

Supplemental Digital Content is available in the text.
Keywords: blood pressure, coronary artery disease, diabetes mellitus, genome-wide association study, risk factors
Background:
Coronary artery disease (CAD) is accelerated in subjects with type 2 diabetes mellitus (T2D).
Methods:
To test whether this reflects differential genetic influences on CAD risk in subjects with T2D, we performed a systematic assessment of genetic overlap between CAD and T2D in 66 643 subjects (27 708 with CAD and 24 259 with T2D). Variants showing apparent association with CAD in stratified analyses or evidence of interaction were evaluated in a further 117 787 subjects (16 694 with CAD and 11 537 with T2D).
Results:
None of the previously characterized CAD loci was found to have specific effects on CAD in T2D individuals, and a genome-wide interaction analysis found no new variants for CAD that could be considered T2D specific. When we considered the overall genetic correlations between CAD and its risk factors, we found no substantial differences in these relationships by T2D background.
Conclusions:
This study found no evidence that the genetic architecture of CAD differs in those with T2D compared with those without T2D.
There is considerable variation in the presentation, severity, and pathology of coronary artery disease (CAD) between subjects with type 2 diabetes mellitus (T2D) and those with no history of diabetes mellitus. Subjects with T2D have more extensive and severe atherosclerosis, suffer more silent infarcts, and are more prone to thrombosis than subjects without diabetes mellitus.1–3 The mechanisms by which T2D accelerates CAD are poorly understood. In principle, the acceleration of CAD in T2D may be attributed to features that jointly predispose subjects to T2D and CAD or to factors intrinsic to the T2D state that increase the risk of CAD, such as hyperglycemia, insulin resistance, and chronic inflammation.4
Predisposition to both CAD and T2D has a substantial genetic component (with ≈163 CAD risk and ≈403 for T2D association signals identified to date in subjects of European Ancestry)5,6 and Mendelian randomization studies support a causal role for T2D in the development of CAD.7–9 A Mendelian randomization study found that the average CAD risk per T2D allele was lower than expected (for the 44 T2D associated variants assessed) compared with the increased risk of CAD attributed to T2D by epidemiological studies.7 This indicated that the T2D associated variants did not account for all the risk of CAD observed in subjects with T2D. Few variants have been associated with both CAD and T2D: a variant near IRS1 was associated with both diseases at genome-wide significance (P≤5×10-8) and 8 other loci at a lower significance level.9 Given that there are few variants jointly associated with CAD and T2D, it is unsurprising that there is sparse evidence for overlapping pathways contributing to both diseases.10
A recent study conducted in the UK Biobank found no evidence of differential effects of CAD risk variants by T2D status. However, in this study, the sample size was relatively small (3968 CAD cases and 11 698 controls).11 Another study found that a genetic risk score constructed from known CAD loci was associated with CAD in subjects with T2D, indicating that variants identified in the general population were predictive of CAD in the context of T2D.12 What has not been systematically addressed in a large sample is whether there is a quantitative or qualitative difference in the pattern of loci influencing risk of CAD among subjects with T2D when compared with those without the condition.
We conducted a comprehensive investigation of genetic differences in the determinants of CAD between subjects with and without T2D in a large sample. The discovery meta-analysis included 66 643 subjects (of whom 27 708 had CAD and 24 259 had T2D), and we sought replication for a subset of variants in a further 117 787 samples (16 694 with CAD; 11 537 subjects with T2D).
Methods
An overview of the study design is illustrated in Figure 1 and the methods are provided in the Data Supplement. The summary statistics have been made available via figshare (10.6084/m9.figshare.7811639). This study made use of data generated from individual studies for which the relevant institutional review board approval had been obtained and all participants consented to inclusion in individual studies.
Figure 1.
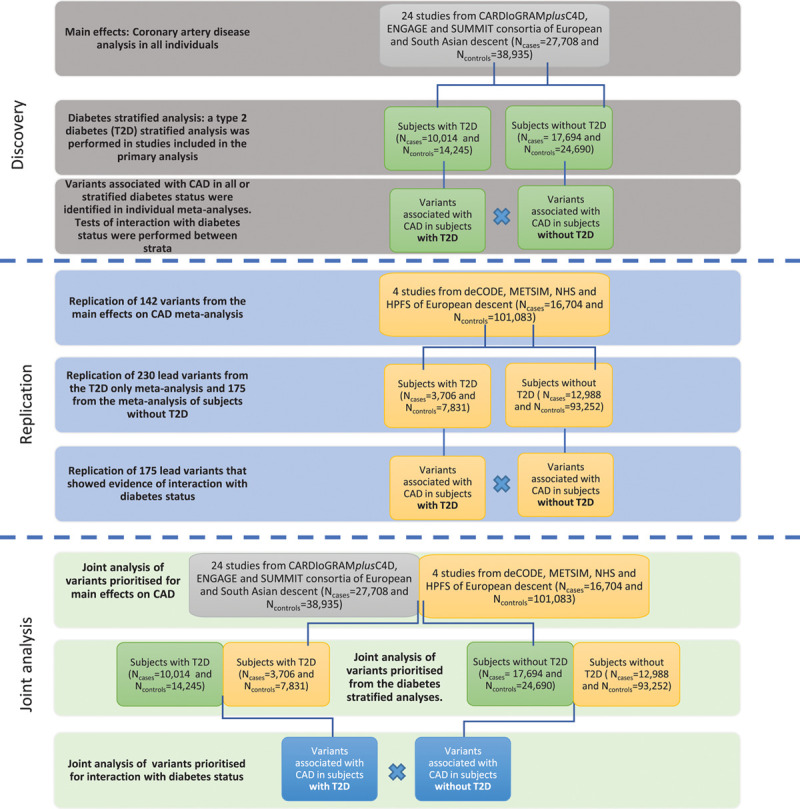
Study design. In the discovery meta-analyses, we performed 4 different meta-analyses of coronary artery disease (CAD): in all individuals irrespective of Type 2 diabetes mellitus (T2D) status; in all individuals corrected for T2D stats; and stratified by T2D status. We examined allelic effects within strata to identify stratum-specific CAD associated variants, and between strata to identify variants that may interact with T2D status to modify the risk of CAD. We selected variants that achieved P<1×10-4 for association with CAD in at least one of the following analyses: all individuals combined regardless of T2D status; subjects with T2D only; subjects without diabetes mellitus; or the interaction analysis. The replication analysis was performed in independent samples using the same study design as the discovery analysis. CARDIoGRAMplusC4D indicates Coronary Artery Disease Genome Wide Replication and Meta-Analysis (CARDIoGRAM) Plus the Coronary Artery Disease (C4D) Genetics; ENGAGE, European Network for Genetic and Genomic Epidemiology; HPFS, Health Professionals Follow-Up Study; METSIM, The Metabolic Syndrome in Men Study; NHS, Nurses’ Health Study; and SUMMIT, Surrogate Markers for Micro- and Macro-Vascular Hard End Points for Innovative Diabetes Tools.
Results
Identification of CAD Cases, CAD Controls, and Subjects With Diabetes Mellitus
This study was performed using full summary statistics from CAD case-control analyses performed separately in subjects with T2D and subjects with no history of diabetes mellitus. The discovery meta-analyses included 27 708 CAD cases (of whom 10 014 had T2D) and 38 935 subjects with no history of CAD (14 245 with T2D) from 23 studies of European descent and one study of South Asian descent, assembled from the CARDIoGRAMplusC4D (Coronary Artery Disease Genome Wide Replication and Meta-Analysis (CARDIoGRAM) Plus the Coronary Artery Disease (C4D) Genetics), ENGAGE (European Network for Genetic and Genomic Epidemiology), and SUMMIT (Surrogate Markers for Micro- and Macro-Vascular Hard End Points for Innovative Diabetes Tools) consortia (Figure 1 and Tables I and II in the Data Supplement). Replication of selected signals was sought in an independent sample of 16 694 CAD cases (3706 with T2D) and 101 093 controls with no history of CAD (7831 with T2D) from 4 studies of European descent with existing genome-wide association study from deCODE, the NHS (Nurses’ Health Study), the METSIM study (The Metabolic Syndrome in Men), and the HPFS (Health Professionals Follow-Up Study; Tables III and IV in the Data Supplement). None of the studies contained overlapping samples.
Main Effects of Variants on CAD
We first set out to identify variants that were associated with CAD in the complete sample set. We performed 2 meta-analyses, the first compared CAD cases to controls without reference to T2D status, whereas the second repeated the analysis adjusted for T2D status. In both analyses, we confirmed many of the previously reported CAD associated loci at genome-wide significance (P≤5×10-8), including SORT1/CELSR2, WDR12, PHACTR1, TCF21, 9p21.3, CXCL12, and ADAMTS7. We selected 142 variants that achieved P≤5×10-4 in either the unadjusted or the T2D- adjusted analyses for replication analyses.
We had access to full summary statistics for the discovery analysis but not from the replication cohorts. We requested summary statistics for selected variants from replication cohorts. Thus, we performed a joint analysis of the estimates from the discovery and replication analyses. In the joint analysis, we expanded the set of known CAD loci detected in this meta-analysis from 7 to 13 reaching genome-wide significance in our dataset (Figure 2A and 2B and Table V in the Data Supplement). For published CAD variants, the risk allele identified in this meta-analysis was the same as the published risk allele for variants associated with CAD P≤1×10-3 (Figure II and Table V in the Data Supplement).5 This reflects, in part, an overlap of samples included in these various analyses (Figure II and Table V in the Data Supplement).
Figure 2.
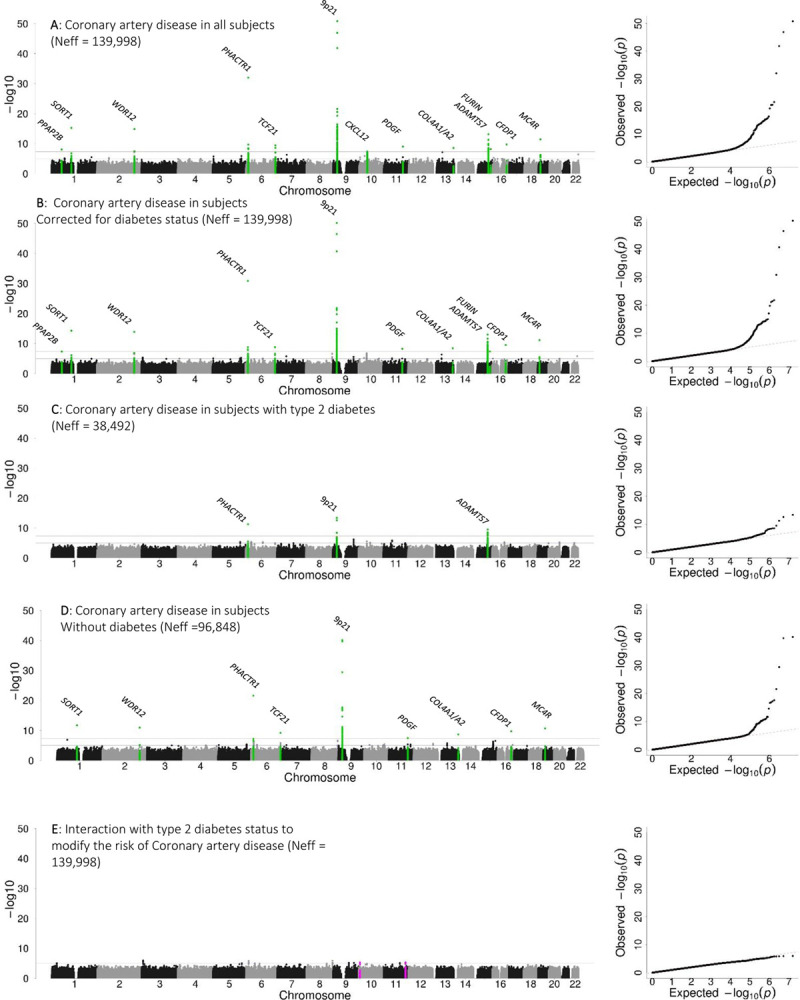
Five genetic association study meta-analyses were performed to investigate the genetic architecture of coronary artery disease (CAD) in the context of Type 2 diabetes mellitus (T2D). Manhattan and QQ plots from (A) a meta-analysis that combined allelic effects on CAD from subjects with T2D status and without diabetes mellitus and (B) corrected for T2D status to identify variants associated with CAD irrespective of T2D status; (C) a meta-analysis of allelic effects on CAD in subjects with T2D to identify loci that may influence the development of CAD in the context of T2D; (D) a meta-analysis of allelic effects on CAD in the absence of diabetes mellitus to identify loci that may influence the development of CAD in the absence of diabetes mellitus; and (E) an interaction analysis to identify loci that may interact with T2D to modify the risk of CAD. The effective sample size was based on the combined discovery and replication sample of 184 250 subjects.
Stratified Analysis
The second approach we used to identify any loci at which CAD risk effects (P≤5×10-8) were influenced by the presence or absence of T2D, involved a T2D-stratified meta-analysis of CAD risk. In the discovery phase of this stratified analysis, 3 known CAD loci reached genome-wide significance: ADAMTS7 in subjects with T2D and 9p21.3 and PHACTR1 in the analysis of subjects without diabetes mellitus (Table V in the Data Supplement). The allelic effects and association signals at the previously reported CAD loci did not show any systematic difference according to T2D background (Figure I in the Data Supplement).
We selected 230 lead variants for replication from the T2D-only analysis and 175 lead variants from the analysis of subjects without diabetes mellitus for replication based on a stratum-specific CAD association of P≤1×10-4. In the joint analysis (discovery and replication), we found no novel CAD risk signals in either stratum (Figure 2C and 2D and Table V in the Data Supplement). Three loci were associated with CAD in subjects with T2D, and these overlapped loci associated with CAD in subjects without diabetes mellitus (Figure 2). The different number of loci associated with CAD by T2D background reflects a difference in power (ie, sample size) to detect associations rather than a systematic difference by T2D background.
Interaction Analysis
In a complementary analysis to the stratified analysis, we performed a T2D interaction analysis (see Methods in the Data Supplement) to identify variants that interacted with T2D status to modify the risk of CAD. We calculated the interaction P values based on summary statistics from the T2D stratified analyses of CAD and not from a meta-analysis of interaction terms. We adopted this approach to maximize the number of samples used to estimate interactive effects (see Methods in the Data Supplement). The interaction analysis was performed by comparing the allelic effects (on the log-odds scale) on CAD risk for each variant between T2D strata. The allelic effects and their associated standard errors for CAD risk estimated in T2D stratified meta-analyses were compared using GWAMA v2.1.13 The smaller the Pinteraction the larger difference in allelic effects on CAD risk by T2D status.
The top interaction in the discovery analysis was represented by rs712755, near GRM7 (Pinteraction=4.6×10-7). This variant had opposing effects on CAD risk dependent on T2D context (effect allele frequency, 0.71, odds ratio [OR]T2D, 0.82 [0.74–0.90], ORNodiabetesmellitus, 1.14 [1.06–1.23]).
We sought replication for 175 loci, including GRM7, with at least modest evidence of interaction with T2D status (Pinteraction ≤1×10-4). We performed a joint interaction meta-analysis of the discovery and replication data and defined replication as a combined (discovery+replication) Pinteraction<2.9×10-4 (0.05/175; that corrects for the number of loci selected for replication) and a joint Pinteraction< discovery Pinteraction. The latter indicates directionally consistent allelic effects by T2D stratum in the discovery and replication stages.
The interaction at GRM7, represented by rs712755, did not replicate (replication Pinteraction=0.36) and none of the other 174 loci met the criteria for replication. Overall, there was no evidence for loci that interacted with T2D status to modify the risk of CAD based on this interaction analysis.
We also examined the known CAD loci for evidence of interaction. Of the 163 known variants for CAD, 161 were present in our data. We applied a Bonferroni correction of Pinteraction≤3.1×10-4 (0.05/161; correcting for the number of known CAD loci). None of the established CAD loci interacted with T2D status to modify the risk of CAD (Table V in the Data Supplement). A variant located near GLUL (rs10911021) had been associated with CAD in subjects with T2D.14 In the current study, rs10911021 showed no association with CAD in subjects with T2D (P=0.54) and had no evidence of interaction with T2D status (Pinteraction=0.46; Figure III in the Data Supplement).
Power to Detect Interactions
A substantial challenge in detecting loci that interact with T2D to modify the risk of CAD is sufficient sample size. Even in this large discovery sample of 66 643 subjects (27 708 with CAD), we had <80% power to detect interactions with at least a 20% difference in allelic odds between strata (ie, ORNodiabetesmellitus, 1.00 versus ORT2D, 1.20) for risk allele frequency>10% at α=1×10-4 (the threshold for replication selection in the interaction analysis; see Methods in the Data Supplement). This was only for interactions where there were opposite allelic effects in strata or where there was a null allelic effect on CAD in one stratum (ie, ORNodiabetesmellitus, 1.00) and a large (ie, ORT2D, 1.20) allelic effect on CAD in the other stratum (Figure IIA and IIB in the Data Supplement). We had little power to detect interactions where allelic effects on CAD were in the same direction in both strata (see Methods and Figure IIC in the Data Supplement). In the replication sample of 117 787 samples (16 694 with CAD) at an α=0.05, we observed similar patterns of power to detect associations with opposing effects by stratum. Thus, we would be unlikely (in this sample size) be able to detect smaller interaction effects or those involving rare alleles.
Genetic Overlap With Risk Factors
We have comprehensively interrogated variants for association with CAD in the context of T2D but not risk factors of both T2D and CAD. There may be a different effect of these risk factors on CAD by T2D context, which may explain some of the increased risk of CAD in subjects with T2D. First, we performed genetic correlation analyses using LDHub (a centralised database of summary-level GWAS results and a web interface for LD score regression) to estimate the overall genetic correlation (based on all variants) between risk factors and CAD separately by T2D background. 15 Subsequently, a heterogeneity test was performed on the risk factor genetic correlation estimates with CAD by T2D background to identify risk factors that may have a variable correlation with CAD based on T2D background. Overall, we found no difference in the genetic correlation between 106 risk factors and CAD by T2D status (Figure IV and Table VI in the Data Supplement).
To investigate this further but only in a subset of variants associated with risk factors at genome-wide significance (P≤5×10−8), we constructed weighted genetic risk scores for seventeen traits related to obesity,16–18 hypertension,19 lipids,20 diabetes mellitus,6,21 glycaemic traits, and insulin resistance.22–28 These genetic risk scores included between 10 and 403 single nucleotide polymorphisms for each phenotype. We tested these for CAD association in the T2D unadjusted (main) analysis, as well as in the T2D-stratified analyses, where we performed a test for heterogeneity for different effects on CAD by T2D background (Methods in the Data Supplement). We adopted a significance threshold of P≤2.9×10-3 that accounted for the 17 genetic risk scores, but not for the multiple CAD associations performed. Genetic risk scores for LDL-C (low-density lipoprotein cholesterol), body mass index, and systolic blood pressure were associated with CAD irrespective of T2D background (Figure V and Table VII in the Data Supplement). Collectively, these analyses provide no evidence to support T2D-stratified differences in CAD risk as conveyed by variants influencing phenotypes known to contribute to CAD development.
Discussion
There is a well-established causal role for T2D in increased risk of CAD. However, this increased risk could not be explained by differences in genetic architecture of CAD between individuals with and without diabetes mellitus. We found no difference in the effects of known CAD loci on the risk of CAD by T2D status. We also found no variants of large effect specifically associated with CAD in the context of T2D. We also found no differences in the effects of risk factors on CAD by T2D background based on analyses that used the genetic variation contributing to these risk factors. Indicating that the genetic variants associated with these risk factors do not have a differential effect on CAD risk by T2D background.
There are many factors that will influence the power to detective genuine interactive effects. Identification of interactive effects requires a large sample size particularly when conducting a genome-wide interaction analysis.29 Even in this study that included 66 643 subjects (considerably larger than previous efforts), we were underpowered to identify variants with small differences in effect on CAD risk by T2D status. If interaction effects do exist, these effects are likely to be modest and only detectable in a much larger sample size.
The accuracy of the phenotype definition will also affect the power to detect interactive effects. Diagnosis of T2D is often contemporaneous to CAD diagnosis and may not reflect the actual onset of diabetes mellitus. We are uncertain of the stage of T2D development when risk of CAD begins to increase. There is evidence of increased vascular risk before the onset of clinically diagnosed T2D.30 Taking this variability into account, we defined CAD cases with T2D as those that had a diagnosis of T2D up to 5 years after a CAD event with no minimum duration of diabetes mellitus. This also allowed us to increase the sample size by including cross-sectional studies for which information on the duration of diabetes mellitus may not be available. We were unable to account for the attenuation of genetic effects due to the misclassification of subjects who may develop CAD and or T2D outside of the study observation period.
This study shows that difference in risk of CAD between subjects with and without T2D cannot be explained by variants of large effect or differences in the genetic variation contributing to known risk factors of either T2D or CAD. There are several other mechanisms, outside the scope of the current study, that could explain some of the increased risk of CAD in subjects with T2D. There could be epigenetic changes induced by some feature of the T2D state. For example, hyperglycemia has been shown to cause epigenetic changes altering gene expression in vascular cells leading to endothelial dysfunction, a hallmark of atherosclerosis.31 Although the evidence for overlapping pathways between CAD and T2D is sparse, treatment of one disease can increase the risk of the other. Statins known to reduce the risk of CAD have been shown to increase the risk of T2D, whereas some thialidazones, used to treat insulin resistance in subjects with T2D, increase the risk of CAD.32 It is likely that the T2D state perturbs or exacerbates some common atherosclerotic processes rather than through T2D background specific genes/pathways to increase the risk of CAD in subjects with T2D.
Sources of Funding
This work was supported by the European Union Framework Programme 7 (FP7/2007-2013) for the Innovative Medicine Initiative (IMI) under grant agreement n° IMI/115006 (the SUMMIT [Surrogate Markers for Micro- and Macro-Vascular Hard End Points for Innovative Diabetes Tools] consortium); Aarno Koskelo Foundation; Academy of Finland (no. 263401; no. 2676882); American Heart Association (13SDG14330006); AstraZeneca; AtheroSysMed (Systems medicine of coronary heart disease and stroke); British Heart Foundation Centre of Research Excellence at Oxford; ERC269045-Gene Target T2D grant; Estonian Research Council (IUT20-60, PUT1660 and PUT1665P); Estonian Center of Genomics/Roadmap II (project No. 2014-2020.4.01.16-0125); European Union (no. 692145; no. 633589; no. 313010; LSHM-CT-2007-037273; no. 201668; 2014-2020.4.01.15-0012;QLG1-CT-2002-00896; EU/QLRT-2001-01254; QLG2-CT-2002-01254 HEALTH-F2-2013-601456); Finnish Foundation for Cardiovascular research; Gentransmed - Centre of Excellence for Genomics and Translational Medicine; German Ministry of Education and Research (no. 01ZX1313A-K); Helsinki University Central Hospital special government funds (TYH7215, TKK2012005, TYH2012209, TYH2014312); Juvenile Diabetes Research Foundation (JDRF, 2-SRA-2014-276-Q-R); National Institute of Diabetes and Digestive and Kidney diseases (NIDDK, 5R01DK106236; U01-DK066134; U01-DK105535; R01DK101478); National Heart, Lung and Blood Institute (NLHBI, R01HL103866); National Institute for Health Research (NIHR); Personalized diagnostics and treatment of high risk coronary artery disease patients (RiskyCAD; 305739); Sigrid Juselius Foundation; Finnish Academy (no. 269517); Finnish Foundation for Cardiovascular Research; Foundation for Strategic Research and Stockholm County Council (560283; 592229); Juho Vainio Foundation; Knut and Alice Wallenberg Foundation; Ministry for Higher Education; Strategic Cardiovascular and Diabetes Programmes of Karolinska Institutet and Stockholm County Council; Swedish Foundation for Strategic Research (SSF; ICA08-0047); Swedish Heart-Lung Foundation; Swedish Research Council (project 8691; 2015-02558; 2016-00598; M-2005-1112 and 2009-2298); Torsten and Ragnar Söderberg Foundation; W.W. Smith Charitable Trust (H1201); Wellcome Trust Institutional strategic support fund; Yrjö Jahnsson Foundation.
Disclosures
Authors have disclosed possible conflicts of interest and have confirmed that these are unrelated to the work described in this article. Dr Ingelsson is a scientific advisor for Precision Wellness. Dr Salomaa has consulted for Novo Nordisk and Sanofi and has ongoing research collaboration with Bayer (all unrelated to the present study). Dr März reports employment with Synlab Holding Deutschland GmbH and has received grants or personal fees from Abbott Diagnostics; Aegerion Pharmaceuticals; AMGEN; AstraZeneca; BASF Pharma Solutions; Danone Research; MSD; Sanofi; Siemens Diagnostics; and Synageva. Dr Colhoun receives research support and honoraria from and is also a member of the advisory panels and speaker’s bureaus for Sanofi Aventis, Regeneron, and Eli Lilly. Dr Colhoun has been a member of Data and Safety Monitoring Board of the Advisory Panel for the CANTOS Trial (Canakinumab. Anti-Inflammatory Thrombosis Outcome Study; Novartis Pharmaceuticals). Dr Colhoun also receives or has recently received nonbinding research support from Roche Pharmaceuticals, Pfizer, Inc, Boehringer Ingelheim, and AstraZeneca. Dr Colhoun is a shareholder of Roche Pharmaceuticals and Bayer. Dr McCarthy has served on advisory panels for Pfizer, NovoNordisk, and Zoe Global, has received honoraria from Merck, Pfizer, Novo Nordisk, and Eli Lilly, and research funding from Abbvie, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Merck, NovoNordisk, Pfizer, Roche, Sanofi Aventis, Servier, and Takeda. As of June 2019, Dr McCarthy is an employee of Genentech and a holder of Roche stock. As of September 2019, Dr van Zuydam is an employee of AstraZeneca. As of 2016, Dr Vlachopoulou is an employee of Medpace. The other authors report no conflicts.
Supplementary Material
Appendix
CARDIoGRAMplusC4D
John Danesh, Jeanette Erdmann, Dongfeng Gu, Jaspal S. Kooner, Robert Roberts, Heribert Schunkert, Themistocles L. Assimes, Stefan Blankenberg, Bernhard O. Boehm, John C. Chambers, Robert Clarke, Rory Collins, George Dedoussis, Paul W. Franks, G. Kees Hovingh, Bong-Jo Kim, Terho Lehtimäki, Ruth McPherson, Markku S Nieminen, Christopher O'Donnell, Samuli Ripatti, Manjinder S Sandhu, Stefan Schreiber, Agneta Siegbahn, Cristen J. Willer, Pierre A. Zalloua
SUMMIT
Michael Mark, Timo Kanninen, Barbara Thorand, Giuseppe Remuzzi, David Dunger, Angela Shore, Ulf Smith, Per-Henrik Groop, Seppo Ylä-Herttuala, Claudio Cobelli, Riccardo Bellazzi, Ele Ferrannini, Carlo Patrono, Pirjo Nuutila, Paul McKeague, Birgit Steckel-Hamann, Li-ming Gan, Everson Nogoceke, Piero Tortoli, Bernd Jablonka, Mary-Julia Brosnan
Nonstandard Abbreviations and Acronyms
- CAD
- coronary artery disease
- CARDIoGRAMplusC4D
- Coronary Artery Disease Genome Wide Replication and Meta-Analysis (CARDIoGRAM) Plus the Coronary Artery Disease (C4D) Genetics
- ENGAGE
- European Network for Genetic and Genomic Epidemiology
- HPFS
- The Health Professionals Follow-Up Study
- LDL-C
- Low-density lipoprotein cholesterol
- METSIM
- The Metabolic Syndrome in Men study
- NHS
- Nurses’ Health Study
- OR
- odds ratio
- SUMMIT
- Surrogate Markers for Micro- and Macro-Vascular Hard End Points for Innovative Diabetes Tools
- T2D
- type 2 diabetes mellitus
A full list of SUMMIT and CARDIoGRAMplusC4D members is given in the Data Supplement
Current address for Dr McCarthy: Genentech, San Francisco, CA.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.119.002769.
References
- 1.Waller BF, Palumbo PJ, Lie JT, Roberts WC. Status of the coronary arteries at necropsy in diabetes mellitus with onset after age 30 years. Analysis of 229 diabetic patients with and without clinical evidence of coronary heart disease and comparison to 183 control subjects. Am J Med. 1980;69:498–506. doi: 10.1016/s0149-2918(05)80002-5 [DOI] [PubMed] [Google Scholar]
- 2.Koistinen MJ. Prevalence of asymptomatic myocardial ischaemia in diabetic subjects. BMJ. 1990;301:92–95. doi: 10.1136/bmj.301.6743.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carr ME. Diabetes mellitus: a hypercoagulable state. J Diabetes Complications. 2001;15:44–54. doi: 10.1016/s1056-8727(00)00132-x [DOI] [PubMed] [Google Scholar]
- 4.El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–2417. doi: 10.1084/jem.20081188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114:1241–1257. doi: 10.1093/cvr/cvy084 [DOI] [PubMed] [Google Scholar]
- 6.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jansen H, Loley C, Lieb W, Pencina MJ, Nelson CP, Kathiresan S, Peloso GM, Voight BF, Reilly MP, Assimes TL, et al. ; CARDIoGRAM consortium. Genetic variants primarily associated with type 2 diabetes are related to coronary artery disease risk. Atherosclerosis. 2015;241:419–426. doi: 10.1016/j.atherosclerosis.2015.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross S, Gerstein HC, Eikelboom J, Anand SS, Yusuf S, Paré G. Mendelian randomization analysis supports the causal role of dysglycaemia and diabetes in the risk of coronary artery disease. Eur Heart J. 2015;36:1454–1462. doi: 10.1093/eurheartj/ehv083 [DOI] [PubMed] [Google Scholar]
- 9.Zhao W, Rasheed A, Tikkanen E, Lee JJ, Butterworth AS, Howson JMM, Assimes TL, Chowdhury R, Orho-Melander M, Damrauer S, et al. ; CHD Exome+ Consortium; EPIC-CVD Consortium; EPIC-Interact Consortium; Michigan Biobank. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49:1450–1457. doi: 10.1038/ng.3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan KH, Huang YT, Meng Q, Wu C, Reiner A, Sobel EM, Tinker L, Lusis AJ, Yang X, Liu S. Shared molecular pathways and gene networks for cardiovascular disease and type 2 diabetes mellitus in women across diverse ethnicities. Circ Cardiovasc Genet. 2014;7:911–919. doi: 10.1161/CIRCGENETICS.114.000676 [DOI] [PubMed] [Google Scholar]
- 11.Fall T, Gustafsson S, Orho-Melander M, Ingelsson E. Genome-wide association study of coronary artery disease among individuals with diabetes: the UK Biobank. Diabetologia. 2018;61:2174–2179. doi: 10.1007/s00125-018-4686-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morieri ML, Gao H, Pigeyre M, Shah HS, Sjaarda J, Mendonca C, Hastings T, Buranasupkajorn P, Motsinger-Reif AA, Rotroff DM, et al. Genetic tools for coronary risk assessment in type 2 diabetes: a cohort study from the ACCORD clinical trial. Diabetes Care. 2018;41:2404–2413. doi: 10.2337/dc18-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mägi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi L, Qi Q, Prudente S, Mendonca C, Andreozzi F, di Pietro N, Sturma M, Novelli V, Mannino GC, Formoso G, et al. Association between a genetic variant related to glutamic acid metabolism and coronary heart disease in individuals with type 2 diabetes. JAMA. 2013;310:821–828. doi: 10.1001/jama.2013.276305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, Hemani G, Tansey K, Laurin C, Pourcain BS, et al. ; Early Genetics and Lifecourse Epidemiology (EAGLE) Eczema Consortium. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017;33:272–279. doi: 10.1093/bioinformatics/btw613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. ; LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, Strawbridge RJ, Pers TH, Fischer K, Justice AE, et al. ; ADIPOGen Consortium; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GEFOS Consortium; GENIE Consortium; GLGC; ICBP; International Endogene Consortium; LifeLines Cohort Study; MAGIC Investigators; MuTHER Consortium; PAGE Consortium; ReproGen Consortium. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–196. doi: 10.1038/nature14132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, Styrkarsdottir U, Gretarsdottir S, Thorlacius S, Jonsdottir I, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. doi: 10.1038/ng.274 [DOI] [PubMed] [Google Scholar]
- 19.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169. doi: 10.1038/ng.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al. ; Type 1 Diabetes Genetics Consortium. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, Lyssenko V, Bouatia-Naji N, Dupuis J, Jackson AU, et al. ; GIANT consortium; MAGIC investigators. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42:142–148. doi: 10.1038/ng.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu CT, Bielak LF, Prokopenko I, et al. ; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Multiple Tissue Human Expression Resource (MUTHER) Consortium. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669. doi: 10.1038/ng.2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, et al. ; DIAGRAM Consortium; GIANT Consortium; Global BPgen Consortium; Anders Hamsten on behalf of Procardis Consortium; MAGIC investigators. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soranzo N, Sanna S, Wheeler E, Gieger C, Radke D, Dupuis J, Bouatia-Naji N, Langenberg C, Prokopenko I, Stolerman E, et al. ; WTCCC. Common variants at 10 genomic loci influence hemoglobin A1© levels via glycemic and nonglycemic pathways. Diabetes. 2010;59:3229–3239. doi: 10.2337/db10-0502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E, Rybin D, Petrie JR, Travers ME, Bouatia-Naji N, Dimas AS, et al. ; DIAGRAM Consortium; GIANT Consortium; MuTHER Consortium; CARDIoGRAM Consortium; C4D Consortium. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes. 2011;60:2624–2634. doi: 10.2337/db11-0415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, et al. ; EPIC-InterAct Consortium; Cambridge FPLD1 Consortium. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49:17–26. doi: 10.1038/ng.3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan J, Mägi R, Strawbridge RJ, Rehnberg E, Gustafsson S, et al. ; DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. doi: 10.1038/ng.2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Leeuwen EM, Smouter FA, Kam-Thong T, Karbalai N, Smith AV, Harris TB, Launer LJ, Sitlani CM, Li G, Brody JA, et al. The challenges of genome-wide interaction studies: lessons to learn from the analysis of HDL blood levels. PLoS One. 2014;9:e109290. doi: 10.1371/journal.pone.0109290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Cai X, Mai W, Li M, Hu Y. Association between prediabetes and risk of cardiovascular disease and all cause mortality: systematic review and meta-analysis. BMJ. 2016;355:i5953. doi: 10.1136/bmj.i5953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, Ziemann M, Karagiannis T, Tonna S, Kowalczyk A, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011;21:1601–1615. doi: 10.1101/gr.116095.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strawbridge RJ, van Zuydam NR. Shared genetic contribution of type 2 diabetes and cardiovascular disease: implications for prognosis and treatment. Curr Diab Rep. 2018;18:59. doi: 10.1007/s11892-018-1021-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.