

ABSTRACT
Myocardial injury in sepsis may be caused by a burst of several inflammatory mediators, leading to vascular endothelial injuries. However, the contribution of neutrophil elastase (NE) to myocardial injury in sepsis is still unknown. We aimed to evaluate whether endotoxemia-induced myocardial injury is associated with NE. Lipopolysaccharide (LPS) was injected intraperitoneally at a dose of 20 mg/kg into granulocyte-colony-stimulating-factor knockout mice (G-CSF-KO), which have few neutrophils, and littermate control mice. The survival rate of G-CSF-KO mice 48 hours after LPS injection was significantly greater than that of control mice. The serum level of troponin I in G-CSF-KO mice was significantly lower than that in control mice. In addition, the concentration of inflammatory cytokine interleukin-6 (IL-6) was significantly decreased 6 and 12 hours after LPS administration compared with that in control mice. Ultrastructural analysis revealed that vascular endothelial structures and the endothelial glycocalyx in G-CSF-KO mice were clearly preserved. Next, mice were injected with 0.2 mg/kg sivelestat (an NE inhibitor) after LPS administration. The survival rate was significantly higher and the serum level of troponin I was lower in sivelestat-injected mice than in control mice, respectively. Furthermore, IL-6 levels were significantly decreased 6 and 12 hours after LPS administration compared with those in control mice. Vascular endothelial structures and the endothelial glycocalyx in sivelestat-treated mice were clearly preserved at the ultrastructural level. In conclusion, NE is significantly associated with myocardial injury in endotoxemia. Inhibition of NE may be a useful tool for the management of endotoxemia.
Keywords: Endothelial cell, myocardial injury, ultrastructure, vascular endothelial injury
INTRODUCTION
Despite the recent advances in intensive care practices, the mortality rate because of sepsis is still approximately 50% (1). Organ failure, including myocardial dysfunction, is among the diagnostic criteria of sepsis (2, 3). Myocardial injury is thought to be one of the critical factors affecting sepsis prognosis. In fact, septic myocardial injury and troponin elevation often occur in patients with sepsis (4). Microcirculatory obstacles in the myocardium are caused by narrowing of cardiac muscle cells and edema of the capillary wall and capillary lumen (5). These findings suggest that septic myocardial injury is caused by microvascular injury.
In terms of pathophysiology, multiple organ failure is caused by microvascular endothelial injury. Glycocalyx is a sugar-protein that coats all healthy vascular endothelial tissue (6–8) and plays an important role in microvascular and endothelial physiology (9–13). In clinical syndromes of systemic inflammation, such as sepsis, major surgery, trauma, ischemia/reperfusion, and prolonged hyperglycemia, diffuse and persistent alterations of glycocalyx are linked to widespread endothelial dysfunction, altered permeability, and impaired oxygen and nutrient delivery to cells (9, 14–16). There is experimental evidence that the thickness and stiffness of endothelial glycocalyx are reduced by lipopolysaccharide (LPS)-induced cytokines (17–20).
Neutrophils are considered to play a pivotal role in aggravation of acute respiratory distress syndrome (ARDS); neutrophil activation and transmigration are hallmark events in the progression of this disease (21). In addition, neutrophil elastase (NE), which is necessary for effective sterilization of phagocytized bacterial and fungal pathogens (22–24), increases alveolocapillary permeability, induces the production of pro-inflammatory cytokines, and enhances neutrophil migration (11, 25, 26). However, to the best of our knowledge, no previous studies have investigated whether neutrophils and NE injure the myocardial endothelial glycocalyx under septic conditions. The aim of this study was to assess whether septic myocardial injury results from changes in glycocalyx structure caused by neutrophils and NE.
MATERIALS AND METHODS
In-vivo animal studies
This study conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication, 8th Edition, 2011) and was approved by the Institutional Animal Research Committee of Gifu University (H30-190, Gifu, Japan). Male granulocyte-colony stimulating factor (G-CSF)-deficient mice (B6; 129P2-Csf3tm1Ard/J) were purchased from Jackson Laboratory (Bar Harbor, ME) and bred as described previously (27). G-CSF genotypes have been previously described (27, 28). G-CSF induces the appearance of colonies containing only granulocytes. G-CSF knockout (G-CSF-KO) mice show chronic neutropenia, granulocyte and macrophage progenitor cell deficiencies, and impaired neutrophil mobilization (27). Although approximately 50% of G-CSF-KO mice die in utero, the surviving mice grow to adulthood (27). As the control, wild-type littermate mice were used. After 16 hours starvation, 9 to 12-week-old male mice were given 20 mg/kg LPS (Sigma Aldrich, St. Louis, MO) intraperitoneally. To assess whether neutrophils cause endothelial injuries in endotoxemia, the NE inhibitor sivelestat was injected intraperitoneally at 3, 6, 9, and 12 hours after LPS administration. The mice were also injected with 0.05 mg/kg buprenorphine subcutaneously after LPS injection. For the control mice, isotonic saline was injected in the same manner. Forty-eight hours after LPS administration, the survival rate was determined. Perfusion fixation and blood sampling from the ophthalmic artery were performed under anesthesia (intraperitoneal mixture of medetomidine hydrochloride 0.3 mg/kg, midazolam 4 mg/kg, and butorphanol tartrate 5 mg/kg). The anesthesia was deemed sufficient if the corneal and hind-paw withdrawal reflex were absent. Then, mice were euthanized by exsanguination from the ophthalmic artery until the lighting reflex was lost before heart specimens were obtained.
Flow cytometry analysis of peripheral neutrophils
Before and at 6 hours after LPS administration, blood samples were collected in the same manner. Samples were prepared as previously described (29).
Cells were stained with the following antibodies (BioLegend, San Diego, CA) for 5 minutes at room temperature: antimouse CD45-PerCP (clone: 30-F11), antimouse CD11b-PE (clone: M1/70), antimouse Ly6G-FITC (clone: 1A8), and antimouse F4/80-Alexa Fluor® 647 (clone: BM8). Neutrophils were defined as the CD45+, CD11b+, Ly6G+, and F4/80− white blood cell population. Flow cytometry was performed on a BD FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ), and data were analyzed using FlowJo software (FlowJo, LLC, Ashland, OR).
Measurement of serum troponin I levels
Serum concentrations of cardiac troponin I were measured 48 hours after LPS injection using a Troponin-I, Mouse Cardiac, ELISA Kit (CTNI-1-HS, Life Diagnostics, Inc., West Chester, PA).
Histological analysis
All surviving mice were euthanized, and their hearts were removed. The excised hearts were cut into 2 transverse slices. The basal specimens were fixed in 10% buffered formalin and embedded in paraffin, after which 4-μm-thick sections were stained with hematoxylin–eosin.
Quantitative assessment of the interstitial area
The area of the interstitial space was quantitatively assessed in ten randomly chosen high-power fields of each hematoxylin–eosin-stained heart specimen using ImageJ software (NIH, Bethesda, MD).
Electron microscopy
Endothelial glycocalyx was detected using electron microscopy as described previously (17–19). To prepare samples for transmission electron microscopy (TEM), each specimen was embedded in epoxy resin. Ultrathin sections (90 nm), stained with uranyl acetate and lead citrate, were then examined using TEM (HT-7700, Hitachi High-Technologies GLOBAL, Tokyo, Japan). For standard electron microscopy, 2.5% glutaraldehyde in 0.1 mol/l phosphate buffer (pH 7.4) without lanthanum nitrate was used as the fixative.
Scoring of lectin-staining intensity
For quantitative analysis of glycocalyx injury, scoring of wheat germ agglutinin (WGA)-staining intensity was performed using a fluorescence microscope (BZ-X810, Keyence, Osaka, Japan) and ImageJ software. After deparaffinization, 4-μm-thick sections were cut and incubated with WGA biotin conjugate (BK-1000, Vector Laboratories, Burlingame, CA). The target proteins were visualized using Alexa Fluor 568 biotinylated secondary antibodies (Invitrogen, Waltham, MA). The intensity of WGA expression was scored manually in 10 high-power fields per sample (n = 6 per sample), in the focal plane.
Measurement of neutrophil elastase and interleukin-6 in serum
Following LPS administration, blood samples were collected from the maxillary artery, allowed to clot at room temperature for 2 hours, and centrifuged at 2000 × g at 4 °C for 20 minutes. The supernatant was collected; this serum was used for measuring NE and IL-6 levels, using ELISA Quantitation Kits for mouse NE (E-EL-M0444, Elabscience, Houston, TX) and IL-6 (cat no. M6000B, R&D Systems, Inc., Minneapolis, MN).
Statistical analysis
Data are presented as means ± SEM. Student's 2-tailed t-test was used for comparing 2 groups, and survival data were analyzed using the log-rank test. Differences were considered statistically significant at P < 0.05. All calculations were performed using Prism (GraphPad, La Jolla, CA).
RESULTS
Profile of granulocyte-colony-stimulating-factor knockout mice
ELISA analysis revealed a G-CSF deficiency in the serum of G-CSF-KO mice (Fig. 1A), confirming the original report (27). Peripheral neutrophil counts obtained using flow cytometry analysis were significantly lower in G-CSF-KO than in wild-type mice before LPS injection (Fig. 1B).
Fig. 1.
Profiles of G-CSF-KO mice.
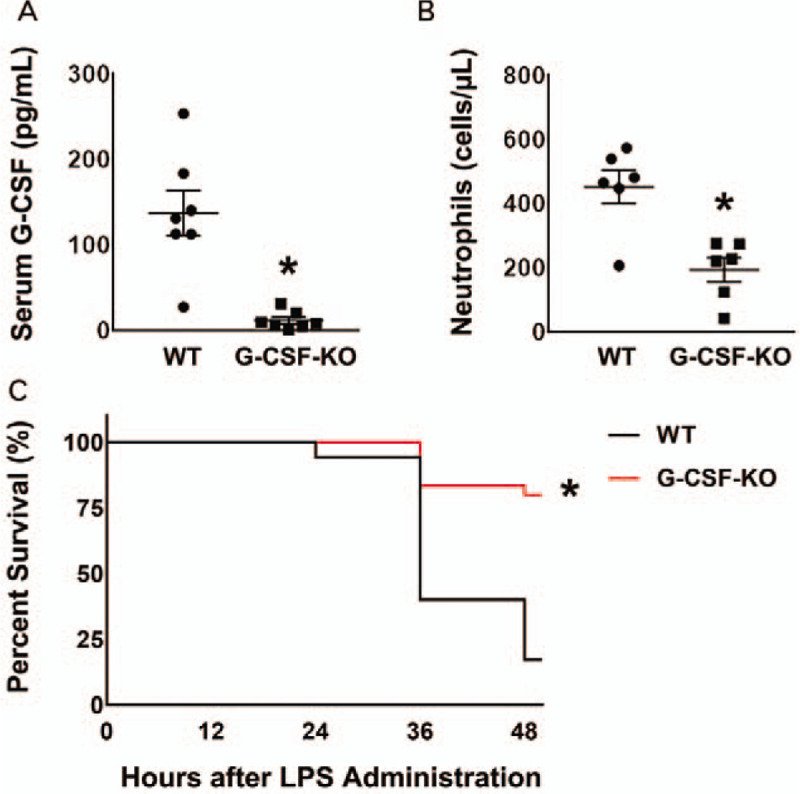
(A) Serum G-CSF concentration: In G-CSF-KO mice (n = 8), serum G-CSF concentration was significantly lower than in wild-type mice (n = 8). ∗P < 0.05 versus wild type. (B) Neutrophil numbers in peripheral blood: in G-CSF-KO mice (n = 6), there were significantly fewer neutrophils than in wild-type mice (n = 6). ∗P < 0.05 versus wild type. (C) Survival rate after LPS administration: The survival rate after LPS administration was significantly higher in the G-CSF-KO mice (24 of 30 mice; 80%) than in the wild-type mice (6 of 35 mice; 17%). ∗P < 0.05 versus wild type.
Granulocyte-colony-stimulating-factor knockout mice in mice with experimental endotoxemia
The survival rate 48 hours after LPS administration was significantly higher (P < 0.05) among G-CSF-KO mice (24 of 30 mice; 80%) than among control mice (6 of 35 mice; 17%) (Fig. 1C).
In wild-type mice, serum NE concentration peaked 6 hours after LPS administration and then decreased gradually. Conversely, serum NE was lower before LPS injection and at 6 and 12 hours after LPS injection, in G-CSF-KO mice than in wild-type mice (Fig. 2A). In addition, 48 hours after LPS administration, peripheral neutrophil counts were markedly lower in G-CSF-KO than in wild-type mice, as they were before LPS administration (Fig. 2B).
Fig. 2.
Profiles of G-CSF-KO mice after LPS administration.
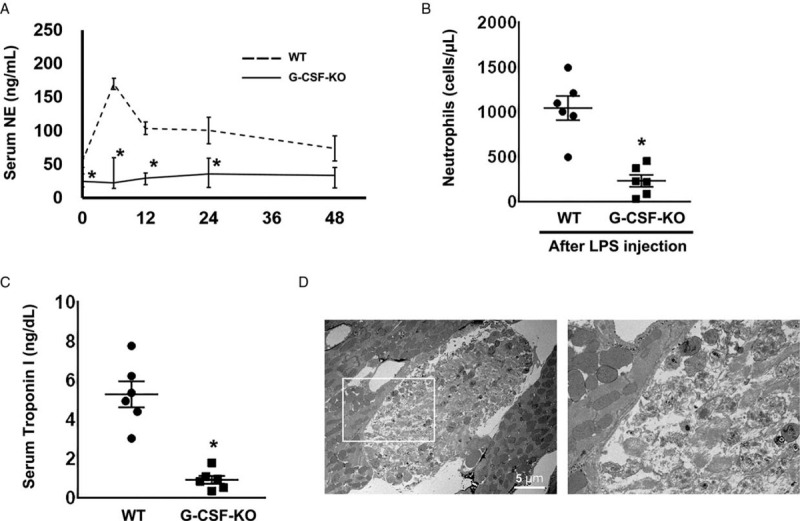
(A) The time course of serum neutrophil elastase (NE) concentration after LPS administration. The NE concentration in G-CSF-KO mice (n = 6 at each time point) was significantly lower than in wild-type mice (n = 6 at each time point), both before and after LPS administration. ∗P < 0.05 versus wild type, at the same time point. (B) Neutrophil numbers in peripheral blood 48 hours after LPS administration: In G-CSF-KO mice (n = 6), neutrophils were significantly less abundant than in the wild-type mice (n = 6). ∗P < 0.05 versus wild type. (C) Serum troponin I concentration: Troponin I concentration was significantly lower in the G-SCFKO mice (n = 6) than in the wild-type mice (n = 6). ∗P < 0.05 versus wild type. (D) The ultrastructure of cardiomyocyte injury in wild-type mice 48 hours after LPS administration. The right panel is the expanded image of the white square in the left panel. There are necrotic cardiomyocytes between healthy cells.
In the wild-type sham mice, serum IL-6 levels reached 106.2 ± 26.1 ng/ml 6 hours after LPS injection and 257.2 ± 43.4 ng/ml 12 hours after injection (see figure, Supplemental Digital Content 1). Serum IL-6 levels returned to baseline by 48 hours after LPS injection. In G-CSF-KO mice, the IL-6 concentration was significantly decreased 6 and 12 hours after LPS administration compared with that in wild-type mice (41.6 ± 6.2 ng/ml and 47.9 ± 8.6 ng/ml, respectively). Moreover, in G-CSF-KO mice, neutrophil infiltration was significantly decreased compared with that in wild-type mice after LPS injection (see figure, Supplemental Digital Content 2).
The serum level of troponin I, a cardiac injury marker, in the G-CSF-KO group was lower than that in the control (0.9 ± 0.2 ng/dl versus 5.3 ± 0.7 ng/dl, P < 0.05, Fig. 2C). Necrotic cardiomyocytes were found among healthy cells in wild-type mouse hearts 48 hours after LPS administration (Fig. 2D).
Endothelial injury was attenuated in granulocyte-colony-stimulating-factor knockout mice
Lectin binds with glycoprotein within endothelial glycocalyx, and it has been reported that WGA allows good visualization of endothelial glycocalyx (30). Therefore, to quantitatively assess endothelial glycocalyx injury, we measured the intensity of WGA staining, because the endothelial glycocalyx is composed of several substances.
Using immunofluorescence microscopy, we showed that endothelial glycocalyx existed on the surface of capillaries (Fig. 3A). In wild-type mice, WGA intensity was lower following LPS administration than it was before administration (Fig. 3B). After LPS injection, WGA intensity was greater in G-CSF-KO mice than in wild-type mice. These results suggest that injury to endothelial glycocalyx in heart capillaries was attenuated in G-CSF-KO mice.
Fig. 3.
WGA intensity score measurement.
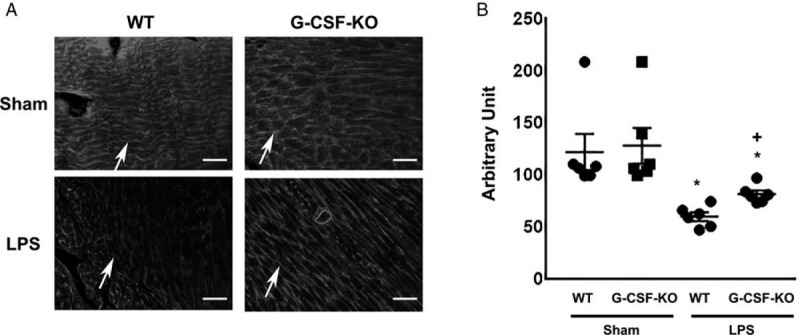
(A) Lectin, a glycocalyx-binding glycoprotein, stained by wheat germ agglutinin (WGA). Arrows indicate lectin-positive glycocalyx. Bars: 50 μm. (B) WGA intensity of wild-type and G-CSF-KO mouse hearts with and without LPS administration. ∗P < 0.05 versus non-LPS injected wild-type mice, +P < 0.05 versus LPS-injected wild-type mice.
In wild-type mice, the percent of interstitial area was significantly increased under endotoxemic conditions compared with that before LPS administration. The percent area was significantly decreased in G-CSF-KO mice compared with that in wild-type mice in endotoxemia (see figure, Supplemental Digital Content 3).
Ultrastructure of endothelial injury under endotoxemia
Although it is convenient to visualize endothelial glycocalyx using lectin, it was impossible to confirm the structure of endothelial glycocalyx. Therefore, electron microscopy analysis was performed using both conventional and lanthanum staining for glycocalyx visualization.
Capillaries in the heart are classified as being continuous. Conventional SEM examination of the lumenal side of the cardiac capillary endothelium showed intracellular tight junctions but no transcellular perforations; endothelial glycocalyx was also undetectable in both wild-type and G-CSF-KO mice under normal conditions (Fig. 4A and B). Although intracellular tight junctions were not detected in the wild-type group under endotoxemia, endothelial injury was not detected in G-CSF-KO mice under endotoxemia (Fig. 4C and D).
Fig. 4.
Ultrastructure of cardiac capillaries in G-CSF-KO mouse hearts after LPS administration.
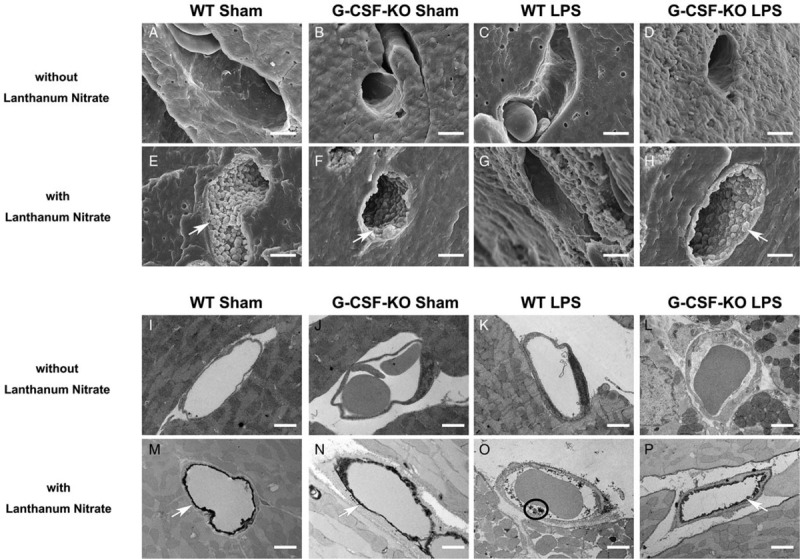
(A–H) Scanning electron microscope and (I–P) transmission electron microscope images of cardiac capillaries: (A–D, I–L) the images without lanthanum nitrate; endothelial glycocalyx was not detected (E–H, M–P). The endothelial glycocalyx was detected using lanthanum nitrate staining. The endothelial glycocalyx formed a continuous structure before LPS administration (E, F, M, and N). Under endotoxemic conditions, the endothelial glycocalyx degraded and separated from endothelial cells. In G-CSF-KO mice, the endothelial glycocalyx maintained a continuous structure on the vascular endothelium (P). White arrows indicate endothelial glycocalyx on the endothelium. Black circle indicates residue of endothelial glycocalyx. Bars: 2 μm.
Lanthanum nitrate staining revealed moss-like or broccoli-like structures on the endothelial cells, which we suspected were endothelial glycocalyx (Fig. 4E–H). Endothelial glycocalyx covered the surface of endothelial cells of both wild-type and G-CSF-KO mice under normal conditions (Fig. 4E and F). Although endothelial glycocalyx was severely injured in the wild-type group after LPS administration, the endothelial glycocalyx structure was substantially maintained in G-CSF-KO mice after LPS administration (Fig. 4G and H).
In the wild-type mice, conventional TEM revealed that the capillary wall became edematous after LPS administration (Fig. 4I and K). Conversely, edema after LPS administration was attenuated in G-CSF-KO mice (Fig. 4G and L).
Bush-like structures were observed on the surface of the endothelial cells of both wild-type and G-CSF-KO mouse hearts (Fig. 4M and N). After LPS injection, vascular endothelial glycocalyx structure was less injured in G-CSF-KO mice than in wild-type mice (Fig. 4O and P). These results were consistent with the immunofluorescence results (Fig. 3).
Neutrophil elastase inhibitor in endotoxemia-induced cardiac injury
To demonstrate the contribution of NE to septic cardiac injury, 0.2 mg/kg sivelestat, an NE inhibitor, was injected intraperitoneally into mice at 3, 6, 9, and 12 hours after LPS administration. At 48 hours after LPS treatment, the survival rate was significantly higher (P < 0.05) among sivelestat-injected mice (81%, 21/26) than among control mice (22%, 6/27) (Fig. 5A). In sivelestat-treated mice, IL-6 levels were significantly decreased to 53.8 ± 22.1 ng/ml at 6 hours after LPS administration and 73.3 ± 9.4 ng/ml at 12 hours after injection compared with those in isotonic saline-treated mice (116.4 ± 22.1 ng/ml and 235.8 ± 73.3 ng/ml, respectively) (see figure, Supplemental Digital Content 4). Moreover, in sivelestat-treated mice, neutrophil infiltration was significantly decreased compared with that in isotonic saline-treated mice after LPS injection (see figure, Supplemental Digital Content 5).
Fig. 5.
Effect of sivelestat treatment after LPS administration.
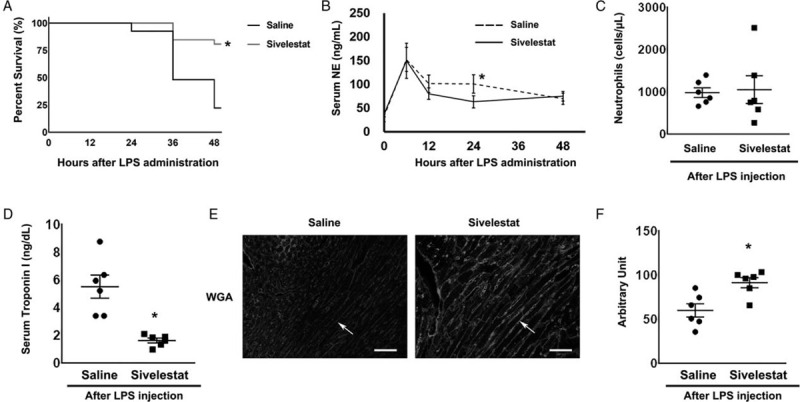
(A) Survival rate after LPS administration: The survival rate after LPS administration was significantly higher in the sivelestat-treated mice (81%, 21/26) than in isotonic saline-treated mice (22%, 6/27). ∗P < 0.05 versus isotonic saline-treated mice. (B) The time course of serum neutrophil elastase (NE) concentration after LPS administration: In sivelestat-treated mice, NE concentration 24 hours after LPS administration was significantly lower than in isotonic saline-treated mice. ∗P < 0.05 versus isotonic saline-treated mice at the same time point. (C) Neutrophil numbers in the peripheral blood 48 hours after LPS administration: There were no significant differences in neutrophil number between sivelestat-treated mice and isotonic saline-treated mice (n = 6 each). (D) Serum troponin I concentration: Troponin I concentration was significantly lower in the sivelestat-treated mice (n = 6) than in isotonic saline-treated mice (n = 6). ∗P < 0.05 versus isotonic saline-treated mice. (E) Lectin, a glycocalyx-binding glycoprotein, stained by wheat germ agglutinin (WGA). Arrows indicate lectin-positive glycocalyx. Bars: 50 μm. (F) The WGA intensity score of isotonic saline-treated and sivelestat-treated mouse hearts 48 hours after LPS administration (n = 6 each). ∗P < 0.05 versus isotonic saline-treated mice.
At 24 hours after LPS administration, serum NE concentration was significantly lower (P < 0.05) in sivelestat-treated mice than in isotonic saline-treated wild-type mice (Fig. 5B). There was no significant difference in neutrophil number between isotonic saline and sivelestat-treated mice (Fig. 5C). In addition, 48 hours after LPS administration, the serum level of troponin I in the sivelestat-treated group was lower than that in isotonic saline-treated mice (1.6 ± 0.2 ng/dl versus 5.5 ± 0.8 ng/dl, P < 0.05, Fig. 5D).
Next, quantitative assessment of endothelial glycocalyx injury was performed to obtain WGA intensity scores. WGA intensity was higher in the sivelestat-treated group than in the isotonic saline-treated group (Fig. 5E and F). These results suggested that endothelial glycocalyx injury after LPS injection was attenuated in sivelestat-treated hearts. The percent of interstitial area was significantly decreased in the sivelestat-treated group compared with that in the isotonic saline-treated group (see figure, Supplemental Digital Content 6).
Ultrastructural analysis revealed that vascular endothelial glycocalyx structure after LPS injection was less injured in sivelestat-treated mice, whereas it was severely injured in isotonic saline-treated mice (Fig. 6). These results suggested that NE plays an important role in microvascular injury in endotoxemia-induced cardiac injury.
Fig. 6.
Ultrastructure of cardiac capillaries in sivelestat-treated mouse hearts after LPS administration.
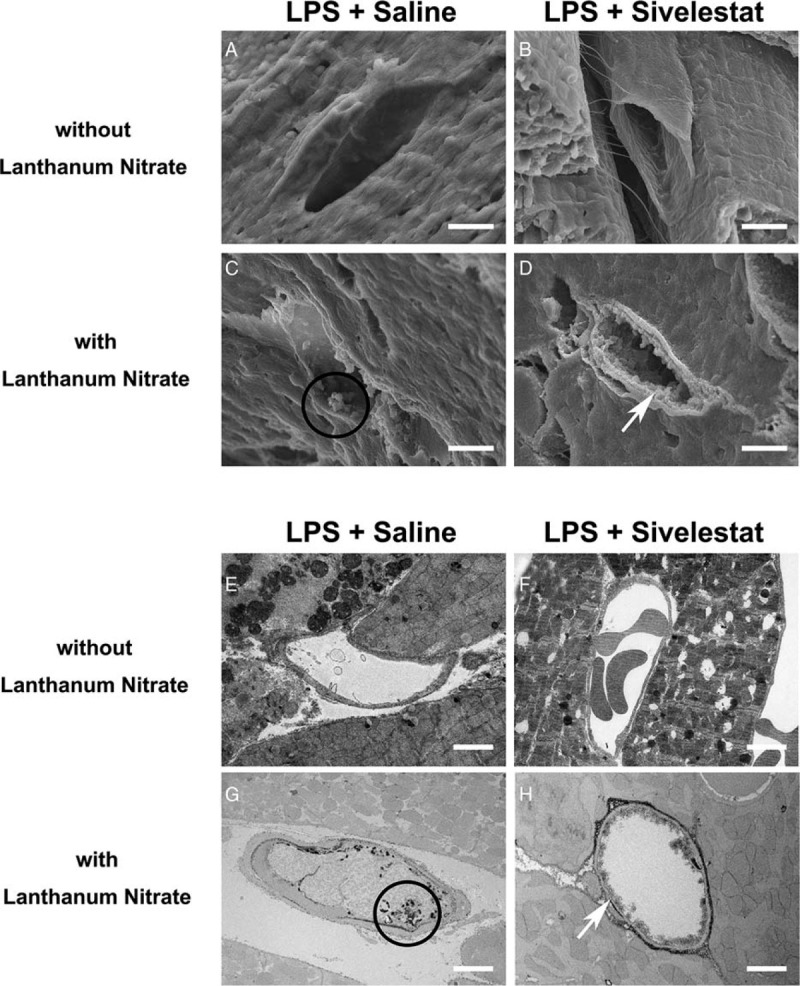
(A–D) Scanning electron microscope and (E–H) transmission electron microscope images of cardiac capillaries: (A, B, E, and F) The images without lanthanum nitrate. Endothelial glycocalyx was not detected. (C, D, G, and H) Endothelial glycocalyx was detected using lanthanum nitrate staining. In sivelestat-treated mice, the endothelial glycocalyx maintained a continuous structure on the vascular endothelium compared with that in isotonic saline-administered mice (C, D, G, and H). White arrows indicate endothelial glycocalyx on the endothelium. Black circle indicates residue of endothelial glycocalyx. Bars: 2 μm.
DISCUSSION
We demonstrated that septic myocardial injury after LPS administration was attenuated in G-CSF-KO mice. Specifically, we showed that the endothelial glycocalyx structure became detached from the capillary lumen under septic conditions induced by LPS injection; and septic myocardial injury was ameliorated in G-CSF-KO mice and in NE inhibitor-treated mice under septic conditions. Neutrophils, therefore, play an important role in septic myocardial injury, and inhibition of NE offers a potential novel means to protect endothelial glycocalyx structure from injury.
Mechanisms of septic myocardial injuries
We found necrotic cardiomyocytes among healthy cells; this finding suggests that septic myocardial injury might result from small impairments of microvascular perfusion. Microvascular injury has several causes, including activation of coagulation, cytokine release, regional ischemia, endothelin upregulation, and impaired coronary microcirculation (31). It has been reported that sepsis causes the mitochondrial ultrastructure to collapse, and in its early stages, increases the blood flow to the myocardium (5).
Several inflammatory cytokines, including tumor necrosis factor alpha (TNF-α) and interleukin 1-beta (IL-1β), are secreted in the early stage of sepsis, mainly from macrophages (32). Previous reports indicate that TNF-α and IL-1β cause septic shock, inducing high cardiac output and low systemic vascular resistance (33, 34). It has also been reported that cardiac function is directly impaired by TNF-α (35–37). This is consistent with the finding that, in granulocyte macrophage-colony stimulating factor-deficient mice, the survival rate under sepsis was significantly better than that in wild-type mice (38). However, the contribution of neutrophils to septic myocardial injury has been unclear. G-CSF-deficient mice are viable, fertile, and superficially healthy, but have chronic neutropenia (27). Collectively, these results suggest that inhibition of NE attenuates endothelial injury under endotoxemia and simultaneously inhibits myocardial injuries.
Neutrophil elastase and endothelial glycocalyx
In healthy vessels, the vascular endothelial glycocalyx covers the endothelial surface and plays an important role in vascular homeostasis (9–13). Under sepsis, the endothelial glycocalyx becomes detached from the capillary vessel wall in the capillaries of several organs (17–19). Proteases and reactive oxygen species derived from neutrophils cause ARDS. NE is one of the most destructive proteases, and it contributes to causing ARDS. NE breaks down the extracellular matrix of elastin, collagen, and proteoglycan; NE is thought to cause ARDS by breaking down the tissue inhibitors of metalloproteinases (endogenous inhibitors of the matrix metalloproteases) and inducing the production of inflammatory cytokines such as IL-8 (39). The components of glycocalyx include cell-bound proteoglycans, glycosaminoglycan side chains, and sialoproteins (16, 40, 41). Therefore, it is considered that glycocalyx constructed by proteoglycan is broken down by NE.
Sivelestat forms an acylated complex with NE at its center and inhibits the enzymatic activity of NE; it is, therefore, referred to as an acylated inhibitor (42). It is known that sivelestat ameliorates multiple-organ failure, including lung failure, under sepsis (43).
The present study shows that septic myocardial injury was attenuated not only in G-CSF-KO mice but also in NE inhibitor-treated mice, via inhibition of endothelial glycocalyx injury. However, in a previous clinical study, intravenous sivelestat had no effect on all-cause mortality or ventilator-free days in patients with heterogeneous acute lung injury (44). One of the reasons for this may be that inhibition of NE affects the ability of neutrophils to penetrate the tissues. If sivelestat can be delivered specifically to the place of injury via new drug delivery systems, sivelestat may be an effective treatment against sepsis. Furthermore, this study revealed that sivelestat decreased neutrophil infiltration in the heart. These results indicate that both NE inhibitor and G-CSF-KO-inhibited activation of neutrophils after LPS injection and that the inhibition of neutrophil activation and possibly NE, rather than NE alone, might play an important role in the beneficial effects of NE inhibitor and G-CSF-KO.
Sepsis is an extremely complicated disease, compared with simple endotoxemia in an experimental model. In the present study, our intention was to focus on the role of neutrophils in endotoxemia. Therefore, we used the mouse model of endotoxemia induced by LPS. Although LPS-injected mice are useful experimental endotoxemia models, they are not infected with the sepsis-causing pathogenic bacteria. Therefore, our study does not accurately reflect clinical conditions. However, the present study showed that neutrophils caused myocardial damage under endotoxemia. On the other hand, IL-6, one of the markers of systemic inflammation, was ameliorated in G-CSF-KO mice and sivelestat-treated mice under endotoxemia, which suggested that inhibition of systemic inflammation may be an important contributor to the protective effects of G-CSF-KO and NE inhibitor. Nonetheless, it remains unknown whether these effects were direct or indirect.
Using G-CSF-KO mice and by pharmacological intervention with NE inhibitor, the present study revealed that the endothelial glycocalyx in the heart is injured to develop endotoxemia-induced myocardial injury, which is pathogenetically associated with neutrophils and NE. Inhibition of NE may thus offer a potential means to attenuate endotoxemic myocardial injury through preservation of endothelial glycocalyx.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Footnotes
Acknowledgments: We thank Yuki Wakida, Yasuko Nogaki and Shoko Kumazaki for technical assistance.
Tetsuya Fukuta and Hideshi Okada: These authors contributed equally to this article.
Conflicts of interest and source of funding: The authors declare that they have no competing interests. This work was supported in part by grants-in-aid for scientific research [19H03756, 19K18347, 19K08499, 19K09410, 18K16511, 18K08914, 18K08884, 17K11569, 16H05497, 16K09509, 16K20381, and 15K10973] from the Ministry of Education, Science and Culture of Japan.
Supplemental digital content is available for this article.
REFERENCES
- 1.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med 35:1244–1250, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, Damske BA, Parrillo JE. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med 100:483–490, 1984. [DOI] [PubMed] [Google Scholar]
- 3.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315:801–810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilhelm J, Hettwer S, Schuermann M, Bagger S, Gerhardt F, Mundt S, Muschik S, Zimmermann J, Amoury M, Ebelt H, et al. Elevated troponin in septic patients in the emergency department: frequency, causes, and prognostic implications. Clin Res Cardiol 103:561–567, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Morisaki H, Bloos F, Keys J, Martin C, Neal A, Sibbald WJ. Compared with crystalloid, colloid therapy slows progression of extrapulmonary tissue injury in septic sheep. J Appl Physiol (1985) 77:1507–1518, 1994. [DOI] [PubMed] [Google Scholar]
- 6.Becker BF, Chappell D, Jacob M. Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol 105:687–701, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Luft JH. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed Proc 25:1773–1783, 1966. [PubMed] [Google Scholar]
- 8.Rehm M, Zahler S, Lötsch M, Welsch U, Conzen P, Jacob M, Becker BF. Endothelial glycocalyx as an additional barrier determining extravasation of 6% hydroxyethyl starch or 5% albumin solutions in the coronary vascular bed. Anesthesiology 100:1211–1223, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care 19:26, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frati-Munari AC. Medical significance of endothelial glycocalyx. Arch Cardiol Mex 83:303–312, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med 363:689–691, 2010. [DOI] [PubMed] [Google Scholar]
- 12.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch 454:345–359, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodcock TE, Woodcock TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth 108:384–394, 2012. [DOI] [PubMed] [Google Scholar]
- 14.Henrich M, Gruss M, Weigand MA. Sepsis-induced degradation of endothelial glycocalix. Sci World J 10:917–923, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Myburgh JA, Mythen MG. Resuscitation fluids. N Engl J Med 369:1243–1251, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Salmon AH, Satchell SC. Endothelial glycocalyx dysfunction in disease: albuminuria and increased microvascular permeability. J Pathol 226:562–574, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Ando Y, Okada H, Takemura G, Suzuki K, Takada C, Tomita H, Zaikokuji R, Hotta Y, Miyazaki N, Yano H, et al. Brain-specific ultrastructure of capillary endothelial glycocalyx and its possible contribution for blood brain barrier. Sci Rep 8:17523, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inagawa R, Okada H, Takemura G, Suzuki K, Takada C, Yano H, Ando Y, Usui T, Hotta Y, Miyazaki N, et al. Ultrastructural alteration of pulmonary capillary endothelial glycocalyx during endotoxemia. Chest 154:317–325, 2018. [DOI] [PubMed] [Google Scholar]
- 19.Okada H, Takemura G, Suzuki K, Oda K, Takada C, Hotta Y, Miyazaki N, Tsujimoto A, Muraki I, Ando Y, et al. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit Care 21:261, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiesinger A, Peters W, Chappell D, Kentrup D, Reuter S, Pavenstädt H, Oberleithner H, Kümpers P. Nanomechanics of the endothelial glycocalyx in experimental sepsis. PLoS One 8:e80905, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abraham E. Neutrophils and acute lung injury. Crit Care Med 31: 4 Suppl: S195–199, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Hagiwara S, Iwasaka H, Togo K, Noguchi T. A neutrophil elastase inhibitor, sivelestat, reduces lung injury following endotoxin-induced shock in rats by inhibiting HMGB1. Inflammation 31:227–234, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Vender RL. Therapeutic potential of neutrophil-elastase inhibition in pulmonary disease. J Investig Med 44:531–539, 1996. [PubMed] [Google Scholar]
- 24.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med 320:365–376, 1989. [DOI] [PubMed] [Google Scholar]
- 25.Miyazaki Y, Inoue T, Kyi M, Sawada M, Miyake S, Yoshizawa Y. Effects of a neutrophil elastase inhibitor (ONO-5046) on acute pulmonary injury induced by tumor necrosis factor alpha (TNFalpha) and activated neutrophils in isolated perfused rabbit lungs. Am J Respir Crit Care Med 157:89–94, 1998. [DOI] [PubMed] [Google Scholar]
- 26.Passi A, Negrini D, Albertini R, De Luca G, Miserocchi G. Involvement of lung interstitial proteoglycans in development of hydraulic- and elastase-induced edema. Am J Physiol 275:L631–635, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 84:1737–1746, 1994. [PubMed] [Google Scholar]
- 28.Kataoka H, Ushiyama A, Kawakami H, Akimoto Y, Matsubara S, Iijima T. Fluorescent imaging of endothelial glycocalyx layer with wheat germ agglutinin using intravital microscopy. Microsc Res Tech 79:31–37, 2016. [DOI] [PubMed] [Google Scholar]
- 29.Merx MW, Weber C. Sepsis and the heart. Circulation 116:793–802, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Kapadia S, Lee J, Torre-Amione G, Birdsall HH, Ma TS, Mann DL. Tumor necrosis factor-alpha gene and protein expression in adult feline myocardium after endotoxin administration. J Clin Invest 96:1042–1052, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okusawa S, Gelfand JA, Ikejima T, Connolly RJ, Dinarello CA. Interleukin 1 induces a shock-like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest 81:1162–1172, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van der Poll T, Romijn JA, Endert E, Borm JJ, Büller HR, Sauerwein HP. Tumor necrosis factor mimics the metabolic response to acute infection in healthy humans. Am J Physiol 261:E457–465, 1991. [DOI] [PubMed] [Google Scholar]
- 33.Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo KS, Banerjee A, Harken AH. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med 27:1309–1318, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Andresen BT, Hill M, Zhang J, Booth F, Zhang C. Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Curr Hypertens Rev 4:245–255, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med 183:949–958, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spight D, Trapnell B, Zhao B, Berclaz P, Shanley TP. Granulocyte-macrophage-colony-stimulating factor-dependent peritoneal macrophage responses determine survival in experimentally induced peritonitis and sepsis in mice. Shock 30:434–442, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawabata K, Hagio T, Matsuoka S. The role of neutrophil elastase in acute lung injury. Eur J Pharmacol 451:1–10, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Ly M, Linhardt RJ. Proteoglycan sequence. Mol Biosyst 8:1613–1625, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paulus P, Jennewein C, Zacharowski K. Biomarkers of endothelial dysfunction: can they help us deciphering systemic inflammation and sepsis? Biomarkers 16: Suppl 1: S11–S21, 2011. [DOI] [PubMed] [Google Scholar]
- 40.Imaki K, Okada T, Nakayama Y, Nagao Y, Kobayashi K, Sakai Y, Mohri T, Amino T, Nakai H, Kawamura M. Non-peptidic inhibitors of human neutrophil elastase: the design and synthesis of sulfonanilide-containing inhibitors. Bioorg Med Chem 4:2115–2134, 1996. [DOI] [PubMed] [Google Scholar]
- 41.Li G, Jia J, Ji K, Gong X, Wang R, Zhang X, Wang H, Zang B. The neutrophil elastase inhibitor, sivelestat, attenuates sepsis-related kidney injury in rats. Int J Mol Med 38:767–775, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeiher BG, Artigas A, Vincent JL, Dmitrienko A, Jackson K, Thompson BT, Bernard G. STRIVE Study Group: neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit Care Med 32:1695–1702, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Morishita K, Takemura G, Tsujimoto A, Kanamori H, Okada H, Chousa M, Ushimaru S, Mikami A, Kawamura I, Takeyama T, et al. Postinfarction cardiac remodeling proceeds normally in granulocyte colony-stimulating factor knockout mice. Am J Pathol 185:1899–1911, 2015. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki K, Inoue S, Kametani Y, Komori Y, Chiba S, Sato T, Inokuchi S, Ogura S. Reduced immunocompetent B cells and increased secondary infection in elderly patients with severe sepsis. Shock 46:270–278, 2016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.