

Abstract
Acute pancreatitis (AP) is one of the most common gastroenterological disorders leading to hospitalization. It has long been debated whether biliary AP, about 30% to 50% of all cases, is induced by bile acids (BAs) when they reach the pancreas via reflux or via the systemic blood circulation.
Besides their classical function in digestion, BAs have become an attractive research target because of their recently discovered property as signaling molecules. The underlying mechanisms of BAs have been investigated in various studies. Bile acids are internalized into acinar cells through specific G-protein–coupled BA receptor 1 and various transporters. They can further act via different receptors: the farnesoid X, ryanodine, and inositol triphosphate receptor. Bile acids induce a sustained Ca2+ influx from the endoplasmic reticulum and release of Ca2+ from acidic stores into the cytosol of acinar cells. The overload of intracellular Ca2+ results in mitochondrial depolarization and subsequent acinar cell necrosis. In addition, BAs have a biphasic effect on pancreatic ductal cells. A more detailed characterization of the mechanisms through which BAs contribute to the disease pathogenesis and severity will greatly improve our understanding of the underlying pathophysiology and may allow for the development of therapeutic and preventive strategies for gallstone-inducedAP.
Key Words/Abbreviations: acinar cells, acute pancreatitis, bile acids, Ca2 +, gallstone, AP - acute pancreatitis, ATP - adenosine triphosphate, BAs - bile acids, [Ca2+]i - intracellular calcium concentration, CCK - cholecystokinin, CDCA - chenodeoxycholic acid, FXR - farnesoid X receptor, Gpbar1 - G-protein–coupled bile acid receptor 1, IL - interleukin, IP3R - inositol triphosphate receptors, NaT - sodium taurocholate, NTCP - NaT cotransporting polypeptide, PI3K - phosphatidylinositol 3-kinase, RyR - ryanodine receptor, SERCA - sarco/endoplasmic reticulum Ca2+, TCA - taurocholic acid, TCDC - taurochenodeoxycholic acid, TLCS - taurolithocholic acid-3-sulfate, TUDCA - tauroursodeoxycholic acid, UDCA - ursodeoxycholic acid
Acute pancreatitis (AP) is one of most common gastroenterological disorders leading to hospital admission with an increasing incidence over the last 20 years.1 Around 10% to 15% of patients suffer from a severe form of the disease with local complications, (multi-)organ failure, and a high mortality. There is still no specific treatment, and management is based on symptomatic and supportive therapy. Migrating gallstones are one of the most common causes for AP, accounting for 30% to 50% of cases2,3 in many countries.
Pancreatitis is believed to begin in pancreatic acinar (exocrine) cells, which are highly susceptible to pathological extracellular stimuli4,5 and in which digestive proteases, initially trypsin, undergo activation.6 The balance between activation7 and degradation8 of digestive enzymes by lysosomal hydrolases appears to determine the extent of cellular injury. Germline mutations in the human trypsinogen (PRSS1) gene9 support the concept of autodigestion as an initiating factor. Whether or not the disease subsequently takes a severe course10 or progresses to chronic pancreatitis11 depends on a variety of factors12 and is hard to predict on admission. Bile and bile acids (BAs) have been implicated in the cellular pathogenesis of pancreatitis.13 Whether and to what extent they are involved will very much affect the search for potential treatment strategies directed against bile BA-mediated events.14
In humans, BAs are synthesized primarily from cholesterol and are conjugated in the liver with glycine or taurine. After being secreted into the duodenum, they are converted to secondary BAs by intestinal bacteria, reabsorbed, and finally recycled via the enterohepatic circulation.13,15,16 In 1848, the first BA, cholic acid, was discovered, and others were subsequently identified as described by Wieland in his Nobel lecture in 1928.17 There have been outstanding advances in the biochemistry and the clinical application of BAs during the last decades.16
It was recently revealed that BAs are not only essential for food digestion but also significantly contribute to either the pathogenesis or the treatment of various gastrointestinal disorders including chronic liver diseases,18,19 disorders of the biliary tract,20 and diabetes mellitus.21
The role of BAs in pancreatitis has been investigated in a number of studies. However, the molecular mechanism of BA-mediated effects is not yet fully understood.3,22 Remaining questions are whether and how BAs enter the acinar cell and which molecular mechanisms are responsible for cellular injury. Here, we review studies that have investigated the role of BAs in pancreatitis and their effect on different cells of the pancreas. Results from both experimental and clinical studies were included.
To this end, an extensive literature search was conducted using the following key words: “bile acids,” “pancreatitis,” “pathogenesis,” “animal experiment,” and “clinical study” in different combinations based on patient, intervention, comparison, outcome model searching strategy.
EFFECTS OF BAs ON PANCREATIC CELLS
Bile acids derive from the cholesterol molecule and are amphiphilic substances being both hydrophilic and lipophilic. The two most abundant primary BAs in humans are cholic acid and chenodeoxycholic acid (CDCA). They are metabolized to the secondary BAs lithocholic acid and deoxycholic acid by intestinal bacteria via 7-dehydroxylase.13 Usually, BAs are conjugated with taurine or glycine to form at least 8 different types of BAs. In rodents, taurine conjugation is predominant; therefore, the major BAs in mouse bile are taurocholic acid (TCA) and taurodeoxycholic acid.23 Bile acids with less hydroxy groups are subjected to sulfation and glucuronidation, which are necessary for their detoxification. Bile acids are biosynthesized via classical (neutral) or alternative (acidic) pathways (Fig. 1). The neutral pathway, which is initiated by CYP7A1, predominates in BA biosynthesis of adults and begins with 7-hydroxylation of cholesterol. The alternative pathway of BA biosynthesis begins via the cytochrome P450 enzyme CYP27A1, followed by oxysterol 7α-hydroxylase.24,25
FIGURE 1.
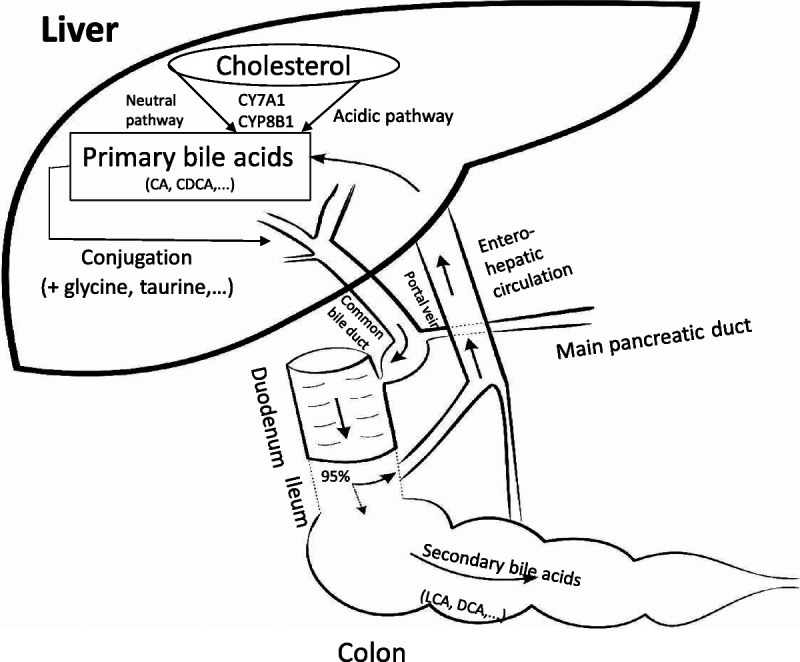
Biosynthesis and conjugation of BAs. Bile acids are primarily synthesized from cholesterol in the liver, by several enzymes, including CYP7A1 and CYP8B1 via an acidic or neutral pathway. Primary BAs are conjugated with taurine, glycine, or nonamino acid. Conjugated BAs are secreted into the bile duct and then enter the intestine via the major duodenal papilla. Most (95%) of BAs are reabsorbed in the terminal ileum and enter the enterohepatic circulation via the portal vein. The remainder will be converted to secondary BAs by bacteria in the colon and absorbed to the blood or discarded through feces. CA, cholic acid; DCA, deoxycholic acid, LCA, lithocholic acid.
Mechanisms by Which BAs Affect Acinar Cells
Effects of BAs on Calcium Signaling and Endoplasmatic Reticulum
Bile-induced acinar cell injury is attributed to both detergent (surfactant) and nondetergent characteristics of BAs. Detergent mechanisms of BAs lead to an elevation of intracellular calcium concentration ([Ca2+]i), mitochondrial membrane depolarization, and a subsequent intracellular adenosine triphosphate depletion and are most often caused by taurolithocholic acid-3-sulfate (TLCS), a monohydroxyl BA. The nondetergent effect of BAs includes an activation of phosphatidylinositol 3-kinase (PI3K) leading to a pathological stimulation of digestive zymogen activation, cellular injury, and death in pancreatic acinar cells.26,27 These damaging effects are mediated by an increase of [Ca2+]i, too, demonstrating that both detergent and nondetergent effects of BAs are acting synergistically. Taurolithocholic acid-3-sulfate initiates Ca2+ transients and calcium signals are localized near secretory granules in the apical region of acinar cells. The ability of BAs to induce a sustained calcium release into the cytosol of pancreatic acinar cells adds an important new aspect to the mechanisms of bile-induced AP.28 In physiological states, the calcium shift into the endoplasmatic reticulum (ER) is mediated by sarco/endoplasmic reticulum Ca2+ (SERCA) ATPase, also termed SERCA pumps. Inhibition of these pumps, that is, by PI3K, facilitates BA-induced Ca2+ responses and increases acinar cell damage. Vice versa, a pharmacological inhibition of PI3K, which can be achieved by LY-294002, activates SERCA, leading to a Ca2+ reuptake into the ER reversing the BA-induced increase of [Ca2+]i.29
During the BA-induced Ca2+ release from the ER, both ryanodine receptors (RyRs) and inositol 1,4,5-trisphosphate receptors (IP3Rs) are activated. Both receptors belong to the family of Ca2+ channels and are located in the ER membrane that will be discussed in the following section (Pancreatic Receptor Proteins and Transporters of BAs).30,31 Sarco/endoplasmic reticulum Ca2+ is found to a minor extent near acidic stores in the secretory granule compartment, that usually contain digestive zymogens.26 A calcium release from this compartment further increases the cytosolic calcium concentration and is most frequently observed when acinar cells are already damaged like in an ischemic situation.32
The G-protein–coupled bile acid receptor 1 (Gpbar1) is a transmembrane cell surface receptor, and its deletion has been associated with a reduced susceptibility to gallstone disease in mice.33 G-protein–coupled bile acid receptor 1–deficient mice also displayed a milder pancreatitis course when exposed to TLCS. This amelioration was mediated by a reduced generation of pathological calcium transients and sustained physiological calcium oscillations despite a pathological stimulus.34
Apparently, one of the main mechanisms how BAs damage acinar cells is based on a pathological increase of [Ca2+]i by different pathways. This pathological [Ca2+]i increase ultimately initiates a premature trypsinogen activation and other digestive protease activation and the necrosis of acinar cells.35
Effects of BAs on Mitochondria and Cytosolic Adenosine Triphosphate Distribution
Besides the ER and secretory granules, BAs directly act on a third subcellular compartment, the mitochondria. Mitochondria are essential for cellular physiology and homeostasis. Maintenance of an intact mitochondrial membrane potential is required for proper cell function. In AP, an impaired mitochondrial function contributes to both apoptosis and necrosis.13 An [Ca2+]i overload induced by BAs causes mitochondrial depolarization that leads to a reduction of adenosine triphosphate (ATP) synthesis in acinar cells. Adenosine triphosphate is an important regulator for cellular maintenance and of physiological reactions, and its depletion causes disturbances in cellular homeostasis. The reduced availability of mitochondrial and cytosolic ATP enhances susceptibility to acinar cell injury.36,37 Moreover, BAs prolong intracellular Ca2+ release resulting in an increase of reactive oxygen species production, impaired production of ATP, apoptosis, and necrosis.38
Effects of BAs on Nonselective Channels and Chemokine Expression
Along with an activation of Ca2+ influx into the cytosol, BAs also depolarize acinar cells by a cationic current through nonselective ion channels.39 These nonselective channels in acinar cells depend on intracellular Ca2+ levels. It is known that acinar cells respond to secretagogs by several ways of membrane potential changes, and one of them is controlled partly by nonselective ion channels located in the basolateral membrane.40 The main cations transported through the channels are Na+ and K+. The action of BAs on nonselective ion currents varies and is most effective with TLCS, to a lesser extent with taurochenodeoxycholic acid (TCDC) and TCA. Already low concentrations of TLCS (10 μM) are sufficient to induce such a cationic current. The depolarization of the transmembrane potential also contributes to the harmful effect of BAs on acinar cells.39
Changes in chemokine expression are observed in pathological conditions including AP.41 Several studies that allow for standardized conditions when investigating chemokine expression profiles have been carried out in vitro. In the rat acinar cell line (AR42J), trypsinogen activation occurred concurrently with an upregulation of 23 proteins and a downregulation of 16 proteins.42 Acinar cells treated with sodium taurocholate (NaT) showed an overexpression of chemokine ligands, including the chemokine (C-C-motif) ligand 2 that was reversed by oxidized 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine.43
Besides activation of transmembrane ion currents, BAs can also be directly transported into the acinar cells, mediated by specific membrane transporters on the luminal (NaT cotransporting polypeptide [NTCP]) and the basolateral side (organic anion transporting polypeptide). Bile acid uptake is dependent on Na+ but can also occur in a Na+-independent pathway.44 In the crystal structure of the apical sodium-dependent BA transporter, TCA was built into the inward-facing perpendicular to the membrane, that is, with the cholesterol ring close to the crossover and the taurine group extending to the intracellular entrance of the cavity.45 Bile acids traverse the cytoplasm of the epithelial cells bound to a BA-binding protein.46,47
Effects of BAs on Ductal and Stellate Cells
Ductal cells are of particular interest when investigating the effects of BAs on the pancreas because this cell type is firstly exposed to BAs during a potential biliary reflux. Interestingly, BAs are able to exert both harmful and beneficial effects on ductal cells depending on the type and concentration. On the one hand, CDCA caused an increase of [Ca2+]i in Capan-1 cells (a tumor cell line), imitating characteristics of pancreatic ductal cells. This shift in [Ca2+]i is a prerequisite for alterations in Cl− and K+ conductance that changes ductal fluid secretion.48 The decrease of fluid flow results in the damage of ductal integrity and ductal cell injury. A limiting factor of these observations is that Capan-1 is an immortalized tumor cell line that makes a translation of results to the in vivo situation quite difficult. Presumably closer to reality are studies conducted in isolated pancreatic ductal cells from animals: high concentrations of CDCA that were added to ductal cells from guinea pigs induced an overload Ca2+ release, which led to ATP depletion and the loss of mitochondrial membrane potential. In contrast, a low concentration of CDCA had a protective effect on the pancreas by stimulating HCO3− secretion.49–51 In addition, the decrease of ductal bicarbonate secretion was also attributed by an inhibition of glycolytic and oxidative metabolism with a consequent depletion of intracellular ATP levels as shown for isolated ductal cells that were exposed to high concentrations of CDCA (1 mM).52 Besides their effects on bicarbonate secretion, BAs also directly change the permeability of the ductal mucosal barrier53; while in a physiological state, the duct cell layer is nearly impermeable to even small molecules, and exposure of specific bile salts increases the permeability to molecules of at least 20,000 Da. Underlying mechanisms are a disruption of tight junctions with swelling of the intercellular space and structural alteration of ductal cell shape.54
Data on the effects of BAs on stellate cells are sparser. The presence of NTCP in stellate cells indicates a Na+-dependent BA uptake mechanism. Once located in the cytoplasm, BA caused cellular damage ultimately leading to necrosis.55 More studies are needed to gain a more comprehensive view on the effects of BAs on ductal and stellate cells.
Pancreatic Receptor Proteins and Transporters of BAs
The distribution of BA receptors in the liver, small intestine, and other organs is well characterized, but there is also expression in the pancreas (Table 1, Fig. 2). As already mentioned previously, the severity of AP was ameliorated in Gpbar1−/− mice after retrograde injection of TLCS but remained unaffected when the cholecystokinin (CCK) analog cerulein was used. This underlines the importance of the Gpbar1 receptor for BA-induced AP.34 Besides its expression near the apical pole of acini, there is also evidence that Gpbar1 is expressed on the luminal side of pancreatic ductal cells, as Capan-1 cells also express this membrane receptor. G-protein–coupled bile acid receptor 1, synonymous to Takeda G-protein–coupled receptor clone 5, stimulates Na+/Ca2+ exchange after exposure to BAs. Bile acids further increase an ATP release and subsequently raises the intracellular [Ca2+] concentration.48 When entering the cell, BAs can interact with intracellularly located receptors. One of them is the nuclear farnesoid X receptor (FXR), which is, besides Gpbar1, the most studied BA-dependent receptor. Farnesoid X receptor is a hormone receptor with high expression in the liver but also in other organs including the pancreatic acinar cell nuclei.56 The role of FXR in AP was studied in FXR−/− mice. Activation and inhibition of FXR signaling are dependent on the type of BA: conjugated BAs can activate the FXR, but CDCA and its converted products taurine- and glycine-β-muricholic acid can inhibit FXR signaling.58 Activation of the FXR suppresses autophagy rendering acinar cells more susceptible to damage caused by cellular stress.56 The role of autophagy in acinar cell homeostasis was already shown in previous studies.59 In addition, TCA and glycochenodeoxycholate increase the expression FXR that consequently reduces autophagy and increases acinar cell death and inflammation.56 In mild AP, induced by the CCK analog cerulein, loss of FXR function did not affect the severity of AP. Moreover, variations in the FXR locus of humans do not seem to affect the susceptibility for pancreatitis.60
TABLE 1.
Receptors Interacting With BAs in Pancreatic Acinar Cell
| Receptor/Transporter | Localization on Acinar Cells | Effect on Severity of BA-Induced AP After Depletion or Overexpression | Candidate of Studies | Type of BA or Salt Used | Author and Publication Year |
|---|---|---|---|---|---|
| Gpbar1 | Apical pole | Reduction (Gpbar1−/− mice) | Isolated mouse acini | TLCS | Perides et al, 201034 |
| FXR | Nuclear | Increase (overexpression of FXR after incubation with BAs) | Human and rat pancreatic cell lines | GCDC and TCA | Zhou et al, 201756 |
| RyR | SERCA | Reduction (pharmacological inhibition) | Isolated mouse acinar cells and whole mice model | TLCS | Husain et al, 201214 |
| IP3Rs | Outer nuclear membrane | Reduction (pharmacological inhibition) | Pancreatic mouse acinar cells | TLCS | Gerasimenko et al, 200626 |
| TRPV1 | Cell membrane | Reduction (Trpv1−/− mice) | Whole mouse model | NaT | Shahid et al, 201557 |
GCDC indicates glycochenodeoxycholate.
FIGURE 2.
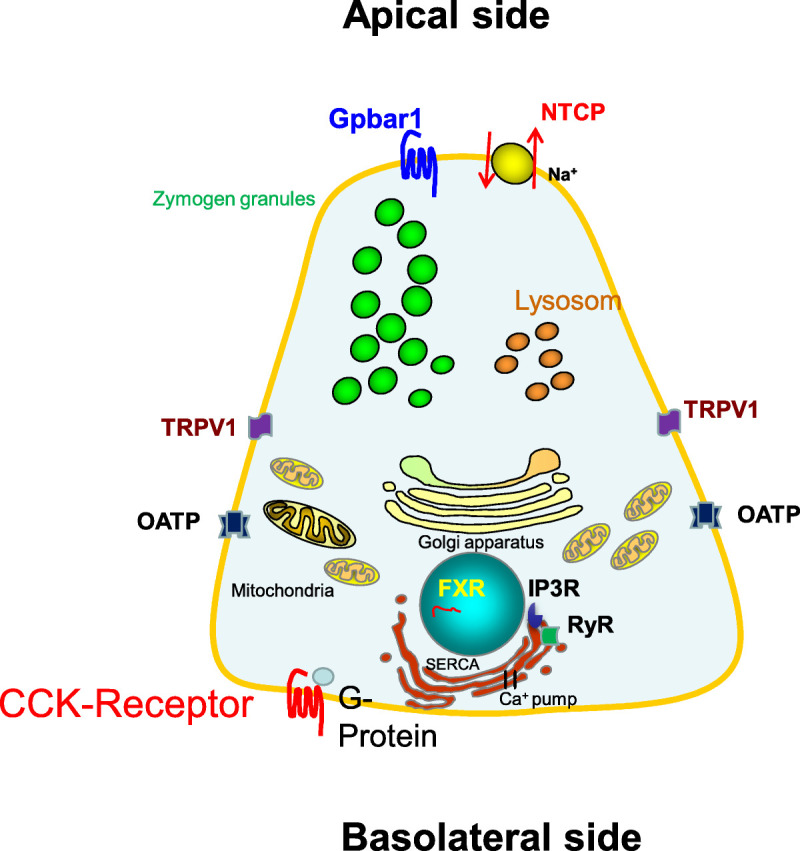
Acinar cell and the relevant BAs transporters and receptors involved in AP. Bile acids can enter the acinar cell via receptors on the membrane such as Gpbar1 and a variety of transporters. They then intracellularly act via FXR in the nuclear, and RyR and IP3Rs at the ER. Bile acids can induce a sustained Ca2+ release from the ER and apical vesicles into the cytosol.
Ryanodine receptors are the largest known ion channels of the cell (>2 MDa) and are located in the SERCA. They exist in three isoforms and are responsible for the release of Ca2+ from intracellular stores. Ryanodine receptors are primarily expressed in skeletal muscles but were also found in human acinar cells.30,61 High concentration of TCA sensitizes the RyRs for 3H-ryanodine binding and trigger a Ca2+ release inside acinar cells. The calcium influx is enabled by an opening of RyRs via an allosteric mechanism, which leads to a Ca2+ leak not only from the ER but also from the zymogen granules in pancreatic acinar cells.62 When the RyRs are inhibited by antagonists, such as dantrolene or ruthenium, BA-induced Ca2+ release in pancreatic acinar cells was significantly reduced.26 Whereas RyRs have a more restricted distribution, IP3Rs are expressed in the outer nuclear membrane of most cells. IP3Rs can be inhibited by caffeine, leading to reduction of Ca2+ release from both the ER and the acidic pool.26 Addition of IP3 to permeabilized pancreatic acinar cells was shown to stimulate the release of Ca2+ from its stores.63
Transient receptor potential vanilloid 1 (TRPV1) is a cellular membrane ion channel, which has been detected in a variety of organs, such as the gastrointestinal tract, brain, lungs, heart, and pancreas.64 In the pancreas, it has been shown that TRPV1 ion channel is expressed in primary sensory nerves, and its activation plays a role in the inflammatory cascade in AP. Potential vanilloid 1 can be activated by leucotrien B4 that is increasingly secreted after exposure to BAs.57 Infusion of leucotrien B4 into the pancreas can induce pancreatic inflammation, and the levels of leucotrien B4 in pancreatic tissue were remarkably higher in rats that underwent ligation of the confluence between common bile duct and main pancreatic duct in comparison with control rats.65 Depletion of TRPV1 ameliorated the severity of disease in BA-induced pancreatitis after retrograde infusion of 2% NaT into the pancreatic duct as observed in TRPV1−/− mice or after coincubation with resiniferatoxin, an excitotoxin that desensitizes TRPV1.57
Types and Concentrations of BAs
Bile acids constitute the main component of bile. They may enter the pancreas by 2 different ways: either by reflux through the pancreatic duct or systemically by transport through the blood stream. Bile acids consist of conjugated and unconjugated components with different hydrophilic properties and a variable ability to enter the pancreatic cells.66 Their concentrations are much higher in the pancreatic duct in currently used experiment models (5%) than in the serum (1–2 μg/mL).67
The effects of BAs on the pancreas either ex vivo on the acinar cell level or in vivo depends on their concentration. At low (micro molar) concentrations, taurine-conjugated BAs induced an intracellular Ca2+ rise in acinar cells, and the most effective of them was TLCS. Other natural BAs such as TCA and taurodeoxycholat are much less effective and require higher concentration than TLCS in triggering calcium signals. Moreover, the required BA concentrations are usually higher for in vivo experiments when BAs are injected retrogradely into the pancreatic duct than after direct incubation using isolated acinar cells. Regarding TLCS, the most commonly used BA, about 6- to 15-fold higher concentrations are needed for animal experiments.34,39,68 TLCS is also a more potent inductor of mitochondrial membrane depolarization than TCDC and TCA. A TLCS concentration of 25 μM results in a membrane depolarization comparable with TCDC at a concentration of 100 μM for the same cellular effect.36 Nevertheless, dihydroxy BAs (such as TCDC, 250 μM) and trihydroxy BAs (such as TCA, 1 mM) are found at higher concentrations in the serum than monohydroxy BAs (eg, TLCS, 25 μM). Therefore, they could substantially contribute to the membrane depolarization or Ca2+ influx in the event of a bile reflux.39
EXPERIMENTAL MODELS FOR BA-INDUCED PANCREATITIS
Duct Obstruction Model
The duct obstruction model mimics gallstone-induced AP. There are various modifications of the ligation site including a separate bile duct or pancreatic duct ligation or a ligation after their fusion as a common biliopancreatic duct. This procedure is mainly done in rodents but was also performed in dogs in the past.69 When the confluence between the bile and pancreatic duct in rats is obstructed by ligation, necrotic AP develops with a clinical presentation resembling multiple organ failure, as is also observed in humans. This model supports the common channel hypothesis of Opie, in which bile reflux enters the pancreas and the pressure rises in the main pancreatic duct. The obstruction of the pancreatic duct seems to be a critical event causing severe necrotizing pancreatitis.70 In some models, an additional bile duct obstruction and reflux of bile into the pancreatic duct is not an important factor because AP occurs irrespective of manipulations of the bile duct.71 The administration of a lithogenic diet consisting of high fat and cholesterol increased the severity of necrotizing pancreatitis. This increase in severity indicates a highly cholesterol-enriched bile with consecutive cholesterol crystal formation that can further damage the pancreas.72 Severity of AP produced by duct ligation varies among animal species (rabbits, rats, or opossums).73,74 In the opossum, duct ligation leads to severe AP with extensive necrosis (50% acinar cell were necrotic).71,75 On the other hand, only 10% acinar cells were necrotic in rats after 1 week after ligation of the common biliopancreatic duct.76 Pancreatic duct ligation in rabbits showed remarkable elevation of pancreatic enzymes and damage of acinar cells, comparable with other experimental models of AP.77 These variable results may be due to differences in the time of organ harvesting after ligation and differences in the surgical technique and protocol.78 These may also be due to species differences in susceptibility to increased pancreatic duct pressure.
Duct Infusion Model
Physiologically, basal pancreatic duct pressure is less than 10 cm H2O, which is equal to 7.36 mm Hg. Perfusion of fluids containing BAs can increase the basal duct pressure several times. Similar changes can result from ductal obstruction caused by spasms of the smooth muscles surrounding the pancreatic duct or shedding of cellular debris. In short, injected BAs increase pancreatic intraductal pressure by different means: one as the consequence of fluid injection and the other due to compression of the duct lumen by the edematous inflammation of the gland.79 There is also an increase in endothelial and capillary permeability after exposure of BAs, resulting in an impairment of capillary blood flow, ischemia, and cellular necrosis.10 The most commonly used BAs to induce severe AP via retrograde infusion are NaT and TCA.68 An intraductal infusion of NaT (0.8 mL, 4%) significantly increased pancreatic duct permeability in rats.80 The impairment of microcirculation was confirmed in models using pigs that received TCA infusions into the pancreatic duct and in which a substantial decrease of pancreatic oxygenation and changes in pancreatic blood flow occurred.81 The course of AP after retrograde BA injection is quite severe, and extensive necrosis is commonly observed with a high mortality mimicking severe AP in humans.82 In addition, the severe inflammation in the duct infusion model is accompanied by nuclear factor–κB activation and proinflammatory cytokine upregulation, indicating a key role of nuclear factor–κB in the development of the inflammatory response in BA-induced AP.83 Bile acids also act on the epithelial ductal cells and modulate production of proinflammatory cytokines, such as interleukin (IL)-8, IL-1, and IL-6. Elevations of IL-6 were observed after retrograde infusion of NaT into the pancreatic duct.46,67,84
RELATED CLINICAL EVIDENCE OF BAs INAP
Located in the retroperitoneal space, the human pancreas is relatively difficult to access for samples by biopsy. In addition, patients with AP usually consult physicians at late stage, when the initial events of AP have already passed. Therefore, investigations that address early pathophysiological events of AP in patients are limited and even rarer when considering biliary pancreatitis. In the majority of patients with gallstone-induced AP (94.4%), gallstones were also detected in their stool. This observation suggests that AP is frequently caused by migrating gallstones with only transient blockage at the ampulla of Vater.2,85 Serum BAs were found to be elevated in all patients within the first 24 hours after the onset of acute abdominal pain, which suggests that a systemic elevation of BAs during the initial stage of biliary AP may play a role.86 After the first day, serum concentrations start to decrease. In patients with AP of other than biliary etiology, the total BA concentrations were lower, suggesting that, apart from bilirubin determination, also serum BA concentrations might be useful for identifying a biliary origin in equivocal cases.87
An interaction of BAs and CCK in humans was demonstrated in patients with a tumor-induced bile duct stenosis who received CDCA added to a liquid test meal. These patients showed a negative feedback control of plasma CCK and had a lower postprandial CCK release.88 Because CCK is known to stimulate enzyme secretion not only in rodent35 but also in human acinar cells,89 the modulation of CCK release by BAs seems to be a relevant event in biliary AP.
Besides the effects of BAs on human acinar cells, there is evidence that BA also acts on human pancreatic stellate cells. Isolated human stellate cells are able to incorporate BAs leading to a release of Ca2+ into the cytosol, similar to acinar cells. This influx of BAs is Na+ dependent and mediated by a sodium-dependent transport protein, the NTCP, which is different from acinar cells, where the sodium-dependent organic anion transporter is presumably the main transporter.55,90
POTENTIAL BENEFICIAL EFFECTS OF BAs AND TARGETED TREATMENT IN AP
Ursodeoxycholic acid (UDCA) is a constituent of human serum, which accounts for 3% of total BAs.91 The role of UDCA, an epimer of CDCA, in AP is still controversial. Although there are reported cases of an increased susceptibility of AP due to UDCA,92 patients who received UDCA after removal of common bile duct stones had a lower risk of stone recurrence and thus a lower risk of acute biliary pancreatitis.93 In view of the protective effects of UDCA in animal experiments, there is currently more evidence for a protective effect of this compound in biliary pancreatitis. One limiting factor is that markedly higher UDCA doses (up to 250 mg/kg body weight) were given to animals in experimental pancreatitis than are usually given to patients.27
Tauroursodeoxycholic acid (TUDCA) is a conjugated derivative of UDCA having protective effects in acute biliary pancreatitis as well. Tauroursodeoxycholic acid reduced activation of intracellular trypsin and caspases, and attenuated ER stress damage. Endoplasmic reticulum stress impairs an unfolded protein response, which balances protein folding demand within organelles in physiological states and is maintained by TUDCA.94 This cytoprotective potential of TUDCA in the exocrine pancreas was shown in single acinar cells and in animal models.95 Further studies in humans may open new perspectives on the use of BAs as therapeutic in pancreatitis.
As mentioned previously, RyR plays an important role in pathological Ca2+ signaling caused by TLCS. Dantrolene, a RyR antagonist, was primarily used as a muscle relaxation agent inhibiting Ca2+ release by binding to RyR. In vivo models showed an amelioration of AP and propose that RyR modulators may provide a therapeutic potential for AP.14 Other activators of RyR are ATP, calmodulin, and cyclic adenine nucleotide cyclic adenosine diphosphate ribose (cADPR). Synthesis of cADPR is dependent on ADP-ribosyl cyclase CD38. Pharmacologic inhibition of cADPR by 8-Br-cADPR reduced the intracellular Ca2+ signals after stimulation by TLCS. On the other hand, CD38-deficient mice were even protected from TLCS-induced pancreatitis, and acinar cell injury was decreased when using nicotinamide, an inhibitor of CD38. This indicates that pharmacologic blockade of CD38 and cADPR could be a therapeutic option in biliary pancreatitis.96 Calcineurin inhibitory peptide was added to prevent BA-induced acinar cell injury because TLCS is able to cause calcineurin activation. Finally, the calcineurin inhibitors limited chymotrypsinogen activation.97 Inhibition of Orai, the principal store-operated calcium entry channel, reduced local damage and systemic features of TCLS-induced AP. As a result, the Orai channel was discussed as a potential treatment target for early AP.98 Recently, a selective inhibition of bromodomain and extraterminal protein, which reduces the interaction between histones and DNA and thus increases gene transcription, reduced pancreatic damage and systemic inflammatory response in BA-induced AP.99
Other potential target proteins are protein kinases. Protein kinases have been implicated in almost every mechanism of AP. Therefore, protein kinases constitute a potential target for AP.Gene therapies based on RNA interference and gene transfection have shown to be successful in the treatment of experimental AP. However, their application in clinical practice is still highly restricted. Further studies are required to develop a specific protein kinase inhibitor for the AP- targeted treatment.100
CONCLUSIONS
Bile acids play an important role in the pathogenesis of AP. Various experimental models mimicking AP have been developed to elucidate the molecular mechanisms by which BAs contribute to AP. Bile acids are internalized into acinar cells through specific receptors and a variety of transporters. Bile acids can induce a sustained Ca2+ influx from the ER at the basal portion or from an acidic pool at the apical part of the acinar cell, from where Ca2+ is ultimately released into the cytosol. The overload of intracellular Ca2+ results in mitochondrial depolarization and subsequent acinar cell necrosis. In pancreatic ductal cells, BAs have a biphasic effect on the fluid secretion in a concentration-dependent manner. Certain BAs seem even to be protective for the pancreas. Further experimental elucidation of the mechanism through which gallstones induce AP and to what extend BA contribute to this process will greatly improve our understanding of the pathophysiology and allow to design therapeutic and preventive strategies for bile-induced AP.
Footnotes
This study was supported by Deutsche Forschungsgemeinschaft (DFG AG 203/4-1, DFG SE 2702/2-1, GRK 1947 A3), the Federal Ministry of Education and Research (BMBF GANI-MED 03IS2061A and BMBF 0314107, 01ZZ9603, 01ZZ0103, 01ZZ0403, 03ZIK012, 03zz0921E), the European Union (EU-FP-7: EPC-TM), V-1-083-VBW-028, PePPP center of excellence MV (ESF/14-BM-A55-0045/16), and the EnErGie/P2 Project (ESF/14-BM-A55-0008/18). Q.T.T. is supported by scholarship from Deutscher Akademischer Austauschdienst.
The authors declare no conflict of interest.
Contributor Information
Quang Trung Tran, Email: tranquangtrung@hueuni.edu.vn; Quangtrung.Tran@med.uni-greifswald.de.
Van Huy Tran, Email: tvhuy@huemed-univ.edu.vn.
Matthias Sendler, Email: matthias.sendler@uni-greifswald.de.
Julia Doller, Email: julia.doller@med.uni-greifswald.de.
Mats Wiese, Email: Mats.Wiese@med.uni-greifswald.de.
Robert Bolsmann, Email: robert.bolsmann@uni-greifswald.de.
Anika Wilden, Email: anika.wilden@uni-greifswald.de.
Juliane Glaubitz, Email: juliane.glaubitz@uni-greifswald.de.
Jana Marielle Modenbach, Email: JanaMarielle.Modenbach@med.uni-greifswald.de.
Franziska Gisela Thiel, Email: thielf@uni-greifswald.de.
Laura L. de Freitas Chama, Email: LauraLeticia.deFreitasChama@med.uni-greifswald.de.
Frank Ulrich Weiss, Email: ulrich.weiss@uni-greifswald.de.
Markus M. Lerch, Email: Markus.Lerch@med.uni-greifswald.de.
REFERENCES
- 1.Peery AF Crockett SD Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology. 2015;149:1731–1741.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acosta JM, Ledesma CL. Gallstone migration as a cause of acute pancreatitis. N Engl J Med. 1974;290:484–487. [DOI] [PubMed] [Google Scholar]
- 3.Lerch MM, Aghdassi AA. The role of bile acids in gallstone-induced pancreatitis. Gastroenterology. 2010;138:429–433. [DOI] [PubMed] [Google Scholar]
- 4.Saluja AK Dawra RK Lerch MM, et al. CCK-JMV-180, an analog of cholecystokinin, releases intracellular calcium from an inositol trisphosphate-independent pool in rat pancreatic acini. J Biol Chem. 1992;267:11202–11207. [PubMed] [Google Scholar]
- 5.Mayerle J Schnekenburger J Krüger B, et al. Extracellular cleavage of E-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology. 2005;129:1251–1267. [DOI] [PubMed] [Google Scholar]
- 6.Halangk W Krüger B Ruthenbürger M, et al. Trypsin activity is not involved in premature, intrapancreatic trypsinogen activation. Am J Physiol Gastrointest Liver Physiol. 2002;282:G367–G374. [DOI] [PubMed] [Google Scholar]
- 7.Lerch MM Saluja AK Dawra R, et al. The effect of chloroquine administration on two experimental models of acute pancreatitis. Gastroenterology. 1993;104:1768–1779. [DOI] [PubMed] [Google Scholar]
- 8.Wartmann T Mayerle J Kähne T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology. 2010;138:726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kereszturi E Szmola R Kukor Z, et al. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum Mutat. 2009;30:575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weidenbach H Lerch MM Gress TM, et al. Vasoactive mediators and the progression from oedematous to necrotising experimental acute pancreatitis. Gut. 1995;37:434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gress TM Müller-Pillasch F Lerch MM, et al. Balance of expression of genes coding for extracellular matrix proteins and extracellular matrix degrading proteases in chronic pancreatitis. Z Gastroenterol. 1994;32:221–225. [PubMed] [Google Scholar]
- 12.Gress T Müller-Pillasch F Elsässer HP, et al. Enhancement of transforming growth factor beta 1 expression in the rat pancreas during regeneration from caerulein-induced pancreatitis. Eur J Clin Invest. 1994;24:679–685. [DOI] [PubMed] [Google Scholar]
- 13.Tazuma S, Takikawa H, eds. Bile Acids in Gastroenterology: Basic and Clinical. Tokyo, Japan: Springer Nature; 2017. [Google Scholar]
- 14.Husain SZ Orabi AI Muili KA, et al. Ryanodine receptors contribute to bile acid-induced pathological calcium signaling and pancreatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1423–G1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann AF, Hagey LR. Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades. J Lipid Res. 2014;55:1553–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieland HO. The chemistry of the bile acids (1928). In: Nobel Foundation. Nobel Lectures, Chemistry 1922–1941. Vol 2. Amsterdam, The Netherlands: Elsevier; 1966:94–102. [Google Scholar]
- 18.Li Y, Lu LG. Therapeutic roles of bile acid signaling in chronic liver diseases. J Clin Transl Hepatol. 2018;6:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottlieb A, Bechmann L, Canbay A. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Ann Hepatol. 2018;17:340–341. [DOI] [PubMed] [Google Scholar]
- 20.Baiocchi L Zhou T Liangpunsakul S, et al. Dual role of bile acids on the biliary epithelium: friend or foe? Int J Mol Sci. 2019;20:1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajani C, Jia W. Bile acids and their effects on diabetes. Front Med. 2018;12:608–623. [DOI] [PubMed] [Google Scholar]
- 22.Niederau C Niederau M Luthen R, et al. Pancreatic exocrine secretion in acute experimental pancreatitis. Gastroenterology. 1990;99:1120–1127. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann AF, Hagey LR, Krasowski MD. Bile salts of vertebrates: structural variation and possible evolutionary significance. J Lipid Res. 2010;51:226–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiang JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol. 2004;40:539–551. [DOI] [PubMed] [Google Scholar]
- 25.Perreault M Bialek A Trottier J, et al. Role of glucuronidation for hepatic detoxification and urinary elimination of toxic bile acids during biliary obstruction. PloS One. 2013;8:e80994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerasimenko JV Flowerdew SE Voronina SG, et al. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J Biol Chem. 2006;281:40154–40163. [DOI] [PubMed] [Google Scholar]
- 27.Katona M Hegyi P Kui B, et al. A novel, protective role of ursodeoxycholate in bile-induced pancreatic ductal injury. Am J Physiol Gastrointest Liver Physiol. 2016;310:G193–G204. [DOI] [PubMed] [Google Scholar]
- 28.Voronina S Longbottom R Sutton R, et al. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol. 2002;540:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer L Gukovskaya AS Penninger JM, et al. Phosphatidylinositol 3-kinase facilitates bile acid-induced Ca2+ responses in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G875–G886. [DOI] [PubMed] [Google Scholar]
- 30.Lanner JT Georgiou DK Joshi AD, et al. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foskett JK White C Cheung KH, et al. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrow SL Voronina SG da Silva Xavier G, et al. ATP depletion inhibits Ca2+ release, influx and extrusion in pancreatic acinar cells but not pathological Ca2+ responses induced by bile. Pflugers Arch. 2008;455:1025–1039. [DOI] [PubMed] [Google Scholar]
- 33.Vassileva G Golovko A Markowitz L, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perides G Laukkarinen JM Vassileva G, et al. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology. 2010;138:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krüger B, Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Clin Pathol. 2000;157:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voronina SG Barrow SL Gerasimenko OV, et al. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem. 2004;279:27327–27338. [DOI] [PubMed] [Google Scholar]
- 37.Voronina SG Barrow SL Simpson AW, et al. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology. 2010;138:1976–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Booth DM Murphy JA Mukherjee R, et al. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140:2116–2125. [DOI] [PubMed] [Google Scholar]
- 39.Voronina SG Gryshchenko OV Gerasimenko OV, et al. Bile acids induce a cationic current, depolarizing pancreatic acinar cells and increasing the intracellular Na+ concentration. J Biol Chem. 2005;280:1764–1770. [DOI] [PubMed] [Google Scholar]
- 40.Siemen D, Hescheler J, eds. Nonselective Cation Channels: Pharmacology, Physiology and Biophysics. Basel, Switzerland: Birkhäuser Verlag; 1993. [Google Scholar]
- 41.Yubero S Ramudo L Manso MA, et al. The role of redox status on chemokine expression in acute pancreatitis. Biochim Biophys Acta. 1792;2009:148–154. [DOI] [PubMed] [Google Scholar]
- 42.Li Z Lu M Chu J, et al. Early proteome analysis of rat pancreatic acinar AR42J cells treated with taurolithocholic acid 3-sulfate. Pancreatology. 2012;12:248–256. [DOI] [PubMed] [Google Scholar]
- 43.Mateu A De Dios I Manso MA, et al. Oxidized phospholipids exert a dual effect on bile acid-induced CCL2 expression in pancreatic acini. Pancreatology. 2017;17:372–380. [DOI] [PubMed] [Google Scholar]
- 44.Kim JY Kim KH Lee JA, et al. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology. 2002;122:1941–1953. [DOI] [PubMed] [Google Scholar]
- 45.Zhou X Levin EJ Pan Y, et al. Structural basis of the alternating-access mechanism in a bile acid transporter. Nature. 2014;505:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hegyi P Maléth J Walters JR, et al. Guts and gall: bile acids in regulation of intestinal epithelial function in health and disease. Physiol Rev. 2018;98:1983–2023. [DOI] [PubMed] [Google Scholar]
- 47.Dawson PA. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol. 2011;169–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kowal JM Haanes KA Christensen NM, et al. Bile acid effects are mediated by ATP release and purinergic signalling in exocrine pancreatic cells. Cell Commun Signal. 2015;13:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venglovecz V Rakonczay Z Jr Ozsvári B, et al. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut. 2008;57:1102–1112. [DOI] [PubMed] [Google Scholar]
- 50.Hegyi P Pandol S Venglovecz V, et al. The acinar-ductal tango in the pathogenesis of acute pancreatitis. Gut. 2011;60:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Venglovecz V, Rakonczay Z, Jr, Hegyi P. The effects of bile acids on pancreatic ductal cells. Pancreapedia. Available at: https://www.pancreapedia.org/reviews/effects-of-bile-acids-on-pancreatic-ductal-cells. Accessed April 7, 2020. [DOI] [PubMed]
- 52.Maléth J Venglovecz V Rázga Z, et al. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut. 2011;60:136–138. [DOI] [PubMed] [Google Scholar]
- 53.Reber HA, Mosley JG. The effect of bile salts on the pancreatic duct mucosal barrier. Br J Surg. 1980;67:59–62. [DOI] [PubMed] [Google Scholar]
- 54.Farmer RC Tweedie J Maslin S, et al. Effects of bile salts on permeability and morphology of main pancreatic duct in cats. Dig Dis Sci. 1984;29:740–751. [DOI] [PubMed] [Google Scholar]
- 55.Ferdek PE Jakubowska MA Gerasimenko JV, et al. Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium-driven bile uptake. J Physiol. 2016;594:6147–6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou X Xie L Bergmann F, et al. The bile acid receptor FXR attenuates acinar cell autophagy in chronic pancreatitis. Cell Death Discov. 2017;3:17027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shahid RA Vigna SR Layne AC, et al. Acinar cell production of leukotriene B4 contributes to development of neurogenic pancreatitis in mice. Cell Mol Gastroenter. 2015;1:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kiriyama Y, Nochi H. The biosynthesis, signaling, and neurological functions of bile acids. Biomolecules. 2019;9:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mareninova OA Hermann K French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nijmeijer RM Schaap FG Smits AJ, et al. Impact of global Fxr deficiency on experimental acute pancreatitis and genetic variation in the FXR locus in human acute pancreatitis. PloS One. 2014;9:e114393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lewarchik CM Orabi AI Jin S, et al. The ryanodine receptor is expressed in human pancreatic acinar cells and contributes to acinar cell injury. Am J Physiol Gastrointest Liver Physiol. 2014;307:G574–G581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Geyer N Diszházi G Csernoch L, et al. Bile acids activate ryanodine receptors in pancreatic acinar cells via a direct allosteric mechanism. Cell Calcium. 2015;58:160–170. [DOI] [PubMed] [Google Scholar]
- 63.Streb H Bayerdörffer E Haase W, et al. Effect of inositol-1,4,5-trisphosphate on isolated subcellular fractions of rat pancreas. J Membr Biol. 1984;81:241–253. [DOI] [PubMed] [Google Scholar]
- 64.Randhawa PK, Jaggi AS. A review on potential involvement of TRPV1 channels in ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther. 2018;23:38–45. [DOI] [PubMed] [Google Scholar]
- 65.Vigna SR Shahid RA Nathan JD, et al. Leukotriene B4 mediates inflammation via TRPV1 in duct obstruction-induced pancreatitis in rats. Pancreas. 2011;40:708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng HY, Chen YC. Role of bile acids in carcinogenesis of pancreatic cancer: an old topic with new perspective. World J Gastroenterol. 2016;22:7463–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le T Eisses JF Lemon KL, et al. Intraductal infusion of taurocholate followed by distal common bile duct ligation leads to a severe necrotic model of pancreatitis in mice. Pancreas. 2015;44:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perides G van Acker GJ Laukkarinen JM, et al. Experimental acute biliary pancreatitis induced by retrograde infusion of bile acids into the mouse pancreatic duct. Nat Protoc. 2010;5:335–341. [DOI] [PubMed] [Google Scholar]
- 69.Jones RS. Effect of insulin on canalicular bile formation. Am J Physiol. 1976;231:40–43. [DOI] [PubMed] [Google Scholar]
- 70.Sendler M Beyer G Mahajan UM, et al. Complement component 5 mediates development of fibrosis, via activation of stellate cells, in 2 mouse models of chronic pancreatitis. Gastroenterology. 2015;149:765–76.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lerch MM Saluja AK Rünzi M, et al. Pancreatic duct obstruction triggers acute necrotizing pancreatitis in the opossum. Gastroenterology. 1993;104:853–861. [DOI] [PubMed] [Google Scholar]
- 72.Yuan Z Zheng J Mei Z, et al. A mouse model of necrotic biliary pancreatitis induced by combining gallstone formation and ligation of the biliary-pancreatic duct. bioRxiv. 2017;158915. [Google Scholar]
- 73.Mooren FCh Hlouschek V Finkes T, et al. Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J Biol Chem. 2003;278:9361–9369. [DOI] [PubMed] [Google Scholar]
- 74.Wan MH Huang W Latawiec D, et al. Review of experimental animal models of biliary acute pancreatitis and recent advances in basic research. HPB (Oxford). 2012;14:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lerch MM, Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology. 2013;144:1180–1193. [DOI] [PubMed] [Google Scholar]
- 76.Kaiser AM Saluja AK Sengupta A, et al. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol. 1995;269:C1295–C1304. [DOI] [PubMed] [Google Scholar]
- 77.Saluja A Saluja M Villa A, et al. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest. 1989;84:1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan YC, Leung PS. Acute pancreatitis: animal models and recent advances in basic research. Pancreas. 2007;34:1–14. [DOI] [PubMed] [Google Scholar]
- 79.Arendt T. Bile-induced acute pancreatitis in cats. Roles of bile, bacteria, and pancreatic duct pressure. Dig Dis Sci. 1993;38:39–44. [DOI] [PubMed] [Google Scholar]
- 80.Plusczyk T Westermann S Rathgeb D, et al. Acute pancreatitis in rats: effects of sodium taurocholate, CCK-8, and Sec on pancreatic microcirculation. Am J Physiol. 1997;272:G310–G320. [DOI] [PubMed] [Google Scholar]
- 81.Kinnala PJ Kuttila KT Grönroos JM, et al. Splanchnic and pancreatic tissue perfusion in experimental acute pancreatitis. Scand J Gastroenterol. 2002;37:845–849. [PubMed] [Google Scholar]
- 82.Kruse P, Hage E, Lasson A. Proteases and protease inhibitors in taurocholate-induced acute pancreatitis in rats. Int J Pancreatol. 1999;25:113–121. [DOI] [PubMed] [Google Scholar]
- 83.Vaquero E Gukovsky I Zaninovic V, et al. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1197–G1208. [DOI] [PubMed] [Google Scholar]
- 84.Laukkarinen JM Van Acker GJ Weiss ER, et al. A mouse model of acute biliary pancreatitis induced by retrograde pancreatic duct infusion of Na-taurocholate. Gut. 2007;56:1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pickartz T, Tran QT, Lerch MM. Three centuries since the discovery of Vater's Papilla. Gut. 2020 Jul 30. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 86.Cucuianu MP Ionescu NG Vulcu V, et al. Transient elevation of serum bile acids during acute pancreatitis. Pancreatology. 1988;3:151–156. [DOI] [PubMed] [Google Scholar]
- 87.Maleszka A Dumnicka P Matuszyk A, et al. The diagnostic usefulness of serum total bile acid concentrations in the early phase of acute pancreatitis of varied etiologies. Int J Mol Sci. 2017;18:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koop I Koop H Gerhardt C, et al. Do bile acids exert a negative feedback control of cholecystokinin release? Scand J Gastroenterol. 1989;24:315–320. [DOI] [PubMed] [Google Scholar]
- 89.Murphy JA Criddle DN Sherwood M, et al. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology. 2008;135:632–641. [DOI] [PubMed] [Google Scholar]
- 90.Geyer J Döring B Meerkamp K, et al. Cloning and functional characterization of human sodium-dependent organic anion transporter (SLC10A6). J Biol Chem. 2007;282:19728–19741. [DOI] [PubMed] [Google Scholar]
- 91.Kotb MA. Molecular mechanisms of ursodeoxycholic acid toxicity & side effects: ursodeoxycholic acid freezes regeneration & induces hibernation mode. Int J Mol Sci. 2012;13:8882–8914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nadir A Nadir F Hassanein T, et al. Acute relapsing pancreatitis induced with ursodeoxycholic acid therapy. J Okla State Med Assoc. 1995;88:295–298. [PubMed] [Google Scholar]
- 93.Yamamoto R Tazuma S Kanno K, et al. Ursodeoxycholic acid after bile duct stone removal and risk factors for recurrence: a randomized trial. J Hepatobiliary Pancreat Sci. 2016;23:132–136. [DOI] [PubMed] [Google Scholar]
- 94.Seyhun E Malo A Schafer C, et al. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G773–G782. [DOI] [PubMed] [Google Scholar]
- 95.Malo A Kruger B Seyhun E, et al. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2010;299:G877–G886. [DOI] [PubMed] [Google Scholar]
- 96.Orabi AI Muili KA Javed TA, et al. Cluster of differentiation 38 (CD38) mediates bile acid-induced acinar cell injury and pancreatitis through cyclic ADP-ribose and intracellular calcium release. J Biol Chem. 2013;288:27128–27137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Muili KA Wang D Orabi AI, et al. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J Biol Chem. 2013;288:570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wen L Voronina S Javed MA, et al. Inhibitors of ORAI1 prevent cytosolic calcium-associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology. 2015;149:481–492.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang W Haynes AC Mukherjee R, et al. Selective inhibition of BET proteins reduces pancreatic damage and systemic inflammation in bile acid- and fatty acid ethyl ester- but not caerulein-induced acute pancreatitis. Pancreatology. 2017;17:689–697. [DOI] [PubMed] [Google Scholar]
- 100.Ma B Wu L Lu M, et al. Differentially expressed kinase genes associated with trypsinogen activation in rat pancreatic acinar cells treated with taurolithocholic acid 3-sulfate. Mol Med Rep. 2013;7:1591–1596. [DOI] [PubMed] [Google Scholar]