

Abstract
Molecular reprogramming in response to chemotherapeutics leads to poor therapeutic outcomes for prostate cancer (PCa). In this study, we demonstrated that CXCR6-CXCL16 axis promotes DTX resistance and acts as a counter-defense mechanism. After CXCR6 activation, cell death in response to DTX was inhibited, and blocking of CXCR6 potentiated DTX cytotoxicity. Moreover, in response to DTX, PCa cells expressed higher CXCR6, CXCL16, and ADAM-10. Furthermore, ADAM-10-mediated release of CXCL16 hyper-activated CXCR6 signaling in response to DTX. Activation of CXCR6 resulted in increased GSK-3β, NF-κB, ERK1/2 phosphorylation, and survivin expression, which reduce DTX response. Finally, treatment of PCa cells with anti-CXCR6 monoclonal antibody synergistically or additively induced cell death with ~1.5-4.5 fold reduction in the effective concentration of DTX. In sum, our data imply that co-targeting of CXCR6 would lead to therapeutic enhancement of DTX, leading to better clinical outcomes for PCa patients.
Keywords: Docetaxel, Prostate cancer, CXCR6, CXCL16, ADAM10
1. INTRODUCTION
Docetaxel (DTX) is commonly used to treat both castration-resistant and hormone-sensitive metastatic prostate cancer (PCa), but it often becomes ineffective during the treatment course [1-3]. The effect of DTX, like other chemotherapeutics, relies on specific drug–target interactions and its capacity to shift the cellular equilibrium from anti-apoptotic towards apoptotic signals. However, in response to DTX, cancer cells activate signaling to overcome its effect. Key signaling pathways involved in overcoming therapeutic response include NF-κB, PI3K/Akt/mTOR, PTEN, Src family kinases (SFKs), and ERK1/2 [4-12]. Subsequently, these effectors modulate the expression and activation of several anti- and pro-apoptotic proteins in PCa [13-15] and diminish the response to DTX.
The association of chemokines and chemokine receptors in tumor progression and metastasis is well established [16-21]. Cancer cells exploit chemokine-mediated signaling to survive and overcome chemotherapeutic effects [16-21], and conversely cytotoxic agents also affect chemokine signaling [22, 23]. CXCR6 and its natural ligand, CXCL16, are associated with various malignancies, including PCa [17, 24-30]. Others and we have shown association of the CXCR6-CXCL16 axis in non-small cell lung cancer; it facilitates cell migration by modulating MMP [17, 31] by activating NF-κB [31]. It also modulates the ERK1/2/RhoA/Cofilin/F-actin pathway, supporting the invasive and metastatic potential of cells [32]. Likewise, in various cancers, the CXCR6-CXCL16 axis influences signaling pathways that regulate growth, survival, angiogenesis, and metastasis, suggesting a role of this axis in disease progression. Earlier, we showed higher CXCR6 expression in PCa cells compared to non-neoplastic cells [24]. CXCL16 has a positive association with the aggressive PCa phenotype and bone metastasis [24]. CXCR6 promotes the metastatic potential of PCa cells by modulating cytoskeletal dynamics [25]. The CXCR6-CXCL16 axis supports PCa growth by promoting AKT/mTOR signaling [33]. It activates NF-κB by promoting IκB degradation via IKK [34], and it is associated with chemoresistance [35]. Here, we examined the effect of DTX on the CXCR6-CXCL16 axis and the impact of the activated CXCR6-CXCL16 axis on DTX-induced cytotoxicity in PCa. We also delineated the CXCL16-induced molecular mechanism that could relate to chemotherapeutic resistance. Our data suggest that the CXCR6-CXCL16 pathway acts as a counter-defense mechanism and that, in PCa, its blocking improves the response to DTX.
2. MATERIAL AND METHODS
2.1. Cell lines and cell culture
Human PCa cell lines (LNCaP, PC3, and DU145) and non-cancerous immortalized human prostate cell line (RWPE-1) were obtained from ATCC. Short tandem repeats genotyping was used out to authenticate the cell lines. PCa cells were maintained in their respective medium supplemented with 10% FBS, 100μg/ml streptomycin, and 100U/ml penicillin (HyClone Laboratories). RWPE-1 cells were grown in keratinocyte serum-free media (K-SFM) supplemented with bovine pituitary extract (0.05mg/ml) and human recombinant EGF (5ng/ml). All cell lines were maintained at 37°C with 5% CO2, and experiments were carried out with K-SFM for RWPE-1 cells and 2% FBS containing RPMI-1640 media for PCa cells.
2.2. Reagents and antibodies
DTX was purchased from Sigma Aldrich. Human CXCL16 was procured from Peprotech. PE-conjugated mouse anti-human CXCR6 antibody, APC-conjugated rat anti-human CXCL16, respective isotype controls (PE-conjugated IgG2b, APC-conjugated IgG2a), anti-phospho-RelA/NF-κB-p65(Ser-529), anti-ADAM10, and CXCL16 Quantikine ELISA kit were purchased from R&D Systems. Phospho-NF-κB-p65(Ser-536), phospho-Erk1/2(Thr-202/Tyr-204), phospho-GSKβ(Ser-9), and anti-survivin antibodies were purchased from CST. FITC-Annexin V/7-AAD apoptosis detection kit and Fc-Block were procured from BioLegend. cDNA Synthesis kit, bicinconic acid kit, and West Pico Chemiluminescent kit were obtained from ThermoFisher Scientific. SYBR-Green Supermix and PVDF membranes were purchased from Bio-Rad. Phosphorylation-specific antibody microarray slides were obtained from Fullmoon Biosystems Inc.
2.3. Cell Viability Assay
Cells (104 cells/well) were cultured in 100μl of media supplemented with 2% FBS in 96-well plates overnight. Cells were then treated with DTX (0-100nM) alone or with CXCL16 (100ng/ml) with or without CXCR6 blockade using anti-CXCR6 for 48h. Cell viability was determined by MTT assays as described previously [36]. All concentrations were tested in triplicates, and the experiment was repeated three times.
2.4. Flow cytometric analysis of cell death and CXCR6/CXCL16 expression in response to DTX
Cells (106) were treated for 48h with the 40nM DTX with or without CXCL16 (100ng/ml) as well as after blocking CXCR6 with anti-CXCR6 monoclonal antibody (1μg/ml). Cells treated with DMSO were used as vehicle controls. Cells were harvested using Accutase and washed thrice with FACS buffer (2% FBS in PBS). Cell death was determined using apoptosis detection kit as described in manufacturer’s protocol.
To determine the effect of DTX on CXCR6 and CXCL16 expression, PCa cells treated with DTX for 48h were incubated with Fc block for 10 min at RT and stained with PE-conjugated anti-CXCR6 for 40 min on ice. Cells were then permeabilized with 0.05% saponin for 30 min and washed with FACS buffer followed by staining with APC-conjugated anti-CXCL16 antibody. Similar approach was taken for isotype control antibody staining. Stained cells were washed with FACS buffer and fixed with 2% paraformaldehyde in PBS for 10 min. Fixed cells were washed with FACS buffer. Fluorescence was acquired using Guava EasyCyte (Millipore) and analyzed by FlowJo 10.0.6 software (Treestar Inc.).
2.5. Gene expression analysis by RT-qPCR
Total RNA from cells (106) treated with or without 40nM DTX for 48h was isolated using Tri-Reagent. Total RNA (1.0μg) was used to generate cDNA using cDNA synthesis kit according to the manufacturer's protocol. CXCR6 and CXCL16 mRNA and, 18S rRNA transcripts were quantified by RT-qPCR using gene-specific primers with SYBR-Green Supermix. Copies of mRNA transcripts of CXCR6 and CXCL16 were represented per million copies of 18S rRNA that used as a standard.
2.6. Soluble CXCL16 ELISA Assay
PCa cells (105/well) were seeded in 6-well plates in 1ml RPMI supplemented with 2% FBS and treated with 40nM DTX for 48h. Conditioned media from DTX-treated or untreated cells were collected, and soluble CXCL16 was quantified as described in our previous publication [17].
2.7. Assessment of CXCL16 induced phosphorylation using a phosphorylation-specific antibody microarray
PC3 cells were treated with CXCL16 (100ng/ml) for 15 min; untreated cells were used as controls. Protein isolation, biotinylation, hybridization, and labeling with streptavidin-conjugated Cy3 were accomplished according to the manufacturer’s instructions. The average signal intensity of six replicate spots for each antibody was normalized to the median signal of the array. Normalized data were used to calculate the fold change in intensities between untreated and CXCL16-treated samples. The ratios of average signal intensities from phospho site-specific (P) and corresponding non-phosphorylated (N) antibodies (P/N ratios) were calculated. A heat map was generated using the CIMminer tool (Genomics and Bioinformatics Group, NIH).
2.8. Western blot analysis
Total proteins from PCa cells (106) treated with DTX (40nM) for 48h and cells stimulated with CXCL16 for 15 min, 30 min, 1h, 2h, and 4h were isolated using RIPA lysis buffer containing protease and phosphatase inhibitors. Protein (50μg/lane) was resolved on 10% SDS-PAGE, transferred to PVDF membrane, then blocked with 5% non-fat milk in PBS with 0.05% Tween-20 for 1h at RT, followed by overnight incubation with primary antibodies (1:1000 dilution) at 4°C. Subsequently, the membranes were washed (3X) and incubated with 1:1000 diluted HRP-conjugated IgG for 1h at RT. β-actin was used as a loading control. Images of protein bands were developed with West Pico Chemiluminescent kit and captured with an ImageQuant (GE Healthcare Life Sciences). To compensate for sample-sample variation densitometric analysis of protein bands were performed using ImageJ analysis software and internal loading control β-actin was used to normalize [37-39].
2.9. Statistical Analyses
All the experiments were performed three times, and results were expressed as mean ± SEM. Wherever suitable, the data were subjected to unpaired two-tailed Student’s t-test, and p<0.05 was considered statistically significant. Statistical analyses for cell viability were done using 2-way Analysis of Variance (ANOVA) followed by Dunnett’s multiple comparisons test. The potential synergistic/additive effect of blockade of the CXCR6-CXCL16 axis on DTX was determined using Calcusyn software as described in our publication [40]. The combination index (CI) quantitatively defines synergism as CI<1, additive effect as CI=1, and antagonism as CI>1, which was calculated using cell viability data and DTX and anti-CXCR6 antibody concentrations.
3. RESULTS
3.1. Activation of the CXCR6-CXCL16 axis reduces Docetaxel-induced PCa cell death
There was a reduction in DTX cytotoxicity when CXCR6 was activated with CXCL16 compared with no activation in PCa cells (LNCaP, PC3 and DU145) but no such difference was observed in RWPE-1 (Fig. 1A). Differences in viability with or without CXCL16 activation was significant in LNCaP and DU145 at 20-, 40-, and 100nM DTX, whereas in PC3 cells such difference was significant at 40 and 100nM DTX. Blocking CXCR6 before CXCL16 and DTX treatment showed significant improvement in DTX response. Reduction in viability of PC3, when CXCR6 was blocked, was significantly high at 20nM DTX. At the same DTX concentration CXCL16 treatment did not show significant change (Fig. 1A). Further, PCa cells treated with most effective concentration (40nM) of DTX showed 55.94%, 39.64% and 52.5% cell death in LNCaP, PC3 and DU145, respectively (Fig.1B). CXCL16 treatment prior to DTX treatment reduced cell death to 38.56% (p≤0.01), 26.18% (p≤0.01) and 27.23% (p≤0.0001) in LNCaP, PC3 and DU145, respectively. Additionally, blocking CXCR6 prior to DTX showed increased cell death of LNCaP (88.97%, p≤0.0001), PC3 (71.42%, p≤0.0001) and DU145 (69.70%, p≤0.0005) compared to DTX treatment alone (Fig. 1B). Such difference was not observed in RWPE-1. These findings show that the CXCR6-CXCL16 axis is involved in DTX-induced cytotoxicity.
Figure 1. Effect of CXCR6 stimulation and blockade on DTX induced cell death.

PCa cells (LNCaP, PC3 and DU145) and non-cancerous immortalized human prostate cell line (RWPE-1) cells treated with different concentration (0-100nM) of DTX ( ) CXCL16 (100ng/ml) + DTX (
) CXCL16 (100ng/ml) + DTX ( ); anti-CXCR6 (1μg/ml) + CXCL16 (100ng/ml) + DTX (
); anti-CXCR6 (1μg/ml) + CXCL16 (100ng/ml) + DTX ( ) for 48h, cell viability assay show effect of CXCR6-CXCL16 axis on DTX (Panel-A). Statistical significance between DTX alone vs. combination treatment group was done using 2-way ANOVA multiple comparison test represented as asterisks (★, p≤0.05; ★★, p≤0.01; ★★★, p≤0.001; ★★★★, p≤0.0001). Apoptosis induced by DTX (40nM), in presence or absence of CXCL16 (100ng/ml), with or without CXCR6 blockade (1μg/ml) was quantified by FACS (Panel-B). DMSO treated cells were used as a vehicle control. Quadrate (Q2) and Q3 show late and early apoptotic cells. Clustered stacked column chart is a quantitative representation of the percentage death in RWPE-1 (■), LNCaP (□), PC3 (■), and DU145 (■) cells (Panel-C). Values are mean ± SEM from three independent experiments. Statistical significance between combination treatment group and DTX alone, was done by Student’s t-test. Statistical difference is represented as ★★, p≤0.01 between DTX alone and CXCL16 + DTX; ★★★★, p≤0.0001 between DTX and anti-CXCR6 + DTX for LNCaP cells; ❖❖, p≤0.01 between DTX alone and CXCL16 + DTX; ❖❖❖❖, p≤0.0001 between DTX and anti-CXCR6 + DTX for PC3 cells and,
) for 48h, cell viability assay show effect of CXCR6-CXCL16 axis on DTX (Panel-A). Statistical significance between DTX alone vs. combination treatment group was done using 2-way ANOVA multiple comparison test represented as asterisks (★, p≤0.05; ★★, p≤0.01; ★★★, p≤0.001; ★★★★, p≤0.0001). Apoptosis induced by DTX (40nM), in presence or absence of CXCL16 (100ng/ml), with or without CXCR6 blockade (1μg/ml) was quantified by FACS (Panel-B). DMSO treated cells were used as a vehicle control. Quadrate (Q2) and Q3 show late and early apoptotic cells. Clustered stacked column chart is a quantitative representation of the percentage death in RWPE-1 (■), LNCaP (□), PC3 (■), and DU145 (■) cells (Panel-C). Values are mean ± SEM from three independent experiments. Statistical significance between combination treatment group and DTX alone, was done by Student’s t-test. Statistical difference is represented as ★★, p≤0.01 between DTX alone and CXCL16 + DTX; ★★★★, p≤0.0001 between DTX and anti-CXCR6 + DTX for LNCaP cells; ❖❖, p≤0.01 between DTX alone and CXCL16 + DTX; ❖❖❖❖, p≤0.0001 between DTX and anti-CXCR6 + DTX for PC3 cells and,  , p≤0.0001 between DTX alone and CXCL16 + DTX;
, p≤0.0001 between DTX alone and CXCL16 + DTX;  , p≤0.0005 between DTX and anti-CXCR6 + DTX for DU145 cells.
, p≤0.0005 between DTX and anti-CXCR6 + DTX for DU145 cells.
3.2. Docetaxel increases CXCR6 and CXCL16 expression in PCa cells
We further determined if DTX affects CXCR6 and CXCL16 expression. PCa cells treated with DTX (40nM) for 48h showed elevated levels of CXCR6 mRNA (LNCaP: 1.5 fold, PC3: ~2-fold, p≤0.05 and DU145: ~3.5-fold, p≤0.05) as compared to untreated cells (Fig. 2A). CXCL16 mRNA was also higher in PCa cells (LNCaP: ~2-fold, p≤0.001; PC3: ~2-fold, p≤0.05 and DU145: ~4.5-fold, p≤0.001) in response to DTX, as compared to the untreated control. No such difference in CXCR6 and CXCL16 mRNA expression was observed in RWPE-1 cell (Fig. 2A and B). Protein levels of CXCR6 (LNCaP: ~1.32-fold, p-≤0.0001; PC3: ~1.8-fold, p≤0.0001 and DU145: ~1.28-fold, p≤0.0001) and CXCL16 (LNCaP and PC3: ~2.1-fold, DU145: ~3-fold, p≤0.0001) were higher in response to DTX compared to untreated controls, but no such difference was observed in non-cancerous immortalized human prostate cell line (Fig. 2C).
Figure 2. CXCR6 and CXCL16 expression in response to DTX in prostate cancer cells.

PCa cells (LNCaP, PC3 and DU145) and non-cancerous immortalized human prostate cell line (RWPE-1) were treated with DTX (40nM). mRNA transcripts in DTX treated (■) or untreated (□) cells was quantified by RT-qPCR and changes in CXCR6 and CXCL16 mRNA transcript copies per million 18S rRNA are shown in Panel-A and – B respectively. Unpaired Students’ t-test was used for statistical analysis. Askterisks (★, p≤0.05; ★★★, p≤0.001) show statistically significant difference between DTX treated and untreated control. CXCR6 and CXCL16 protein expression in PCa cells and non-cancerous immortalized human prostate cell line (RWPE-1) cells in response to DTX (40nM) treatment by flow cytometric analysis (Panel-C). Histograms show CXCR6 or CXCL16 expression with (■) or without (□) DTX treatment. Inset bar graphs are quantitative comparison of mean fluorescence intensities (MFI) of CXCR6 or CXCL16 expression in DTX treated (■) and untreated cells (□). MFI shown are after subtracting MFI of isotype controls. Asterisk (★, p≤0.0001) shows statistically significant difference between DTX treated and untreated control.
3.3. Docetaxel promotes cleavage of membrane-bound CXCL16 by modulating ADAM-10 via NF-κB and activates CXCR6 signaling
The elevated response to DTX after blocking CXCR6 without exogenous CXCL16 implies that DTX activates CXCR6 signaling. Hence, soluble CXCL16 (sCXCL16) was quantified in DTX-treated conditioned media. Significantly (p≤0.05) higher levels of sCXCL16 were present in DTX-treated conditioned media as compared to controls (Fig. 3A). Additionally, ADAM-10 protein, which cleaves membrane-bound CXCL16, was elevated in DTX-treated PCa cells compared to controls. DU145 cells showed the highest increase in ADAM10 (~3.5-fold, p≤0.001) followed by LNCaP (~2.5-fold, p≤0.001) and PC3 (~1.8-fold, p≤0.01) cells (Fig. 3B).
Figure 3. Docetaxel induced cleavage of CXCL16, ADAM-10 expression and NF-κB activation in prostate cancer cells.

ELISA showing soluble CXCL16 levels in conditioned media of LNCaP, PC3, DU145 treated with DTX (40nM) for 48h compared to untreated control (Panel-A). Values are mean ± SEM from three independent experiments. Multiple t-test using the Holm-Sidak method was used to test the significance, asterisks (★, p≤0.05) show statistically significant difference between treated and untreated control. Representative western blot images showing ADAM-10 expression in LNCaP, PC3 and DU145 48h after DTX (40nM) treatment (Panel-B). Quantified band intensity represents fold change in ADAM-10 expression after normalization with β-actin in the bar graph. Asterisks (★★, p≤0.01, ★★★, p≤0.001, ★★★★, p≤0.0001) show statistically significant difference between treated and untreated control. Phosphorylation of NF-κB at Ser-536 (Panel-C) and Ser-529 (Panel-D) in PCa cells (LNCaP, PC3 and DU145) treated with DTX (40nM) for 48h compared to control. Asterisks (★, p≤0.05, ★★, p≤0.01, ★★★, p≤0.001) statistically significant show difference between treated and untreated control.
Since activation of NF-κB is linked with promotion of CXCL16 cleavage [41-44] we examined NF-κB activation in response to DTX. There was greater phosphorylation of NF-κB p65(Ser-536/Ser-529) in DTX-treated PC3 cells compared to controls (Fig. 3C and D). After DTX treatment, DU145 cells did not show any change in phosphorylation at either of these serine residues (Fig. 3C and D). However, LNCaP cells showed lower phosphorylation at Ser-536 with no variation in phosphorylation at Ser-529 compared to controls (Fig. 3C and D).
3.4. CXCR6-CXCL16 alters molecular signatures responsible for overcoming DTX response by increasing survival and decreasing apoptotic molecules
We investigated CXCL16-triggered molecular signaling that could be responsible for supporting cell survival and resisting apoptosis. Based on the antibody microarray, CXCL16 treatment showed phosphorylation of GSK-3β, NF-κB, and p44/42 MAPK (ERK1/2) (Fig. 4A). There was ~2-fold increase in phosphorylation of GSK-3β(Ser-9), NF-κB-p65(Ser-529), and Erk1/2(Thr-202/Tyr-204) in CXCL16-treated PC3 cells compared to untreated controls. Increases in phosphorylation of NF-κB-p65(Thr-254) (~1.2-fold) and NF-κB-p105/50(Ser-893) (~1.3-fold) were also evident, albeit marginal. GSK-3α(Ser-21) did not show a significant increase in phosphorylation. These were validated by western blot analysis of PCa cells (Fig. 4B-E). Similar to the antibody array, there was a significant increase in GSK-3β(Ser-9) phosphorylation in CXCL16-treated PC3 cells at 15 min, 30 min, 1h, 2h and 4h (Fig. 4B). However, LNCaP and DU145 cells showed oscillatory pattern in GSK-3β(Ser-9) phosphorylation (Fig.4B). A similar pattern was observed for NF-κB-p65(Ser-529) in CXCL16-treated LNCaP cells compared to untreated cells (Fig. 4D). However, CXCL16-treated PC3 cells showed elevated NF-κB-p65(Ser-529) phosphorylation from 15 min to 1h, which decreased thereafter. Phosphorylation of NF-κB-p65(Ser-529) in CXCL16-treated DU145 cells increased after 1h (Fig. 4D). Additionally, a time-dependent increase in NF-κB-p65(Ser-536) phosphorylation was observed in CXCL16-treated PC3 and DU145 cells. LNCaP cells did not show significant changes (Fig. 4E). Further, CXCL16 induced ERK1/2(Thr-202/Tyr-204) phosphorylation in PC3 and DU145 cells, showing a significant increase at 30 min and, in LNCaP cells, at 15 min (Fig. 4C). In LNCaP cells, survivin expression increased immediately after CXCR6 stimulation; PC3 and DU145 cells showed delayed induction (Fig.4B). These results demonstrate that, in PCa cells, the CXCR6-CXCL16 axis regulates pro-survival signals.
Figure 4. CXCL16 induced molecular signals responsible for undermining cytotoxic effect of DTX on PCa cells.

Heat map shows phosphorylation status of signaling molecules in PCa cells 15 minutes after CXCL16 treatment (Panel-A). Phosphorylated to unphosphorylated protein (P/N) ratios were calculated from the intensity values. Intensity of red represents increase and green represents decrease in phosphorylation as shown in the intensity bar below the hear map. Change in phosphorylation status of GSK-3β(Ser-9), ERK1/2(Thr-202/Tyr-204), NF-κB-p65(Ser-529), NF-κB-p65(Ser-536) are shown in Panel-B, -C, -D and E; change in survivin expression in LNCaP, PC3 and DU145 cells treated with CXCL16 is also shown in Panel-B. Band intensity was quantified by imageJ and normalization with β-actin. Fold change in expression or phosphorylation is shown in bar graph. Asterisks represent statistically significant difference (★, p≤0.05, ★★, p≤0.01, ★★★, p≤0.001) between treated and untreated control. Same loading control was used for the blots, which were stripped and re-probed with another antibody.
3.5. CXCR6 blockade reduces the effective concentration of DTX and enhances its cytotoxic activity
To establish if CXCR6 blockade has a synergistic or additive effect on DTX, median-effect plots and dose-effect curves were generated by Calcusyn software using MTT assay data (Fig. 5). In PC3 cells, the cytotoxic effects of various DTX concentrations (20-, 40-, and 100nM) when combined with anti-CXCR6 (1μg/ml) were significantly greater than the effect of DTX alone (Fig. 5). Analysis of similar dose-effect curves for LNCaP and DU145 cells showed greater effects only at 20- and 100nM concentrations of DTX. For PC3 cells, the combination index (CI) for DTX/anti-CXCR6 treatment, at multiple concentrations of DTX (20-, 40-, and 100nM), was below CI=1, indicating a synergistic association between the two drugs (Table 1). Strong synergism was observed at 20- and 40nM DTX, and there was moderate synergism at 100nM DTX. Similarly, LNCaP cells showed strong synergism at 20- and 40nM DTX (Table 1). However, CI values for DU145 cells represented nearly additive effects at 20- and 40nM DTX, when cells were CXCR6 was blocked (Table 1). Correlation of CI values with synergistic, additive, or antagonistic association between two drugs is represented in Supplementary Table 1.
Figure 5. Effect of CXCR6 blockade on DTX response.

PCa cells were treated with increasing concentrations of DTX (20, 40 and 100nM) either alone or in combination with anti-CXCR6 (1μg/ml) for 48h. Calcusyn software was used to generate dose-effect curve, median-effect plots and combination index (CI).
Table I.
Combination index (CI) and dose reduction index (DRI) values for prostate cancer cells treated with different concentration of DTX and fixed concentration of anti-CXCR6 antibody.
| Cell line | DTX (nM) |
αCXCR6 (ng/mL) |
Fa1 | CI2 | DTX alone (nM) |
DRI |
|---|---|---|---|---|---|---|
| LNCaP | 20 | 1000 | 0.3526 | 0.699 | 33.44 | 1.672 |
| 40 | 1000 | 0.5137 | 0.513 | 82.84 | 2.071 | |
| 100 | 1000 | 0.5325 | 1.115 | 91.85 | 0.919 | |
| PC3 | 20 | 1000 | 0.5928 | 0.315 | 203.22 | 10.161 |
| 40 | 1000 | 0.5741 | 0.456 | 183.21 | 4.580 | |
| 100 | 1000 | 0.5739 | 0.785 | 183 | 1.830 | |
| DU145 | 20 | 1000 | 0.4059 | 1.052 | 34.15 | 1.708 |
| 40 | 1000 | 0.4964 | 0.998 | 65.28 | 1.632 | |
| 100 | 1000 | 0.5836 | 1.143 | 121.52 | 1.215 |
represents fraction of cells affected
represents combination index (CI), a quantitative representation of the pharmacological interactivity between DTX (D1) and anti-CXCR6 (D2) antibody, which factors in both the potency (Dm) and the shape of the dose-effect curve. CI is derived from the formula CI = (D)1/(Dx)1 + (D)2(Dx)2 + (D)1(D)2/(Dx)1(Dx)2, where (Dx)1 and (Dx)2 are the concentrations for Drug 1 (DTX) and Drug 2 (anti-CXCR6 antibody) separately, resulting in X% inhibition, and (D)1 and (D)2 the concentrations of the respective drugs in combination, resulting in the same percentage inhibition. The CI quantitatively defines synergism as CI<1, additive effect as CI=1 and antagonism as CI>1. The dose-reduction index (DRI) is a measure of how much the dose of each drug in a synergistic combination may be reduced at a given effect level compared with the doses for each drug alone, and is calculated by the equation (DRI)Drug A = (Dx)Drug A/(D)Drug A.
Further, the increase in DTX cytotoxicity with CXCR6 blockade was quantified as the drug reduction index (DRI) for all PCa cells (Table 1). The DTX concentration required to show ~50% cytotoxic effect (Fa= 0.5) was reduced by ~4-fold, ~2-fold, and ~1.6-fold for PC3, LNCaP and DU145 cells, respectively (Table 1). Overall, these findings show that CXCR6 blockade enhances efficacy of DTX either synergistically or in an additive manner.
DISCUSSION
DTX, often used to treat advanced PCa with palliative intent, provides modest survival benefits in castration-resistant disease and moderate survival benefits in hormone-sensitive disease, but these effects are associated with toxicities. Therefore, optimizing the therapeutic index by developing tolerable and synergistic combinations is imperative. Under chemotherapeutic pressure, cancer cells thrive either by developing a resistant phenotype [45, 46] or by clonal expansion of a resistant phenotype [47]. Hence, a better understanding of DTX-induced molecular changes is needed to develop effective treatment options for advanced PCa.
Clinical trials conducted on high-risk, localized and metastatic PCa showed differential therapeutic outcomes in response to DTX [48-52]. Inclusion of DTX with androgen-deprivation therapy increases overall survival of patients with metastatic, hormone-sensitive PCa [48]. The combination is less effective for metastatic, castration-resistant, and early non-metastatic cancer [49-52]. Thus, different states of PCa exhibit contrasting sensitivity to DTX and underlying molecular signatures could serve as putative prognostic biomarkers for DTX response. In this regard, our laboratory has shown higher expression of CXCR6 in PCa cells compared to immortalized prostate epithelial cells as well as increases in CXCL16-driven metastatic potential [25]. Our data showed reduced cytotoxic potential of DTX after CXCL16 treatment; blocking CXCR6 with anti-CXCR6 prior to DTX treatment enhanced the response to DTX. Additionally, in response to DTX, our data also showed increase in CXCR6 and CXCL16 in PCa cells, but not in non-cancerous immortalized prostate cells. This implies that PCa cells reduce the DTX response by overexpressing CXCR6 and CXCL16.
Signaling initiated by membrane-bound CXCL16 is anti-oncogenic whereas the cleaved or soluble form (sCXCL16) is more pro-oncogenic [17, 25, 53]. Higher levels of sCXCL16 in the DTX-treated conditioned media confirmed the autocrine activation CXCR6-CXCL16 signaling, suggesting that DTX affects PCa outcomes negatively via CXCR6. ADAM-10 that was elevated in response to DTX regulates the bioavailability of CXCL16 and TNF-α [41, 54-56]. TNF-α and CXCL16 can mutually upregulate each other [34, 41, 57, 58]; however, this regulation requires involvement of NF- κB that, together with Stat3, controls cytokines- and chemokine- encoding genes [59, 60]. Our data showed phosphorylation of NF-κB-p65(Ser-536, Ser-529) in response to DTX. Phosphorylation of these sites leads to transcriptional activation of NF-κB. DTX-induced transcriptional activation of NF-κB increases HIF-1α [43, 61] (Summarized in Fig. 6) and activates X-box-binding protein (XBP)-1 [62, 63]. Both HIF-1α and XBP-1 are inducers of ADAM10 [42]. This could be the case for PC3 cells, which showed increased NF-κB-p65(Ser-529/536) phosphorylation in response to DTX. CXCL16 treatment also enhanced NF-κB phosphorylation, implying that the DTX-induced NF-κB-p65 activation could be via activation of the CXCR6-CXCL16 axis. The time-dependent oscillations in NF-κB-p65(Ser-529/536) phosphorylation induced by CXCL16 could have important implications in controlling the dynamics of target gene transcription [64]. Unlike phosphorylation of NF-κB-p65(Ser-529), phosphorylation at Ser-536 serves a dual role of promoting its transactivation as well as its proteasomal degradation [65]. This suggests a CXCL16-regulated feedback mechanism for NF-κB activation in PCa cells. External stimuli such as TNFα and IL-1β induce NF-κB-p65(Ser-529) phosphorylation via CKII, which increases its transcriptional activity [66-68]. TNFα could also cause IKKβ-mediated phosphorylation of NF-κB-p65(Ser-536), leading to its enhanced transcriptional output [69]. These results corroborate that DTX treatment turns on a positive feed-forward loop between CXCL16-NF-κB-TNF-α via ADAM-10, thereby increasing CXCR6 signaling by cleaving membrane-bound CXCL16.
Figure 6. Model showing contribution of CXCR6-CXCL16 axis in overcoming the effect of DTX:
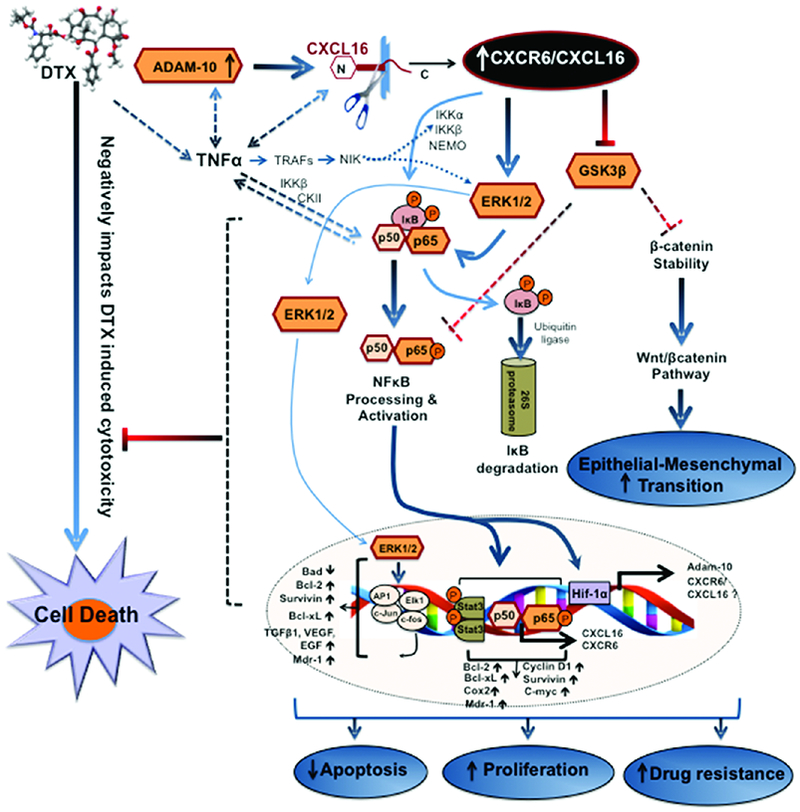
In response to DTX, prostate cancer cells up-regulate CXCR6, CXCL16 and ADAM-10, which also promotes CXCL16 cleavage. These changes together hyper-activate CXCR6-CXCL16 axis. TNFα, which could be regulated by ADAM-10 and CXCL16, leads to NF-κB activation by TRAFs (TNF-receptor associated factors) and NIK (NF-κB-inducing kinase). NIK also activates IKKα and Erk1/2. Activation of IKK complex leads to phosphorylation of IκB and enhances its proteasomal degradation. Free NF-κB subunits are then phosphorylated and translocate to the nucleus where they form homo- or heterodimers and increase cxcr6, cxcl16, and adam-10. Hyper-activated CXCR6-CXCL16 signaling a) supports epithelial to mesenchymal transition via Wnt/βcatenin pathway by affecting GSK3β activation and β-catenin stability and b) decreases apoptosis and enhances proliferation by affecting ERK1/2 activation, processing and trans-activation of NFκB resulting in over-expression of survivin as well as other anti-apoptotic and pro-survival molecules in concert with Stat3.
Activated NF-κB exerts its oncogenic potential by inhibiting apoptosis [70, 71] and supporting cell proliferation [72], migration, and invasion [73] (Fig. 6). Further, NF-κB, along with Stat3, regulates anti-apoptotic and cell cycle genes, including C-myc, cyclin D1, Bcl-2, Bcl-xL, COX2, and survivin (Summarized in Fig. 6), which are also related to chemo-resistance [35, 74-77]. It also inhibits p53-mediated transcriptional activation [78]. NF-κB also regulates the expression of multi-drug resistance gene, mdr1 [74, 79], and is involved in resistance to chemotherapy [80]. Thus activation of NF-κB, as observed after DTX as well as CXCL16 treatment, could reduce the response to DTX in PCa.
Our data also show that CXCR6 activates ERK1/2, which promotes tumor progression and drug resistance [81, 82] by regulating the Bcl-2 family of proteins, reducing p53 activity, and regulating ABCB1 expression [82-85]. Further, ERK1/2 phosphorylation confers chemoresistance by increasing HIF-1α-dependent activation of the ABCG2 drug transporter [86]. Our data showed a time-dependent increase in GSK-3β(Ser-9) phosphorylation following CXCL16 treatment. Phosphorylation of GSK-3β(Ser-9) confers cisplatin resistance by stabilizing p53 in ovarian cancer [87]. It also regulates the kinase activity of GSK-3β. Dephosphorylated GSK-3β inhibits pro-survival factors and activates pro-apoptotic transcription factors [88-90]. However, phosphorylated GSK-3β allows stabilization and nuclear translocation of β-catenin and thereby regulates various tumorigenic effects by activating Wnt/β-catenin pathways [91-95]. Further, dephosphorylated GSK-3β increases CRE transcriptional activity necessary for cell differentiation and suppression of cell growth [88]. Therefore, for cells to proliferate extensively, the level of dephosphorylated GSK-3β should be low. The constitutively higher phosphorylation of GSK-3β(Ser-9), at all time-points, in CXCL16-treated PC3 cells could be attributed to its higher aggressive behavior compared to LNCaP and DU145 cells. Thus, DTX activates CXCR6 by inducing CXCL16 release and makes cancer cells more resistant to death.
One of the pleiotropic roles of p-NF-κB-p65(Ser-529/536), p-ERK1/2, and p-GSK-3β(Ser-9) is modulation of expression and subcellular redistribution of survivin [96-98]. Expression of survivin varied temporally and in a cell-specific manner after CXCL16 treatment of PCa cells. In PC3 cells, survivin expression decreased initially followed by an increase, suggesting that the inhibitory role of p-NF-κB-p65(Ser-536) pre-dominated after CXCR6 activation but was subsequently subverted by p-GSK-3β(Ser-9). However, survivin expression in LNCaP and DU145 cells coordinated with the activation status of the above-mentioned upstream effector molecules, which could be independent of p-NF-κB-p65(Ser-536). This point needs further investigation. This is a notable finding, as, in addition to having roles in apoptosis, mitosis, and angiogenesis, survivin is involved in resistance to various drugs [99-105].
Our data show that DTX and anti-CXCR6 antibody had a synergistic anti-cancer effect on PC3 and LNCaP cells and an additive effect on DU145 cells. Since the effect of blocking CXCR6-CXCL16 signaling before DTX treatment was synergistic for p53-null (PC3) and wild-type (LNCaP) PCa cells but additive for p53-mutated (DU145) cells, it appears that the effects of this combination treatment depended on the differential CXCR6 signaling supporting cell survival and apoptosis. Nonetheless, blocking CXCR6 has potential to improve the therapeutic efficacy of DTX in PCa.
In summary, our current findings show that the CXCR6-CXCL16 axis supports PCa cell survival and inhibits apoptosis and, that PCa cells reprogram their cellular machinery to overcome the response to DTX by activating CXCR6-CXCL16. Since this axis was not affected in non-cancerous immortalized prostate cells by DTX targeting of CXCR6 could reduce the effective DTX dose and therefore reduce toxicity. Our work explicitly points out the relevance of CXCR6-CXCL16 in therapeutic outcomes for PCa.
Supplementary Material
Highlights:
Prostate cancer cells overexpress CXCR6 and CXCL16 in response to DTX.
DTX induces ADAM-10 and increases CXCL16 cleavage in prostate cancer cells.
Prostate cancer cells overcome DTX effects by hyperactivating CXCL6-CXCL16 axis.
Prostate cancer cells modulate NF-kB, GSK-3β, ERK1/2 and survivin via CXCR6.
Blocking CXCR6-CXCL16 signaling improves DTX cytotoxicity in prostate cancer cells.
Acknowledgement
This study was supported in part by the funds (SC1 CA180212, UO1 CA179701, R21 CA169716 and U54 CA118638) from NCI and Morehouse School of Medicine flow cytometry core supported by the NIMHD 5U54MD007602. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Authors would also like to acknowledge Don Hill, scientific editor of U54 partnership for editing this manuscript.
Abbreviations:
- DTX
Docetaxel
- PCa
Prostate Cancer
- CI
Combination Index
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
No potential conflicts of interest to disclose.
References
- [1].Seruga B, Ocana A, Tannock IF, Drug resistance in metastatic castration-resistant prostate cancer, Nature reviews. Clinical oncology, 8 (2011) 12–23. [DOI] [PubMed] [Google Scholar]
- [2].Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA, T. Investigators, Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer, N Engl J Med, 351 (2004) 1502–1512. [DOI] [PubMed] [Google Scholar]
- [3].Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, Wong YN, Hahn N, Kohli M, Cooney MM, Dreicer R, Vogelzang NJ, Picus J, Shevrin D, Hussain M, Garcia JA, DiPaola RS, Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer, N Engl J Med, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dieterle A, Orth R, Daubrawa M, Grotemeier A, Alers S, Ullrich S, Lammers R, Wesselborg S, Stork B, The Akt inhibitor triciribine sensitizes prostate carcinoma cells to TRAIL-induced apoptosis, International journal of cancer, 125 (2009) 932–941. [DOI] [PubMed] [Google Scholar]
- [5].Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J, SRC family kinase activity is up-regulated in hormone-refractory prostate cancer, Clinical cancer research : an official journal of the American Association for Cancer Research, 15 (2009) 3540–3549. [DOI] [PubMed] [Google Scholar]
- [6].Fizazi K, The role of Src in prostate cancer, Annals of oncology : official journal of the European Society for Medical Oncology / ESMO, 18 (2007) 1765–1773. [DOI] [PubMed] [Google Scholar]
- [7].Chen X, Thakkar H, Tyan F, Gim S, Robinson H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N, Srivastava RK, Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer, Oncogene, 20 (2001) 6073–6083. [DOI] [PubMed] [Google Scholar]
- [8].Thakkar H, Chen X, Tyan F, Gim S, Robinson H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N, Srivastava RK, Pro-survival function of Akt/protein kinase B in prostate cancer cells. Relationship with TRAIL resistance, The Journal of biological chemistry, 276 (2001) 38361–38369. [DOI] [PubMed] [Google Scholar]
- [9].Cho MC, Choi HS, Lee S, Kim BY, Jung M, Park SN, Yoon DY, Epiregulin expression by Ets-1 and ERK signaling pathway in Ki-ras-transformed cells, Biochemical and biophysical research communications, 377 (2008) 832–837. [DOI] [PubMed] [Google Scholar]
- [10].Vaira V, Lee CW, Goel HL, Bosari S, Languino LR, Altieri DC, Regulation of survivin expression by IGF-1/mTOR signaling, Oncogene, 26 (2007) 2678–2684. [DOI] [PubMed] [Google Scholar]
- [11].Garcia GE, Nicole A, Bhaskaran S, Gupta A, Kyprianou N, Kumar AP, Akt-and CREB-mediated prostate cancer cell proliferation inhibition by Nexrutine, a Phellodendron amurense extract, Neoplasia (New York, N.Y.), 8 (2006) 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kumar AP, Bhaskaran S, Ganapathy M, Crosby K, Davis MD, Kochunov P, Schoolfield J, Yeh IT, Troyer DA, Ghosh R, Akt/cAMP-responsive element binding protein/cyclin D1 network: a novel target for prostate cancer inhibition in transgenic adenocarcinoma of mouse prostate model mediated by Nexrutine, a Phellodendron amurense bark extract, Clinical cancer research : an official journal of the American Association for Cancer Research, 13 (2007) 2784–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, Kitada S, Reed JC, Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers, The American journal of pathology, 148 (1996) 1567–1576. [PMC free article] [PubMed] [Google Scholar]
- [14].Hughes C, Murphy A, Martin C, Sheils O, O'Leary J, Molecular pathology of prostate cancer, Journal of clinical pathology, 58 (2005) 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Downward J, How BAD phosphorylation is good for survival, Nature cell biology, 1 (1999) E33–35. [DOI] [PubMed] [Google Scholar]
- [16].Kapur N, Mir H, Clark Iii CE, Krishnamurti U, Beech DJ, Lillard JW, Singh S, CCR6 expression in colon cancer is associated with advanced disease and supports epithelial-to-mesenchymal transition, British journal of cancer, 114 (2016) 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mir H, Singh R, Kloecker GH, Lillard JW Jr., Singh S, CXCR6 expression in non-small cell lung carcinoma supports metastatic process via modulating metalloproteinases, Oncotarget, 6 (2015) 9985–9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Singh R, Gupta P, Kloecker GH, Singh S, Lillard JW Jr., Expression and clinical significance of CXCR5/CXCL13 in human nonsmall cell lung carcinoma, International journal of oncology, 45 (2014) 2232–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Singh R, Stockard CR, Grizzle WE, Lillard JW Jr., Singh S, Expression and histopathological correlation of CCR9 and CCL25 in ovarian cancer, International journal of oncology, 39 (2011) 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Johnson-Holiday C, Singh R, Johnson E, Singh S, Stockard CR, Grizzle WE, Lillard JW Jr., CCL25 mediates migration, invasion and matrix metalloproteinase expression by breast cancer cells in a CCR9-dependent fashion, International journal of oncology, 38 (2011) 1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gerber PA, Hippe A, Buhren BA, Muller A, Homey B, Chemokines in tumor-associated angiogenesis, Biological chemistry, 390 (2009) 1213–1223. [DOI] [PubMed] [Google Scholar]
- [22].Chen W, Qin Y, Wang D, Zhou L, Liu Y, Chen S, Yin L, Xiao Y, Yao XH, Yang X, Ma W, Chen W, He X, Zhang L, Yang Q, Bian X, Shao ZM, Liu S, CCL20 triggered by chemotherapy hinders the therapeutic efficacy of breast cancer, PLoS biology, 16 (2018) e2005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chow MT, Luster AD, Chemokines in cancer, Cancer immunology research, 2(2014) 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hu W, Zhen X, Xiong B, Wang B, Zhang W, Zhou W, CXCR6 is expressed in human prostate cancer in vivo and is involved in the in vitro invasion of PC3 and LNCap cells, Cancer science, 99 (2008) 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Singh R, Kapur N, Mir H, Singh N, Lillard JW Jr., Singh S, CXCR6-CXCL16 axis promotes prostate cancer by mediating cytoskeleton rearrangement via Ezrin activation and alphavbeta3 integrin clustering, Oncotarget, 7 (2016) 7343–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee JT, Lee SD, Lee JZ, Chung MK, Ha HK, Expression analysis and clinical significance of CXCL16/CXCR6 in patients with bladder cancer, Oncology letters, 5 (2013) 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML, Demaria S, Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells, Journal of immunology (Baltimore, Md. : 1950), 181 (2008) 3099–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wente MN, Gaida MM, Mayer C, Michalski CW, Haag N, Giese T, Felix K, Bergmann F, Giese NA, Friess H, Expression and potential function of the CXC chemokine CXCL16 in pancreatic ductal adenocarcinoma, International journal of oncology, 33 (2008) 297–308. [PubMed] [Google Scholar]
- [29].Ou DL, Chen CL, Lin SB, Hsu CH, Lin LI, Chemokine receptor expression profiles in nasopharyngeal carcinoma and their association with metastasis and radiotherapy, The Journal of pathology, 210 (2006) 363–373. [DOI] [PubMed] [Google Scholar]
- [30].Gutwein P, Schramme A, Sinke N, Abdel-Bakky MS, Voss B, Obermuller N, Doberstein K, Koziolek M, Fritzsche F, Johannsen M, Jung K, Schaider H, Altevogt P, Ludwig A, Pfeilschifter J, Kristiansen G, Tumoural CXCL16 expression is a novel prognostic marker of longer survival times in renal cell cancer patients, European journal of cancer (Oxford, England : 1990), 45 (2009) 478–489. [DOI] [PubMed] [Google Scholar]
- [31].Luo Q, Lin H, Ye X, Huang J, Lu S, Xu L, Trim44 facilitates the migration and invasion of human lung cancer cells via the NF-kappaB signaling pathway, International journal of clinical oncology, 20 (2015) 508–517. [DOI] [PubMed] [Google Scholar]
- [32].Xiao G, Wang X, Wang J, Zu L, Cheng G, Hao M, Sun X, Xue Y, Lu J, Wang J, CXCL16/CXCR6 chemokine signaling mediates breast cancer progression by pERK1/2-dependent mechanisms, Oncotarget, 6 (2015) 14165–14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang J, Lu Y, Wang J, Koch AE, Zhang J, Taichman RS, CXCR6 induces prostate cancer progression by the AKT/mammalian target of rapamycin signaling pathway, Cancer research, 68 (2008) 10367–10376. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [34].Chandrasekar B, Bysani S, Mummidi S, CXCL16 signals via Gi, phosphatidylinositol 3-kinase, Akt, I kappa B kinase, and nuclear factor-kappa B and induces cell-cell adhesion and aortic smooth muscle cell proliferation, The Journal of biological chemistry, 279 (2004) 3188–3196. [DOI] [PubMed] [Google Scholar]
- [35].Voboril R, Hochwald SN, Li J, Brank A, Weberova J, Wessels F, Moldawer LL, Camp ER, MacKay SL, Inhibition of NF-kappa B augments sensitivity to 5-fluorouracil/folinic acid in colon cancer, The Journal of surgical research, 120 (2004) 178–188. [DOI] [PubMed] [Google Scholar]
- [36].Mosmann T, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, Journal of immunological methods, 65 (1983) 55–63. [DOI] [PubMed] [Google Scholar]
- [37].Ghosh R, Gilda JE, Gomes AV, The necessity of and strategies for improving confidence in the accuracy of western blots, Expert review of proteomics, 11 (2014) 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Janes KA, An analysis of critical factors for quantitative immunoblotting, Science signaling, 8 (2015) rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rocha-Martins M, Njaine B, Silveira MS, Avoiding pitfalls of internal controls: validation of reference genes for analysis by qRT-PCR and Western blot throughout rat retinal development, PloS one, 7 (2012) e43028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Carriere PP, Kapur N, Mir H, Ward AB, Singh S, Cinnamtannin B-1 inhibits cell survival molecules and induces apoptosis in colon cancer, International journal of oncology, 53 (2018) 1442–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, Hartmann D, Sedlacek R, Dietrich S, Muetze B, Schuster B, Kallen KJ, Saftig P, Rose-John S, Ludwig A, The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10, Journal of immunology (Baltimore, Md. : 1950), 172 (2004) 6362–6372. [DOI] [PubMed] [Google Scholar]
- [42].Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, Graham CH, Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide, Cancer research, 71 (2011) 7433–7441. [DOI] [PubMed] [Google Scholar]
- [43].van Uden P, Kenneth NS, Rocha S, Regulation of hypoxia-inducible factor-1alpha by NF-kappaB, The Biochemical journal, 412 (2008) 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ainbinder E, Revach M, Wolstein O, Moshonov S, Diamant N, Dikstein R, Mechanism of rapid transcriptional induction of tumor necrosis factor alpha-responsive genes by NF-kappaB, Molecular and cellular biology, 22 (2002) 6354–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH, ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma, Cancer research, 65 (2005) 4320–4333. [DOI] [PubMed] [Google Scholar]
- [46].Matsumoto Y, Takano H, Fojo T, Cellular adaptation to drug exposure: evolution of the drug-resistant phenotype, Cancer research, 57 (1997) 5086–5092. [PubMed] [Google Scholar]
- [47].McDermott M, Eustace AJ, Busschots S, Breen L, Crown J, Clynes M, O'Donovan N, Stordal B, In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies, Frontiers in oncology, 4 (2014) 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, Wong YN, Hahn N, Kohli M, Cooney MM, Dreicer R, Vogelzang NJ, Picus J, Shevrin D, Hussain M, Garcia JA, DiPaola RS, Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer, The New England journal of medicine, 373 (2015) 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Oudard S, Latorzeff I, Caty A, Miglianico L, Sevin E, Hardy-Bessard AC, Delva R, Rolland F, Mouret L, Priou F, Beuzeboc P, Gravis G, Linassier C, Gomez P, Voog E, Muracciole X, Abraham C, Banu E, Ferrero JM, Ravaud A, Krakowski I, Lagrange JL, Deplanque G, Zylberait D, Bozec L, Houede N, Culine S, Elaidi R, Effect of Adding Docetaxel to Androgen-Deprivation Therapy in Patients With High-Risk Prostate Cancer With Rising Prostate-Specific Antigen Levels After Primary Local Therapy: A Randomized Clinical Trial, JAMA oncology, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA, Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer, The New England journal of medicine, 351 (2004) 1502–1512. [DOI] [PubMed] [Google Scholar]
- [51].Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr., Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED, Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer, The New England journal of medicine, 351 (2004) 1513–1520. [DOI] [PubMed] [Google Scholar]
- [52].Qi WX, Fu S, Zhang Q, Guo XM, Efficacy and toxicity of molecular targeted therapies in combination with docetaxel for metastatic castration-resistant prostate cancer: a meta-analysis of phase III randomized controlled trials, Journal of chemotherapy (Florence, Italy), 27 (2015) 181–187. [DOI] [PubMed] [Google Scholar]
- [53].Fang Y, Henderson FC Jr., Yi Q, Lei Q, Li Y, Chen N, Chemokine CXCL16 expression suppresses migration and invasiveness and induces apoptosis in breast cancer cells, Mediators of inflammation, 2014 (2014) 478641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hikita A, Tanaka N, Yamane S, Ikeda Y, Furukawa H, Tohma S, Suzuki R, Tanaka S, Mitomi H, Fukui N, Involvement of a disintegrin and metalloproteinase 10 and 17 in shedding of tumor necrosis factor-alpha, Biochemistry and cell biology = Biochimie et biologie cellulaire, 87 (2009) 581–593. [DOI] [PubMed] [Google Scholar]
- [55].Lunn CA, Fan X, Dalie B, Miller K, Zavodny PJ, Narula SK, Lundell D, Purification of ADAM 10 from bovine spleen as a TNFalpha convertase, FEBS letters, 400 (1997) 333–335. [DOI] [PubMed] [Google Scholar]
- [56].Mezyk-Kopec R, Bzowska M, Stalinska K, Chelmicki T, Podkalicki M, Jucha J, Kowalczyk K, Mak P, Bereta J, Identification of ADAM10 as a major TNF sheddase in ADAM17-deficient fibroblasts, Cytokine, 46 (2009) 309–315. [DOI] [PubMed] [Google Scholar]
- [57].Tas F, Duranyildiz D, Argon A, Oguz H, Camlica H, Yasasever V, Topuz E, Serum levels of leptin and proinflammatory cytokines in advanced-stage nonsmall cell lung cancer, Medical oncology (Northwood, London, England), 22 (2005) 353–358. [DOI] [PubMed] [Google Scholar]
- [58].Derin D, Soydinc HO, Guney N, Tas F, Camlica H, Duranyildiz D, Yasasever V, Topuz E, Serum levels of apoptosis biomarkers, survivin and TNF-alpha in nonsmall cell lung cancer, Lung cancer (Amsterdam, Netherlands), 59 (2008) 240–245. [DOI] [PubMed] [Google Scholar]
- [59].Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R, Enkemann S, Jove R, Haura EB, Stat3 regulates genes common to both wound healing and cancer, Oncogene, 24 (2005) 3397–3408. [DOI] [PubMed] [Google Scholar]
- [60].Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR, Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB, Genes & development, 21 (2007) 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gorlach A, Bonello S, The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison, The Biochemical journal, 412 (2008) e17–19. [DOI] [PubMed] [Google Scholar]
- [62].Mhaidat NM, Thorne R, Zhang XD, Hersey P, Involvement of endoplasmic reticulum stress in Docetaxel-induced JNK-dependent apoptosis of human melanoma, Apoptosis : an international journal on programmed cell death, 13 (2008) 1505–1512. [DOI] [PubMed] [Google Scholar]
- [63].Kim S, Joe Y, Kim HJ, Kim YS, Jeong SO, Pae HO, Ryter SW, Surh YJ, Chung HT, Endoplasmic reticulum stress-induced IRE1alpha activation mediates cross-talk of GSK-3beta and XBP-1 to regulate inflammatory cytokine production, Journal of immunology (Baltimore, Md. : 1950), 194 (2015) 4498–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR, Oscillations in NF-kappaB signaling control the dynamics of gene expression, Science (New York, N.Y.), 306 (2004) 704–708. [DOI] [PubMed] [Google Scholar]
- [65].Lawrence T, Bebien M, Liu GY, Nizet V, Karin M, IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation, Nature, 434 (2005) 1138–1143. [DOI] [PubMed] [Google Scholar]
- [66].Wang D, Baldwin AS Jr., Activation of nuclear factor-kappaB-dependent transcription by tumor necrosis factor-alpha is mediated through phosphorylation of RelA/p65 on serine 529, The Journal of biological chemistry, 273 (1998) 29411–29416. [DOI] [PubMed] [Google Scholar]
- [67].Wang D, Westerheide SD, Hanson JL, Baldwin AS Jr., Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II, The Journal of biological chemistry, 275 (2000) 32592–32597. [DOI] [PubMed] [Google Scholar]
- [68].Bird TA, Schooley K, Dower SK, Hagen H, Virca GD, Activation of nuclear transcription factor NF-kappaB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit, The Journal of biological chemistry, 272 (1997) 32606–32612. [DOI] [PubMed] [Google Scholar]
- [69].Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W, IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain, The Journal of biological chemistry, 274 (1999) 30353–30356. [DOI] [PubMed] [Google Scholar]
- [70].Beg AA, Baltimore D, An essential role for NF-kappaB in preventing TNF-alpha-induced cell death, Science (New York, N.Y.), 274 (1996) 782–784. [DOI] [PubMed] [Google Scholar]
- [71].Wang CY, Mayo MW, Baldwin AS Jr., TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB, Science (New York, N.Y.), 274 (1996) 784–787. [DOI] [PubMed] [Google Scholar]
- [72].Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG, NF-kappaB and cell-cycle regulation: the cyclin connection, Cytokine & growth factor reviews, 12 (2001) 73–90. [DOI] [PubMed] [Google Scholar]
- [73].Huang DB, Chen YQ, Ruetsche M, Phelps CB, Ghosh G, X-ray crystal structure of proto-oncogene product c-Rel bound to the CD28 response element of IL-2, Structure (London, England : 1993), 9 (2001) 669–678. [DOI] [PubMed] [Google Scholar]
- [74].Godwin P, Baird AM, Heavey S, Barr MP, O'Byrne KJ, Gately K, Targeting nuclear factor-kappa B to overcome resistance to chemotherapy, Frontiers in oncology, 3 (2013) 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H, Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors, Cancer cell, 15 (2009) 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Pahl HL, Activators and target genes of Rel/NF-kappaB transcription factors, Oncogene, 18 (1999) 6853–6866. [DOI] [PubMed] [Google Scholar]
- [77].El-Rayes BF, Ali S, Ali IF, Philip PA, Abbruzzese J, Sarkar FH, Potentiation of the effect of erlotinib by genistein in pancreatic cancer: the role of Akt and nuclear factor-kappaB, Cancer research, 66 (2006) 10553–10559. [DOI] [PubMed] [Google Scholar]
- [78].Webster GA, Perkins ND, Transcriptional cross talk between NF-kappaB and p53, Molecular and cellular biology, 19 (1999) 3485–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bentires-Alj M, Barbu V, Fillet M, Chariot A, Relic B, Jacobs N, Gielen J, Merville MP, Bours V, NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells, Oncogene, 22 (2003) 90–97. [DOI] [PubMed] [Google Scholar]
- [80].Montagut C, Tusquets I, Ferrer B, Corominas JM, Bellosillo B, Campas C, Suarez M, Fabregat X, Campo E, Gascon P, Serrano S, Fernandez PL, Rovira A, Albanell J, Activation of nuclear factor-kappa B is linked to resistance to neoadjuvant chemotherapy in breast cancer patients, Endocrine-related cancer, 13 (2006) 607–616. [DOI] [PubMed] [Google Scholar]
- [81].McCubrey JA, Steelman LS, Franklin RA, Abrams SL, Chappell WH, Wong EW, Lehmann BD, Terrian DM, Basecke J, Stivala F, Libra M, Evangelisti C, Martelli AM, Targeting the RAF/MEK/ERK, PI3K/AKT and p53 pathways in hematopoietic drug resistance, Advances in enzyme regulation, 47 (2007) 64–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Balmanno K, Cook SJ, Tumour cell survival signalling by the ERK1/2 pathway, Cell death and differentiation, 16 (2009) 368–377. [DOI] [PubMed] [Google Scholar]
- [83].Katayama K, Yoshioka S, Tsukahara S, Mitsuhashi J, Sugimoto Y, Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein, Molecular cancer therapeutics, 6 (2007) 2092–2102. [DOI] [PubMed] [Google Scholar]
- [84].Kisucka J, Barancik M, Bohacova V, Breier A, Reversal effect of specific inhibitors of extracellular-signal regulated protein kinase pathway on P-glycoprotein mediated vincristine resistance of L1210 cells, General physiology and biophysics, 20 (2001) 439–444. [PubMed] [Google Scholar]
- [85].McCubrey JA, Abrams SL, Ligresti G, Misaghian N, Wong EW, Steelman LS, Basecke J, Troppmair J, Libra M, Nicoletti F, Molton S, McMahon M, Evangelisti C, Martelli AM, Involvement of p53 and Raf/MEK/ERK pathways in hematopoietic drug resistance, Leukemia, 22 (2008) 2080–2090. [DOI] [PubMed] [Google Scholar]
- [86].He X, Wang J, Wei W, Shi M, Xin B, Zhang T, Shen X, Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1alpha and contributes to chemoresistance in pancreatic cancer cells, Cancer biology & therapy, 17 (2016) 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Cai G, Wang J, Xin X, Ke Z, Luo J, Phosphorylation of glycogen synthase kinase-3 beta at serine 9 confers cisplatin resistance in ovarian cancer cells, International journal of oncology, 31 (2007) 657–662. [PubMed] [Google Scholar]
- [88].Frame S, Cohen P, Biondi RM, A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation, Molecular cell, 7 (2001) 1321–1327. [DOI] [PubMed] [Google Scholar]
- [89].Kim L, Kimmel AR, GSK3, a master switch regulating cell-fate specification and tumorigenesis, Current opinion in genetics & development, 10 (2000) 508–514. [DOI] [PubMed] [Google Scholar]
- [90].Ding VW, Chen RH, McCormick F, Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling, The Journal of biological chemistry, 275 (2000) 32475–32481. [DOI] [PubMed] [Google Scholar]
- [91].Mulholland DJ, Dedhar S, Wu H, Nelson CC, PTEN and GSK3beta: key regulators of progression to androgen-independent prostate cancer, Oncogene, 25 (2006) 329–337. [DOI] [PubMed] [Google Scholar]
- [92].Venkov CD, Link AJ, Jennings JL, Plieth D, Inoue T, Nagai K, Xu C, Dimitrova YN, Rauscher FJ, Neilson EG, A proximal activator of transcription in epithelial-mesenchymal transition, The Journal of clinical investigation, 117 (2007) 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Frame S, Cohen P, GSK3 takes centre stage more than 20 years after its discovery, The Biochemical journal, 359 (2001) 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Miller JR, Moon RT, Signal transduction through beta-catenin and specification of cell fate during embryogenesis, Genes & development, 10 (1996) 2527–2539. [DOI] [PubMed] [Google Scholar]
- [95].Peifer M, Polakis P, Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus, Science (New York, N.Y.), 287 (2000) 1606–1609. [DOI] [PubMed] [Google Scholar]
- [96].Ai Z, Yin L, Zhou X, Zhu Y, Zhu D, Yu Y, Feng Y, Inhibition of survivin reduces cell proliferation and induces apoptosis in human endometrial cancer, Cancer, 107 (2006) 746–756. [DOI] [PubMed] [Google Scholar]
- [97].Li J, Xing M, Zhu M, Wang X, Wang M, Zhou S, Li N, Wu R, Zhou M, Glycogen synthase kinase 3beta induces apoptosis in cancer cells through increase of survivin nuclear localization, Cancer letters, 272 (2008) 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lin J, Guan Z, Wang C, Feng L, Zheng Y, Caicedo E, Bearth E, Peng JR, Gaffney P, Ondrey FG, Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways, Clinical cancer research : an official journal of the American Association for Cancer Research, 16 (2010) 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Notarbartolo M, Cervello M, Dusonchet L, Cusimano A, D'Alessandro N, Resistance to diverse apoptotic triggers in multidrug resistant HL60 cells and its possible relationship to the expression of P-glycoprotein, Fas and of the novel anti-apoptosis factors IAP (inhibitory of apoptosis proteins), Cancer letters, 180 (2002) 91–101. [DOI] [PubMed] [Google Scholar]
- [100].Wang L, Zhang GM, Feng ZH, Down-regulation of survivin expression reversed multidrug resistance in adriamycin-resistant HL-60/ADR cell line, Acta pharmacologica Sinica, 24 (2003) 1235–1240. [PubMed] [Google Scholar]
- [101].Esteve PO, Chin HG, Pradhan S, Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells, The Journal of biological chemistry, 282 (2007) 2615–2625. [DOI] [PubMed] [Google Scholar]
- [102].IAppleyard MV, O'Neill MA, Murray KE, Paulin FE, Bray SE, Kernohan NM, Levison DA, Lane DP, Thompson AM, Seliciclib (CYC202, R-roscovitine) enhances the antitumor effect of doxorubicin in vivo in a breast cancer xenograft model, International journal of cancer, 124 (2009) 465–472. [DOI] [PubMed] [Google Scholar]
- [103].Liu F, Xie ZH, Cai GP, Jiang YY, The effect of survivin on multidrug resistance mediated by P-glycoprotein in MCF-7 and its adriamycin resistant cells, Biological & pharmaceutical bulletin, 30 (2007) 2279–2283. [DOI] [PubMed] [Google Scholar]
- [104].Lu J, Tan M, Huang WC, Li P, Guo H, Tseng LM, Su XH, Yang WT, Treekitkarnmongkol W, Andreeff M, Symmans F, Yu D, Mitotic deregulation by survivin in ErbB2-overexpressing breast cancer cells contributes to Taxol resistance, Clinical cancer research : an official journal of the American Association for Cancer Research, 15 (2009) 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lv YG, Yu F, Yao Q, Chen JH, Wang L, The role of survivin in diagnosis, prognosis and treatment of breast cancer, Journal of thoracic disease, 2 (2010) 100–110. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.