

Abstract
Introduction:
Kikuchi's disease (KD) is a rare form of necrotizing lymphadenitis that rarely occurs in association with hemophagocytic lymphohistiocytosis (HLH) in children.
Patient concerns:
We report the case of a 4-year-5-month-old boy who suffered from fever, cervical lymphadenopathy, pancytopenia, hypertriglyceridemia, splenomegaly, low NK cell activity.
Diagnoses:
A diagnosis of KD with HLH was made based on the results of biopsy of cervical lymph node and HLH-2004 trial guidelines.
Interventions:
The patient was treated with corticosteroids, cyclosporine, etoposide, continuous hemodiafiltration (HDF), and plasma exchange (PE).
Outcomes:
He showed a complete response to therapy, and his condition gradually improved. He was discharged on day 45 after admission due to his good recovery status.
Conclusion:
HLH can be associated with KD, especially in childhood, and may have an aggressive clinical course. Continuous HDF and PE and chemotherapy should be reserved for those patients who fail to respond to IVIG and corticosteroids.
Keywords: continuous hemodiafiltration, hemophagocytic lymphohistiocytosis, Kikuchi's disease, plasma exchange
1. Introduction
Kikuchi's disease (KD), also called histiocytic necrotizing lymphadenitis, is a benign, self-limiting disease, characterized by fever, cervical lymphadenopathy and leukopenia and can be accompanied by other symptoms such as a skin rash, hepatomegaly, abdominal pain, and weight loss.[1] The most common symptoms associated with hemophagocytic lymphohistiocytosis (HLH) are unremitting fever, splenomegaly, and pancytopenia. HLH is characterized by a hyperinflammatory state due to uncontrolled T-cell, macrophage and histiocyte activation, accompanied by excessive cytokine production. This rare condition is almost universally fatal unless it is promptly recognized and treated.[2] It seems that HLH associated with KD that occurs during childhood may have a less aggressive clinical course and better prognosis than in adults,[3] but sometimes it may still be fatal.[4] Here, we report a case of KD associated with HLH in a 4-year, 5-month-old boy and review the literature on HLH and KFD in children and adults.
2. Case report
A 4-year, 5-month-old boy was admitted to hospital with a history of fever and cough for more than 20 days. His highest temperature recorded was 40.4°C. A rash and muscle pain accompanied the fever, and the rash subsided when his body temperature turned normal. He was treated with antibiotics at a local hospital with no clinical improvement. He was admitted to the inpatient floor with persistent fever and cough. His physical exam was normal except for multiple tender lymph nodes on the left lateral side of his neck, which ranged in size from 0.8 to 1.2 cm. The liver was approximately 3.0 cm below the ribs.
Initial laboratory studies revealed a hemoglobin level of 9.6 g/dl, a leukocyte count of 9.63 × 109/L, with 72.8% neutrophils and 5.5% monocytes, a platelet count of 303 × 109/L, a C-reactive protein level of 149.81 mg/L, a serum ferritin level of more than 1500 ng/ml (normal range, 15–152 ng/ml). Most of his serum chemistry test results were normal, including aspartate aminotransferase, alanine aminotransferase, bilirubin, electrolytes, albumin, creatinine, and blood urea nitrogen. Serologic tests for Epstein-Barr virus were negative, and a T-SPOT was negative for tuberculosis. Blood and sputum cultures were also negative. Rheumatological tests, including antinuclear antibody, deoxyribonucleic acid antibody, and rheumatoid factor were all negative. Bone marrow and cerebrospinal fluid were normal. Echocardiography and chest computed tomography revealed no obvious abnormalities.
On the third day of hospitalization, the patients body temperature returned to normal. However, on the 7th day, the patient developed a fever (up to 42°C) and a maculopapular rash over his neck and back. The rash became more severe and pruritic and involved the patients entire body. The patient developed bilateral cervical lymph node enlargement of approximately 1.5 to 3.0 cm, diarrhea accompanied by abdominal pain, and edema. The patient was treated with intravenous immunoglobulin (IVIG, 2 g/kg) without improvement. A biopsy of the right cervical lymph node performed on the 11th day showed scattered fibrin deposition, a large amount of nuclear debris and large mononuclear cell aggregates in the necrotic area, with no evidence of malignancy. The immunohistochemical results showed CD163 (2 +) (Fig. 1). Histopathology and immunohistochemical results were suggestive of necrotizing lymphadenitis. The patient had continued hyperthermia, and his highest body temperature recorded was 40°C. He was treated with methylprednisolone (2 mg/kg, q8 hour). During treatment on the 12th day, the patients clinical condition improved. His temperature normalized, the rash gradually subsided, and the edema slightly subsided. However, his laboratory parameters worsened. On the 14th day, the patient developed acute respiratory failure required ventilator-assisted ventilation. On the 15th day, the patient developed pancytopenia, and a complete blood cell count showed leukopenia (white blood cell count=3.68 × 109/L, with 41.2% neutrophils, and 47.5% lymphocyte), a hemoglobin level of 6.5 g/dl, and a platelet count of 8 × 109/L). Blood tests showed elevated D-dimers of 21.12, concentration of fibrin degradation products was 65.15 μg/ml, and a decreased fibrinogen of 60 mg/dl. The patients condition quickly worsened, and eventually DIC developed. Aspartate aminotransferase levels were 738 IU/L, alanine aminotransferase levels were 138.7 IU/L (normal < 40 IU/L) and lactate dehydrogenase (LDH) levels were 136 1U/L. The patients fever period was more than 7 days and he had pancytopenia, hypertriglyceridemia, elevated ferritin levels, reduced NK cell activity, and splenomegaly (7.0 cm below the ribs). The patient was diagnosed as HLH based on the published criteria from the HLH-2004 trial. On the 16th day, the patient developed capillary leak syndrome. The patient was then started on chemotherapy (early-stage dexamethasone, cyclosporine and etoposide for 8 weeks, according to the HLH-2004 treatment guidelines) with continuous HDF (98 hours) and PE treatment twice. He showed a complete response to the therapy, and his condition gradually improved. He was discharged on day 45 after admission due to his good recovery status. The patient had another 26 days of chemotherapy, and there was no recurrence 17 months after discharge.
Figure 1.
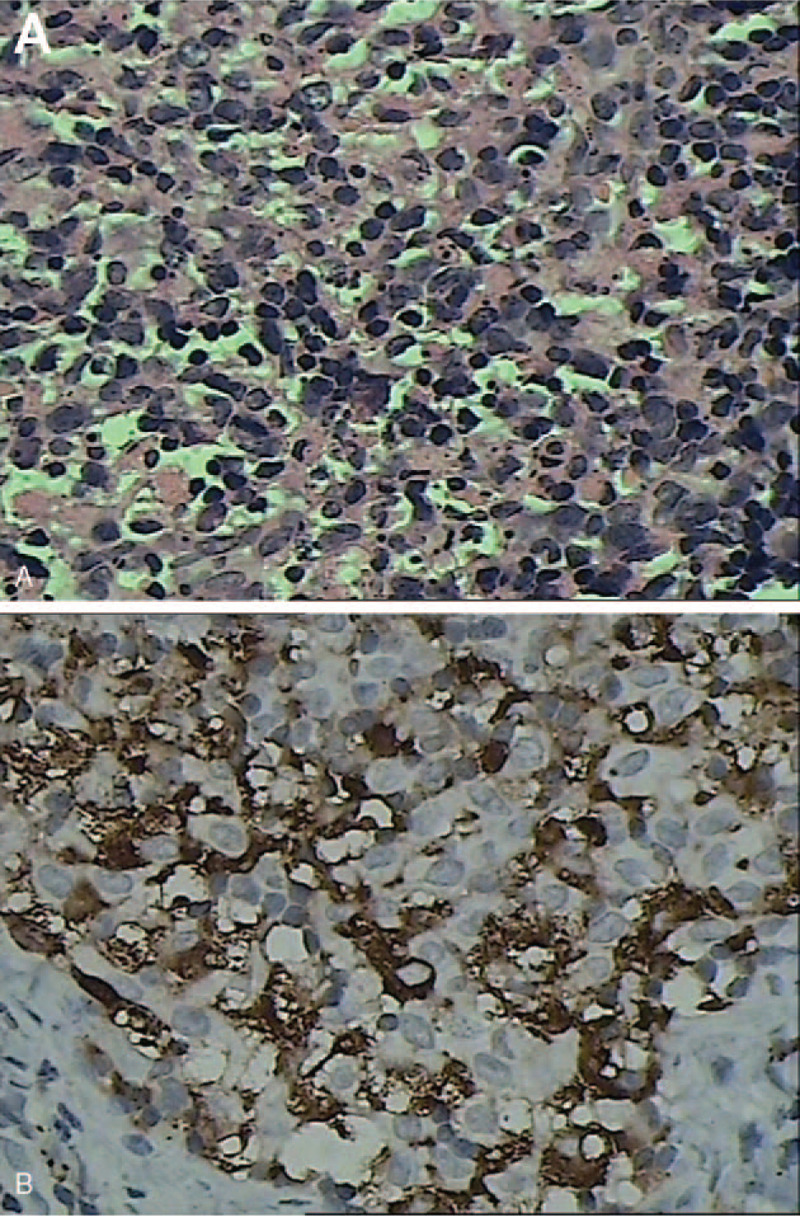
(A) Multiple focal necrotic areas and significant phagocytosis (H&E, X400). (B) Immunohistochemical stains showing CD163-positive histiocytes (X400).
3. Discussion
KD usually occurs in young women and adolescents and occurs commonly in Asia, although it has a worldwide distribution.[5] It commonly manifests as a prolonged high fever, cervical lymphadenopathy, and leukopenia. The female-to-male ratio is between 1.1:1 and 2.75:1.[6] Infectious agents including Epstein-Barrvirus, human herpes viruses 6 and 8, parvovirus,[7] parainfluenza, paramyxoviruses, and dengue have been reported to play causative roles,[8] but their roles have not been confirmed. However, in this patient, there was no clear basis for pathogen infection. The pathogenesis of KD is not clear. It is believed that there is an immune response of T cells and histiocytes to an inciting agent. Cellular destruction is thought to be due to apoptotic cell death mediated by CD8 T lymphocytes.[9] KD may not be caused by a single factor; the incidence of viral infection and autoimmune responses may be related and lead to damage of the body's immune balance, ultimately causing immune-mediated disease or hypersensitivity disease-like changes that manifest as KD.[10]
HLH is a heterogenic syndrome, due to uncontrolled T cell, macrophage and histiocyte activation, hyperinflammatory state accompanied by excessive cytokine production.[2] The most common symptoms are fever, hepatosplenomegaly, pancytopenia, infiltration of various organs by histiocytes, and high serum ferritin levels. This condition has been described in association with an autosomal recessive familial syndrome,[11] viral and other infections, and various malignant diseases,[12] autoimmune and metabolic disorders. Delays in therapy with HLH may lead to irreversible multiorgan failure, the treatment and prognosis of KD associated with HLH is unclear.[3] The KD and HLH may be parts of a continuum of a single clinical condition, rather than representing separate entities for the etiology and clinical features between KD and HLH are some overlaps.[13]
We therefore performed a literature review and summarized 18 such cases in children not including the present case and 9 cases in adult (Tables 1 and 2). The median age at diagnosis was 13 years for children and 31 years for adult respectively, and the male-to-female ratio was 1: 1 and 5:4 among children and adults respectively. Lymph node involvement was cervical (n = 10 and 4), auxiliary (n = 5 and 1) and generalized (n = 4 and 5) in children and adults respectively. All children and adults showed cytopenia and increased serum ferritin and LDH levels. The mean ferritin and LDH levels elevated (7047.7 ng/ml and 25965.5 ng/ml, 1855 IU/L and 1805.2 IU/L, respectively) in children and adults, which were higher than in KD patients without HLH.[8] Only about 2% splenomegaly and 3% hepatomegaly are seen in KD,[18] but splenomegaly was observed in 7 (38.9%) and 4 (44.4%) cases, hepatomegaly was observed in 6 (33.3%) and 1 (11.1%) cases for children and adults of HLH-associated KD, respectively. In contrast to the benign course of KD, for 2 (11.1%) children and 3 (33.3%) adult patients HLH-associated KFD died. Recurrence was observed in only 3 children.
Table 1.
18 cases of HLH associated KD in children.
| case | Age (y)/Gender | Lymph node | Splenomegaly | Hepatomegaly | WBC (∗10^9/L) | Hb (g /dl) | Plt (∗10^9/L) | LDH (IU/L) | SF (ng/ml) | Pathogen | Associated diseases | Treatment | Outcome | Literature |
| 1 | 13/F | C | (−) | (−) | 0.41 | 9.2 | 19 | NA | 14955 | IVIG, mPDN, VP16, DEX | improved | [3] | ||
| 2 | 0.75/M | G (C,I,MD,R,PE) | NA | NA | 18.2 | 6.9 | 43 | 1615 | 3090 | NA | improved | [14] | ||
| 3 | 12/M | C | (−) | (−) | 2.5 | 10.5 | 220 | 1105 | 1003 | PDN | recurrence | [4] | ||
| 4 | 14/M | C,A,I | (−) | (−) | 3.2 | 9.1 | 169 | 682 | 2541 | EBV | IVIG, ACV, DEX, VP16 | recurrence | [4] | |
| 5 | 5/F | G (C,A,I,P | (+) | (+) | 1.8 | 6.7 | 110 | 1540 | 3371 | DIC | PDN, VP16, DEX | died | [4] | |
| 6 | 14/F | C | (−) | (−) | 2.4 | 9.8 | 108 | 627 | 472 | IVIG, DEX,VP16, CyA | improved | [4] | ||
| 7 | 8/M | G (C,A,I,M) | (+) | (+) | 3 | 11.6 | 105 | 1308 | 1168 | EBV | PDN | improved | [4] | |
| 8 | 16/F | C,I,A | (−) | (−) | 1.5 | NA | NA | 1682 | 9329 | PDN, mPDN, CyA | improved | [15] | ||
| 9 | 16/M | C | (+) | (−) | 1.24 | 12.6 | 130 | NA | 892.9 | EBV | NA | improved | [16] | |
| 10 | 13/M | A,I | (+) | (+) | 1.7 | NA | 135 | NA | NA | PDN | recurrence | [17] | ||
| 11 | 16/F | A | NA | NA | 0.8 | 8.9 | 214 | > 2150 | 777 | Dengue virus | corticosteroids | improved | [8] | |
| 12 | 14/M | C | (−) | (+) | 1.45 | 12 | 98 | 1238 | 128 | IVIG,PDN | improved | [18] | ||
| 13 | 10/F | C | NA | NA | 1.4 | 9.7 | 219 | 852 | 1083 | IVIG,PDN | improved | [18] | ||
| 14 | 1/F | G | (+) | (−) | 4.1 | 5.8 | 89 | 1317 | 41500 | JIA | IVIG, mPDN, PDN, CyA, MTX | improved | [19] | |
| 15 | 15/F | C | (+) | (+) | 1.4 | 7.5 | 147 | 1941 | 2500 | PVB19 | PDN | improved | [20] | |
| 16 | 17/F | C | NA | NA | 3.1 | 9.9 | normal | 1573 | >1000 | IVIG | improved | [13] | ||
| 17 | 6/M | A | NA | NA | 3.2 | 124 | 238 | 8340 | 35500 | RSV | MOF | PDN, VP16 and DEX, cyA | improved | [21] |
| 18 | 1.75/M | (+) | (+) | (+) | NA | NA | 3 | NA | 500 | JMML | PDN,6-mercaptopurine | died | [22] |
Table 2.
Cases of HLH associated KD in adults.
| Case | Age (y)/Gender | Lymph node | Splenomegaly | Hepatomegaly | WBC (∗10^9/L) | Hb (g/dl) | Plt (∗10^9/L) | LDH (IU/L) | SF (ng/ml) | Pathogen | Associated diseases | Treatment | Outcome | Literature |
| 1 | 21/M | C | (−) | (−) | 2 | 11.8 | 58 | 436 | NA | Sweet's syndrome | PDN | improved | [23] | |
| 2 | 24/F | C | NA | NA | 2.6 | 9 | 23 | NA | NA | EBV | Pregnancy, DIC, ARNF, ARDS | IVIG | died | [24] |
| 3 | 35/F | C | NA | NA | NA | 7.8 | 67 | NA | 10024 | pregnancy, ALF, PPE | mPDN,VP16,DEX | died | [25] | |
| 4 | 54/F | G (C,A,MD,I) | NA | NA | 3.7 | 5.7 | NA | 2420 | 47147 | LCN | NA | improved | [26] | |
| 5 | 50/M | G (C,A,I) | (+) | (−) | 2.04 | 8.4 | 96 | 3056 | NA | SLE, AORF,SS | mPDN | died | [27] | |
| 6 | 36/F | G (A,PEL,CH,AB) | (+) | (+) | 13.4 | 7.4 | 53 | 3056 | 40000 | EBV, PVB19 | SS,ARF | DEX, VP16 | HNL recurrence | [28] |
| 7 | 21/M | G (C,A,R,I) | NA | NA | 1.6 | 10 | 142 | 752 | 6691 | SPTL | VP16, PND, Mpdn | improved | [29] | |
| 8 | 30/M | G (C,A,I,P,M) | (+) | (−) | 1.5 | 12.5 | 130 | 1111 | NA | PND | improved | [30] | ||
| 9 | 40/M | C,A | (+) | (−) | 2.7 | 13.2 | 82 | NA | NA | naproxen | improved | [31] |
Epstein-Barr virus (n = 3), parvovirus B19 (n = 1), dengue virus (n = 1) and respiratory syncytial virus (n = 1) infections were documented in these children. Among adults, 1 patient was infected with Epstein-Barr virus, 1 patient was infected with Epstein-Barr virus and parvovirus B19.
HLH-associated KFD has a potentially fatal outcome, and early treatment is required. In the reviewed cases, the treatment in children included corticosteroids (n = 15), IVIG (n = 7), cyclosporine (n = 4) and etoposide (n = 5). The treatment in adults included corticosteroids (n = 6), IVIG (n = 1) and etoposide (n = 3). Among children, 2 patients died, with one case associated with Juvenile myelomonocytic leukemia, and the other one with DIC. Among adults, 3 of the 9 cases were fatal. 2 cases were pregnant women with 1 pregnant woman with DIC and other with organ function failure. The last case was an older man with SLE and organ failure. DIC and multiple organ failure (MOF) are both causes of death in children and adults.
For our patient, we performed a lymph node biopsy for diagnosis. KD usually has a benign course and is self-limiting, but sometimes it may be fatal with HLH or if these patients develop DIC.[27,32] No effective or standard unified treatment has been established for this disease. Usually, supportive treatment alone is sufficient; corticosteroids and IVIG may be used for severe cases of KD.[33,34] The treatment and prognosis of childhood KD associated with hemophagocytic syndrome remains unclear. According to Kim YM,[3] childhood KD is more frequently associated with hemophagocytic syndrome. Mahadeva U[21] reported that treatment with corticosteroids, VP16, and cyclosporine for a young boy with KD and HLH led to rapid resolution of symptoms. Chen[18] reported treatment of a patient with IVIG (2 g/kg) and prednisolone (2 mg/kg/day), led to a good recovery. But from our reviews, the patients with DIC or MOF died, despite treatment with corticosteroids or VP16 or IVIG.
Since the excessive activation of T cells and macrophages may lead to “cytokine storm”, the inhibition of inflammation by blocking this cytokine storm may be an effective therapeutic strategy for HLH. As an adjuvant therapy, continuous HDF can recover organ function by reducing cytokine levels.[35] Additionally, considering that HLH may lead to acute MOF, the efficacy of continuous HDF may be considerable in pediatric patients with MOF.[36] Demirkol et al[37] reported that patients with secondary hemophagocytic syndrome were successfully treated with PE, IVIG, and methylprednisolone.
Our patient with KD and hemophagocytic syndrome had a relatively malignant progression of disease. Administration of IVIG and corticosteroids failed to prevent further progression of disease. The patient developed acute respiratory failure, DIC, and capillary leak syndrome. Methylprednisolone treatment was initiated, followed by dexamethasone, cyclosporine, etoposide, continuous HDF and PE, and the patient recovered uneventfully. Chemotherapy, continuous HDF and PE may be one of the ways to treat children with severe condition.
In conclusion, although KD usually has a benign course, is self-limiting and, in most cases, administration of IVIG and corticosteroids yield satisfactory results. When KD is associated with HLH, DIC, MOF, it may be fatal. Continuous hemodiafiltration, plasma exchange and chemotherapy should be reserved for those patients who fail to respond to IVIG and corticosteroids. While the body temperature of patients with KD improves after steroid therapy, the condition may still worsen if pancytopenia develops. Higher levels of serum ferritin and LDH and hepatosplenomegaly are more frequently observed in patients with HLH-associated KD than those in patients with typical KD.
Author contributions
Conceptualization: Wei Duan, Hai-Yan Luo.
Formal analysis: Wei Duan.
Investigation: Wei Duan.
Methodology: Wei Duan, Zheng-Hui Xiao.
Resources: Wei Duan, Zheng-Hui Xiao, Long-Gui Yang.
Supervision: Hai-Yan Luo.
Validation: Hai-Yan Luo.
Writing – original draft: Wei Duan.
Writing – review & editing: Hai-Yan Luo.
Footnotes
Abbreviations: HLH = lymphohistiocytosis; IVIG = intravenous immunoglobulin, KD = Kikuchi's disease, LDH = lactate dehydrogenase, MOF = multiple organ failure.
How to cite this article: Duan W, Xiao ZH, Yang LG, Luo HY. Kikuchi's disease with hemophagocytic lymphohistiocytosis: a case report and literature review. Medicine. 2020;99:51(e23500).
Written informed consent was obtained from the patient's parents for publication of this case report and accompanying images.
The authors report no conflicts of interest.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
ACV = acyclovir, ALF = acute liver failure, AORF = acute olguric renal, ARDS = acute respiratory distress syndrome, ARF = acute respiratory failure, ARNF = acute renal failure, CyA = cyclosporine A, DEX = dexamethazone, EBV = Epstein-Barr virus, IVIG = intravenous immunoglobulin, JIA = systemic juvenile idiopathic arthritis, JMML = juvenile myelomonocytic leukemia, LCN = liver cell necrosis, LN = lymph node, mPDN = methylprednisolone, MTX = methotrexate, NA = not available, PDN = prednisolone, PPE = proximal pulmonary embolus, PVB19 = parvovirus B19, RSV = respiratory syncytial virus, SLE = systemic lupus erythematosus, SPTL = subcutaneous panniculitis-like T-cell lymphoma, SS = septic shock, VP16 = Etoposide. The affected lymph nodes are described as follows: A = axillary, AB = abdomen, C = cervical, CH = chest, G = generalized, I = inguinal, M = mesenteric, MD = mediastinal, P = para-aortic, PEL = pelvis, R = retroperitoneum.
ACV = acyclovir, ALF = acute liver failure, AORF = acute olguric renal, ARDS = acute respiratory distress syndrome, ARF = acute respiratory failure, ARNF = acute renal failure, CyA = cyclosporine A, DEX = dexamethazone, EBV = Epstein-Barr virus, IVIG = intravenous immunoglobulin, JIA = systemic juvenile idiopathic arthritis, JMML = juvenile myelomonocytic leukemia, LCN = liver cell necrosis, LN = lymph node, mPDN = methylprednisolone, MTX = methotrexate, NA = not available, PDN = prednisolone, PPE = proximal pulmonary embolus, PVB19 = Parvovirus B19, RSV = respiratory syncytial virus, SLE = Systemic lupus erythematosus, SPTL = subcutaneous panniculitis-like T-cell lymphoma, SS = septic shock, VP16 = Etoposide. The affected lymph nodes are described as follows: A = axillary, AB = abdomen, C = cervical, CH = chest, G = generalized, I = inguinal, M = mesenteric, MD = mediastinal, P = para-aortic, PEL = pelvis, R = retroperitoneum.
References
- [1].Jung IY, Ann HW, Kim JJ, et al. The incidence and clinical characteristics by gender differences in patients with Kikuchi-Fujimoto disease. Medicine (Baltimore) 2017;96:e6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tothova Z, Berliner N. Hemophagocytic syndrome and critical illness: new insights into diagnosis and management. J Intensive Care Med 2015;30:401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim YM, Lee YJ, Nam SO, et al. Hemophagocytic syndrome associated with Kikuchi's disease. J Korean Med Sci 2003;18:592–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lim GY, Cho B, Chung NG. Hemophagocytic lymphohistiocytosis preceded by Kikuchi disease in children. Pediatr Radiol 2008;38:756–61. [DOI] [PubMed] [Google Scholar]
- [5].Stephan L, Elanine SJ. Philip AP, David GP. Histiocytoses. Principles and practice of pediatric oncology. Baltimore: Williams &Wilkins; 1997;739-740. [Google Scholar]
- [6].Kuo TT. Kikuchi's disease (histiocytic necrotizing lymphadenitis). A clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology, and DNA ploidy. Am J Surg Pathol 1995;19:798–809. [DOI] [PubMed] [Google Scholar]
- [7].Kim TY, Ha KS, Kim Y, et al. Characteristics of Kikuchi-Fujimoto disease in children compared with adults. Eur J Pediatr 2014;173:111–6. [DOI] [PubMed] [Google Scholar]
- [8].Sykes JA, Badizadegan K, Gordon P, et al. Simultaneous acquired self-limited hemophagocytic lymphohistiocytosis and Kikuchi necrotizing lymphadenitis in a 16-year-old teenage girl: a case report and review of the literature. Pediatr Emerg Care 2016;32:792–8. [DOI] [PubMed] [Google Scholar]
- [9].Ura H, Yamada N, Torii H, et al. Histiocytic necrotizing lymphadenitis (Kikuchi's disease): the necrotic appearance of the lymph node cells is caused by apoptosis. J Dermatol 1999;26:385–9. [DOI] [PubMed] [Google Scholar]
- [10].Nomura Y, Takeuchi M, Yoshida S, et al. Phenotype for activated tissue macrophages in histiocytic necrotizing lymphadenitis. Pathol Int 2009;59:631–5. [DOI] [PubMed] [Google Scholar]
- [11].Perry MC, Harrison EG, Jr, Burgert EO, Jr, et al. Report of two cases and clinicopathologic review. Cancer 1976;38:209–18. [DOI] [PubMed] [Google Scholar]
- [12].Linn YC, Tien SL, Lim LC, et al. Haemophagocytosis in bone marrow aspirate--a review of the clinical course of 10 cases. Acta Haematol 1995;94:182–91. [DOI] [PubMed] [Google Scholar]
- [13].Kelly J, Kelleher K, Khan MK, et al. A case of haemophagocytic syndrome and Kikuchi-Fujimoto disease occurring concurrently in a 17-year-old female. Int J Clin Pract 2000;54:547–9. [PubMed] [Google Scholar]
- [14].Kim HA, Im SA, Chung NG, et al. Disseminated Kikuchi disease associated with hemophagocytic syndrome in an infant: whole-body MRI. Indian J Pediatrics 2011;78:616–9. [DOI] [PubMed] [Google Scholar]
- [15].Mara L, Laura S, Ilaria A, et al. Kikuchi-Fujimoto disease in children: two case reports and a review of the literature. Italian J Pediatrics 2018;44:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee HY, Huang YC, Lin TY, et al. Primary epstein-barr virus infection associated with Kikuchi's disease and hemophagocytic lymphohistiocytosis: a case report and review of the literature. J Microbiol Immun Infect 2010;43:253–7. [DOI] [PubMed] [Google Scholar]
- [17].Lin YW, Horiuchi H, Ueda I, et al. Recurrent hemophagocytic lymphohistiocytosis accompanied by Kikuchi's disease. Leukemia Lymphoma 2007;48:2447–51. [DOI] [PubMed] [Google Scholar]
- [18].Chen JS, Chang KC, Cheng CN, et al. Childhood hemophagocytic syndrome associated with Kikuchi's disease. Haematologica 2000;85:998–1000. [PubMed] [Google Scholar]
- [19].Ramanan AV. Systemic juvenile idiopathic arthritis, Kikuchi\”s disease and haemophagocytic lymphohistiocytosis--is there a link? Case report and literature review. Rheumatology 2003;42:596–8. [DOI] [PubMed] [Google Scholar]
- [20].Yufu Y, Matsumoto M, Miyamura T, et al. Parvovirus B19-associated haemophagocytic syndrome with lymphadenopathy resembling histiocytic necrotizing lymphadenitis (Kikuchi's disease). Br J Haematol 1997;96:868–71. [DOI] [PubMed] [Google Scholar]
- [21].Mahadeva U, Allport T, Bain B, et al. Haemophagocytic syndrome and histiocytic necrotising lymphadenitis (Kikuchi's disease). J Clin Pathol 2000;53:636–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gerritsen A, Lam K, Schneider EM, et al. An exclusive case of juvenile myelomonocytic leukemia in association with Kikuchi\"s disease and hemophagocytic lymphohistiocytosis and a review of the literature. Leukemia Res 2006;30:1299–303. [DOI] [PubMed] [Google Scholar]
- [23].Koga T, Takano K, Horai Y, et al. Sweet's syndrome complicated by Kikuchi's disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Internal Med 2013;52:1839–43. [DOI] [PubMed] [Google Scholar]
- [24].Chmait RH, Meimin DL, Koo CH, et al. Hemophagocytic syndrome in pregnancy. Obstetrics Gynecol 2000;95:1022–4. [DOI] [PubMed] [Google Scholar]
- [25].Giard JM, Decker KA, Lai JC, et al. Acute liver failure secondary to hemophagocytic lymphohistiocytosis during pregnancy. Acg Case Rep J 2016;3:e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ubels FL. Expanding the clinical spectrum of self-limiting, rare Kikuchi disease - A case with overwhelming multi-organ involvement. Netherlands J Med 2017;75:112–6. [PubMed] [Google Scholar]
- [27].Kampitak T. Fatal Kikuchi-Fujimoto disease associated with SLE and hemophagocytic syndrome: a case report. Clin Rheumatol 2008;27:1073–5. [DOI] [PubMed] [Google Scholar]
- [28].Gowarty J, Oda J, Cable C. Hemophagocytic lymphohistiocytosis. Proc (Bayl Univ Med Cent) 2018;31:350–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Notaro E, Shustov A, Chen X, et al. Kikuchi–Fujimoto disease associated with subcutaneous panniculitis-like T-cell lymphoma. Am J Dermatopathol 2016;38:e77–80. [DOI] [PubMed] [Google Scholar]
- [30].Nishiwaki M, Hagiya H, Kamiya T. Kikuchi-Fujimoto disease complicated with reactive hemophagocytic lymphohistiocytosis. Acta Medica Okayama 2016;70:383–8. [DOI] [PubMed] [Google Scholar]
- [31].Khan FY, Morad NA, Fawzy Z. Kikuchi's disease associated with hemophagocytosis. Chang Gung Med J 2007;30:370–3. [PubMed] [Google Scholar]
- [32].Uslu E, Gurbuz S, Erden A, et al. Disseminated intravascular coagulopathy caused by Kikuchi-Fujimoto disease resulting in death: first case report in Turkey. Int Med Case Rep J 2014;7:19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol 2007;26:50–4. [DOI] [PubMed] [Google Scholar]
- [34].Noursadeghi M, Aqel N, Gibson P, et al. Successful treatment of severe Kikuchi's disease with intravenous immunoglobulin. Rheumatology (Oxford) 2006;45:235–7. [DOI] [PubMed] [Google Scholar]
- [35].Cui Y, Zhang YC, Kang YL, et al. High-volume hemofiltration in critically ill patients with secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome: a prospective study in the PICU. Pediatr Crit Care Med 2016;17:e437–43. [DOI] [PubMed] [Google Scholar]
- [36].Shiga H, Kikuchi Y, Hattori N, et al. Special considerations in continuous hemodiafiltration with critically ill pediatric patients. Contrib Nephrol 2010;166:158–66. [DOI] [PubMed] [Google Scholar]
- [37].Demirkol D, Yildizdas D, Bayrakci B, et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care 2012;16:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]