

Abstract
Leukotrienes (LT) are lipid mediators of the inflammatory response that are linked to asthma and atherosclerosis. LT biosynthesis is initiated by 5-lipoxygenase (5-LOX) with the assistance of the substrate-binding 5-LOX-activating protein at the nuclear membrane. Here, we contrast the structural and functional consequences of the binding of two natural product inhibitors of 5-LOX. The redox-type inhibitor nordihydroguaiaretic acid (NDGA) is lodged in the 5-LOX active site, now fully exposed by disordering of the helix that caps it in the apo-enzyme. In contrast, the allosteric inhibitor 3-acetyl-11-keto-beta-boswellic acid (AKBA) from frankincense wedges between the membrane-binding and catalytic domains of 5-LOX, some 30 Å from the catalytic iron. While enzyme inhibition by NDGA is robust, AKBA promotes a shift in the regiospecificity, evident in human embryonic kidney 293 cells and in primary immune cells expressing 5-LOX. Our results suggest a new approach to isoform-specific 5-LOX inhibitor development through exploitation of an allosteric site in 5-LOX.
The enzyme 5-lipoxygenase (5-LOX) initiates the biosynthesis of leukotrienes (LTs), potent mediators of the inflammatory response, with a two-step reaction that starts with oxygenation of the substrate arachidonic acid (AA) to generate the intermediate 5-S-hydroperoxyeicosatetraenoic acid (5-S-HPETE)1–3. A second hydrogen abstraction converts 5-S-HPETE to LTA4 (ref.4). These reactions occur at the nuclear membrane, where 5-LOX acquires substrate from its partner 5-LOX-activating protein (FLAP), a membrane-embedded AA-binding trimer5,6. The presence of FLAP ensures that the two-step reaction goes to completion7,8. Both FLAP and 5-LOX have been identified as drug targets, and inhibitors that target FLAP can effectively shut down LT synthesis in a cellular context9–11.
Mammals express multiple LOXs12; each isoform of the Fe2+ enzymes generates a distinct product from the common substrate AA by positioning the central carbon of one of the three substrate pentadienes for hydrogen abstraction13,14. The free radical generated by the catalytic machinery is oxygenated on the antarafacial site of the substrate, at a position two carbons from the site of attack. Thus, 5-LOX must attack at C(7) to generate the 5-hydroperoxy isomer of AA (that is, 5-S-HPETE). Although LOX isoforms may differ with respect to the pentadiene flanked by the catalytic Fe2+ and O2 pocket (such that the resulting oxygenation can yield 5-, 12- or 15-HPETE), the core active site that cradles the targeted pentadiene is invariant15,16. The high level of conservation presents a challenge to the development of 5-LOX-specific inhibitors.
Here, we report crystal structures of human Stable-5-LOX, which harbors mutations that prolong enzyme activity and improve solubility and stability17, with two well-recognized natural product inhibitors: NDGA, a nonselective antioxidant that keeps the active site iron in the resting inactive Fe2+ state18 and AKBA, a noncompetitive inhibitor of 5-LOX19,20. AKBA, a pentacyclic triterpene acid, is a bioactive ingredient of frankincense with well-documented anti-inflammatory properties21–23. Mechanistic details of the inhibitor modes of action may reveal new vulnerabilities to be exploited for modulation of LT biosynthesis and thus, open new avenues for isoform-specific inhibitor design. While the redox inhibitor NDGA induces substantial structural disorder in the peptide regions that shield the active site, the AKBA-binding allosteric site is wedged between the membrane-binding and catalytic domains. The limited conformational changes induced by AKBA have distal structural consequences at the active site, some 30 Å away.
Cell-free assays of 5-LOX activity do not fully recapitulate its cellular activity24. In intact cells, the 5-LOX/FLAP interaction is essential for the two-step reaction to go to completion, while under cell-free conditions the presence of FLAP is inconsequential. We therefore monitored enzyme activity in the presence of these inhibitors in both human embryonic kidney 293 (HEK293) cells and primary immune cells that express 5-LOX and FLAP. We find that while the cellular inhibition data for NDGA are predicted by cell-free activity data, the results with AKBA suggest that the compound invokes a shift of the regiospecificity of 5-LOX. Our data suggest that AKBA-induced allosteric modulation of 5-LOX activity in the cellular context leads not only to diminished LT levels, but alters the regiospecificity toward a 12/15-lipoxygenating enzyme.
Results
Crystal structure of Stable-5-LOX with NDGA.
The crystal structure of Stable-5-LOX, which lacks membrane insertion loops and harbors stabilizing mutations, revealed the canonical LOX fold: an amino-terminal β-barrel membrane-binding domain and a larger α-helical domain that harbors the catalytic iron, which is located at the base of a U-shaped hydrophobic cavity. While the active site cavity in 5-LOX is ‘corked’ by the side chains Phe177 and Tyr181, the core features of the characteristic U-shaped cavity are apparent16,17. Conserved amino acids from two helices help define the active site: helix-α2, which in 5-LOX contributes the ‘cork’ that blocks access to the catalytic iron, and the ‘arched helix’, which shelters the active site and contributes an invariant Leu (5-LOX, 414) that appears to have a role in positioning the substrate pentadiene for attack.
NDGA is one of several plant polyphenols that inhibit LOXs due to antioxidant properties25. These compounds are thought to bind in the active site and prevent the conversion of Fe2+ to Fe3+ to activate the enzyme24. The crystal structure of Stable-5-LOX in complex with NDGA was determined to 2.71 Å resolution (Fig. 1a,b and Supplementary Table 1) with X-ray data collected from crystals soaked in 500 μM inhibitor. These crystals were isomorphous with those for Stable-5-LOX in the absence of inhibitor. Substantial portions of the peptide chain could not be modeled due to the lack of interpretable electron density, but density consistent with one catechol ring and the linker region of NDGA was visible in a 3σ omit map (Supplementary Fig. 1) in one monomer in the asymmetric unit and allowed its positioning for further refinement of the cocrystal structure. The missing peptide regions include amino acids 173–214, which contain helix-α2 and the active-site corking residues Phe177 and Tyr181. There is additional disorder that begins at the apex of the arched helix (416–429) that spans the active site and provides an invariant Leu to clamp the targeted pentadiene of AA for attack. Amino acids 294–303 are also disordered, but their displacement from positions in the apo structure appears to be a downstream consequence of active-site remodeling induced by the rigid inhibitor.
Fig. 1 |. NDGA induces disorder at the active site of Stable-5-LOX.
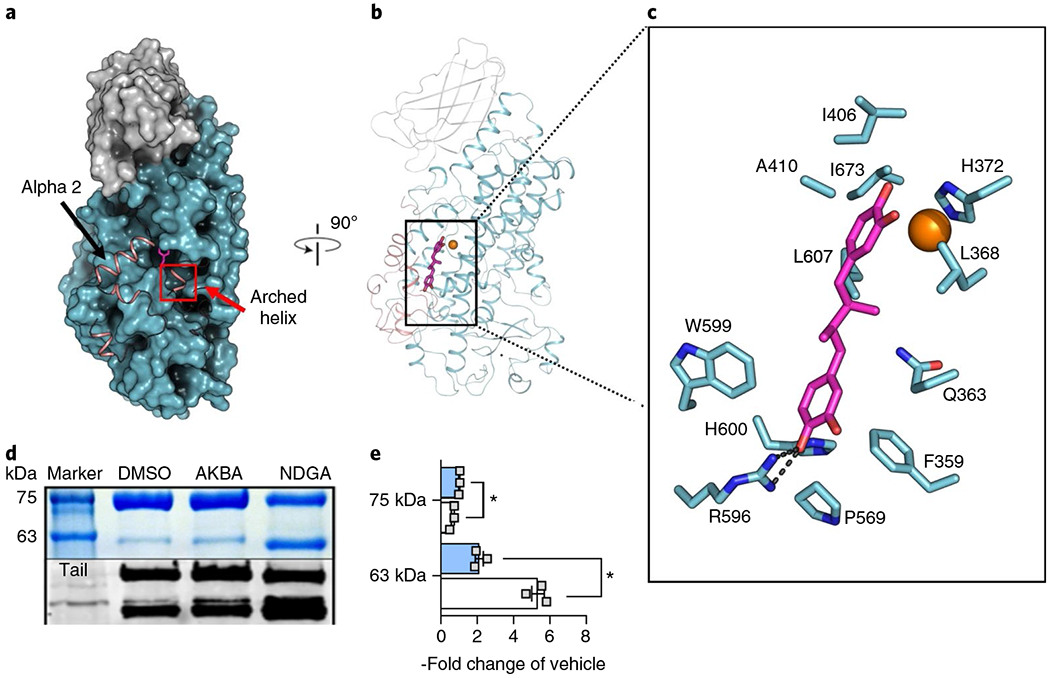
a, A surface rendering of Stable-5-LOX with the amino-terminal domain in gray and catalytic domain in cyan. Electron density is not observed for the polypeptide in the salmon ribbon, modeled from the apo structure (3O8Y). NDGA (magenta stick rendering) is visible in an open cavity. b, The corresponding ribbon diagram rotated 90° with the catalytic Fe as an orange sphere. c, NDGA (C, magenta; O, red) and surrounding amino acids (C, cyan; N, blue; O, red) within 4 Å of the inhibitor, as identified with LigPlot v.2.1 (ref. 49). A single polar contact is provided by R596. d, Proteolysis of Stable-5-LOX in the absence and presence of inhibitors. DMSO as vehicle, AKBA or NDGA were incubated with Stable-5-LOX and the samples were subjected to limited proteolysis using pepsin. The SDS gel (above, see also Supplementary Fig. 3) and western blot (below) of proteolysis products reveal a 63 kD fragment recognized by an antibody to the 5-LOX C-terminus, consistent with cleavage in the region of helix αlpha2. e, Bar graph (+AKBA, blue; + NDGA, white) of band intensity data shown as the fold change of vehicle control from three independent experiments, Student’s paired t-test, *P < 0.05 AKBA versus NDGA.
The NDGA-induced remodeling exposes a deep ravine in the catalytic domain. NDGA tenuously bridges this ravine, tethered only by Arg596, with nominal protein-ligand contacts (Fig. 1a,c). One catechol ring sits above the iron and blocks access to the open position of its coordination sphere. His372, one of the iron ligands, is the only side chain that may be close enough to H-bond to it (3.3 Å). At the opposite end, Arg596 is positioned to H-bond with a catechol hydroxyl (2.8 Å, Fig. 1c and Supplementary Fig. 2) of the ring first observed in the omit map. There are limited van der Waals contacts between the protein and the ligand, and the linker that joins the catechol rings is solvent exposed. The presence of NDGA is incompatible with the closed structure of Stable-5-LOX. Thus, the corking residues Phe177 and Tyr181 must move out of the way to make room for a catechol ring to sit above the iron. Likewise, at the opposite end Phe421 and Asn425, which sit atop the innermost prong of the putative U-shaped cavity that accommodates the AA carboxyl, are no longer visible in the electron density. The second catechol has pushed them out of the way and triggered disorder in this region, the roof of the active site.
It is difficult to envision how substrate might enter the active site of Stable-5-LOX without a conformational change in the α2-corking segment26. In an effort to corroborate the extensive disordering induced by NDGA in the crystal structure, we asked whether the enzyme is more susceptible to proteolysis in the presence of NDGA. On incubation of the enzyme (78 kD) with NDGA and pepsin we observed a fragment of ~63 kD that results from cleavage of the amino-terminal region, as it is recognized by an antibody generated against the C-terminal 12 amino acids (Fig. 1d and Supplementary Fig. 3)27. This fragment is consistent with loss of a 15 kD fragment, placing the cut site near helix-α2. In triplicate experiments the 63 kD fragment is detected at ~5 × (5.4 ± 0.5) the intensity observed in the presence of carrier only (Fig. 1e). In contrast, a significant increase in susceptibility to proteolysis is not observed when the substrate AA is included. Nor does the LOX inhibitor caffeic acid, which has a single catechol group and is roughly half the size of NDGA, increase proteolytic susceptibility (Supplementary Fig. 3). NDGA is bulky and constrained; it is highly likely that nonnative conformational changes are necessary to accommodate it in a U-shaped active site. Moreover, the presence of NDGA, but not of AKBA, significantly lowers the temperature at which Stable-5-LOX unfolds in a florescence-based thermal denaturation assay28 (47.6 ± 1.5, 52.1 ± 0.4, 55.8 ± 0.2 °C for +NDGA, + AKBA, + DMSO carrier only, respectively). AKBA is almost twice the size of NDGA (512.7 versus 302.4 Da), but does not induce extensive disordering of the enzyme structure, as described next. Nor did AKBA dramatically increase the enzyme susceptibility to protease, yielding a 63 kD band only ~2 × (2.1 ± 0.4) the intensity.
Crystal structure of Stable-5-LOX with AKBA.
The crystal structure of 5-LOX in the presence of AKBA was determined with data obtained from crystals that had been soaked in 160 μM inhibitor. The electron density map revealed a bulky volume of density (Supplementary Fig. 4), consistent with a pentacyclic triterpene, wedged between the two domains in one monomer in the asymmetric unit. After manual rebuilding of the four molecules in the asymmetric unit and positioning AKBA into the additional density, the model was refined to 3.0 Å resolution (Fig. 2 and Supplementary Table 1).
Fig. 2 |. AKBA is wedged between the two domains of Stable-5-LOX.
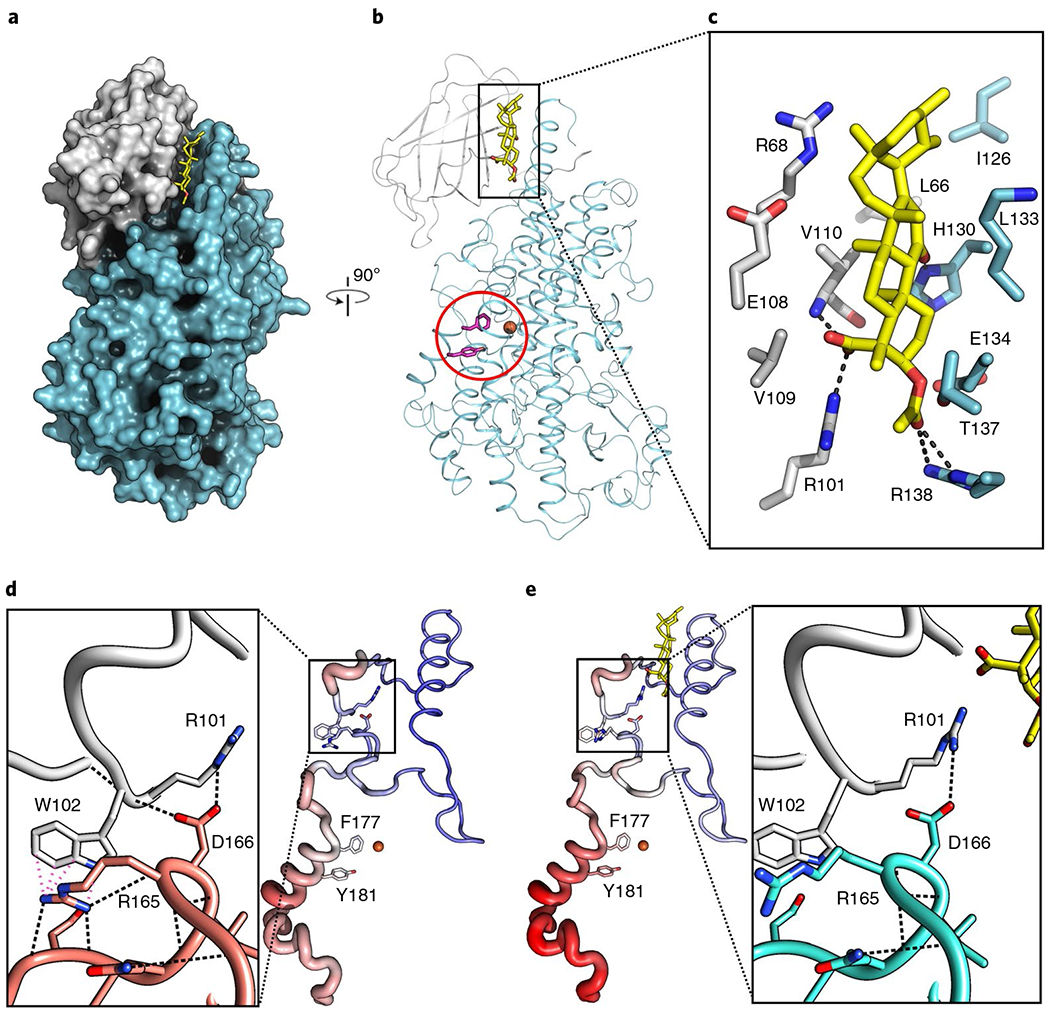
a, A surface rendering of Stable-5-LOX, with the amino-terminal domain in gray and catalytic domain in cyan. AKBA (stick rendering, C, yellow; O, red) lies in a crevice between the two domains. b, The corresponding ribbon diagram, rotated 90°, with the catalytic Fe as an orange sphere and the amino acids that close off the active site in stick rendering (magenta, Phe177, Tyr181). c, Stick rendering of amino acids that form the AKBA-binding site. d,e, Detail of the domain interface in Stable-5-LOX (3O8Y) (d) and the AKBA-bound structure (e). H-bond and cation–π interactions according to Chimera50 are shown as black and pink dashed lines, respectively. The loss of these interactions may contribute to increased B factors in helix-α2, which covers the 5-LOX active site. Tube renderings are of amino acids 100–210 in the apo-(d) and AKBA-occupied structures (e). Tube diameter increases with increasing B factor, accompanied by coloring changes from blue (cool) to red (hot).
AKBA lies lengthwise in a deep groove between the amino-terminal and catalytic domains (Fig. 2a,b). The A ring of AKBA, which carries carboxy and acetoxy groups, is innermost in a distorted chair conformation. The C(3) acetoxy group is in a distorted equatorial position and makes polar contacts with Arg101, Thr137 and Arg138, and the main chain of Val110 (Fig. 2c and Supplementary Fig. 5). The AKBA carboxylate-Arg101 charge pair is shielded by the side chain of Val109. His130 makes a close contact to the keto group on ring C. Leu66, Val110, Ile126 and the methylene carbons of Lys133 contribute hydrophobic surfaces to the groove. The groove is just short of being deep enough to fully obscure the sides of AKBA, and one edge is surface exposed. Roughly 80% of the accessible surface area of AKBA is buried in the complex. For the most part, there are only modest adjustments in side chain positions in the AKBA-bound structure, as a deep groove at the domain interface is a consistent feature of LOXs. A notable displacement induced by the binding of AKBA is disruption of a cation–π interaction between Trp102 of the amino-terminal domain and Arg165 of the catalytic domain (Fig. 2d,e). This highly conserved cation–π interaction has been proposed to mediate interdomain communication29. In addition, another interdomain restraint that modulates activity (the charge pair Arg101, Asp166, ref. 30) is disturbed by the presence of AKBA.
While the conformational changes induced by AKBA appear to be minimal, the loss of the cation–π interaction disrupted intramolecular contacts in the catalytic domain, specifically in the region that includes the aromatic ‘cork’ (Phe177, Tyr181) that seals the active site. One can see in Fig. 2d,e that the polypeptide segment from 100–210 displays more thermal motion, as defined by crystallographic B factors, in AKBA-bound Stable-5-LOX than in the apo-enzyme. The B factors in the segments in question in the latter (three independent copies) are at most 11% higher than the overall protein B factor. The corresponding region in the AKBA-bound copy displays B factors 34% higher. This same pattern of high B factors in the AKBA-bound molecule was observed regardless of the refinement strategy used, leading us to infer that the observed increase in mobility is a consequence of the presence of AKBA. Wedged between the catalytic and membrane-binding domains, AKBA is positioned to affect interdomain mobility and any conformational changes that require it.
AKBA-inhibition of Stable-5-LOX activity was assessed with mutants designed to test the binding site revealed in the crystal structure. Arg101 and His130 are positioned to interact with the AKBA carboxylate and C-ring keto group, respectively, and replacement of these residues with Ala did not compromise enzyme activity; yet both Ala mutants were more sensitive to AKBA than the progenitor protein (Supplementary Fig. 6). In stopped-flow assay conditions when 5-HPETE production is monitored, Stable-5-LOX retains 75% activity at 50 μM AKBA, while the H130A and R101A mutants have ~50% activity or less. While one might expect that loss of amino acids that make polar contacts with the ligand will lead to diminished inhibitor susceptibility, the results are indeed consistent with the X-ray structure. As mentioned above, AKBA binding disrupted an interdomain salt-link between Arg101 and Asp166. Thus, although AKBA has lost a contact site with the R101A variant, there is no need to break the interdomain salt-link, which was previously shown to restrain enzyme activity31. An increased susceptibility for the H130A substitution is likely a consequence of the close contact between the AKBA C-ring keto group and the side chain; replacement of His130 by Ala creates a spacious aqueous cavity that alleviates steric repulsion. Consistent with the binding cavity depicted in Fig. 3a, both mutations alter sensitivity to AKBA. However, they are not essential for AKBA to settle into the cavernous interdomain crevice. Note that a similarly prepared surface rendering for the NDGA site (Fig. 3b) illustrates the absence of a well-defined cavity complementary in shape to that inhibitor.
Fig. 3 |. The AKBA- and NDGA- binding sites in 5-LOX.
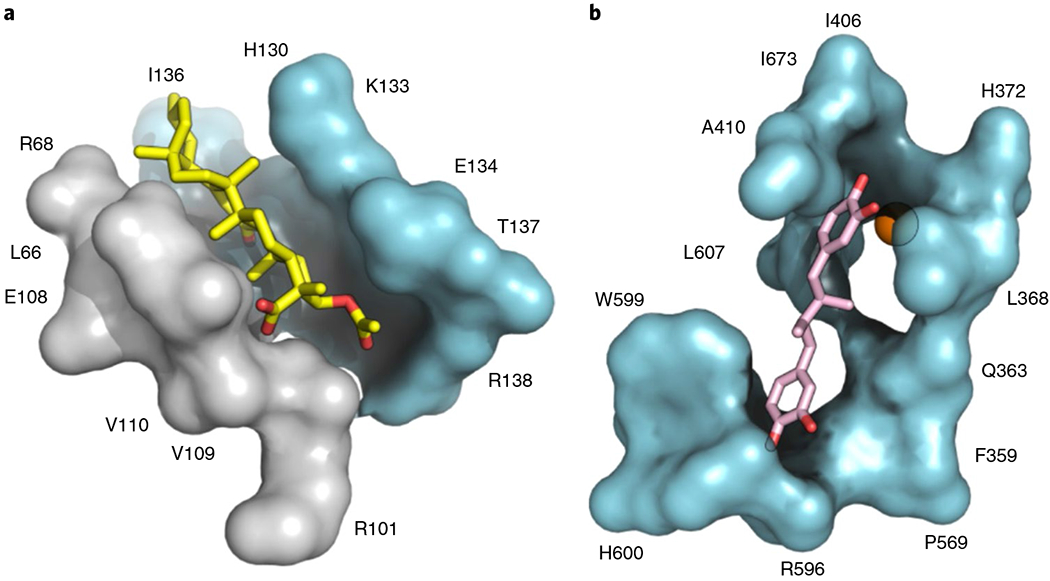
a, AKBA (stick; C, yellow and O, red) binds lengthwise in an interdomain crevice of 5-LOX, and while not fully encapsulated makes multiple hydrophobic and polar contacts along the interface and is poised to affect interdomain flexibility. b, The cavity of 5-LOX that houses NDGA (stick; C, pink and O red) is open to solvent and does not effectively envelop the inhibitor.
AKBA invokes a regioselectivity shift of 5-LOX.
We next analyzed how NDGA and AKBA might modulate the activity of human recombinant 5-LOX by monitoring the formation of 5-hydroxyeicosatetraenoic acid (5-HETE, derived from 5-HPETE) and trans-isomers of LTB4 (derived from the breakdown of LTA4) as classical enzymatic products formed from AA. The formation of these products from 10 μM AA by isolated human recombinant 5-LOX was concentration-dependently inhibited by AKBA (10–50 μM) as well as by NDGA (0.1–10 μM) (Fig. 4), with more marked efficiency by NDGA. We also observed 12- and 15-HETE as very minor byproducts of the 5-LOX reaction. AKBA caused a significant increase in the formation of 12-HETE with the most prominent effect at 10 μM, while levels of 15-HETE were not affected. In contrast, NDGA inhibited the generation of 12- and 15-HETE in a concentration-dependent manner (Fig. 4).
Fig. 4 |. AKBA modulates the enzymatic activity of purified human 5-LOX in a cell-free assay.
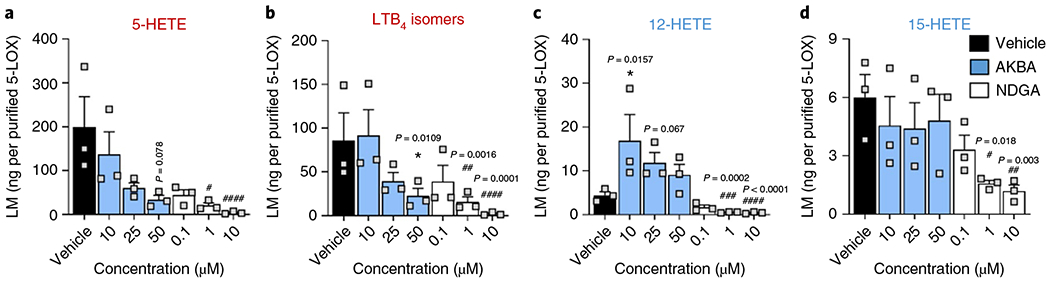
Purified human recombinant 5-LOX (0.5 μg ml−1 PBS pH 7.4 plus 1 mM EDTA) was preincubated with increasing concentrations of AKBA (blue), NDGA (white) or with vehicle (0.1% DMSO, black) on ice for 15 min. The samples were incubated with 2 mM CaCl2 and 10 μM AA at 37 °C. a-d, After 10 min, the reaction was stopped on ice and formed 5-LOX products (lipid mediators (LM) 5-HETE (a); LTB4 isomers (b); 12-HETE (c) and 15-HETE (d)) were analyzed by UPLC-MS/MS. Data obtained in n = 3 independent experiments were log-transformed for statistical analysis; *P < 0.05 AKBA versus vehicle control; #P < 0.05, ##P < 0.01, ###P < 0.001 NDGA versus vehicle control; ANOVA and Dunnett’s multiple comparisons test.
Several LT biosynthesis inhibitors display divergent efficiencies in cells as compared to cell-free 5-LOX activity assays, seemingly due to the complex regulation of 5-LOX in the cellular context2,31. To fully evaluate the impact of NDGA and AKBA on 5-LOX activity in a cellular environment, formation of 5-LOX-derived LTB4, LTB4 isomers and 5-HETE were analyzed in HEK293 cells stably coexpressing 5-LOX and FLAP32 by using an ultrahigh-performance liquid chromatography–tandem mass spectroscopy (UPLC–MS/MS) approach33. HEK293 cells do not express appreciable amounts of 5-, 12- or 15-LOX enzymes per se (Supplementary Fig. 7) and do not produce notable LOX-derived lipid mediators (LMs)32. NDGA (1 μM) potently inhibited the formation of all 5-LOX products in HEK293 cells transfected with 5-LOX and FLAP and stimulated with Ca2+-ionophore A23187 plus 1 μM of exogenous AA for 15 min (Fig. 5a). Also, AKBA (25 μM) significantly reduced formation of all 5-LOX products, especially LTA4-derived LTB4 and its isomers, which is in agreement with IC50 values < 10 μM observed for cell-free assays with human recombinant 5-LOX and human neutrophils34. As observed for isolated 5-LOX, concomitant with the loss of the canonical 5-LOX products, we found a significant increase in 12-HETE, but no significant modulation of 15-HETE biosynthesis, with AKBA (Fig. 5a). In contrast, NDGA did not cause an increase in 12-HETE or 15-HETE formation. To determine whether the modulatory effects of AKBA are unique to 5-LOX, we analyzed the LM profiles of AKBA-treated HEK293 cells expressing platelet-type 12-LOX, 15-LOX-1 or 15-LOX-2. Here, AKBA did not affect LM formation of these LOXs and the AKBA-induced production of 12-HETE was solely evident for 5-LOX-expressing cells (Fig. 5b). Again, NDGA failed to increase 12-HETE formation regardless of the LOX isoform expressed. Thus, the allosteric regulation of 5-LOX by AKBA appears to be isoform-specific and exclusive for AKBA. Moreover, FLAP does not play a principal role in either the inhibition of LT formation or stimulation of 12-HETE production by AKBA, as LM analysis of HEK293 cells expressing only 5-LOX revealed the same pattern of modulation: diminished LT and 5-HETE levels but increased 12-HETE production (Supplementary Table 2). Finally, we found that the iron-ligand 5-LOX-selective inhibitor zileuton (3 μM) was able to prevent AKBA-induced 12-HETE formation in HEK293 cells expressing 5-LOX and FLAP (Supplementary Fig. 8), consistent with 5-LOX as the source of 12-HETE production. Note that both zileuton and AKBA alone only partially inhibited the formation of 5-LOX products, but their combination completely abolished 5-LOX activity.
Fig. 5 |. AKBA modulates LM formation in 5-LOX-expressing HEK293 cells.
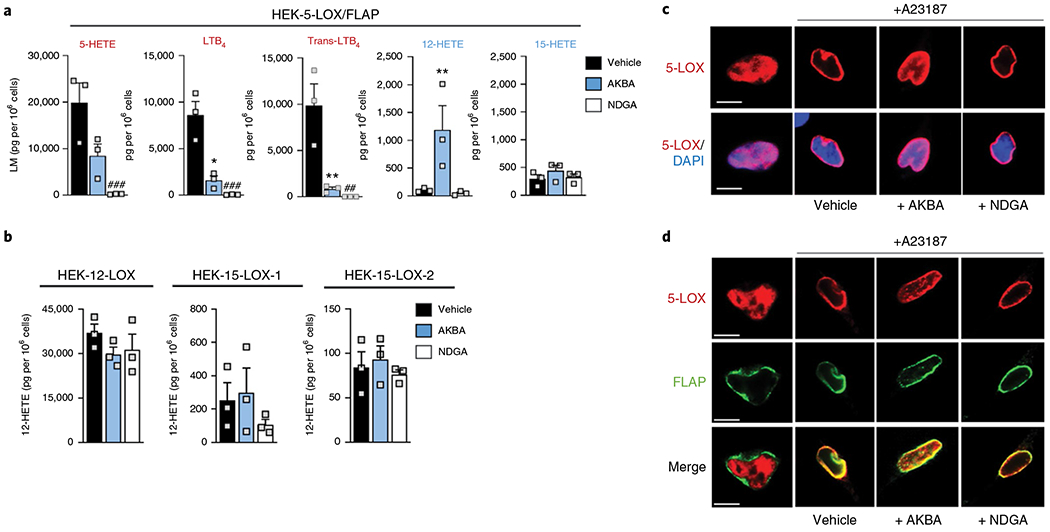
Stably transfected HEK293 cells (1 × 106) were pretreated with 25μM AKBA (blue), 1 μM NDGA (white) or vehicle (0.1% DMSO, black) for 5 min before stimulation with 2.5 μM A23187 plus 1 μM AA for another 15 min at 37°C. Cell supernatant was subjected to solid-phase extraction and UPLC-MS/MS analysis, and LM are shown in pg per 1 × 106 cells of n = 3 independent experiments. a, Effect of AKBA and NDGA on the LM profile of 5-LOX and FLAP-expressing cells. Data were log-transformed for statistical analysis *P < 0.05, **P < 0.01 AKBA versus vehicle control; #P < 0.05, ##P < 0.01, ###P < 0.001 NDGA versus vehicle control, ANOVA and Dunnett’s multiple comparisons test. b, Effect of AKBA and NDGA on 12-HETE biosynthesis by other LOX isoforms expressing cells. c,d, 5-LOX translocation in HEK293 expressing 5-LOX (c) or 5-LOX and FLAP (d). Cells were preincubated for 5 min with 25 μM AKBA, 1 μM NDGA or vehicle (0.1% DMSO) and then stimulated with 2.5 μM A23187 for another 15 min. Then, cells were fixed, permeabilized and incubated with antibodies against 5-LOX (red) and FLAP (green). Scale bars, 10μm. Results shown for one single cell are representative for approximately 100 individual cells analyzed in n = 3 independent experiments.
Since 5-LOX nuclear translocation and interaction with FLAP determines LT formation, and certain LT biosynthesis inhibitors interfere with these processes10,32, we studied how NDGA and AKBA would affect 5-LOX subcellular redistribution and colocalization with FLAP. A23187-induced 5-LOX translocation from the nucleoplasm to the nuclear membrane in HEK293 cells expressing 5-LOX and cells coexpressing 5-LOX and FLAP was impaired by AKBA but not by NDGA (Fig. 5c,d).
To corroborate the 5-LOX-modulatory features of AKBA in (patho-)physiologically relevant human innate immune cells, we used bacteria-challenged human neutrophils and monocyte-derived macrophages (MDM) that are prominent sources for LT production35,36. Neutrophils were preincubated with AKBA or NDGA and then challenged with pathogenic E. coli (serotype O6:K2:H1; ratio 1:50) for LM biosynthesis. Note that in contrast to HEK293 cells where single, selected LOXs were stably expressed, in neutrophil preparations endogenous 12-LOX and 15-LOX enzymes are present that generate 12-HETE and 15-HETE on cell stimulation. AKBA caused a similar 5-LOX modulation pattern in bacteria-stimulated neutrophils as compared to HEK293 cells: 5-HETE and LTB4 levels were significantly reduced while 12-HETE was significantly increased and 15-HETE biosynthesis was slightly elevated (Fig. 6a). As expected, NDGA blocked formation of 5-LOX-derived products and reduced 12-HETE/15-HETE production (Supplementary Table 3). Next, we investigated the effects of AKBA on LOX activities in human MDM with a pro-inflammatory M1 phenotype that markedly express 5-LOX/FLAP but also small amounts of 12/15-LOXs36. Preincubation of these M1-like MDM with AKBA clearly decreased LTB4 formation evoked by pathogenic E. coli (serotype O6:K2:H1; ratio 1:50). Again, as for neutrophils, AKBA (10 μM) stimulated 12-HETE biosynthesis in these MDM (Fig. 6b).
Fig. 6 |. Effects of AKBA on LM formation in human neutrophils and M1-like MDM activated with E. coli.
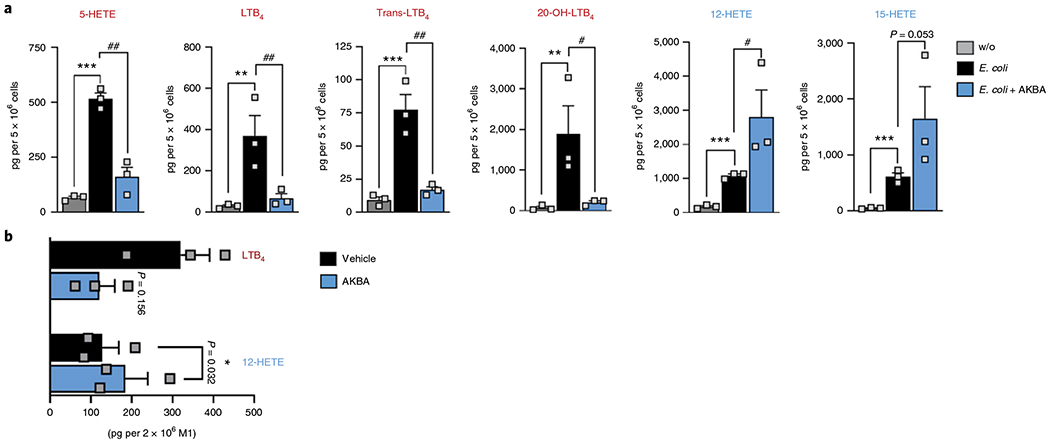
Cells were preincubated with 10 μM AKBA or vehicle (0.1% DMSO) for 5 min before stimulation with E. coli (multiplicity of infection, 50) for 90 min at 37°C. Formed LM were isolated by solid-phase extraction and analyzed by UPLC-MS/MS. a, Data are shown as pg per 5 × 106 neutrophils as a bar chart of n = 3 independent experiments. Data were log-transformed for statistical analysis. **P < 0.01, ***P < 0.001, E. coli-stimulated cells versus unstimulated control; #P < 0.05, ##P < 0.01, AKBA versus vehicle in E. coli-stimulated cells; ANOVA + Dunnett’s multiple comparisons test. w/o, without. b, LTB4 and 12-HETE levels are shown as pg per 2 × 106 M1-like MDM in a bar chart of n = 3 independent experiments. Data were log-transformed for statistical analysis, *P < 0.05 AKBA versus vehicle in E. coli-stimulated MDM, Student’s paired t-test.
A possible consequence of a shift in regiospecificity.
5-LOX contributes to the biosynthesis of some of the specialized proresolving mediators (SPM) that encompass lipoxins, resolvins, maresins and protectins that promote resolution of inflammation3,36. SPM biosynthesis from AA and from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) also requires oxygenations by 12- and/or 15-LOXs36–38. The fact that 5-LOX can produce 12-HETE begs the question of whether AKBA might promote SPM formation by provoking a change in regiospecificity of 5-LOX toward a 12/15-lipoxygenating enzyme. Although 5-LOX-rich neutrophils, monocytes and macrophages may serve as suitable test systems, these cells express 12/15-LOXs, a feature that complicates their use to address the effect of an AKBA-induced product shift. To explore whether AKBA might promote SPM formation by 5-LOX, we monitored SPM formation in HEK293 cells transfected with 5-LOX and FLAP, which express neither notable amounts of 12- nor 15-LOX (Supplementary Fig. 7). As predicted, AKBA increased A23187-induced formation of some SPM in 5-LOX/FLAP-expressing HEK293 cells supplemented with AA, EPA and DHA (Supplementary Fig. 9). While overall SPM formation is very low, the change in the LM distribution profile is striking: the levels of LM that require C5 oxygenation by 5-LOX (for example, LTB4, LXA4, RvE1) are decreased by AKBA. In contrast, the levels of SPM that require the equivalent of a 12/15-lipoxygenation (MaR1, PD1, RvD1, RvD2 and RvD5) are increased by AKBA.
Discussion
These studies have revealed the structural basis for the distinct actions of the inhibitors NDGA and AKBA on human 5-LOX. Consistent with the proposed antioxidant mechanism of LOX inhibition25, NDGA was observed to bind in close proximity to the catalytic iron where it induced disordering of helical segments that contribute to the active site cavity. Access to the catalytic center requires a conformational change, but that provoked by the binding of NDGA is not likely to mimic that of the substrate. NDGA renders the enzyme susceptible to limited proteolysis that generates a C-terminal fragment consistent with cleavage in the vicinity of the helical segment that ‘corks’ the closed active site. Given the extensive disorder induced by NDGA (Fig. 1) and the limited contacts it makes with the protein (Fig. 3b), the structure of this enzyme–inhibitor complex does not reveal a pathway for engineering isoform-specific variations of the natural product. In contrast, AKBA is an allosteric modulator of 5-LOX that binds in a deep groove at the interface of the membrane-binding and catalytic domains (Fig. 3a). While it is not a potent inhibitor, it is specific for the 5-LOX isoform and the structure of the enzyme–inhibitor complex reveals ligand–protein contacts that might be optimized. The polar groups of AKBA are positioned to form H-bonds with the side chains of His130, Arg101 and Thr137, as well as with main chain atoms. The model is entirely consistent with the structure–activity relationship defined by Sailer et al.39 who demonstrated the importance of the C-ring keto group and A-ring carboxylate of AKBA. Subsequent work reported that a bulky reactive group appended to the A ring can be cross-linked to 5-LOX20. We see in our structure that this group can extend AKBA lengthwise and penetrate deeper into a cave-like gap between the two domains, as the domain interface is not tightly packed.
A similar interdomain groove is present in the homologous enzymes 15-LOX-1 (refs. 40,41) and 15-LOX-2 (ref. 42). 15-LOX-1 conserves two of three H-bonding residues (Supplementary Table 4), however, placement of the last β strand of its amino-terminal domain restricts the volume of the groove. In 15-LOX-2, each of the H-bond donors differs (Gln108 for Arg101, Gln136 for His130, Ala144 for Thr137) and the groove is constricted at the C-ring by a charge pair that spans it (Arg68, Glu140). Accordingly, we did not observe inhibition by AKBA at equivalent concentrations in HEK293 cells expressing 15-LOX-2 or 15-LOX-1.
The active site of 5-LOX is fully encapsulated in the crystal structure and the substrate entry path is unknown17. Moreover, the transformation of AA to LTA4 is a two-step reaction, requiring an active site that must accommodate both substrate and intermediate (that is, 5-HPETE). In addition, to convert endogenously supplied AA in the cell, 5-LOX must recognize its helper protein FLAP43–45 and interact with the nuclear membrane32. This expansive repertoire of molecular interactions may require more ‘flexibility’ in 5-LOX when compared to other more stable LOX homologs that seemingly do not interact with protein partners. Crystal structures of 8R-LOX with and without substrate revealed only minor conformational differences between the two states15,46. As with most animal LOX, 8R-LOX does not require a helper protein and performs a single catalytic reaction.
Our results suggest that when AKBA is bound to 5-LOX, the regiospecificity of the enzyme is compromised, particularly when AA is delivered to 5-LOX by FLAP in a cellular context. The simplest explanation for the observed decrease in 5-LOX products in the presence of AKBA is that the inhibitor hinders substrate access to the catalytic site of 5-LOX by stabilizing a closed conformation. Yet the concomitant 5-LOX-derived appearance of 12-HETE in the presence of AKBA, and to a lesser extent of 15-HETE, suggests that substrate does enter the active site and that AKBA provokes a change in regiospecificity. Can AKBA affect two seemingly disparate processes—substrate access and product specificity? By combining information from open-cavity LOX structures a robust model for a common U-shaped active site has been proposed16,26 and can be extrapolated to 5-LOX. For 5-LOX to adopt the ‘open’ structure an aromatic ‘cork’ must reposition. Upstream on the capping helix lies Arg165, which makes a cation–π interaction with Trp102 of the membrane-binding domain. This highly conserved cation–π interaction, a communication link between the two domains29, is disrupted in the AKBA-5-LOX structure. Given its role in corking the active site, subtle differences vicinal to helix-α2 could affect active site access and the enzyme’s interaction with FLAP45, which presumably must ‘deliver’ the substrate to an open cavity. Moreover, it can affect the regiospecificity of the enzyme by modulating the flexibility of this segment that must undergo a conformational change for substrate to be positioned at the catalytic machinery. The successive reactions catalyzed by 5-LOX (conversion of AA to 5-HPETE, which is subsequently transformed to LTA4) are both initiated by H abstraction from a pentadiene: in the first step 5-LOX attacks C(7), and in the second C(10)47. The 12-HPETE isomer would be generated if 5-LOX were to attack C(10) first. The presence of AKBA may simply allow a conformation of the active site in which C(10) is positioned between the active site iron and the O2 pocket that lie at the base of the ‘U-shaped’ site. Alternatively, the interaction with AKBA may compromise the active site in a way that both enzyme activity and regiospecificity are impaired. In either case, the AKBA allosteric site establishes a prototype for the design of inhibitors that function by restricting 5-LOX from accessing its most productive conformation(s) for generation of LT and downgrade the enzyme to a labile, low-fidelity LOX. Exploitation of the AKBA-binding groove may be a potent approach for isoform-specific modulation of activity and the loss of regiospecificity may provide a beneficial therapeutic strategy.
Allosteric inhibition of 5-LOX by AKBA occurred in the cell-free and in the cellular context, but the modulation of the regiospecificity and thus 12-/15-HETE production was more pronounced in intact cells. This may suggest that components such as 5-LOX-interacting proteins (for example, FLAP, coactosin-like protein, actin), diacylglycerols, membranes or regulatory mechanisms such as phosphorylation2,31,48 govern the allosteric impact of AKBA on regiospecificity. The use of HEK293 cells stably transfected with select LOXs confirmed that 5-LOX is the isoform responsible for producing 12-HETE in presence of AKBA, and is supported by the inhibitory effect of the 5-LOX inhibitor zileuton on AKBA-mediated elevation of 12-HETE. Moreover, results from human neutrophils and M1-like MDM with endogenous LOXs confirm that AKBA-induced 12-HETE formation occurs under pathophysiologically relevant conditions. As 12/15-LOX activities are required for the synthesis of SPM36–38, it was tempting to speculate that AKBA could also promote SPM formation in 5-LOX-rich neutrophils, monocytes or macrophages by provoking a change in regiospecificity of 5-LOX toward a 12/15-lip oxygenating enzyme. Since these leukocytes also express 12/15-LOXs, this might obscure AKBA-induced signals. Thus, more detailed research and better-defined experimental settings are necessary to address AKBA-induced SPM formation in primary cells and in vivo. Nevertheless, employing 5-LOX/FLAP-transfected HEK293 cells devoid of 12/15-LOXs represents a valuable model for pilot studies, and indeed yielded hints for elevated SPM formation in presence of AKBA. Notably, SPM that require the equivalent of 12/15-lipoxygenation were increased while LM that are oxygenated at C5 were suppressed by AKBA, as expected.
In conclusion, we have provided crystal structures of inhibitor-bound 5-LOX and these revealed the mechanistic basis for the divergent actions of the active site inhibitor NDGA and the allosteric inhibitor AKBA. Allosteric modulation of human 5-LOX by AKBA not only inhibited the formation of classical 5-LOX products but provoked a change in regiospecificity toward a 12-lipoxygenating enzyme, particularly in the cellular environment. Our findings may be key to the development of innovative, efficient, LOX-isoform-selective modulators with new and favorable pharmacological profiles. In particular, the availability of small molecules capable of promoting the LM class switch from pro-inflammatory LT to anti-inflammatory SPM might permit new strategies for therapeutic intervention with inflammation: AKBA may serve as a prototype for compounds with such properties.
Methods
Protein expression and purification.
Stable-5-LOX was expressed and purified as previously described17. Briefly, Rosetta 2 (DE3) cells transformed with a pET14 plasmid carrying Stable-5-LOX DNA were cultured in Terrific Broth. Cells were gathered and frozen at −80 °C. The cell pellets were resuspended with Bugbuster protein extraction reagent and lysed with a French Pressure cell press. Protein was purified by immobilized metal (Co2+) affinity chromatography followed by size exclusion chromatography with HisTrap and Superdex-200 columns, respectively, fitted on an ÄKTA-FPLC (GE LifeSciences). Stable-5-LOX was purified in 1 d and only enzyme eluting at a volume consistent with a monomeric molecular weight was used.
Crystallization.
While extensive efforts to crystallize Stable-5-LOX in the presence of the inhibitors were undertaken, we were not able to obtain cocrystals of the complexes. However, soaking experiments were ultimately optimized to generate these cocrystal structures. Despite collecting at least ten data sets for each of the ligands, we never achieved full substitution of either inhibitor. Stable-5-LOX crystallizes with two monomers in the asymmetric unit and differences in the packing environments appear to preclude full substitution in the crystallographically independent monomers. Protein crystals were grown by sitting-drop vapor diffusion at 295 K by mixing 1 μl of monomeric protein (10 mg ml−1) and 2 μl reservoir solution containing 8–12% Tacsimate, pH 6.0 (Hampton Research). Crystals typically appear within 5–10 d. Single crystals of Stable-5-LOX were transferred to a new sitting-drop vapor-diffusion plate with 70% Tacsimate (pH 6.0) mother liquor. These crystals were then soaked with 70% Tacsimate (pH 6.0) supplemented with 160 μM AKBA or 500 μM NDGA for 18 h. Crystals were frozen in liquid nitrogen before data collection.
Structure determination.
Crystals were screened at 100 K at the Protein Crystallography beamline at the Center for Advanced Microstructure and Devices at Louisiana State University. Diffraction data were collected at 100 K with a wavelength of 0.979 Å at the NE-CAT beamline 24-IC-C for the Stable-5-LOX crystal soaked with AKBA and at the NE-CAT beamline 24-IC-E for the Stable-5-LOX crystal soaked with NDGA. NE-CAT beamlines 24-IC-C and E are located at the Advanced Photon Source. Data were processed with XDS51 and scaled using Aimless52 in the Rapid Automated Processing of Data software suite, which implements a CC1/2 (Pearson’s correlation coefficient calculated with randomly generated half data sets) of 0.3 or better to set the resolution limit. Data were processed in space group P21 (unit cell parameters: a = 76.71, b = 204.36, c = 48.15 Å, β = 99.91°) with the unit cell isomorphous to PDB 3O8Y for the NDGA data set; however, the unit cell size doubled in volume and the number of monomers in the asymmetric unit increased from two to four in AKBA-soaked Stable-5-LOX crystals (unit cell parameters: a = 76.87, b = 203.72, c = 110.44 Å, β = 109.71°).
Model building and phase refinement for 5-LOX-NDGA.
A monomer of Stable-5-LOX (3O8Y) was stripped of water, hydrogens and the amino-terminal histidine tag. Phaser was used in Phenix for molecular placement and two molecules were positioned in the asymmetric unit. The solution was refined in Phenix. refine53 using rigid body restraints. In the resulting map the real-space correlation coefficients for several peptide regions that encapsulate the active site were below 0.5. The composite omit map with annealing was generated to remove model bias and verify electron density around these particular peptide regions. When we included all peptide regions in Phenix.refine cycles with translation-libration-screw and noncrystallographic symmetry (NCS) restraints, the quality of the electron density map was poor and unconvincing in these same ‘missing’ regions. Peptide regions 173–214, 294–303 and 416–429 from chain B had average B factors that were two to three times greater than overall average. These peptide regions, along with their counterparts in chain A, could not be modeled. Despite the disorder in the active-site region, electron density consistent with NDGA was observed (Supplemental Fig. 1) and the inhibitor was added to the model. The coordinates of the refined model and data have been deposited with PDB ID 6N2W. Final Ramachandran favored, allowed and outliers are 95.5, 4.5 and 0%, respectively.
Model building and phase refinement for 5-LOX-AKBA.
A monomer of Stable-5-LOX (3O8Y) for a molecular replacement search was prepared as above for the AKBA-5-LOX diffraction data. Molecular replacement with Phaser54 positioned four molecules in the asymmetric unit. The solution was refined in Phenix. refine initially using rigid body restraints. Subsequently, B factors for each amino acid group were also refined, along with translation-libration-screw rotation parameters. Each chain consisted of three groups: (5–112, 113–187 and 188–673). Initially, torsion-angle NCS restraints were used to improve model quality and after multiple cycles of refinement, a positive difference density (Fo–Fc, Supplementary Fig. 2) consistent with a bulky pentacyclic terpenoid was revealed in chain B between the β-barrel and catalytic domain. Consequently, further refinements did not include NCS restraints.
AKBA parameters were built in Phenix.elbow from the structure data file of AKBA in Pubchem. In this crystal structure of AKBA (deposited as CCDC 138321) the C(3) acetoxy group and C(4) carboxyl are positioned in a trans-axial arrangement55. However, the electron density we observed is consistent with a distorted chair conformation of the A ring with the C(3) acetoxy group in a bent axial and C(4) carboxyl in a full axial position. The real-space correlation coefficient of AKBA is 0.81, while the overall correlation coefficient of monomer B is 0.83. For comparison, the catalytic irons in the four molecules have real-space correlation coefficients of 0.73, 0.76, 0.81 and 0.86. AKBA does not make contacts with neighboring monomers in the unit cell.
When data quality was assessed in Phenix.xtriage, a warning of pseudosymmetry was indicated56, presumably due to the simple doubling of the unit cell in the AKBA-soaked crystal. The resulting pseudosymmetry (four monomers in the asymmetric unit rather than two) cannot easily be distinguished from a doubling error in a unit cell dimension. Given that ligand binding has been shown to induce changes in unit cell parameters and/or symmetry in other soaking experiments57,58, we felt it prudent to use the lower symmetry unit cell as defined in data processing with four independent monomers. The final r.m.s.d. between the two pairs of monomers found in the AKBA-soaked crystal conforms to the criteria established by Zwart et al. for distinguishing between pseudosymmetry and true crystallographic symmetry56. When comparing the two electron density maps, AKBA was clearly visible in only one of the four molecules (chain B) in the asymmetric unit and refined to a real-space correlation coefficient equivalent to that of the protein. Coordinates and X-ray data can be accessed with PDB ID 6NCF. Final Ramachandran favored, allowed and outliers are 95.6, 4.0 and 0.3%, respectively.
Limited proteolysis of enzyme:inhibitor complexes.
Monomeric Stable-5-LOX was incubated with equimolar concentrations of pepsin (5 μM, Sigma) in 0.1 M sodium phosphate buffer pH 6.0 at 37 °C for 30 min. Pepsin is most active at pH ~2, and equimolar enzyme is required at pH 6.0, the pH of the crystallization conditions. Vehicle (DMSO) or small molecule inhibitor (200 μM) was incubated for 10 min before pepsin was added. The reaction was stopped by adding standard 4× SDS–PAGE loading buffer with 1 μM pepstatin. Samples were boiled for 5 min before application to 10% acrylamide SDS–PAGE gels. For western blot analysis, the gels were blotted on to a polyvinylidene difluoride membrane with a Bio-Rad Trans-Blot system according to the manufacturer’s instructions. The primary antibody was a rabbit antibody raised to a peptide corresponding to the last 12 amino acids of the 5-LOX enzyme27 and it is highly sensitive, leading to saturation of signal below the level of protein applied to the gels. The secondary antibody used was goat anti-rabbit Alexa Fluor 647. Western blots were imaged with a Typhoon 9410 imager using channel 670 30 BP. Standard quantification calculations of SDS–PAGE band intensities triplicate experiments were performed in Photoshops CS5 by first creating a selection box around the 75- or 63-kD band from each experiment. The integrated intensity difference was calculated for each band along with an area for background subtraction. Incubations with AKBA or NDGA were compared to vehicle for a relative increase in band intensity.
Melting temperature assays.
Differential scanning fluorimetry assays were performed according to the protocol described by Ericsson et al.28. Stable-5-LOX was diluted to 1 μM in a final volume of 100 μl in 0.1 M sodium phosphate buffer pH 6.0 with 5× SYPRO Orange protein stain (Invitrogen). Samples were aliquoted in triplicate volumes of 30 μl in a 96-well reaction plate. The plate was subjected to a linear thermal shift from 5 to 94 °C in 1 °C increments in a QuantStudio 7 Flex Real-Time PCR System (ThermoFisher Scientific). The SYBR Green cutoff filter was used for fluorescence detection of denaturing protein-SYPRO Orange binding. The resulting data were exported and processed using the DMAN software by the Hofmann laboratory59 where the sigmoidal part of the curve was averaged for each triplicate and fit to a four parameter sigmoidal equation. The melting temperatures reported are the results of multiple independent experiments, each in triplicate with similar trends seen in different buffering systems.
Preparation of Stable-5-LOX mutants.
Stable-5-LOX mutants L66D, R101A, H130A, R596A and H130Y were constructed using the QuikChange II XL site-directed mutagenesis kit (Agilent). All mutations were verified by sequencing. The proteins were expressed and purified according to the Stable-5-LOX protocol described above. Only the variants that carried the R596A, H130A and R101A substitutions were expressed at levels equivalent to Stable-5-LOX. H130A and R101A had enzyme activities equivalent or greater than that of the progenitor enzyme. R101D has previously been shown to have an elevated enzyme activity31, which we confirmed here. The L66D, H130Y and R596A variants had no detectable enzyme activity. Enzyme activity for H130A and R101A was examined with an Applied Photophysics Model SX17MV stopped-flow spectrometer operating in the absorbance mode at 20 °C. Product formation was monitored at 238 nm with a cell path length of 0.2 cm. The enzyme concentration was 500 nM in phosphate-buffered saline (PBS). Initial velocity measurements were performed in triplicate with 10 μM AA (Cayman Chemical) in the absence and presence of AKBA at 25, 50, 75 and 100 μM.
Activity assay of human recombinant 5-LOX.
Human recombinant 5-LOX was expressed in E. coli BL21 transformed with pT3-5-LO plasmid (provided by O. Radmark, Karolinska Institutet, Stockholm, Sweden) at 30 °C overnight as described before60. Cells were lysed in lysis buffer containing triethanolamine (50 mM, pH 8.0), EDTA (5 mM), phenylmethanesulfonylfluoride (1 mM), soybean trypsin inhibitor (60 μg ml−1), dithiothreitol (2 mM) and lysozyme (1 mg ml−1) and homogenized by sonification (3 × 15 s). 5-LOX was purified from the 40,000g supernatant (20 min, 4 °C) using an ATP-agarose column and diluted with PBS pH 7.4 containing 1 mM EDTA. Aliquots corresponding to 0.5 μg 5-LOX were preincubated with the indicated concentrations of AKBA or NDGA or with vehicle (0.1% DMSO) on ice for 15 min, and then incubated with 10 μM AA in presence of 2 mM CaCl2 for another 10 min at 37 °C. The reaction was stopped with 2 ml ice-cold methanol containing the deuterium-labeled internal standards d8-5S-HETE and d4-LTB4 (500 pg, each). LM analysis by UPLC–MS/MS was performed as described in the section LM analysis using UPLC–MS/MS.
LM formation in stably transfected HEK293 cells.
HEK293 cells expressing FLAP and/or 5-LOX were selected by 200 μg ml−1 hygromycin B and/or 400 μg ml−1 geneticin, respectively, as described elsewhere32. Cells expressing 12-LOX32 as well as 15-LOX-1 and 15-LOX-2 using the plasmids pCMV6_15-LOX-1 (Origene, NM_001140) and pcDNA3.1/neom (+)_15-LOX-2 were selected by 400 μg ml−1 geneticin. Stably transfected HEK293 cells (1 × 106 cells per ml) were preincubated with 25 μM AKBA or 1 μM NDGA or with vehicle (0.1% DMSO) in PBS pH 7.4 plus 1 mM CaCl2 for 5 min. LM biosynthesis was initiated by addition of 2.5 μM A23187 plus 1 μM AA and terminated after 15 min at 37 °C. For analysis of SPM biosynthesis, HEK293 cells were incubated with 25 μM AKBA or vehicle plus 1 μM AA, 1 μM EPA and 1 μM DHA for 90 min before stimulation with 2.5 μM A23187 for another 10 min at 37 °C. The reactions were stopped by addition of two volumes of ice-cold methanol containing 10 μl of deuterium-labeled internal standards (200 nM d8-5S-HETE, d4-LTB4, d5-LXA4, d5-RvD2, d4-PGE2 and 10 μM d8-AA) to facilitate quantification. Deuterated and nondeuterated LM standards were from Cayman Chemical/Biomol GmbH. LM analysis by UPLC–MS/MS was performed as described in the section LM analysis using UPLC–MS/MS.
LM formation in neutrophils, monocytes and macrophages.
Leukocyte concentrates from freshly withdrawn peripheral blood of healthy adult donors were provided by the Institute of Transfusion Medicine at the University Hospital Jena, Germany. Venous blood was collected in heparinized tubes (16 I.U. heparin per ml blood) from fasted (12 h) adult (18–65 years) male and female registered healthy volunteers, with informed written consent. These subjects donated blood every 8–12 weeks, had no apparent infections, inflammatory conditions or current allergic reactions (according to previous physical inspection by a clinician) and had not taken antibiotics or anti-inflammatory drugs for at least 10 d before blood collection. The experimental protocol was approved by the ethical committee of the University Hospital Jena. All methods were performed in accordance with the relevant guidelines and regulations. Human Neutrophils and peripheral blood mononuclear cells were isolated using dextran sedimentation and Ficoll-Histopaque 1077-1 (Sigma-Aldrich) centrifugation and subjected to hypotonic lysis of erythrocytes. Monocytes were separated by adherence to culture flasks for 1 h at 37 °C. Subsequently, monocytes were differentiated for 6 d and polarized toward M1-like MDM. In brief, MDM were generated by incubating monocytes with 20 ng ml−1 GM-CSF (Peprotech) for 6 d in RPMI 1640 supplemented with 10% fetal calf serum, 2 mmol l−1 l-glutamine (Biochrom/Merck) and penicillin-streptomycin (Biochrom/Merck), followed by polarization using 100 ng ml−1 LPS and 20 ng ml−1 INF-γ (Peprotech) for another 48 h. Human neutrophils (5 × 106 cells per ml) or M1-like MDM (2 × 106 cells per ml) were preincubated for 5 min with indicated concentrations of AKBA or NDGA or with vehicle (0.1% DMSO) in PBS pH 7.4 plus 1 mM CaCl2. LM biosynthesis was initiated by addition of freshly grown E. coli (serotype O6:K2:H1; ratio, 1:50 (neutrophils/MDM, E. coli). After 90 min at 37 °C, supernatants were transferred to 2 ml of ice-cold methanol containing 10 μl of deuterium-labeled internal standards (200 nM d8-5S-HETE, d4-LTB4, d5-LXA4, d5-RvD2, d4-PGE2 and 10 μM d8-AA) and subjected to UPLC–MS/MS analysis.
LM analysis using UPLC–MS/MS.
Samples were kept at −20 °C for 60 min and then centrifuged (1,200g, 4 °C, 10 min). Solid-phase C18 cartridges were equilibrated with 6 ml methanol before addition of 6 ml H2O. To the samples, 9 ml acidified H2O (pH 3.5, HCl) was added and then loaded onto the conditioned C18 columns that were subsequently washed with 6 ml H2O, followed by 6 ml n-hexane. The LM were eluted with 6 ml of methyl formate. Samples were brought to dryness using an evaporation system (TurboVap LV, Biotage) and immediately resuspended in methanol-water (50:50 vol/vol) for UPLC–MS/MS automated injections. The UPLC–MS/MS system consisted of an Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm2, Waters) and an Acquity UPLC (Waters) as well as a QTRAP 5500 mass spectrometer (Sciex) equipped with an electrospray ionization source33. The QTrap 5500 was operated in negative ionization mode using scheduled multiple reaction monitoring (MRM) coupled with information-dependent acquisition and an enhanced product ion scan. The scheduled MRM window was 90 s, and each LM parameter was optimized individually. LM analysis, using an MRM method with diagnostic ion fragments and identification, was performed as described previously36.
Immunofluorescence microscopy.
HEK293 cells expressing 5-LOX or 5-LOX and FLAP were seeded onto polylysine-coated glass coverslips at 0.2 × 106 cells per ml. After 48 h at 37 °C and 5% CO2 the culture medium was removed and PBS pH 7.4 plus 1 mM CaCl2 was used for further incubations. Cells were preincubated for 5 min with 25 μM AKBA or vehicle (0.1% DMSO) before stimulation with 2.5 μM A23187 for another 15 min. The reaction was stopped by fixation with 4% paraformaldehyde solution and acetone for 3 min at 4 °C, followed by 0.25% Triton X-100 for 10 min at room temperature for permeabilization of cells before blocking with normal goat serum 10% (50062Z, ThermoFisher). Samples were incubated with mouse monoclonal anti-5-LOX antibody, 1:100 (610695, BD Bioscience) and rabbit polyclonal anti-FLAP antibody, 5 μg ml−1 (ab85227, Abcam) at 4 °C overnight. The next day, Alexa Fluor 488 goat anti-rabbit IgG (H + L), 1:500 (A11034, ThermoFisher) and Alexa Fluor 555 goat anti-mouse IgG (H + L); 1:500 (A21424, ThermoFisher) were added for 30 min at room temperature in the dark. Nuclear DNA was stained with ProLong Gold Antifade Mountant with DAPI (15395816, ThermoFisher). Samples were analyzed by a Zeiss Axiovert 200 M microscope, and a Plan Neofluar ×40/1.30 Oil (DIC III) objective (Carl Zeiss). An AxioCam MR camera (Carl Zeiss) was used for image acquisition.
Western blot analysis of LOXs and FLAP in HEK293 cells.
Cell lysates of HEK293 cells corresponding to 2 × 106 cells were separated on 10% polyacrylamide gels for 5-LOX, 12-LOX, 15-LOX-1 and 15-LOX-2 or on a 16% polyacrylamide gels for FLAP, and blotted onto nitrocellulose membranes (Amersham Protran Supported 0.45 μm nitrocellulose, GE Healthcare). The membranes were incubated with the following primary antibodies: rabbit polyclonal anti 5-LOX, 1:1,000 (by Genscript, Piscataway to a peptide with the C-terminal 12 amino acids of 5-LOX: CSPDRIPNSVAI); mouse monoclonal anti-15-LOX-1, 1:500 (ab119774, Abcam); rabbit polyclonal anti-15-LOX-2, 1:500 (ab23691, Abcam); rabbit polyclonal anti-FLAP antibody, 5 μg ml−1 (ab85227, Abcam); polyclonal rabbit anti-12-LOX, 1:200 (NBP2-29941, Novus Biologicals) and rabbit polyclonal anti-β-actin, 1:1,000 (4967S, Cell Signaling). Immunoreactive bands were stained with IRDye 800CW Goat anti-Mouse IgG (H+L), 1:10,000 (926-32210, LI-COR Biosciences), IRDye 800CW Goat anti-Rabbit IgG (H+L), 1:15,000 (926 32211, LI-COR Biosciences) and/or IRDye 680LT Goat anti-Mouse IgG (H+L), 1:40,000 (926-68020, LI-COR Biosciences) and visualized by an Odyssey infrared imager (LI-COR Biosciences).
Statistical analyses.
Results are expressed as mean ± standard error of the mean (s.e.m.) of n observations, where n represents the number of experiments with separate donors, HEK293 cell cultures or purified 5-LOX, performed on different days, as indicated. Analyses of data were conducted using GraphPad Prism 8 software. A two-tailed paired t-test was used for comparison of two groups. For multiple comparison, one-way analysis of variance (ANOVA) with Dunnett’s post hoc tests were applied as indicated. The criterion for statistically significant is P < 0.05.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work was funded in part by grants to M.E.N. (nos. NIH HL107887 and AHA 16GRNT31000010, and the NIH P50AT002776 seed grant, and the Louisiana Governor’s Biotechnology Initiative) and O.W. (Deutsche Forschungsgemeinschaft (DFG, the German Research Foundation), project no. 316213987, SFB 1278 PolyTarget (project nos. A04 and C02), CRC 1127 ChemBioSys (project no. A04) and Free State of Thuringia and the European Social Fund (2016 FGR 0045)). J.G. received a Carl Zeiss postdoctoral stipend. Preliminary X-ray data were collected at the Center for Advanced Microstructures and Devices (Baton Rouge). We thank the staff at the Center for Advanced Microstructures and Devices for screening and data collection of macromolecular crystals at the Protein Crystallography beamline. The work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (grant no. P30 GM124165). The Eiger 16M detector on 24-ID-E beam line is funded by a NIH-ORIP HEI grant (no. S10OD021527). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357.
Footnotes
Online content
Any Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-020-0544-7.
Data availability
Coordinates and structure factors (6N2W, 6NCF) are available at the Protein Bank, www.rcsb.org.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-020-0544-7.
References
- 1.Haeggstrom JZ & Funk CD Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev 111, 5866–5898 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Radmark O, Werz O, Steinhilber D & Samuelsson B 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim Biophys. Acta 1851, 331–339 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimizu T et al. Characterization of leukotriene A4 synthase from murine mast cells: evidence for its identity to arachidonate 5-lipoxygenase. Proc. Natl Acad. Sci. USA 83, 4175–4179 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon RAF et al. Requirement of a 5-lipoxygenase-activating protein for leukotriene synthesis. Nature 343, 282–284 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Ferguson AD et al. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science 317, 510–512 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Vickers PJ, Deluca C, Wong E & Abramovitz M The effect of 5-lipoxygenase-activating protein (FLAP) on substrate utilization by 5-lipoxygenase. Adv. Exp. Med Biol 400A, 145–151 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Abramovitz M et al. 5-lipoxygenase-activating protein stimulates the utilization of arachidonic acid by 5-lipoxygenase. Eur. J. Biochem 215, 105–111 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Evans JE, Ferguson AD, Mosley RT & Hutchinson JH What’s all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharm. Sci 29, 72–78 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Werz O, Gerstmeier J & Garscha U Novel leukotriene biosynthesis inhibitors (2012-2016) as anti-inflammatory agents. Expert Opin. therapeutic Pat 27, 607–620 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Pettersen D, Davidsson O & Whatling C Recent advances for FLAP inhibitors. Bioorg. Med Chem. Lett 25, 2607–2612 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Funk CD, Chen XS, Johnson EN & Zhao L Lipoxygenase genes and their targeted disruption. Prostaglandins Other Lipid Mediat. 68-69, 303–312 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Schneider C, Pratt DA, Porter NA & Brash AR Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem. Biol 14, 473–488 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brash AR Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem 274, 23679–23682 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Neau DB et al. Crystal structure of a lipoxygenase in complex with substrate: the arachidonic acid-binding site of 8R-lipoxygenase. J. Biol. Chem 289, 31905–31913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newcomer ME & Brash AR The structural basis for specificity in lipoxygenase catalysis. Protein Sci. 24, 298–309 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilbert NC et al. The structure of human 5-lipoxygenase. Science 331, 217–219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bokoch GM & Reed PW Evidence for inhibition of leukotriene A4 synthesis by 5,8,11,14-eicosatetraynoic acid in guinea pig polymorphonuclear leukocytes. J. Biol. Chem 256, 4156–4159 (1981). [PubMed] [Google Scholar]
- 19.Safayhi H, Sailer ER & Ammon HP Mechanism of 5-lipoxygenase inhibition by acetyl-11-keto-beta-boswellic acid. Mol. Pharm 47, 1212–1216 (1995). [PubMed] [Google Scholar]
- 20.Sailer ER, Schweizer S, Boden SE, Ammon HP & Safayhi H Characterization of an acetyl-11-keto-beta-boswellic acid and arachidonate-binding regulatory site of 5-lipoxygenase using photoaffinity labeling. Eur. J. Biochem 256, 364–368 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Poeckel D & Werz O Boswellic acids: biological actions and molecular targets. Curr. Med. Chem 13, 3359–3369 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Abdel-Tawab M, Werz O & Schubert-Zsilavecz M Boswellia serrata: an overall assessment of in vitro, preclinical, pharmacokinetic and clinical data. Clin. Pharmacokinet 50, 349–369 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Sturner KH et al. A standardised frankincense extract reduces disease activity in relapsing-remitting multiple sclerosis (the SABA phase Ila trial). J. Neurol. Neurosurg. Psychiatry 89, 330–338 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Werz O & Steinhilber D Development of 5-lipoxygenase inhibitors–lessons from cellular enzyme regulation. Biochem Pharm. 70, 327–333 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Kemal C, Louis-Flamberg P, Krupinski-Olsen R & Shorter AL Reductive inactivation of soybean lipoxygenase 1 by catechols: a possible mechanism for regulation of lipoxygenase activity. Biochemistry 26, 7064–7072 (1987). [DOI] [PubMed] [Google Scholar]
- 26.Mitra S, Bartlett SG & Newcomer ME Identification of the substrate access portal of 5-lipoxygenase. Biochemistry 54, 6333–6342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schexnaydre EE et al. A 5-lipoxygenase-specific sequence motif impedes enzyme activity and confers dependence on a partner protein. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 543–551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ericsson UB, Hallberg BM, Detitta GT, Dekker N & Nordlund P Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem 357, 289–298 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Eek P et al. Structure of a calcium-dependent 11R-lipoxygenase suggests a mechanism for Ca2+ regulation. J. Biol. Chem 287, 22377–22386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakonjac Ryge M et al. A mutation interfering with 5-lipoxygenase domain interaction leads to increased enzyme activity. Arch. Biochem Biophys 545, 179–185 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Werz O Inhibition of 5-lipoxygenase product synthesis by natural compounds of plant origin. Planta Med. 73, 1331–1357 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Gerstmeier J, Weinigel C, Barz D, Werz O & Garscha U An experimental cell-based model for studying the cell biology and molecular pharmacology of 5-lipoxygenase-activating protein in leukotriene biosynthesis. Biochim Biophys. Acta 1840, 2961–2969 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Werner M et al. Targeting biosynthetic networks of the proinflammatory and proresolving lipid metabolome. FASEB J. 33, 6140–6153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siemoneit U et al. On the interference of boswellic acids with 5-lipoxygenase: mechanistic studies in vitro and pharmacological relevance. Eur. J. Pharm 606, 246–254 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Surette ME, Palmantier R, Gosselin J & Borgeat P Lipopolysaccharides prime whole human blood and isolated neutrophils for the increased synthesis of 5-lipoxygenase products by enhancing arachidonic acid availability: involvement of the CD14 antigen. J. Exp. Med 178, 1347–1355 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Werz O et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun 9, 59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng B et al. Maresin biosynthesis and identification of maresin 2, a new anti-inflammatory and pro-resolving mediator from human macrophages. PLoS ONE 9, e102362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carion TW et al. Immunoregulatory role of 15-lipoxygenase in the pathogenesis of bacterial keratitis. FASEB J. 32, 5026–5038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sailer ER et al. Acetyl-11-keto-beta-boswellic acid (AKBA): structure requirements for binding and 5-lipoxygenase inhibitory activity. Br. J. Pharm 117, 615–618 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gillmor SA, Villasenor A, Fletterick R, Sigal E & Browner MF The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct. Biol 4, 1003–1009 (1997); erratum 5, 242 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Choi J, Chon JK, Kim S & Shin W Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins 70, 1023–1032 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Kobe MJ, Neau DB, Mitchell CE, Bartlett SG & Newcomer ME The structure of human 15-lipoxygenase-2 with a substrate mimic. J. Biol. Chem 289, 8562–8569 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandal AK et al. The membrane organization of leukotriene synthesis. Proc. Natl Acad. Sci. USA 101, 6587–6592 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandal AK et al. The nuclear membrane organization of leukotriene synthesis. Proc. Natl Acad. Sci. USA 105, 20434–20439 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerstmeier J et al. 5-Lipoxygenase-activating protein rescues activity of 5-lipoxygenase mutations that delay nuclear membrane association and disrupt product formation. FASEB J. 30, 1892–1900 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neau DB et al. The 1.85 A structure of an 8R-lipoxygenase suggests a general model for lipoxygenase product specificity. Biochemistry 48, 7906–7915 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy RC & Gijon MA Biosynthesis and metabolism of leukotrienes. Biochem J. 405, 379–395 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Flamand N, Luo M, Peters-Golden M & Brock TG Phosphorylation of serine 271 on 5-lipoxygenase and its role in nuclear export. J. Biol. Chem 284, 306–313 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laskowski RA & Swindells MB LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 51, 2778–2786 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Pettersen EF et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Kabsch WXDS Acta Crystallogr. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schweizer S, Eichele K, Ammon HP & Safayhi H 3-Acetoxy group of genuine AKBA (acetyl-11-keto-beta-boswellic acid) is alpha-configurated. Planta Med. 66, 781–782 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Zwart PH et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol 426, 419–435 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Dauter Z, Li M & Wlodawer A Practical experience with the use of halides for phasing macromolecular structures: a powerful tool for structural genomics. Acta Crystallogr. D Biol. Crystallogr 57, 239–249 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Parsons S Introduction to twinning. Acta Crystallogr. D Biol. Crystallogr 59, 1995–2003 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Wang CK, Weeratunga SK, Pacheco CM & Hofmann A DMAN: a Java tool for analysis of multi-well differential scanning fluorimetry experiments. Bioinformatics 28, 439–440 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Fischer L, Szellas D, Radmark O, Steinhilber D & Werz O Phosphorylation- and stimulus-dependent inhibition of cellular 5-lipoxygenase activity by nonredox-type inhibitors. FASEB J. 17, 949–951 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.