vaccine, RotaTeq. Vaccine 2009;27(suppl 6):G72–G81. [DOI] [PubMed] [Google Scholar]
- 60.Ward RL, Bernstein DI. Rotarix: a rotavirus vaccine for the world. Clin Infect Dis 2009;48(2):222–228. [DOI] [PubMed] [Google Scholar]
- 61.Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4+ T cells in immunity to viruses. Nat Rev Immunol 2012;12(2):136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rivino L, Kumaran EAP, Jovanovic V, et al. Differential targeting of viral components by CD4+ versus CD8+ T lymphocytes in dengue virus infection. J Virol 2013;87(5):2693–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manca F, Kunkl A, Fenoglio D, Fowler A, Sercarz E, Celada F. Constraints in T-B cooperation related to epitope topology on E. coli beta-galactosidase. I. The fine specificity of T cells dictates the fine specificity of antibodies directed to conformation-dependent determinants. Eur J Immunol 1985;15(4):345–350. [DOI] [PubMed] [Google Scholar]
- 64.Lanzavecchia A Antigen-specific interaction between T and B cells. Nature 1983;314(6011):537–539. [DOI] [PubMed] [Google Scholar]
- 65.Sette A, Moutaftsi M, Moyron-Quiroz J, et al. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity 2008;28(6):847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363 (5):411–422. [DOI] [PubMed] [Google Scholar]
- 67.Banchereau J, Palucka a K. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 2005;5(4):296–306. [DOI] [PubMed] [Google Scholar]
- 68.Larché M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat Med 2005;11(suppl 4):S69–S76. [DOI] [PubMed] [Google Scholar]
- 69.Wen F, Esteban O, Zhao H. Rapid identification of CD4+ T-cell epitopes using yeast displaying pathogen-derived peptide library. J Immunol Methods 2008;336(1):37–44. [DOI] [PubMed] [Google Scholar]
- 70.Yee CM, Zak AJ, Hill BD, Wen F. The coming age of insect cells for manufacturing and development of protein therapeutics. Ind Eng Chem Res 2018;57(31):10061–10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.