

Abstract

Visceral leishmaniasis (VL) is a parasitic infection that results in approximately 26 000–65 000 deaths annually. The available treatments are hampered by issues such as toxicity, variable efficacy, and unsuitable dosing options. The need for new treatments is urgent and led to a collaboration between the Drugs for Neglected Diseases initiative (DNDi), GlaxoSmithKline (GSK), and the University of Dundee. An 8-hydroxynaphthyridine was identified as a start point, and an early compound demonstrated weak efficacy in a mouse model of VL but was hampered by glucuronidation. Efforts to address this led to the development of compounds with improved in vitro profiles, but these were poorly tolerated in vivo. Investigation of the mode of action (MoA) demonstrated that activity was driven by sequestration of divalent metal cations, a mechanism which was likely to drive the poor tolerability. This highlights the importance of investigating MoA and pharmacokinetics at an early stage for phenotypically active series.
Introduction
According to the World Health Organization (WHO), neglected tropical diseases (NTDs) affect in excess of 1 billion people, the majority being in the most impoverished areas of the world.1 Of these NTDs, visceral leishmaniasis (VL) remains one of the most challenging to treat. VL is caused by infection with the protozoan parasites Leishmania donovani and Leishmania infantum, which are transmitted through the bite of female phlebotomine sand flies.2 Following infection, parasites reside predominantly in the liver, spleen, and bone marrow, and if left untreated, the disease is invariably fatal. Although it is challenging to precisely determine the number of people affected by VL, WHO estimates suggest that 50 000–90 000 new infections result in an annual death toll of between 26 000 and 65 000, with the vast majority of cases occurring in India, Bangladesh, Sudan, Ethiopia, and Brazil.3
Although there are a number of available treatments, all suffer from major issues that limit their use. Miltefosine, the only orally available treatment, is teratogenic so can only be prescribed to women of childbearing age alongside contraception, and also shows other side effects, as well as variable efficacy.4 Liposomal amphotericin B (Ambisome) is widely effective in Asia, but its use is limited by the need for intravenous (iv) administration which requires hospitalization, and it also requires a cold-chain for storage. An alternative aminoglycoside antibiotic paromomycin is much less expensive but demonstrates variable efficacy and requires a long course of painful intramuscular injections.5 Finally, pentavalent antimonials, such as sodium stibogluconate, have been widely used since the 1940s, but they are cardiotoxic, require parenteral administration, and are largely ineffective in large areas of India due to high levels of drug resistance. Although new compounds have recently entered the development pipeline,6 including two preclinical candidates developed within this collaboration (DDD853651/GSK3186899 and DDD1305143/GSK3494245) as well as the Novartis compound GNF6702, which have been disclosed in recent publications,7 there is still an urgent need for new treatments with improved safety profiles, more straightforward administration, lower costs, and also with alternate modes of action.
One of the major challenges for drug discovery for VL is the lack of robustly validated drug targets in Leishmania spp. For this reason, to identify suitable start points for drug discovery, compound libraries are screened directly against parasites in vitro, leading to phenotypically active compounds with unknown mechanisms of action. An additional confounding factor in attempts to identify chemical start points is the fact that in the human host, parasites are found within macrophages, where they reside within a parasitophorous vacuole. Therefore, relevant high-content screens, suitable for high-throughput screening (HTS), require culture of parasites in macrophages, typically differentiated THP-1 cells.8 For any compounds to be identified as hits in these screens, they would be required to have suitable physicochemical properties to cross a number of cell membranes, across various pH gradients, as well as having antiparasitic activity. As a result, hit rates are extremely low in these assays, typically below 0.1%.9 Nevertheless, compound series that are active in this assay and can be developed to give suitable properties for in vivo dosing have a high likelihood of success in rodent models of VL.
Using this intramacrophage assay, an HTS screen of a GlaxoSmithKline (GSK) collection of 1.8 M compounds was performed against L. donovani.10 The hits identified were screened in secondary antiparasitic assays, assessed for nonspecific cytotoxicity in HepG2 cells, clustered, and filtered based on favorable physicochemical properties, resulting in the identification of 33 chemical series and 75 singletons. One of the identified series was exemplified by 1 (Table 1), an 8-hydroxynaphthyridine, which was shown to have potency against the parasite (pEC50 = 6.5) with ∼100-fold selectivity over the human THP-1 cell line (pEC50 = 4.5). This compared favorably to the current treatments, amphotericin (pEC50 = 6.7) and miltefosine (pEC50 = 5.4). Upon further profiling, 1 was shown to have reasonable aqueous (aq) solubility (219 μM) but low stability in mouse liver microsomes (Cli = 18 mL/min/g). On the basis of this, 1 was selected as a suitable start point for a hit-to-lead program.
Table 1.

Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes. Data are the result of at least four independent replicates, and standard deviations are ≤0.4.
Cli is mouse liver microsomal intrinsic clearance.
Aq solubility is kinetic aqueous solubility.
ND means not determined.
The principal aim of this work was to identify analogues of 1 with a suitable profile for dosing in a mouse efficacy model of VL as rapidly as possible, to demonstrate that the series had the potential to progress into lead-optimization. Therefore, the initial chemistry program focused on understanding the structure–activity relationship (SAR) of the series, with an aim of identifying compounds with improved solubility and metabolic stability, as well as suitable potency for in vivo studies. Our targets were to achieve pEC50 > 5.8, aqueous solubility >200 μM, and mouse liver microsomal clearance of <5.0 mL/min/g, as these criteria had been used previously to identify chemical series likely to have in vivo efficacy.12
Results and Discussion
Lack of knowledge regarding the molecular target of compound 1 made optimization challenging, with no guide as to the potential pharmacophore, or which vectors were most likely to positively influence activity. We therefore focused on utilizing tractable chemistry that would facilitate a rapid exploration of SAR. Also, to maintain good solubility and hopefully improve metabolic stability, we aimed to reduce, or at least maintain, the Log D of the initial analogues. This led us to focus on the triazole substituent, as well as the 5-position of the naphthyridine as initial points for exploration.
SAR of 7-Triazolyl Analogues
Variations to the benzyl substituent of the triazole (Table 2), including substitutions on the phenyl position (exemplified by 2), or on the methylene (exemplified by 3), led to a ∼10-fold loss in potency compared to 1, although 3 did show an improvement in metabolic stability. A truncated analogue 4 was inactive, but we were encouraged by its improved solubility and metabolic stability. We thus replaced the 4-chlorophenyl group of 1 with more polar substituents, with the aim of regaining in vitro potency while maintaining a favorable absorption, distribution, metabolism, and excretion (ADME) profile. Morpholine-substituted 5 and pyrrolidinone-substituted 6 were synthesized and indeed proved to be both soluble and metabolically stable, although both compounds were essentially inactive. Switching to an amide as an isosteric replacement for the triazole was also investigated, and the matched pairs (7vs1) showed similar levels of potency, although the amide did not appear to show any clear advantage over the triazole as its mouse liver microsomal clearance was still not below the targeted 5 mL/min/g.
Table 2.
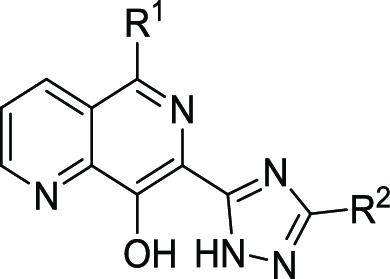
Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes. Data are the result of at least three independent replicates, and standard deviations are ≤0.4.
Cli is mouse liver microsomal intrinsic clearance.
Aq solubility is kinetic aqueous solubility.
ND means not determined.
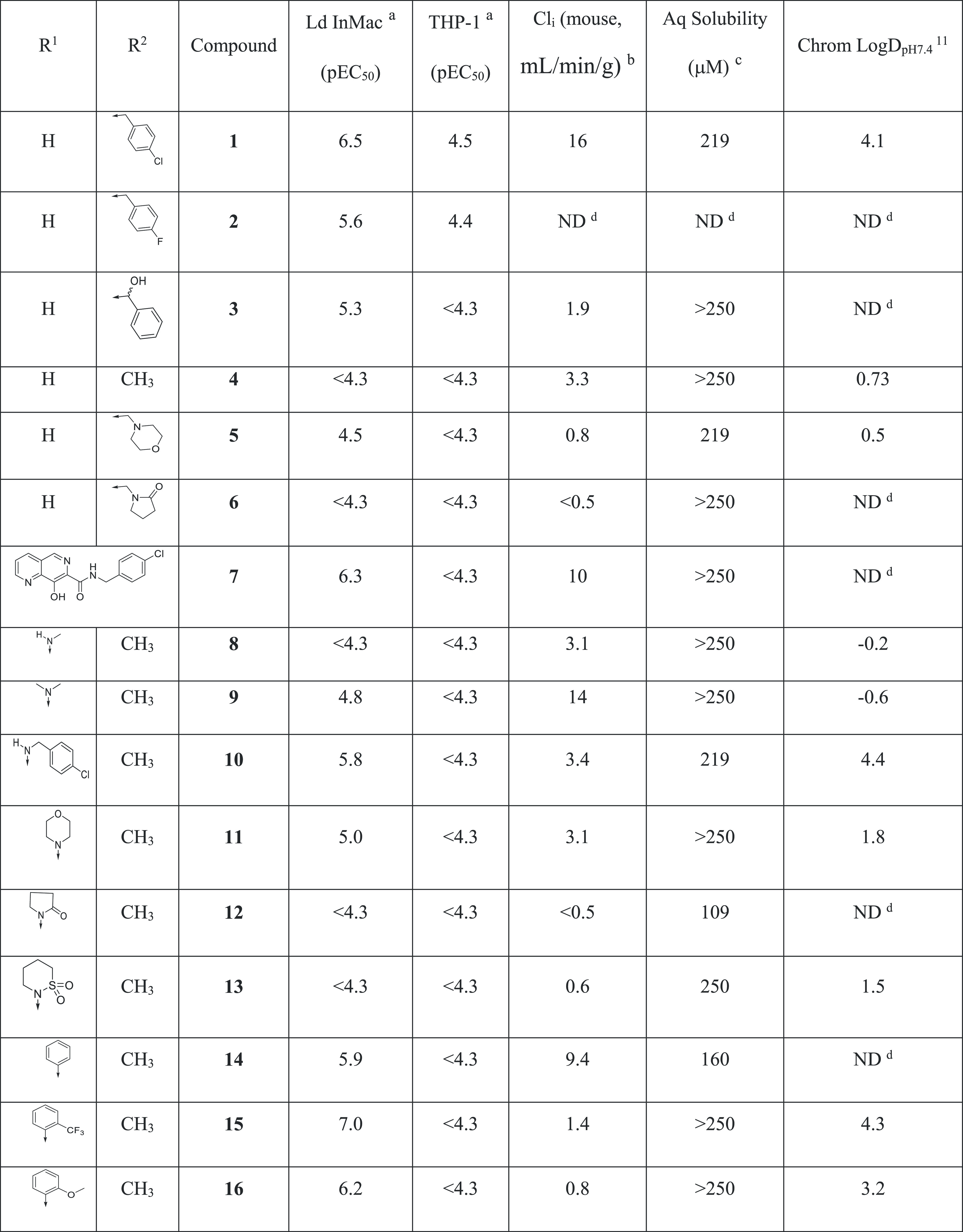
We next switched attention to the naphthyridyl 5-position. Initially, nitrogen-linked analogues were investigated. While both methylamine 8 and dimethylamine 9 were essentially inactive, larger amines such as p-chlorobenzylamine 10 and morpholine 11 both had pEC50 values above 5, with good solubility and low clearance. Cyclic amides, such as 12, proved to be inactive, as did sultam 13, presumably due to the reduced electron density in the aromatic ring. Compound 13 was of particular interest as 5-sultam-substituted naphthyridines had been previously reported in a series of integrase inhibitors and were shown to impart very good pharmacokinetics (PK) properties.13 Indeed, the lead compound from this series progressed as far as phase II clinical trials (compound 30; Table 5). Further exploration of N-linked analogues failed to deliver compounds with the necessary potency for progression to in vivo studies, so we switched our focus to carbon-linked analogues. Interestingly, the unsubstituted phenyl analogue 14 showed reasonable potency (pEC50 = 5.9), and further analogues showed ortho-substitution to be beneficial, with the o-trifluoromethyl analogue 15 giving a significant increase in antiparasitic activity (pEC50 = 7.0), possibly driven by the increased lipophilicity, and the o-methoxy analogue 16 showing a good balance of potency, stability, and solubility (pEC50 = 6.2, Cli = 1.4 mL/min/g and aqueous solubility >250 μM). Replacement of phenyl by aromatic heterocycles (such as pyridyl or pyrazolyl) was also explored, as was substitution on the other side of the naphthyridine (2-, 3-, and 4-positions), but these changes led to only weakly active compounds (data not shown).
Table 5.
Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes. Data are the result of at least three independent replicates, and standard deviations are ≤0.4.
Cli is mouse liver microsomal intrinsic clearance.
Aq solubility is kinetic aqueous solubility.
ND means not determined.
Profiling of Compound 16
Compounds 15 and 16 both showed a promising balance of potency, solubility, and metabolic stability. Because of having higher metabolic stability (in mouse liver microsomes) and lower Chrom Log D, compound 16 was progressed into a VL in vivo efficacy study, carried out in our previously described VL mouse model.7 Mice were dosed orally with the standard antileishmanial drug miltefosine, or with 16 dosed intraperitoneal (ip) two times daily for 5 days post infection (although 16 had a suitable profile for oral dosing, we elected to dose ip to maximize exposure and increase our chances of demonstrating in vivo proof of concept for the series). Parasite load was determined in the livers of animals 3 days after cessation of treatment, and parasite burden was expressed in Leishman Donovan units (LDUs, the mean number of amastigotes per liver cell × mg weight of liver). The blood exposure of compound 16 was also determined in dosed animals on days 1 and 5 to better understand the PK/pharmacodynamics (PD) relationship of the series. According to our project criteria, a compound needs to reduce parasite burden by >70% before being considered suitable for progression to lead-optimization, while a reduction of >95% would be considered suitable for a preclinical development candidate.12
In the study, miltefosine behaved as expected, reducing parasite levels by >99% at 30 mg/kg qd. After twice daily ip dosing at 50 mg/kg, compound 16 reduced parasite burden in mouse liver by 46%. This provided an early proof of concept for this series but fell short of our target of >70% parasite reduction. Upon examining the blood samples taken on days 1 and 5, it was clear that 16 was rapidly cleared from blood, with unbound concentrations of compound exceeding EC99 only during the first hour post-dose (Table 3). Further examination of the samples revealed the presence of glucuronidated adducts of 16, suggesting secondary metabolism as the key driver of the low exposure.
Table 3. Blood Levels of 16, Measured on Days 1 and 5 of a Mouse Efficacy Study, Dosing ip at 50 mg/kg b.i.d.
| AUC(0–t) (μM min) |
Cmax (μM) |
Tmax (h) |
|||
|---|---|---|---|---|---|
| day 1 | day 5 | day 1 | day 5 | day 1 | day 5 |
| 1236 | 606 | 25.2 | 4.2 | 0.5 | 0.5 |
Glucuronidation is a means of increasing water solubility of small molecules, facilitating their elimination from the body in urine. It involves transfer of the glucuronic acid component of uridine diphosphate glucuronic acid to a suitable substrate, catalyzed by UDP-glucuronosyltransferase (UGT), and occurs mainly in the liver.14 Glucuronidation occurs at nucleophilic sites such as R–OH, R–NH2, or R–COOH, which can be present in the small molecule, or generated via phase I metabolism. Due to the low rates of microsomal clearance of 16, alongside our in vivo data, we surmised that glucuronidation was occurring on the parent compound, most likely at the phenolic OH or the triazole N–H. Also, the observed reduction of parasite load, despite the high in vivo clearance, suggested that reducing glucuronidation to increase the duration of exposure above EC99 would be a key strategy to progress the series toward lead-optimization.
As a means of measuring glucuronidation in vitro, 16 was assessed in a mouse liver hepatocyte assay. Unfortunately, 16 showed similar stability to that seen in mouse liver microsomes (0.9 mL/min/g in microsomes vs 1.4 mL/min/g in hepatocytes) with negligible amounts of the glucuronide adduct being observed. This suggested that there was little involvement of the hepatic UDP-glucuronosyltransferase in the in vivo phase II metabolism. This result made series progression challenging, as there was no way to determine the potential impact of glucuronidation without running an in vivo study, limiting our understanding of the SAR surrounding the observed secondary metabolism.
One possible strategy to identify compounds with improved metabolic stability would be to reduce lipophilicity. This had been shown previously to be a potential strategy for reducing glucuronidation;15 however, as shown in Figure 1, this was very challenging within this compound series. Looking at measured log D (Chrom Log D7.4),11 analogues with Chrom Log D7.4 values below 3 were generally only weakly active, with pEC50’s above 5.8 only being achieved where Chrom Log D7.4 was greater than 3. In our experience, this is a common problem when trying to optimize series phenotypically, where increasing potency without increasing lipophilicity is very challenging; this highlights a key advantage of running structure-enabled programs. Because of this, alternative approaches to improving metabolic stability were required.
Figure 1.
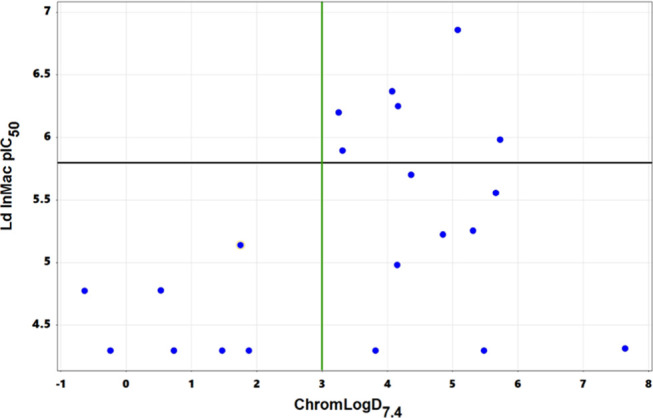
Chrom Log D7.4vs intramacrophage potency (Ld InMac pEC50) for all compounds presented. The green line represents Chrom Log D7.4 = 3; the black line represents Ld InMac pEC50 = 5.8.
SAR of the Naphthyridine Core
Our initial approach to reduce potential phase II metabolism within the series was therefore to investigate the SAR around the phenolic hydroxyl group and the triazole. Understanding which of these features was important for antiparasitic activity might enable us to synthesize potent analogues with lower potential for glucuronidation. As shown in Table 4, removal or methylation of the naphthyridine 8-hydroxyl group led to a loss of antiparasitic activity compared to 16 (17 and 18, respectively), although 18 was close to the targeted pEC50 of 5.8. Also, replacement of triazole with oxadiazole to remove the nucleophilic N–H led to a loss of activity (19). Synthesis of other analogues to explore the naphthyridine SAR proved extremely challenging within the triazole subseries, so to more rapidly address this, we switched attention to the bioisosteric replacement of triazole with amide; this change had previously been seen to have limited effect on potency (e.g., 1vs7) and allowed much more straightforward synthesis of the analogues of interest. Thus, deletion of the naphthyridine N-6 of 7 led to a compound that was toxic to the host THP-1 cells, and deletion of naphthyridine N-1 led to loss of antiparasitic activity (20 and 21, respectively). Moving to a scaffold that trapped the phenolic OH as a carbonyl removed all antiparasitic activity despite the parent amides having pEC50 values >6.0 (comparing 22 to 28 and 23 to 27). Finally, we examined Raltegravir (24), an inhibitor of human immunodeficiency virus (HIV) integrase marketed as a treatment for HIV.16 As shown in Figure 2, 24 contains the key acceptor–donor–acceptor binding motif identified within the naphthyridine series. Although 24 was inactive in our in vitro Leishmania assays, we surmised that transferring the known SAR of the naphthyridine core onto the Raltegravir scaffold could be a viable strategy to regain activity. Unfortunately, as exemplified by 25, none of the analogues based on this scaffold were active. From this round of synthesis, we concluded that we were unlikely to identify active compounds without the acceptor–donor–acceptor binding motif of the 7-substituted-1,6-naphthyridin-8-ol core.
Table 4.
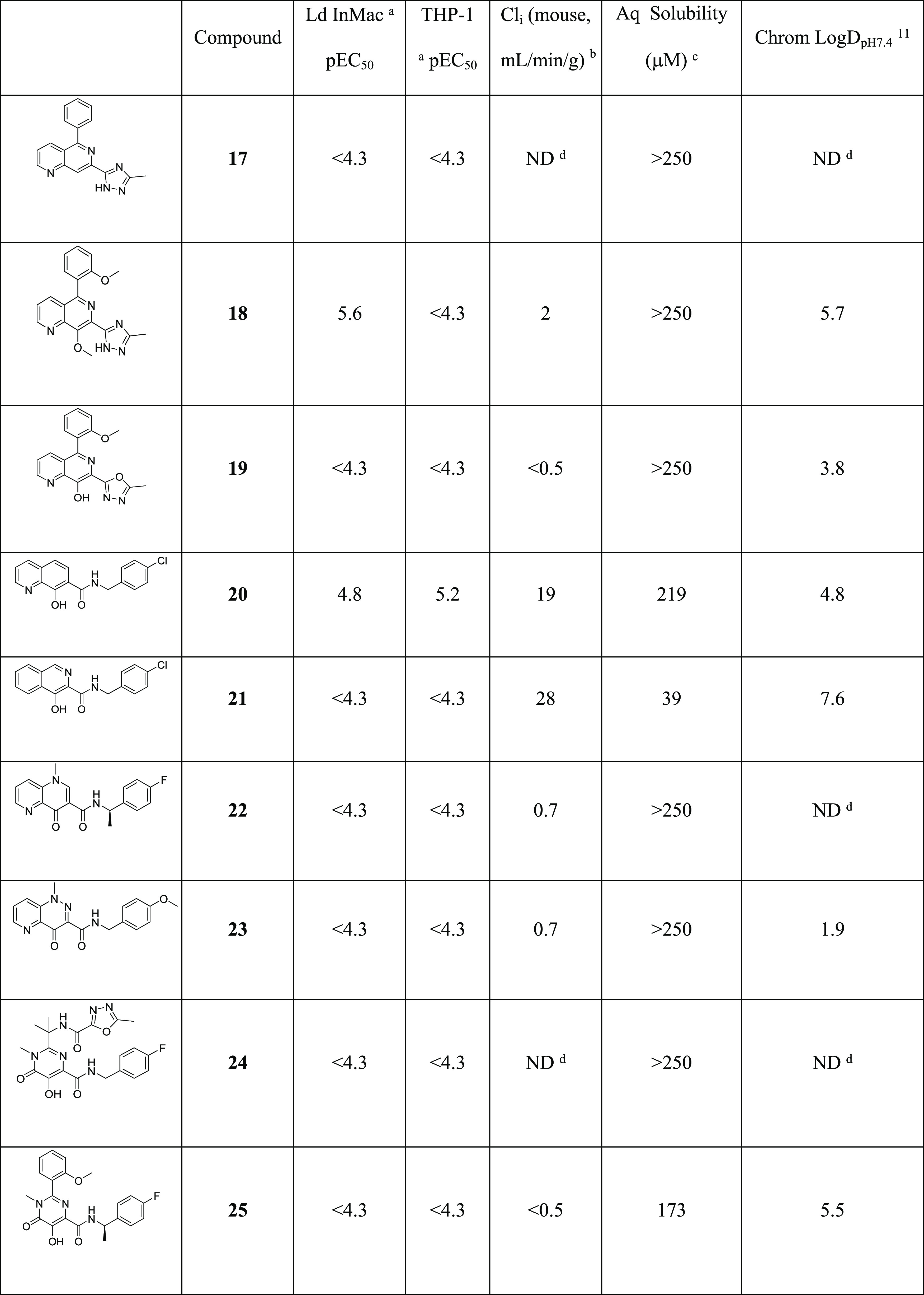
Ld InMac is the intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes. Data are the result of at least three independent replicates, and standard deviations are ≤0.4.
Cli is mouse liver microsomal intrinsic clearance.
Aq solubility is kinetic aqueous solubility.
ND means not determined.
Figure 2.

Highlighting the acceptor–donor–acceptor motifs of triazole analogue 1, amide analogue 7, and Raltegravir 24.
SAR of 7-Carboxamide Analogues
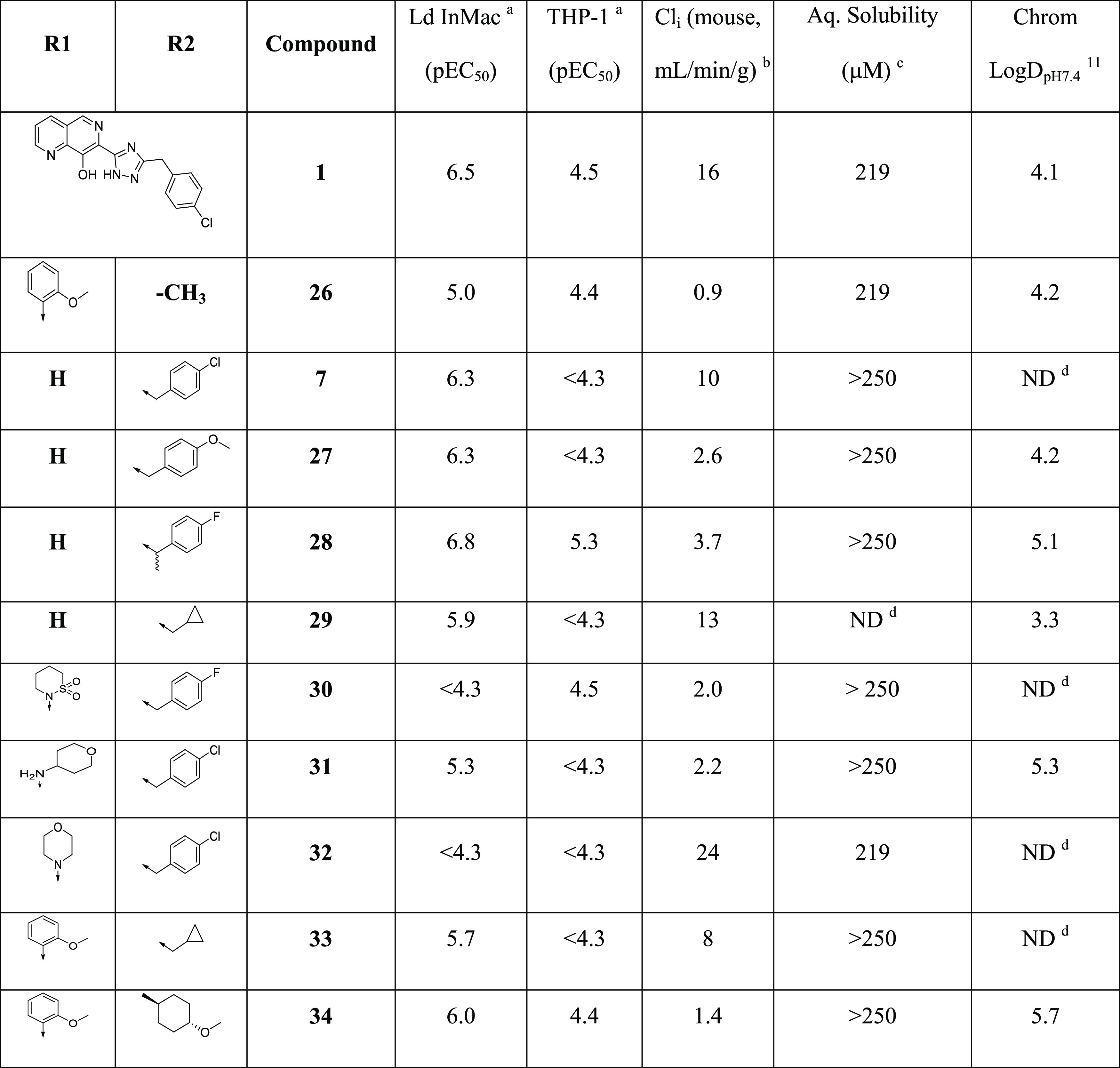
Previous SAR demonstrated that replacing the triazole with an amide led to compounds such as 7 that retained antiparasitic activity. Since 7 itself was metabolically unstable (Cli = 10 mg/mL/g), we became interested in transferring the SAR from the triazole subseries (e.g., 16) to investigate its impact on metabolic stability, with a particular focus on glucuronidation. To this end, we synthesized 26 (Table 5) as a direct analogue of triazole 16. Although it did not show sufficient potency for progression into efficacy studies, it had reasonable aqueous solubility (219 μM) and good metabolic stability (Cli = 0.9 mL/min/g) and was therefore progressed into a mouse PK study to assess the extent of in vivo glucuronidation. After dosing (50 mg/kg ip) and analyzing the metabolites generated (Figure 3), there was little evidence of glucuronidation, with the major metabolism observed being hydroxylation. As significant quantities of parent were still present 8 h post-dose, this supported a strategy of switching to the amide series to reduce phase II metabolism and improve in vivo exposure.
Figure 3.
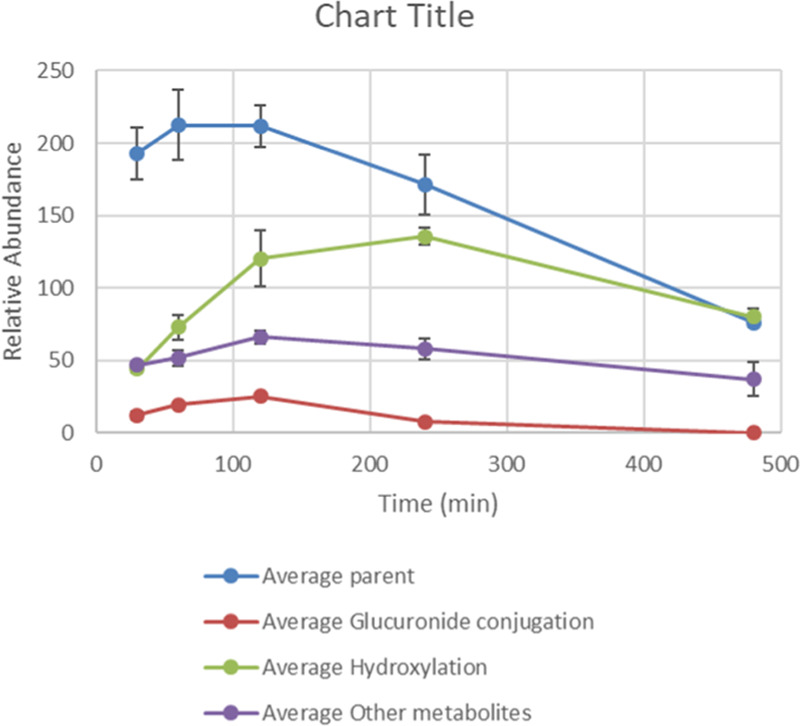
PK profile of 26, showing levels of parent, glucuronide adduct, major hydroxylated metabolite, and other minor metabolites. Data are based on two replicates, with error bars representing the range of the data from the two runs. Blood/water (30 μL, 1:2) were collected following 50 mg/kg ip administration to Balb C mice at prescribed time points; 90 μL of acetonitrile (ACN) was then added to each sample and the samples were centrifuged for 5 min at 7000 rpm; 90 μL of supernatant was removed and added to 50 μL of Milli-Q water before ultrahigh-performance liquid chromatography–quadrupole time-of-flight (UPLC Q-TOF) analysis.
With this in mind, we further explored the SAR of the amide subseries, as shown in Table 5. We synthesized a set of benzyl amides, where 4-methoxy analogue 27 and α-methyl-4-fluoro analogue 28 both gave very encouraging profiles, meeting progression criteria in terms of potency, metabolic stability, and clearance. Alternatively, nonaromatic amides were explored, and although none were identified with suitable profiles for progression, cyclopropylmethyl analogue 29 did demonstrate reasonable potency. Due to its impressive in vitro potency, we selected 28 for progression into an in vivo PK study. However, upon dosing (50 mg/kg ip), the compound proved to be toxic, rapidly giving symptoms (within 3.5 h) requiring termination of the experiment.
We noted that a related compound from Merck, L-870,810 (30),17 was reported as a clinical candidate targeting HIV integrase, which progressed as far as phase II clinical trials. Compound 30 was inactive in our in vitro efficacy assays, but the report, alongside the in vivo data for 26, suggested that compounds with substitutions in the naphthyridine 5-position could have suitable profiles for in vivo studies. Also, introducing substituents into the 5-naphthyridyl position had been a successful strategy for improving metabolic stability and potency in the triazole subseries. We therefore investigated a range of 5-substituted naphthyridyl analogues (Table 5). THP-amine 31 and morpholine 32 lost potency compared to parent compound 7, so we switched attention back to 5-phenylnaphthyridines. Previously identified groups were combined (the 4-methoxyphenyl of 16 and the cyclopropylmethyl amide of 29), leading to 33, which unfortunately did not deliver the expected increase in potency. Further combinations of nonaromatic amides with differently substituted 5-phenylnaphthyridines were synthesized, and while changing the phenyl substituent gave flat SAR and no advantage over previous compounds, exploration of the amide led to 34 with a trans-4-methoxycyclopropylamide. Compound 34 gave a good balance of potency, solubility, and microsomal stability and was therefore selected for a mouse PK study. Disappointingly, when dosed at 50 mg/kg ip, the mice again displayed the similar symptoms as with compound 28, and the study was terminated after 60 min.
Profiling of 16, 28, and 34
Due to the encouraging results within the series (16 showing low-level efficacy and 26 giving a good PK profile), we were keen to understand the origins of the observed toxicity of 28 and 34. As previously mentioned, compound 30 progressed as far as phase II clinical trials as an HIV integrase inhibitor, and although it was inactive against VL, we were keen to assess whether there was scope to progress our related series further. To this end, two studies were conducted in parallel; a screen against receptors with known links to toxicity and investigation of mode of action (MoA).
First, the compounds which were poorly tolerated in mice, 28 and 34, were screened against a panel of >30 receptors with known links to in vivo toxicity (GSK-enhanced cross-screen panel (eXP)).18 Compound 28 gave a pIC50 value of 5.4 against monoamine oxidase A, highlighting a slight risk of drug–drug interactions and possible side effects, and a pIC50 value of 4.9 in a phenotypic cell health assay, suggesting possible effects on mitochondrial integrity that could lead to an increased risk of hepatotoxicity. Compound 34 also showed potency in the cell health assay, alongside activity in a bile salt export pump (BSEP) assay (pIC50 value of 4.8, hepatotoxicity risk) and a phospholipidosis assay. From this, it was not clear whether the effects seen in the receptor screen were related to the observed in vivo toxicity.
Alongside these screens, mode-of-action studies were initiated focusing on 10, 16, and 28, as representatives of both the triazole and amide subseries. We were particularly interested in confirming that the compounds inhibited a shared target, identifying off-target effects, and understanding the source of the observed toxicity. The results of these studies have been reported previously and demonstrated that the compounds act as nonspecific chelators of divalent metal cations, in particular Zn2+, Fe2+, and Cu2+, and that this property is likely responsible for their antiparasitic activity.19 Indeed, the propensity of these compounds to nonspecifically chelate divalent cations may well explain the observed in vivo toxicity associated with this series.
Chemistry
To access the required analogues, a number of different approaches were needed, as illustrated in scheme 1–3. Compounds 1 and 2 were previously reported,20 and synthesis of the remaining 7-triazolyl-8-hydroxy naphthyridines started from 7-cyanonaphthyridine 35(20) (Scheme 1), where cyclization with substituted hydrazides under acidic conditions led to compounds 3–6. To access 5-substituted analogues, 35 could be brominated with N-bromosuccinimide (NBS) to give 36 and subsequently cyclized to give 37, which was treated with a suitable amine to give 8–11. For 12 and 13, protection of the phenol proved to be necessary. Hence, 36 could be tosyl-protected to give 38, followed by either Buchwald–Hartwig coupling to introduce the pryrrolidinone (12) or copper coupling with 1,2-thiazinane 1,1-dioxide to introduce the sultam (13). Alternatively, 36 could be directly coupled with phenylboronic acid to give 14, or benzyl-protected to give 39, which could be coupled to give 15 and 16.
Scheme 1.
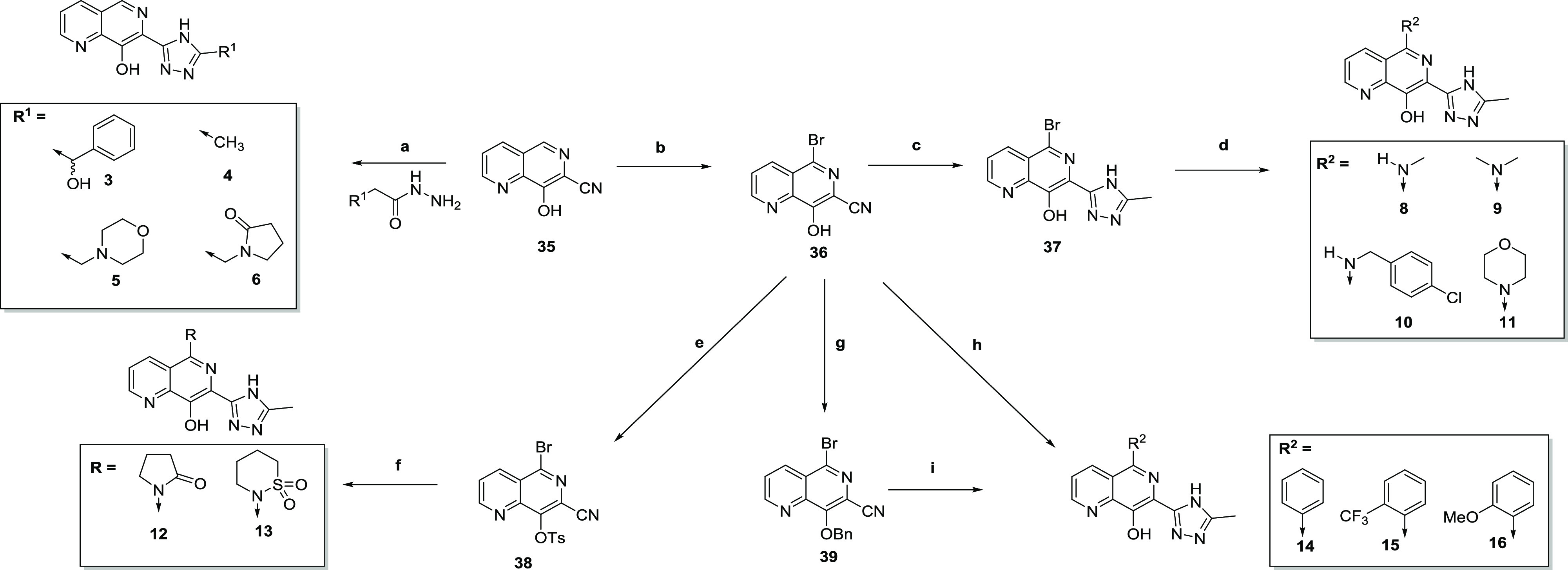
Scheme 3.

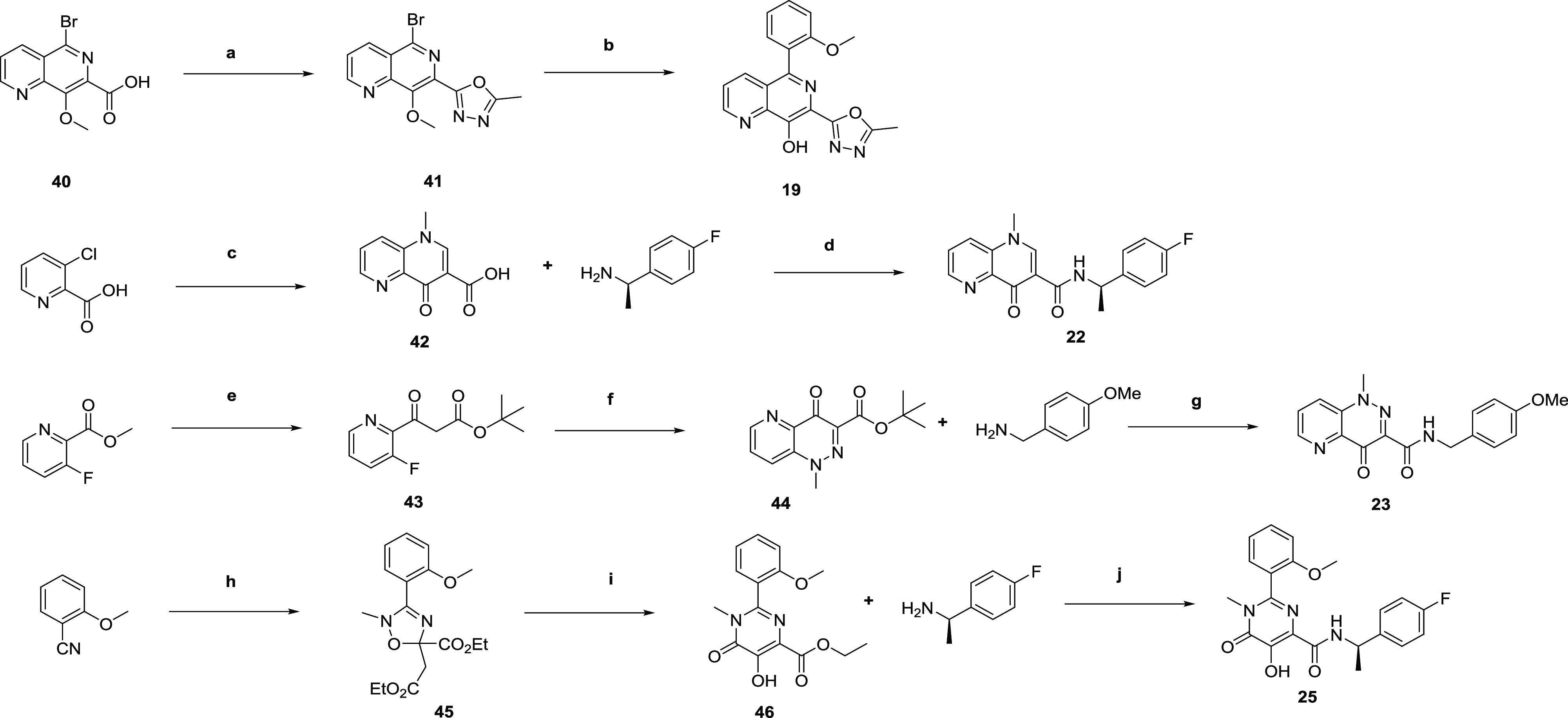
To fully explore the SAR around the naphthyridine ring, and also to introduce alternative heterocycles to replace the triazole, a number of analogues required bespoke synthesis. Analogues 17, 18, 20, and 21 were synthesized according to established procedures and are described in the Supporting Information, with the synthesis of 19, 22, 23, and 25 highlighted in Scheme 2. Thus, 5-bromo-8-methoxy-1,6-naphthyridine-7-carboxylic acid 40 was cyclized with acetylhydrazide to give 41, followed by Suzuki coupling and deprotection of the 8-methoxy group to give oxadiazole 19. Compound 22 was synthesized from 3-chloropicolinic acid via conversion to the acid chloride, condensation with ethyl 3-(dimethylamino)acrylate, cyclization with methylamine, and ester hydrolysis to give 42. This was then coupled with 4-fluoro-α-(R)-methylbenzylamine to give 22. To access 23, methyl 3-fluoropicolinate was condensed with tert-butyl acetate to give 43, which was treated with 4-acetamidobenzenesulfonyl azide (ABSA), PBu3, then 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)/iodomethane to give cyclized 44, which was subsequently hydrolyzed and coupled with 4-methoxybenzylamine to give 23. Finally, 25 was synthesized according to a previously published route such that 2-cyanoanisole was treated with N-methylhydroxylamine and cyclized to 45. Thermal rearrangement gave 46, which was treated with 4-fluoro-α-(R)-methylbenzylamine to give 25.21
Scheme 2.
7-Carboxamide analogues were synthesized from the corresponding ester 47 according to Scheme 3. Amides 7 and 27–29 were synthesized directly from the ester by treating with the relevant amine at high temperature. To access the desired 6-functionalized analogues, 47 was brominated with NBS to give 48, tosyl-protected to give 49, and converted to sultam 30.22 Alternatively, 48 could be treated with 4-chlorobenzylamine to give 51a, then coupled with another amine to give 31 and 32. Compound 49 could also be coupled with 2-methoxyphenylboronic acid and hydrolyzed to give 50, which was coupled with trans-4-methoxycyclohexylamine to give 34. Alternatively, 48 could be treated directly with a relevant amine to give 51b and 51c, with subsequent Suzuki coupling with 2-methoxyphenylboronic acid giving 26 and 33.
Conclusions
To identify new compound series with the potential to be developed as new therapeutics for VL, a collection of 1.8 M compounds from the GSK corporate collection was screened against L. donovaniin vitro. One hit series identified from this, exemplified by 1, was selected for a hit-to-lead program. In vivo studies of an early compound, 16, demonstrated that the series had the potential to reduce parasite burden, but that glucuronidation was a potential barrier to series progression. Scaffold hopping from the core triazole to an amide was a key strategy for progressing the series, leading to 28 with a very good in vitro profile. Dosing of 28 identified an issue with toxicity for the series and further chemistry failed to identify compounds that did not carry this liability. MoA studies suggested that the antiparasitic activity, and the toxicity, was likely driven by chelation of divalent metal cations. Based on these findings, we concluded that attempting to develop compounds within this series that would separate antiparasitic activity from inherent toxicity would be extremely challenging and unlikely to succeed. With this in mind, work on the series was halted. This demonstrates the importance of understanding the mode of action from a very early stage in the drug discovery process, when working to progress phenotypically active hit compounds.
Experimental Section
Chemistry
Chemicals and solvents were purchased from Aldrich Chemical Company, Fluka, ABCR, VWR, Acros Organics, Fluorochem, and Alfa Aesar and were used as received. Air- and moisture-sensitive reactions were carried out under an inert atmosphere of argon in oven-dried glassware. Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (layer 0.20 mm silica gel 60 with fluorescent indicator UV254, from Merck). Developed plates were air-dried and analyzed under a UV lamp (UV254/365 nm). Flash column chromatography was performed using prepacked silica gel cartridges (230–400 mesh, 40–63 μm, from SiliCycle) using a Teledyne ISCO CombiFlash Companion, or CombiFlash Retrieve. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance DPX 500 spectrometer (1H at 500.1 MHz, 13C at 125.8 MHz). Chemical shifts (δ) are expressed in parts per million (ppm) recorded using the residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad (b), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hz. Liquid chromatography–mass spectrometry (LC–MS) analyses were performed with either an Agilent HPLC 1100 series connected to a Bruker Daltonics MicrOTOF or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole LC/MS, where both instruments were connected to an Agilent diode array detector. Mobile phase was water/acetonitrile + 0.1% HCOOH, or water/acetonitrile + 0.1% NH3; linear gradient, 80:20–5:95 over 3.5 min and then held for 1.5 min; flow rate, 0.5 mL/min. All intermediates had a measured purity ≥90%, and all assay compounds had a measured purity of ≥95%, as determined using this analytical LC–MS system (total ion current (TIC) and UV). High-resolution electrospray measurements were performed on a Bruker Daltonics MicrOTOF mass spectrometer. Microwave-assisted chemistry was performed using a Biotage Initiator Microwave Synthesizer.
7-(3-(Hydroxy(phenyl)methyl)-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-8-ol (3)
35 (100 mg, 0.58 mmol) and 2-hydroxy-2-phenylacetohydrazide (288 mg, 1.74 mmol) in 1,4-dioxane (1.5 mL)/acetic acid (0.5 mL) were heated in microwave at 140 °C for 2 h. The mixture was cooled to RT, and the resulting solid was collected, washed with 1,4-dioxane, and dried under vacuum to give 3 (43 mg, 0.13 mmol, 22%). 1H NMR (dimethyl sulfoxide (DMSO)-d6): δ 14.90 (s, 1H), 12.29 (bs, 1H), 9.15 (s, 1H), 9.02 (s, 1H), 8.63–8.56 (m, 1H), 7.82–7.74 (m, 1H), 7.56–7.50 (m, 2H), 7.42–7.24 (m, 3H), 6.23 (bs, 1H), 5.91 (bs, 1H); m/z [M + H]+ calcd for C17H14N5O2, 320.1147; found, 320.1146.
7-(3-Methyl-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-8-ol (4)
35 (30 mg, 0.16 mmol) and acetylhydrazide (58.4 mg, 0.79 mmol) in 1,4-dioxane (2 mL)/acetic acid (0.2 mL) were heated in microwave at 200 °C for 15 min. The mixture was cooled, solvent-evaporated, and the crude material was purified by mass-directed prep. HPLC to give 4 (10 mg, 0.04 mmol, 27%). 1H NMR (DMSO-d6): δ 14.74 (s, 1H), 12.45 (s, 1H), 9.18–9.15 (m, 1H), 9.04 (s, 1H), 8.63–8.58 (m, 1H), 7.83–7.76 (m, 1H), 2.45 (s, 3H); 13C NMR (DMSO-d6): δ 158.8, 158.5, 154.5, 149.5, 143.9, 141.9, 136.7, 125.6, 124.8, 12.8; m/z [M + H]+ calcd for C11H10N5O, 228.0885; found, 228.0886.
7-(3-(Morpholinomethyl)-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-8-ol (5)
35 (150 mg, 0.88 mmol) and 2-(4-morpholinyl)acetohydrazide (419 mg, 2.63 mmol) in 1,4-dioxane (3 mL)/acetic acid (0.3 mL) were heated in microwave at 180 °C for 2 h. The resulting solution was evaporated, cyclohexane (3 × 10 mL) was added, and evaporated. Acetone (15 mL) was added, and the resulting solid was collected by filtration, washed with MeOH, and dried to give 5 (105 mg, 0.34 mmol, 38%). 1H NMR (DMSO-d6): δ 14.83 (s, 1H), 9.18–9.16 (m, 1H), 9.04 (s, 1H), 8.63–8.60 (m, 1H), 8.15 (s, 1H), 7.82–7.78 (m, 1H), 3.73 (s, 2H), 3.63–3.58 (m, 4H), 2.60–2.54 (m, 4H, under solvent peak); m/z [M + H]+ calcd for C15H17N6O2, 313.1413; found, 313.1411.
1-((5-(8-Hydroxy-1,6-naphthyridin-7-yl)-1H-1,2,4-triazol-3-yl)methyl)pyrrolidin-2-one (6)
6 was synthesized by an analogous method to 3, from 35 (100 mg, 0.58 mmol) and 2-(2-oxopyrrolidin-1-yl)acetylhydrazide (273 mg, 1.74 mmol). The crude material was purified by mass-directed prep. HPLC to give 6 (70 mg, 0.15 mmol, 26%). 1H NMR (DMSO-d6): δ 9.20–9.17 (m, 1H), 9.06 (s, 1H), 8.65–8.61 (m, 1H), 7.83–7.80 (m, 1H), 4.61 (s, 2H), 3.47–3.44 (m, 2H), 2.34–2.29 (m, 2H), 2.04–1.95 (m, 2H); m/z [M + H]+ calcd for C15H15N6O2, 311.1256; found, 311.1260.
N-(4-Chlorobenzyl)-8-hydroxy-1,6-naphthyridine-7-carboxamide (7)
A mixture of 47 (51 mg, 0.25 mmol) and 4-chlorobenzylamine (701 mg, 0.5 mmol) in EtOH (4 mL) was stirred at 80 °C for 18 h. The hot reaction mixture was poured into a solution of acetic acid (0.5 mL) in water (4 mL). After stirring for 10 min, cold water (15 mL) was added and stirred at RT for 40 min. The resulting solid was collected, washed with water, and dried to give 7 (42 mg, 0.13 mmol, 53%). 1H NMR (DMSO-d6): δ 13.67 (s, 1H), 9.18–9.16 (m, 1H), 8.93 (bs, 1H), 8.61 (d, J = 7.7 Hz, 1H), 7.86–7.82 (m, 1H), 7.44–7.39 (m, 4H), 4.55 (d, J = 6.5 Hz, 2H). 13C NMR (DMSO-d6): δ 170.1, 154.6, 142.6, 138.4, 136.5, 132.0, 129.9, 128.8, 127.0, 126.0, 125.4, 42.1; m/z [M + H]+ calcd for C16H13N3O2Cl, 314.0696; found, 314.0695.
7-(3-Methyl-1H-1,2,4-triazol-5-yl)-5-(methylamino)-1,6-naphthyridin-8-ol (8)
A mixture of 37 (100 mg, 0.33 mmol), methylamine (327 μL, 0.653 mmol, 2 N in THF), and DIPEA (0.171 mL, 0.980 mmol) in NMP (2 mL) was stirred at 180 °C for 18 h in a sealed tube. Further, methylamine (327 μL, 0.653 mmol, 2 N in THF) was added and stirring was continued for further 18 h. The solvent was evaporated, DCM (3 mL) was added, and the resulting solid was collected and purified by mass-directed prep. HPLC to give 8 (21 mg, 0.82 mmol, 25%). 1H NMR (DMSO-d6): δ 14.06 (s, 1H), 10.97 (s, 1H), 8.94–8.91 (m, 1H), 8.54 (d, J = 7.9 Hz, 1H), 7.57–7.51 (m, 1H), 7.29 (bs, 1H), 2.97 (d, J = 3.8 Hz, 3H), 2.33 (s, 3H); m/z 257.2 [M + H]+.
5-(Dimethylamino)-7-(3-methyl-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-8-ol (9)
A mixture of 37 (100 mg, 0.33 mmol), dimethylamine (0.327 mL, 0.653 mmol, 2 M in THF), and DIPEA (171 μL, 0.98 mmol) in NMP (2 mL) was stirred at 180 °C for 18 h in a sealed tube. Further dimethylamine (0.327 mL, 0.653 mmol, 2 M in THF) was added and stirring was continued for a further 18 h. The solvent was evaporated, and the crude residue was purified by flash chromatography (0–50% EtOAC/EtOH (3:1)/cyclohexanes) to give 9 (34 mg, 0.13 mmol, 38%). 1H NMR (DMSO-d6): δ 14.20 (s, 1H), 11.47 (bs, 1H), 8.99–8.96 (m, 1H), 8.46 (d, J = 8.2 Hz, 1H), 7.61–7.56 (m, 1H), 2.93 (s, 6H), 2.35 (s, 3H); m/z [M + H]+ calcd for C13H15N6O, 271.1307; found, 271.1309.
5-((4-Chlorobenzyl)amino)-7-(3-methyl-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-8-ol (10)
10 was synthesized by an analogous method to 8 from 37 (50 mg, 0.15 mmol), 4-chlorobenzylamine (42 mg, 0.29 mmol), and DIPEA (57 mg, 0.44 mmol) to give 10 (30 mg, 0.074 mmol, 50%). 1H NMR (DMSO-d6): δ 14.18 (bs, 1H), 11.12 (bs, 1H), 9.04–9.01 (m, 1H), 8.75–8.72 (m, 1H), 7.90–7.85 (m, 1H), 7.67–7.62 (m, 1H), 7.54–7.50 (m, 2H), 7.37–7.33 (m, 2H), 4.89 (s, 2H), 2.43 (s, 3H); 13H NMR (DMSO-d6): δ 158.8, 154.9, 153.5, 149.3, 144.5, 141.2, 140.6, 132.4, 131.4, 130.1, 128.4, 122.7, 121.2, 116.1, 43.4, 14.0; m/z [M + H]+ calcd for C18H16N6OCl, 367.1074; found, 367.1088.
7-(3-Methyl-1H-1,2,4-triazol-5-yl)-5-morpholino-1,6-naphthyridin-8-ol (11)
A mixture of 37 (50 mg, 0.16 mmol), DIPEA (90 μL, 0.49 mmol), and morpholine (28 μL, 0.32 mmol) in NMP (0.5 mL) was heated in microwave to 200 °C for 1 h. After cooling, the crude mixture was purified by prep. HPLC to give 11 (31 mg, 0.073 mmol, 46%) as the TFA salt. 1H NMR (DMSO-d6): δ 9.13 (dd, J = 1.6, 4.2 Hz, 1H), 8.59 (dd, J = 1.6, 8.5 Hz, 1H), 7.77–7.73 (m, 1H), 3.90–3.85 (m, 4H), 3.34–3.30 (m, 4H), 2.52 (s, 3H, under DMSO signal); m/z 313.2 [M + H]+.
1-(8-Hydroxy-7-(3-methyl-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-5-yl)pyrrolidin-2-one (12)
To a solution of pyrrolidin-2-one (0.068 mL, 0.89 mmol) in 1,4-dioxane (7 mL) was added 38 (300 mg, 0.74 mmol), caesium carbonate (484 mg, 1.48 mmol), palladium acetate (3 mg, 0.015 mmol), and Xantphos (17 mg, 0.03 mmol), and the mixture was heated at 65 °C for 18 h. Water (50 mL)/HCl (2 M, two drops) was added, extracted with DCM (2 × 50 mL), and the combined organics were dried over Na2SO4, filtered, and solvent-evaporated. The crude residue (120 mg, 0.30 mmol) in DMF (0.3 mL) was added to a solution of sodium methoxide (1.5 mL, 0.74 mmol, 0.5 M in MeOH) and heated to 50 °C for 5 min, cooled to room temperature, and stirred for 15 min. Acetic acid (0.033 mL, 0.59 mmol) and water (3 mL) were added dropwise, and the resulting suspension was stirred for 2 h, poured into water (20 mL), and the pH adjusted to approximately 5 by addition of HCl (1 M, 0.1 mL). DCM (20 mL) was added, and the organic layer was dried over Na2SO4, filtered, and concentrated to afford crude 8-hydroxy-5-(2-oxopyrrolidin-1-yl)-1,6-naphthyridine-7-carbonitrile (57 mg, 0.22 mmol, 25% over two steps). To this crude intermediate in a mixture of acetic acid (0.1 mL) and 1,4-dioxane (1 mL) was added acetic hydrazide (50 mg, 0.67 mmol), and the mixture was heated in microwave to 180 °C for 2 h. Further, acetic hydrazide (50 mg, 0.67 mmol) was added and heated in microwave to 180 °C for 2 h. The solvent was evaporated, and the crude material was purified by prep. HPLC to give 12 (5 mg, 0.016 mmol, 7%). 1H NMR (DMSO-d6): δ 14.48 (bs, 1H), 12.25 (bs, 1H), 9.18–9.15 (m, 1H), 8.39–8.35 (m, 1H), 7.78–7.73 (m, 1H), 4.15–4.08 (m, 2H), 2.66–2.60 (m, 2H), 2.49 (s, 3H, under solvent peak), 2.32–2.23 (m, 2H); m/z 311.1 [M + H]+.
2-(8-Hydroxy-7-(3-methyl-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-5-yl)-1,2-thiazinane 1,1-dioxide (13)
To a solution of 38 (300 mg, 0.74 mmol) in 1,4-dioxane (7 mL) were added 1,2-thiazinane 1,1-dioxide (120 mg, 0.89 mmol), caesium carbonate (484 mg, 1.48 mmol), Pd2(dba)3 (13 mg, 0.015 mmol), and Xantphos (17 mg, 0.03 mmol), and the resulting mixture was stirred at 65 °C for 1 h. The mixture was poured into water (50 mL)/HCl (2 M, two drops) and extracted with DCM (2 × 50 mL). The combined organics were dried over Na2SO4, filtered, and solvent-evaporated. The crude mixture in DMF (0.6 mL) was added to a solution of sodium methoxide (2.7 mL, 1.36 mmol, 0.5 M in MeOH), and the resulting solution heated to 50 °C for 5 min, cooled to room temperature, and stirred 15 min. An acetic acid (0.06 mL, 1.09 mmol)/water (5.5 mL) mixture was added dropwise, and the resulting suspension was stirred for 2 h, poured into water (20 mL), and the pH was adjusted to approximately 5 by addition of HCl (1 M, 0.1 mL). DCM (20 mL) was added, and the organic layer was dried over Na2SO4, filtered, and concentrated to afford crude 5-(1,1-dioxido-1,2-thiazinan-2-yl)-8-hydroxy-1,6-naphthyridine-7-carbonitrile (190 mg, 0.53 mmol, 71% over two steps). To this crude material in a mixture of acetic acid (0.1 mL) and 1,4-dioxane (1 mL) was added acetic hydrazide (139 mg, 1.83 mmol), and the mixture was stirred in microwave at 180 °C for 2 h, concentrated to dryness, and purified by mass-directed prep. HPLC to give 13 (35 mg, 0.1 mmol, 16%). 1H NMR (DMSO-d6): δ 14.45 (bs, 1H), 12.41 (bs, 1H), 9.09–9.06 (m, 1H), 8.51–8.45 (m, 1H), 7.78–7.70 (m, 1H), 4.02–3.97 (m, 1H), 3.89–3.81 (m, 1H), 3.70–3.61 (m, 2H), 3.38–3.31 (m, 2H), 3.09–3.06 (m, 1H), 2.23 (s, 3H), 1.57–1.48 (m, 1H); m/z [M + H]+ calcd for C15H17N6O3S, 361.1083; found, 361.1079.
7-(3-Methyl-1H-1,2,4-triazol-5-yl)-5-phenyl-1,6-naphthyridin-8-ol (14)
36 (75 mg, 0.39 mmol), 4,4,5,5-tetramethyl-2-phenyl-1,3,2-dioxaborolane (122 mg, 0.60 mmol), potassium phosphate tribasic (191 mg, 0.90 mmol), and Pd(dppf)Cl2·CH2Cl2 (12 mg, 0.015 mmol) in DMF (1.5 mL)/water (0.5 mL) were heated in microwave at 130 °C for 1 h. EtOAc (10 mL) and 20% aqueous NaCl (10 mL) were added, and the organic layer was separated, washed with brine, and solvent-evaporated to give crude intermediate. To this crude material in 1,4-dioxane (2 mL)/acetic acid (0.2 mL) was added acetylhydrazide (667 mg, 0.90 mmol) and heated in microwave to 200 °C for 45 min. The resulting mixture was filtered to remove solid, solvent-evaporated, and purified by mass-directed prep. HPLC to give 14 (3 mg, 0.009 mmol, 3%). 1H NMR (DMSO-d6): δ 13.50 (bs, 1H), 9.15 (s, 1H), 8.37 (d, J = 8.3 Hz, 1H), 7.76–7.66 (m, 3H), 7.63–7.52 (m, 4H), 2.48 (s, 3H, under DMSO signal); m/z 304.1 [M + H]+.
7-(3-Methyl-1H-1,2,4-triazol-5-yl)-5-(2-(trifluoromethyl)phenyl)-1,6-naphthyridin-8-ol (15)
A mixture of 39 (250 mg, 0.74 mmol), 2-(trifluoromethyl)benzeneboronic acid (279 mg, 1.47 mmol), sodium carbonate (234 mg, 2.21 mmol), and tetrakis(triphenylphosphine)palladium(0) (42 mg, 0.037 mmol) in water (3 mL)/1,4-dioxane (9 mL), was heated in microwave at 120 °C for 1 h. The mixture was partitioned between DCM (15 mL) and sat. NH4Cl (30 mL), the phases were separated, and the aqueous layer was further extracted with DCM (3 × 10 mL). The combined organics were dried over Na2SO4, filtered, and concentrated, and the resulting solid was triturated with ether to give a mixture of 8-(benzyloxy)-5-(2-(trifluoromethyl)phenyl)-1,6-naphthyridine-7-carbonitrile and 8-hydroxy-5-(2-(trifluoromethyl)phenyl)-1,6-naphthyridine-7-carbonitrile (0.150 g). The crude mixture was taken up in acetic acid (0.1 mL)/1,4-dioxane (1 mL), acetic hydrazide (46 mg, 0.62 mmol) was added, and heated in microwave at 180 °C for 8 h. Further, acetic hydrazide (0.031 g, 0.416 mmol) was added and heating was continued for 17 h. The solvent was evaporated, and the crude mixture was dissolved in 5% MeOH/DCM (20 mL), washed with water (1 × 50 mL), and the aqueous phase was extracted with further DCM (1 × 30 mL). The combined organics were washed with water, dried over Na2SO4, filtered, and concentrated to give a brown oil, which was precipitated with a mixture of ether and acetone. The resulting solid was further triturated with ether to give a brown pale solid, which was dried under vacuum to give 15 (60 mg, 0.16 mmol, 22% over two steps). 1H NMR (CDCl3): δ 9.23–9.20 (m, 1H), 7.96–7.89 (m, 2H), 7.77–7.69 (m, 2H), 7.57–7.47 (m, 2H), 2.59 (s, 3H); m/z 372.1 [M + H]+.
5-(2-Methoxyphenyl)-7-(3-methyl-1H-1,2,4-triazol-5-yl)-1,6-naphthyridin-8-ol (16)
A mixture of 39 (700 mg, 2.06 mmol), 2-methoxyphenylboronic acid (626 mg, 4.12 mmol), sodium carbonate (655 mg, 6.18 mmol), and tetrakis(triphenylphosphine)palladium(0) (120 mg, 0.103 mmol) in 1,4-dioxane (9 mL)/water (3 mL) was heated at 120 °C in microwave for 1.5 h, poured into water (15 mL)/brine (5 mL), and extracted with EtOAc (20 mL). The organics were dried over MgSO4, filtered, and concentrated. The crude was dissolved in acetic acid (0.5 mL)/1,4-dioxane (5 mL), acetic hydrazide (0.23 g, 3.10 mmol) was added, and stirred at 180 °C in microwave for 2 h. Further, acetic hydrazide (0.23 g, 3.10 mmol) was added and the mixture was heated in microwave at 180 °C for 6 h. The resulting solution was concentrated and taken up in 9:1 DCM/MeOH (15 mL), washed with water (10 mL), brine (5 mL), dried over Na2SO4, filtered, and concentrated. The resulting solid was triturated with ether (10 mL) and acetonitrile (10 mL) to give 16 (165 mg, 0.50 mmol, 24%). 1H NMR (MeOD): δ 9.09–9.05 (m, 1H), 8.09–8.05 (m, 1H), 7.68–7.63 (m, 1H), 7.58–7.53 (m, 1H), 7.50–7.46 (m, 1H), 7.22–7.13 (m, 2H), 3.71 (s, 3H), 3.32 (s, 3H); m/z [M + H]+ calcd for C18H16N5O2, 334.1304; found, 334.1294.
5-(2-Methoxyphenyl)-7-(5-methyl-1,3,4-oxadiazol-2-yl)-1,6-naphthyridin-8-ol (19)
41 (80 mg, 0.25 mmol), 2-methoxyphenylboronic acid (76 mg, 0.50 mmol), sodium carbonate (79 mg, 0.747 mmol), and tetrakis(triphenylphosphine)palladium(0) (14 mg, 0.012 mmol) in a mixture of water (1 mL)/1,4-dioxane (3 mL) were stirred at 120 °C overnight. The reaction mixture was concentrated to dryness, and the crude residue was purified by flash chromatography (0–70% EtOH/EtOAc (1:3)/cyclohexane) to give 2-(8-methoxy-5-(2-methoxyphenyl)-1,6-naphthyridin-7-yl)-5-methyl-1,3,4-oxadiazole (86 mg, 0.25 mmol). 1H NMR (DMSO-d6): δ 9.16–9.14 (m, 1H), 7.95–7.92 (m, 1H), 7.69–7.65 (m, 1H), 7.51–7.46 (m, 1H), 7.32–7.29 (m, 1H), 7.18–7.15 (m, 1H), 7.08–7.04 (m, 1H), 4.19 (s, 3H), 3.58 (s, 3H), 2.53 (s, 3H). To a solution of 2-(8-methoxy-5-(2-methoxyphenyl)-1,6-naphthyridin-7-yl)-5-methyl-1,3,4-oxadiazole (68 mg, 0.195 mmol) in CH3CN (2 mL) were added trimethylchlorosilane (37 μL, 0.293 mmol) and sodium iodide (44 mg, 0.293 mmol) and heated to reflux overnight. The crude residue was poured into water (30 mL) and extracted with DCM (50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The crude residue was triturated with MeOH (5 mL), and the resulting solid was collected by filtration to give 19 (29 mg, 0.087 mmol, 44%). 1H NMR (DMSO-d6): δ 9.19 (dd, J = 1.5, 4.0 Hz, 1H), 7.99 (dd, J = 1.5, 8.6 Hz, 1H), 7.77 (dd, J = 4.2, 8.5 Hz, 1H), 7.58–7.53 (m, 1H), 7.40–7.37 (m, 1H), 7.23 (d, J = 8.1 Hz, 1H), 7.15 (dd, J = 7.3, 7.3 Hz, 1H), 3.66 (s, 3H), 2.62 (s, 3H). 13C NMR (DMSO-d6): δ 164.1, 163.6, 157.2, 153.9, 150.1, 149.6, 141.9, 136.7, 131.7, 131.1, 126.9, 125.1, 124.5, 122.9, 121.2, 111.9, 55.8, 11.1; m/z [M + H]+ calcd for C18H15N4O3, 335.1144; found, 335.1155.
(R)-N-(1-(4-Fluorophenyl)ethyl)-1-methyl-4-oxo-1,4-dihydro-1,5-naphthyridine-3-carboxamide (22)
To 42 (100 mg, 0.47 mmol) in DMF (2 mL) were added 4-fluoro-α-(R)-methylbenzylamine (71 mg, 0.51 mmol) and DIPEA (180 mg, 1.40 mmol). Propylphosphonic anhydride (444 mg, 1.4 mmol, 50% solution in EtOAc) was added dropwise, and the mixture was stirred for 2 h, poured into sat NaHCO3 (10 mL), and extracted into EtOAc (3 × 10 mL). The combined organics were washed with brine, dried, evaporated, and the crude material was purified by column chromatography (0–20% MeOH/DCM) to give 22 (13 mg, 0.38 mmol, 8%). 1H NMR (DMSO-d6): δ 10.49 (d, J = 7.8 Hz, 1H), 8.88–8.85 (m, 2H), 8.35–8.31 (m, 1H), 7.89–8.85 (m, 1H), 7.46–7.40 (m, 2H), 7.21–7.15 (m, 2H), 5.24–5.15 (m, 1H), 4.02 (s, 3H), 1.51 (d, J = 6.9 Hz, 3H); m/z 326.1 [M + H]+.
N-(4-Methoxybenzyl)-1-methyl-4-oxo-1,4-dihydropyrido[3,2-c]pyridazine-3-carboxamide (23)
To crude 43 (8 g, 33.4 mmol) in MeCN (130 mL) at 0 °C were added triethylamine (6 g, 60 mmol) and 4-acetamidobenzenesulfonyl azide (8.8 g, 36.8 mmol), allowed to warm to RT, and stirred for 18 h. The crude mixture was filtered to remove solid, and the filtrate was concentrated and purified by column chromatography (pet. ether/EtOAc, 3:1) to give tert-butyl 2-diazo-3-(3-fluoropyridin-2-yl)-3-oxopropanoate (8.1 g, 30.5 mmol, 91%). This material was dissolved in THF (80 mL), tributylphosphine (6.854 g, 33.6 mmol) was added, stirred for 15 min, and quenched by addition of water (5.5 mL). The solvent was evaporated, and the residue was purified by flash chromatography (pet. ether/EtOAc 3:1) to give tert-butyl-3-(3-fluoropyridin-2-yl)-2-hydrazono-3-oxopropanoate (9 g, quant.). This material was dissolved in MeCN (180 mL), and DBU (10.7 g, 10.7 mmol) and methyl iodide (5 g, 35.3 mmol) were added and stirred at RT for 18 h. The mixture was quenched with sat. aq NH4Cl, extracted into DCM, and the organics were concentrated and purified by flash chromatography (1% MeOH in DCM) to give crude 44, which was used without further purification (2.8 g, 10.7 mmol, 32%); m/z 206.0 [M + H – tBu]+. To a solution of 44 (2.8 g, 10.7 mmol) in DCM (100 mL) was added TFA (15.7 g, 139 mmol) and stirred for 18 h. Ether was added and evaporated and further ether added. The resulting solid was collected and dried to give crude 1-methyl-4-oxo-1,4-dihydropyrido[3,2-c]pyridazine-3-carboxylic acid (1.6 g, 73%), which was used without purification.
A mixture of the crude material (300 mg, 1.46 mmol), HOBt (237 mg, 1.75 mmol), EDCI (420 mg, 2.19 mmol), and DIPEA (378 mg, 2.92 mmol) in DCM (12 mL) was stirred for 30 min, a solution of (4-methoxyphenyl)methanamine (241 mg, 1.75 mmol) in DCM (12 mL) added, and stirred for 16 h. The mixture was concentrated and purified by prep. HPLC to give 23 (419 mg, 1.29 mmol, 88%). 1H NMR (DMSO-d6): δ 9.87–9.84 (m, 1H), 8.93 (d, J = 4.2 Hz, 1H), 8.43 (d, J = 8.8 Hz, 1H), 7.96–7.94 (m, 1H), 7.31 (d, J = 8.1 Hz, 2H), 6.91 (d, J = 8.2 Hz, 2H), 4.50 (d, J = 5.8 Hz, 2H), 4.24 (s, 3H), 3.75 (s, 3H); 13C NMR (DMSO-d6): δ 169.3, 162.0, 158.8, 150.1, 141.3, 140.3, 139.0, 131.4, 129.2, 128.9, 127.0, 114.3, 55.5, 44.9, 42.3; m/z [M + H]+ calcd for C17H17N4O3, 325.1295; found, 325.1305.
(R)-N-(Fluoro(4-methoxyphenyl)methyl)-5-hydroxy-2-(2-methoxyphenyl)-1-methyl-6-oxo-1,6-dihydropyrimidine-4-carboxamide (25)
To a suspension of 46 (240 mg, 0.79 mmol) in toluene (5 mL), (R)-4-fluoro-α-methylbenzylamine (549 mg, 3.94 mmol) was added. The resulting solution was heated to reflux overnight. The solvent was evaporated, and the residue was purified by prep. HPLC to give 25 (145 mg, 46% yield). 1H NMR (DMSO-d6): δ 12.55–12.48 (m, 1H), 9.07–9.02 (m, 1H), 7.56–7.50 (m, 1H), 7.47–7.40 (m, 3H), 7.20–7.09 (m, 4H), 5.17 (dd, J = 8.0, 15.3 Hz, 1H), 3.81 (d, J = 7.3 Hz, 3H), 3.17 (d, J = 3.0 Hz, 3H), 1.50 (dd, J = 2.7, 6.9 Hz, 3H); m/z 398.2 [M + H]+.
8-Hydroxy-5-(2-methoxyphenyl)-N-methyl-1,6-naphthyridine-7-carboxamide (26)
To a mixture of 51a (72 mg, 0.25 mmol), (2-methoxyphenyl)boronic acid (77 mg, 0.51 mmol), and potassium phosphate tribasic (162 mg, 0.77 mmol) in 1,4-dioxane (2 mL) was added Pd(dppf)Cl2.DCM (12 mg, 0.015 mmol) in water (0.5 mL) and heated in microwave at 140 °C for 1 h. After cooling, EtOAc (9 mL) and 20% aqueous NaCl (10 mL) were added and the organics were separated, dried over MgSO4, evaporated, and the crude material was purified by column chromatography (0–8% MeOH/DCM) to give 26 (25 mg, 0.077 mmol, 30%). 1H NMR (DMSO-d6): δ 9.17–9.15 (m, 1H), 9.02–8.98 (m, 1H), 7.95–7.92 (m, 1H), 7.75–7.72 (m, 1H), 7.58–7.54 (m, 1H), 7.44–7.42 (m, 1H), 7.25–7.22 (m, 1H), 7.18–7.15 (m, 1H), 3.68 (s, 3H), 2.91 (d, J = 4.9 Hz, 3H). 13C NMR (DMSO-d6): δ 170.3, 157.2, 154.1, 148.6, 142.5, 136.4, 132.0, 131.0, 127.1, 125.9, 125.1, 121.2, 111.8, 55.8, 26.3; m/z [M + H]+ calcd for C17H16N3O3, 310.1192; found, 310.1196.
8-Hydroxy-N-(4-methoxybenzyl)-1,6-naphthyridine-7-carboxamide (27)
A mixture of 47 (100 mg, 0.50 mmol) and 4-methoxybenzylamine (134 mg, 0.98 mmol) in EtOH (20 mL) was heated to reflux overnight. To the hot mixture was added water (30 mL) and acetic acid (2 mL), cooled to RT, solvent-evaporated, and the crude material was purified by mass-directed prep. HPLC to give 27 (84 mg, 0.27 mmol, 54%). 1H NMR (DMSO-d6): δ 13.69 (bs, 1H), 9.83 (bs, 1H), 9.19–9.14 (m, 1H), 8.93–8.85 (m, 1H), 8.60 (d, J = 8.3 Hz, 1H), 7.83–7.79 (m, 1H), 7.35–7.32 (m, 2H), 6.92–6.89 (m, 2H), 4.49 (d, J = 6.4 Hz, 2H), 3.75 (s, 3H). 13C NMR (DMSO-d6): δ 169.8, 158.8, 154.9, 154.4, 142.5, 136.5, 131.3, 129.5, 127.0, 126.1, 125.4, 114.2, 55.5, 42.1; m/z [M + H]+ calcd for C17H16N3O3, 310.1192; found, 310.1192.
N-(1-(4-Fluorophenyl)ethyl)-8-hydroxy-1,6-naphthyridine-7-carboxamide (28)
A mixture of 47 (300 mg, 1.4 mmol) and 4-fluoro-α-methylbenzylamine (383 mg, 2.7 mmol) in EtOH (12 mL) was heated in microwave at 120 °C for 2 h. After cooling to 50 °C, water (10 mL) and acetic acid (1 mL) were added and the mixture was cooled to RT. Further, water (10 mL) and acetic acid (1 mL) were added, the mixture was stirred for 40 min, and the resulting solid was collected, washed with water, and dried to give 28 (280 mg, 0.89 mmol, 65%). 1H NMR (DMSO-d6): δ 13.69 (s, 1H), 9.66–9.60 (m, 1H), 9.18–9.15 (m, 1H), 8.93 (s, 1H), 8.64–8.61 (m, 1H), 7.85–7.81 (m, 1H), 7.57–7.51 (m, 2H), 7.20–7.15 (m, 2H), 5.31–5.24 (m, 1H), 1.59 (d, J = 7.0 Hz, 3H). 13C NMR (DMSO-d6): δ 169.2, 161.6 (d, J = 242 Hz), 154.9, 154.6, 142.4, 140.5, 136.5, 128.9, (d, J = 8.1 Hz), 127.0. 125.9, 125.4, 115.5 (d, J = 21.3 Hz), 48.2, 22.1; m/z [M + H]+ calcd for C17H15N3O2F, 312.1148; found, 312.1139.
N-(Cyclopropylmethyl)-8-hydroxy-1,6-naphthyridine-7-carboxamide (29)
29 was synthesized by an analogous method to 28 from 47 (100 mg, 0.50 mmol) and aminomethylcyclopropane (70 mg, 0.98 mmol) to give 29 (23 mg, 0.09 mmol, 19%). 1H NMR (DMSO-d6): δ 13.92 (bs, 1H), 9.38 (bs, 1H), 9.18–9.16 (m, 1H), 8.92 (s, 1H), 8.64–8.60 (m, 1H), 7.86–7.81 (m, 1H), 3.26 (t, J = 6.6 Hz, 2H), 1.19–1.11 (m, 1H), 0.49–0.45 (m, 2H), 0.34–0.29 (m, 2H). 13C NMR (DMSO-d6): δ 169.8, 154.8, 154.5, 142.5, 142.3, 136.4, 126.9, 126.2, 125.2, 43.6, 11.3, 3.8; m/z 244.1 [M + H]+.
N-(4-Fluorobenzyl)-5-(1,1-dioxido-1,2-thiazinan-2-yl)-8-hydroxy-1,6-naphthyridine-7-carboxamide (30)
To a solution of 49 (365 mg, 0.835 mmol) in 1,4-dioxane (8.4 mL), 1,2-thiazinane 1,1-dioxide (135 mg, 1.00 mmol), caesium carbonate (544 mg, 1.67 mmol), Pd2(dba)3 (15 mg, 0.017 mmol), and Xantphos (19 mg, 0.033 mmol) were added, and the resulting mixture was stirred at 65 °C for 2.5 h. The reaction was partitioned between DCM (25 mL) and sat. NH4Cl (50 mL), the phases were separated, and the aqueous phase was extracted with further DCM (1 × 25 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated to give a crude material, which was dissolved in dichloromethane and purified by flash chromatography (0–30% ethyl acetate/ethanol (3:1)/cyclohexane) to give methyl 5-(1,1-dioxido-1,2-thiazinan-2-yl)-8-(tosyloxy)-1,6-naphthyridine-7-carboxylate, which was used without further purification. To a suspension of the crude material (0.070 g, 0.142 mmol) in isopropanol (1.4 mL), a solution of lithium hydroxide (20 mg, 0.47 mmol) in water was added, and the resulting suspension was heated at 60 °C overnight and then stirred at room temperature overnight. 2 M HCl was added to adjust the pH to approximately 2, and the mixture was concentrated to give crude 5-(1,1-dioxido-1,2-thiazinan-2-yl)-8-hydroxy-1,6-naphthyridine-7-carboxylic acid. To a suspension of the crude material (108 mg, 0.33 mmol) in DMF (3.3 mL), 4-fluorobenzylamine (0.045 mL, 0.40 mmol), HOBt (61 mg, 0.40 mmol), and DIPEA (0.088 mL, 0.50 mmol) were added, and the resulting solution was purged with nitrogen and cooled to 0 °C. EDCI (96 mg, 0.50 mmol) was added, and the solution was stirred at room temperature for 8 h and then at 50 °C overnight. Further, HOBt (61 mg, 0.40 mmol), EDCI (96 mg, 0.50 mmol), and 4-fluorobenzylamine (0.046 mL, 0.40 mmol) were added, and the resulting solution was stirred at 50 °C overnight. After cooling, the reaction was partitioned between EtOAc (20 mL) and sat. NH4Cl (50 mL), the layers were separated, and the aqueous phase was extracted with further EtOAc (1 × 20 mL). The combined organics were dried over Na2SO4, filtered, and concentrated to give a crude oil, which was purified by preparative HPLC to give 30 (11 mg, 0.025 mmol, 7% over three steps). 1H NMR (DMSO-d6): δ 9.15 (bs, 1H), 8.61–8.59 (m, 1H), 7.84–7.80 (m, 1H), 7.49–7.43 (m, 2H), 7.37–7.25 (m, 1H), 7.18–7.12 (m, 2H), 7.11–7.03 (m, 1H), 4.66 (d, J = 6.0 Hz, 2H), 3.89–3.86 (m, 2H), 3.49–3.45 (m, 2H), 2.36–2.28 (m, 3H), 2.02–1.99 (m, 1H); m/z 431.0 [M + H]+; m/z [M + H]+ calcd for C20H20N4O4SF, 431.1189; found, 431.1192.
N-(4-Chlorobenzyl)-8-hydroxy-5-((tetrahydro-2H-pyran-4-yl)amino)-1,6-naphthyridine-7-carboxamide (31)
To a solution of DIPEA (747 μL, 0.428 mmol) in NMP (1.43 mL) were added 4-aminotetrahydropyran (103 μL, 0.998 mmol) and 51c (56 mg, 0.143 mmol), and the resulting solution was heated in microwave at 180 °C for 5 h. The reaction was partitioned between EtOAc (15 mL) and a saturated solution of NaHCO3 (60 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 30 mL). The organic phases were combined, dried over Na2SO4, filtered, concentrated, and the crude material was purified by preparative HPLC to afford 31 (10 mg, 17%). 1H NMR (DMSO-d6): δ 12.43 (s, 1H), 9.07–9.02 (m, 2H), 8.78 (dd, J = 1.5, 8.6 Hz, 1H), 7.69 (dd, J = 4.3, 8.3 Hz, 1H), 7.43–7.35 (m, 4H), 7.00 (d, J = 7.6 Hz, 1H), 4.58 (d, J = 6.3 Hz, 2H), 4.47–4.39 (m, 1H), 3.92–3.88 (m, 2H), 3.55–3.47 (m, 2H), 1.97–1.90 (m, 2H), 1.62–1.50 (m, 2H); m/z 413.17 [M + H]+.
N-(4-Chlorobenzyl)-8-hydroxy-5-morpholino-1,6-naphthyridine-7-carboxamide (32)
A mixture of 51c (30 mg, 0.07 mmol) and morpholine (312 mg, 0. 36 mmol) in NMP (0.5 mL) was heated in microwave at 150 °C for 1 h. The crude solution was purified by mass-directed prep. HPLC to give 32 (10 mg, 0.024 mmol, 33%). 1H NMR (DMSO-d6): δ 13.14 (bs, 1H), 9.45–9.32 (m, 1H), 9.16–9.07 (m, 1H), 8.62–8.53 (m, 1H), 7.81–7.70 (m, 1H), 7.45–7.38 (m, 4H), 4.62–5.53 (m, 2H), 3.92–3.81 (m, 4H), 3.30–3.23 (m, 4H, under water signal); m/z 399.1, 401.1 [M + H]+.
N-(Cyclopropylmethyl)-8-hydroxy-5-(2-methoxyphenyl)-1,6-naphthyridine-7-carboxamide (33)
33 was synthesized by an analogous method to 26 from 51b (40 mg, 0.14 mmol) and 2-methoxyphenylboronic acid (43 mg, 0.28 mmol) to give 33 (20 mg, 0.05 mmol, 38%). 1H NMR (DMSO-d6): δ 13.51 (s, 1H), 9.17–9.15 (m, 1H), 8.28–8.23 (m, 1H), 8.00–7.96 (m, 1H), 7.58–7.44 (m, 3H), 7.21–7.17 (m, 1H), 7.12–7.09 (m, 1H), 3.74 (s, 3H), 3.40–3.35 (m, 2H), 1.16–1.08 (m, 1H), 0.61–0.56 (m, 2H), 0.36–0.311 (m, 2H); m/z 350.2 [M + H]+.
8-Hydroxy-N-(trans-4-methoxycyclohexyl)-5-(2-methoxyphenyl)-1,6-naphthyridine-7-carboxamide (34)
To a solution of 50 (750 mg, 2.53 mmol) in DMF (17 mL) were added trans-4-methoxycyclohexanamine (491 mg, 3.80 mmol), DIPEA (1.33 mL, 7.59 mmol), and propylphosphonic anhydride solution (T3P) (4.83 g, 7.59 mmol) and stirred at RT for 2 h and then at 50 °C for 18 h. After cooling, the reaction mixture was partitioned between water (70 mL) and EtOAc (30 mL), the layers were separated, and the pH of the aqueous layer was adjusted to approximately 5 with 1 M NaOH. The aqueous layer was extracted with EtOAc (2 × 30 mL), and the combined organics were washed with sat. NH4Cl (1 × 40 mL), dried over Na2SO4, filtered, and concentrated. The resulting solid was triturated with ether (3 × 5 mL) and dried to give 34 (580 mg, 1.42 mmol, 56% yield). 1H NMR (DMSO-d6): δ 13.88 (s, 1H), 9.14–9.12 (m, 1H), 8.67–8.64 (m, 1H), 7.90–7.88 (m, 1H), 7.72–7.70 (m, 1H), 7.57–7.52 (m, 1H), 7.32–7.40 (m, 1H), 7.22–7.13 (m, 2H), 3.92–3.84 (m, 1H), 3.64 (s, 3H), 3.23 (s, 3H), 3.11–3.05 (m, 1H), 2.05–1.99 (m, 2H), 1.85–1.83 (m, 2H), 1.63–1.54 (m, 2H), 1.27–1.17 (m, 2H); 13C NMR (DMSO-d6): δ 169.2, 157.2, 154.3, 154.1, 148.6, 142.4, 136.4, 131.9, 131.1, 127.0, 125.9, 121.2, 111.8, 78.1, 55.8, 55.6, 48.1, 30.8, 29.8; m/z [M + H]+ calcd for C23H26N3O4, 408.1923; found, 408.1913.
5-Bromo-8-hydroxy-1,6-naphthyridine-7-carbonitrile (36)
To a solution of 35 (1.10 g, 6.43 mmol) in DCM (64 mL), NBS (1.37 g, 7.71 mmol) was added and stirred for 2 h. Water (100 mL) was added, the phases were separated, and the aqueous layer was extracted with 5% MeOH/DCM (3 × 40 mL). The combined organics were washed with water (4 × 100 mL), dried over Na2SO4, filtered, and concentrated to give 36 as a yellow solid (1.35 g, 5.42 mmol, 84%). 1H NMR (DMSO-d6): 9.30–9.28 (m, 1H), 8.64–8.61 (m, 1H), 8.06–8.02 (m, 1H); δ m/z 250.0, 252.0 [M + H]+.
5-Bromo-7-(5-methyl-4H-1,2,4-triazol-3-yl)-1,6-naphthyridin-8-ol (37)
To 36 (750 mg, 2.85 mmol) in 1,4-dioxane (6 mL)/acetic acid (2 mL) was added acetohydrazide (666 mg, 9.0 mmol) and heated in microwave at 200 °C for 0.5 h. Upon cooling, the resulting solid was collected, washed with 1,4-dioxane, and dissolved in water. 1 M HCl was added to adjust the pH to approximately 4–5, and the resulting solid was collected, washed with water, and dried in vacuo to give 37 (460 mg, 1.35 mmol, 45%). 1H NMR (DMSO-d6): δ 14.14 (bs, 1H), 8.83–8.82 (m, 1H), 8.27–8.24 (m, 1H), 7.63–7.60 (m, 1H), 2.31 (s, 3H); m/z 305.9, 307.9 [M + H]+.
5-Bromo-7-cyano-1,6-naphthyridin-8-yl 4-methylbenzenesulfonate (38)
To a suspension of 36 (500 mg, 2.0 mmol) in DCM (15 mL) were added triethylamine (418 μL, 3.00 mmol) and tosyl chloride (457 mg, 2.4 mmol) and heated in a sealed tube at 40 °C for 3 h. After cooling, sat. NaHCO3 (25 mL) was added, the layers were separated, and the aqueous layer was extracted with DCM (30 mL). The combined organics were dried over Na2SO4, filtered, concentrated, and purified by flash chromatography (0–80% EtOAc/cyclohexane) to give 38 (867 g, 1.66 mmol, 83%); 1H NMR (DMSO-d6): δ 9.01–9.00 (m, 1H), 8.62–8.60 (m, 1H), 7.92–7.89 (m, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.3 Hz, 2H), 2.36 (s, 3H); m/z 406.0. 408.0 [M + H]+.
8-(Benzyloxy)-5-bromo-1,6-naphthyridine-7-carbonitrile (39)
To a solution of 38 (2.47 g, 9.9 mmol) in THF (60 mL) at 0 °C was added triphenylphosphine (3.89 g, 14.8 mmol) and benzyl alcohol (1.12 mL, 10.9 mmol), stirred for 15 min, and diisopropyl azodicarboxylate (2.91 mL, 14.82 mmol) was added dropwise. The resulting solution was stirred at 0 °C for 2 h, allowed to warm to room temperature, and stirred overnight. The solution was poured into water (150 mL) and extracted with EtOAc (150 mL). The organic layer was dried over Na2SO4, filtered, concentrated, and purified using column chromatography (0–25% EtOAc/cyclohexane) to give a white solid, which was triturated with a mixture of Et2O and cyclohexane (1:1, 20 mL) to give 39 (0.5 g, 1.47 mmol, 15%). 1H NMR (DMSO-d6): δ 9.40–9.37 (m, 1H), 8.72–8.68 (m, 1H), 8.08–8.04 (m, 1H), 7.53–7.48 (m, 2H), 7.43–7.35 (m, 3H), 5.86 (s, 2H); m/z 338.1, 340.1 [M + H]+.
2-(5-Bromo-8-methoxy-1,6-naphthyridin-7-yl)-5-methyl-1,3,4-oxadiazole (41)
To a solution of EDCI (335 mg, 1.75 mmol) in DCM (15 mL) at 0 °C were added DIPEA (0.509 mL, 2.91 mmol) and HOBt (268 mg, 1.75 mmol) and stirred at 0 °C for 10 min. Acethydrazide (130 mg, 1.75 mmol) and 40 (330 mg, 1.17 mmol) were added, and the solution was stirred for 18 h. Water (25 mL) and DCM (25 mL) were added, and the aqueous layer was extracted with DCM (2 × 15 mL). The combined organics were dried over Na2SO4, filtered, and concentrated to give crude N′-acetyl-5-bromo-8-methoxy-1,6-naphthyridine-7-carbohydrazide (170 mg, 43%) which was used without purification. Triphenylphosphine (247 mg, 0.944 mmol), carbon tetrachloride (0.182 mL, 1.89 mmol), and triethylamine (0.132 mL, 0.944 mmol) were added to a mixture of the crude material (160 mg, 0.472 mmol) in CH3CN (4 mL) and stirred at 60 °C for 2 h. After cooling, the reaction mixture was concentrated and purified by flash chromatography (0–70% EtOH/EtOAc 1:3/cyclohexane) to give 41 (0.08 g, 53% yield). 1H NMR (DMSO-d6): δ 9.34–9.33 (m, 1H), 8.76–8.73 (m, 1H), 8.00–7.97 (m, 1H), 4.25 (s, 3H), 2.66 (s, 3H); m/z 321.0, 323.0 [M + H]+.
1-Methyl-4-oxo-1,4-dihydro-1,5-naphthyridine-3-carboxylic Acid (42)
To 3-chloropyridine-2-carboxylic acid (1 g, 6.3 mmol) in DCM (40 mL) was added oxalyl chloride (1.6 g, 12.7 mmol) and DMF (one drop) and stirred for 2 h. The solvent was evaporated, and THF (20 mL) was added. In parallel, to 3-ethoxy-3-oxo-propanoic acid (1.31 g, 10.2 mmol) in THF (20 mL) at −78 °C was added BuLi (4.3 mL, 2.5 M in hexanes, 10.8 mmol) dropwise over 10 min. To this was added the acid chloride solution dropwise, and the mixture was stirred at −78 °C for 30 min, warmed to −30 °C, and stirred again for 30 min, and then quenched by pouring into a mix of ice/1 M HCl. The aqueous layer was extracted with EtOAc (3 × 20 mL), and the combined organics were washed with sat NaHCO3, 1 M HCl, and brine, dried, and evaporated. The crude material (480 mg, 1.9 mmol) and DMF·DMA (2.49 g, 2.1 mmol) in DMF (2 mL) were heated in microwave at 100 °C for 1 h and solvent-evaporated. Water (5 mL) was added and washed with EtOAc (2 × 5 mL). The combined organics were washed with brine, dried, and solvent-evaporated. Ether (4 mL)/EtOH (1 mL) and methylamine (33% in EtOH, 60 mg, 1.9 mmol) were added and stirred for 1 h, and the resulting solid was collected, taken up in DMF (4 mL), potassium carbonate (424 mg, 3.0 mmol) was added, and the mixture was stirred in a sealed tube at 100 °C overnight. The solvent was evaporated, 1 M NaOH (1 mL)/MeOH (1 mL) was added, and the mixture was stirred for 2 h. The resulting solid was collected, suspended in water (5 mL), and acidified with acetic acid. The resulting solid was collected, washed with water, and dried under vacuum to give 42 (151 mg, 0.70 mmol, 69% yield). 1H NMR (DMSO-d6): δ 15.32 (bs, 1H), 9.11 (bs, 1H), 8.98–8.96 (m, 1H), 8.48–8.44 (m, 1H), 8.00–7.97 (m, 1H), 4.11 (s, 3H); m/z 205.1 [M + H]+.
tert-Butyl 3-(3-Fluoropyridin-2-yl)-3-oxopropanoate (43)
To LDA (33 mL, 65.8 mmol, 2 M in THF) at −78 °C was added tert-butyl acetate (9.5 g, 82.2 mmol), stirred for 45 min, and methyl 3-fluoropicolinate (5.1 g, 32.9 mmol) in THF (30 mL) was added dropwise. The reaction mixture was stirred for 2 h, quenched at −78 °C by addition of pet. ether and water, and warmed to RT. The layers were separated, and the aqueous phase was extracted with pet. ether. The combined organics were dried over Na2SO4, filtered, and evaporated to give crude 43, which was used in the synthesis of 23 without purification (8 g, quant.); m/z 205.1 [M + H]+.
Ethyl 5-(2-Ethoxy-2-oxoethyl)-3-(2-methoxyphenyl)-2-methyl-2,5-dihydro-1,2,4-oxadiazole-5-carboxylate (45)
To a mixture of 2-methoxybenzonitrile (500 mg, 3.76 mmol) and N-methylhydroxylamine hydrochloride (627 mg, 7.51 mmol) in water (3.8 mL)/EtOH (1.9 mL) was added sodium carbonate (438 mg, 4.13 mmol), and the mixture was stirred at 80 °C for 28 h. The solvent was evaporated, DCM was added, stirred for 18 h, and the mixture was filtered. The solid was discarded, and the filtrate was evaporated to give crude N-hydroxy-2-methoxy-N-methylbenzimidamide (742 mg, 4.12 mmol), which was used in the next step without purification. To this crude material in EtOH (10 mL) was added diethyl acetylenedicarboxylate (771 mg, 4.53 mmol), stirred for 1 h, and the solvent was evaporated. The crude material was purified by flash chromatography (0–80% EtOAc/cyclohexane) to give 45 (880 mg, 2.5 mmol, 61%). 1H NMR (DMSO-d6): δ 7.57–7.53 (m, 1H), 7.49–7.46 (m, 1H), 7.18–7.16 (m, 1H), 7.07–7.03 (m, 1H), 4.20–4.16 (m, 2H), 4.08 (q, J = 7.0 Hz, 2H), 3.83 (s, 3H), 3.27–3.23 (m, 1H), 2.97–2.93 (m, 1H), 2.91 (s, 3H), 1.24–1.16 (m, 6H); m/z 351.2 [M + H]+.
Ethyl 5-Hydroxy-2-(2-methoxyphenyl)-1-methyl-6-oxo-1,6-dihydropyrimidine-4-carboxylate (46)
A solution of 45 (880 mg, 2.51 mmol) in p-xylene (5.38 mL) was heated to reflux for 2 days. Hexane (5 mL) was added, the resulting mixture was sonicated, and the resulting suspension was stored at 0 °C for 3 days. The solid was collected, washed with cyclohexane, and dried to give 46 (540 mg, 1.78 mmol, 41%). 1H NMR (DMSO-d6): δ 10.45 (s, 1H), 7.56–7.51 (m, 1H), 7.37–7.34 (m, 1H), 7.19–7.17 (m, 1H), 7.11–7.07 (m, 1H), 4.28 (q, J = 6.9 Hz, 2H), 3.82 (s, 3H), 3.20 (s, 3H), 1.27 (t, J = 7.0 Hz, 3H); m/z 305.1 [M + H]+.
Methyl 5-Bromo-8-hydroxy-1,6-naphthyridine-7-carboxylate (48)
To 47 (800 mg, 3.9 mmol) in DCM (20 mL) was added NBS (837 mg, 4.7 mmol) and stirred for 2 h. The resulting solid was collected, washed with DCM, and dried under vacuum to give 46 (1.09 g, 3.65 mmol, 93% yield). 1H NMR (DMSO-d6): δ 11.54 (bs, 1H), 9.27–9.25 (m, 1H), 8.60–8.57 (m, 1H), 8.01–7.99 (m, 1H), 3.95 (s, 3H); m/z 283.0. 285.0 [M + H]+.
Methyl 5-Bromo-8-(tosyloxy)-1,6-naphthyridine-7-carboxylate (49)
To a suspension of 48 (1.3 g, 4.59 mmol) in DCM (31 mL), triethylamine (0.96 mL, 6.89 mmol) and tosyl chloride (1.05 g, 5.51 mmol) were added, and the suspension was heated at 40 °C for 2 days. A saturated solution of NaHCO3 (50 mL) was added, and the layers were separated. The organic layer was dried over Na2SO4, filtered, concentrated, and purified by flash chromatography (0–80% EtOAc/cyclohexane) to give 49 (0.98 g, 2.3 mmol, 49%). 1H NMR (DMSO-d6): δ 8.95–8.92 (m, 1H), 8.55–8.53 (m, 1H), 7.84–7.81 (m, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.3 Hz, 2H), 3.67 (s, 3H), 2.32 (s, 3H); m/z 437.0, 439.0 [M + H]+.
8-Hydroxy-5-(2-methoxyphenyl)-1,6-naphthyridine-7-carboxylic Acid (50)
To a suspension of 49 (19.75 g, 45.2 mmol) in 1,4-dioxane (339 mL) were added (2-methoxyphenyl)boronic acid (13.73 g, 90 mmol), aqueous sodium carbonate (14.36 g, 136 mmol, 113 mL of water), and tetrakis(triphenylphosphine)palladium(0) (2.61 g, 2.26 mmol), purged with N2, and stirred at 60 °C for 18 h. After cooling, the mixture was partitioned between DCM (200 mL) and water (1.2 L), and the organic phase was dried over Na2SO4, filtered, and concentrated. The crude intermediate was purified by flash chromatography (0–100% EtOAc/ethanol (5:1)/cyclohexane) to give methyl 5-(2-methoxyphenyl)-8-(tosyloxy)-1,6-naphthyridine-7-carboxylate (16 g, 34.5 mmol), which was used without further purification. To a solution of the crude material (16 g, 34.5 mmol) in DMF (34.4 mL) was added sodium methoxide in MeOH (172 mL, 86 mmol, 0.5 N), stirred at 50 °C for 5 min, cooled to RT, and stirred for a further 15 min. Acetic acid (3.94 mL, 68.9 mmol)/water (130 mL) was added, stirred for 30 min, and the resulting solid was collected and washed with 1:1 MeOH/H2O (2 × 50 mL). The crude solid was taken up in THF (193 mL)/MeOH (97 mL), a solution of lithium hydroxide monohydrate (2.43 g, 58.0 mmol) in water (50 mL) was added, and the resulting suspension was stirred at 60 °C overnight. After cooling, pH was adjusted to approximately 4–5 by addition of 2 M HCl, and the resulting solid was collected, washed with 1:1 MTBE/cyclohexane, and dried to give 50 (7.65 g, 26 mmol, 89%). 1H NMR (DMSO-d6): δ 8.99–8.97 (m, 1H), 7.79–7.76 (m, 1H), 7.57–7.54 (m, 1H), 7.52–7.47 (m, 1H), 7.34–7.32 (m, 1H), 7.18–7.15 (m, 1H), 7.13–7.08 (m, 1H), 3.64 (s, 3H).
5-Bromo-N-(4-chlorobenzyl)-8-hydroxy-1,6-naphthyridine-7-carboxamide (51a)
51a was synthesized by an analogous method to 51b from 48 (120 mg, 0.40 mmol) and 4-chlorobenzylamine (57 mg, 9.40 mmol) to give 51a (75 mg, 0.18 mmol, 45%). 1H NMR (DMSO-d6): δ 13.70 (bs, 1H), 9.75 (bs, 1H), 9.24–9.21 (m, 1H), 8.59–8.57 (m, 1H), 7.97–7.94 (m, 1H), 7.42–7.41 (m, 4H), 4.56 (d, J = 6.4 Hz, 2H); m/z 391.8, 393.8 [M + H]+.
5-Bromo-8-hydroxy-N-methyl-1,6-naphthyridine-7-carboxamide (51b)
To 48 (1.3 g, 4.6 mmol) in EtOH (25 mL) was added methylamine (33% in EtOH, 2.2 mL, 23 mmol) and stirred at 100 °C overnight. The hot mixture was poured into water (40 mL)/acetic acid (10 mL), allowed to cool to RT, stirred for 1 h, and the resulting solid was collected, washed with water, and dried under vacuum to give 51b (1.18 g, 3.96 mmol, 86% yield). 1H NMR (DMSO-d6): δ 13.98 (bs, 1H), 9.24–9.06 (m, 2H), 8.59–8.55 (m, 1H), 7.96–7.92 (m, 1H), 2.90 (d, J = 2.9 Hz, 3H); m/z 282.0, 284.0 [M + H]+.
5-Bromo-N-(cyclopropylmethyl)-8-hydroxy-1,6-naphthyridine-7-carboxamide (51c)
51c was synthesized by an analogous method to 51b from 48 (400 mg, 1.4 mmol) and cyclopropylmethanamine (150 mg, 2.1 mmol) to give 51c (392 mg, 1.16 mmol, 82%). 1H NMR (DMSO-d6): δ 14.00 (bs, 1H), 9.28–9.22 (m, 2H), 8.62–8.58 (m, 1H), 8.00–7.95 (m, 1H), 3.26–3.23 (m, 2H), 1.18–1.12 (m, 1H), 0.51–0.45 (m, 2H), 0.34–0.30 (m, 2H); m/z 322.0 [M + H]+.
Intramacrophage L. donovani Assay
This assay was conducted as previously described,8 except for compound exposure time, which was 96 h instead of 72 h.
Kinetic Aqueous Solubility Assessment and Intrinsic Clearance Experiments
These assays were conducted as previously described.23
Chrom Log DpH7.4
This assay was conducted as previously described.11
In vivo Mouse Efficacy Studies
In vivo studies were carried out as previously described.12
Ethical Statements
Mouse and Rat Pharmacokinetics
All animal studies were ethically reviewed and carried out in accordance with Animals (Scientific Procedures) Act 1986 and the GSK/Dundee University Policy on the Care, Welfare, and Treatment of Animals.
In Vivo Efficacy
All regulated procedures, at the University of Dundee, on living animals were carried out under the authority of a project license issued by the Home Office under the Animals (Scientific Procedures) Act 1986, as amended in 2012 (and in compliance with EU Directive EU/2010/63). License applications will have been approved by the University’s Ethical Review Committee (ERC) before submission to the Home Office. The ERC has a general remit to develop and oversee policy on all aspects of the use of animals on University premises and is a subcommittee of the University Court, its highest governing body.
Acknowledgments
Funding for this work was provided by Wellcome (nos. 092340 and 100476). The authors thank DNDi and their team for their support and scientific discussions. They also thank Gina MacKay, Darren Edwards, and Dan Fletcher for performing HRMS and for assistance with performing other NMR and MS analyses; Raul Fernandez Velasco and the Galchimia chemistry team for synthesizing key compounds; and Alastair Pate, Francesco Gastaldello, and James Burkinshaw for data management.
Glossary
Abbreviations
- VL
visceral leishmaniasis
- DNDi
Drugs for Neglected Diseases initiative
- GSK
GlaxoSmithKline
- WHO
World Health Organization
- NTD
neglected tropical disease
- THP-1
human monocytic cell line derived from an acute monocytic leukemia patient
- Ld InMac
intramacrophage assay carried out in THP-1 cells with L. donovani amastigotes
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00705.
The authors declare the following competing financial interest(s): The following authors have shares in GlaxoSmithKline: J.M.F., P.G.W., M.M., K.D.R., and T.J.M. The other authors declare no competing interests.
Supplementary Material
References
- World Health Organization (WHO) List of Neglected Tropical Diseases; World Health Organization: Geneva, 2018. http://www.who.int/neglected_diseases/diseases/en/ (accessed April 23, 2020). [Google Scholar]
- Field M. C.; Horn D.; Fairlamb A. H.; Ferguson M. A. J.; Gray D. W.; Read K. D.; De Rycker M.; Torrie L. S.; Wyatt P. G.; Wyllie S.; Gilbert I. H. Anti-trypanosomatid Drug Discovery: an Ongoing Challenge and a Continuing Need. Nat. Rev. Microbiol. 2017, 15, 217–231. 10.1038/nrmicro.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (WHO) Leishmaniasis Fact Sheet; World Health Organization: Geneva, 2019. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed April 23, 2020). [Google Scholar]
- Sundar S.; Olliaro P. L. Miltefosine in the treatment of leishmaniasis: Clinical Evidence for Informed Clinical Risk Management. Ther. Clin. Risk Manage. 2007, 3, 733–740. [PMC free article] [PubMed] [Google Scholar]
- Sundar S.; Jha T. K.; Thakur C. P.; Sinha P. K.; Bhattacharya S. K. Injectable Paromomycin for Visceral Leishmaniasis in India. N. Engl. J. Med. 2007, 356, 2571–2581. 10.1056/NEJMoa066536. [DOI] [PubMed] [Google Scholar]
- Drugs for Neglected Diseases initiative (DNDi) Portfolio. https://www.dndi.org/diseases-projects/leishmaniasis/leish-portfolio/ (accessed April 23, 2020).
- a Wyllie S.; Thomas M.; Patterson S.; Crouch S.; De Rycker M.; Lowe R.; Gresham S.; Urbaniak M. D.; Otto T. D.; Stojanovski L.; Simeons F. R. C.; Manthri S.; MacLean L. M.; Zuccotto F.; Homeyer N.; Pflaumer H.; Boesche M.; Sastry L.; Connolly P.; Albrecht S.; Berriman M.; Drewes G.; Gray D. W.; Ghidelli-Disse S.; Dixon S.; Fiandor J. M.; Wyatt P. G.; Ferguson M. A. J.; Fairlamb A. H.; Miles T. J.; Read K. D.; Gilbert I. H. Cyclin-Dependent Kinase 12 is a Drug Target for Visceral Leishmaniasis. Nature 2018, 560, 192–197. 10.1038/s41586-018-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wyllie S.; Brand S.; Thomas M.; De Rycker M.; Chung C.; Pena I.; Bingham R. P.; Bueren-Calabuig J. A.; Cantizani J.; Cebrian D.; Craggs P. D.; Ferguson L.; Goswami P.; Hobrath J.; Howe J.; Jeacock L.; Ko E.-J.; Korczynska J.; MacLean L.; Manthri S.; Martinez M. S.; Mata-Cantero L.; Moniz S.; Nühs A.; Osuna-Cabello M.; Pinto E.; Riley J.; Robinson S.; Rowland P.; Simeons F. R. C.; Shishikura Y.; Spinks D.; Stojanovski L.; Thomas J.; Thompson S.; Viayna Gaza E.; Wall R. J.; Zuccotto F.; Horn D.; Ferguson M. A. J.; Fairlamb A. H.; Fiandor J. M.; Martin J.; Gray D. W.; Miles T. J.; Gilbert I. H.; Read K. D.; Marco M.; Wyatt P. G. Preclinical Candidate for the Treatment of Visceral Leishmaniasis that Acts Through Proteasome Inhibition. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 9318–9323. 10.1073/pnas.1820175116. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Khare S.; Nagle A. S.; Biggart A.; Lai Y. H.; Liang F.; Davis L. C.; Barnes S. W.; Mathison C. J. N.; Myburgh E.; Gao M.-Y.; Gillespie J. R.; Liu X.; Tan J. L.; Stinson M.; Rivera I. C.; Ballard J.; Yeh V.; Groessl T.; Federe G.; Koh H. X. Y.; Venable J. D.; Bursulaya B.; Shapiro M.; Mishra P. K.; Spraggon G.; Brock A.; Mottram J. C.; Buckner F. S.; Rao S. P. S.; Wen B. G.; Walker J. R.; Tuntland T.; Molteni V.; Glynne R. J.; Supek F. Proteasome Inhibition for Treatment of Leishmaniasis, Chagas Disease and Sleeping Sickness. Nature 2016, 537, 229–233. 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rycker M.; Hallyburton I.; Thomas J.; Campbell L.; Wyllie S.; Joshi D.; Cameron S.; Gilbert I. H.; Wyatt P. G.; Frearson J. A.; Fairlamb A. H.; Gray D. W. Comparison of a High-Throughput High Content Intracellular Leishmania donovani Assay with an Axenic Amastigote Assay. Antimicrob. Agents Chemother. 2013, 57, 2913–2922. 10.1128/AAC.02398-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don R.; Ioset J.-R. Screening Strategies to Identify New Chemical Diversity for Drug Development to Treat Kinetoplastid Infections. Parasitology 2014, 141, 140–146. 10.1017/S003118201300142X. [DOI] [PubMed] [Google Scholar]
- Peña I.; Pilar Manzano M.; Cantizani J.; Kessler A.; Alonso-Padilla J.; Bardera A. I.; Alvarez E.; Colmenarejo G.; Cotillo I.; Roquero I.; de Dios-Anton F.; Barroso V.; Rodriguez A.; Gray D. W.; Navarro M.; Kumar V.; Sherstnev A.; Drewry D. H.; Brown J. R.; Fiandor J. M.; Julio Martin J. New Compound Sets Identified from High Throughput Phenotypic Screening against Three Kinetoplastid Parasites: an Open Resource. Sci. Rep. 2015, 5, 8771 10.1038/srep08771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting Physical in Drug Discovery II: the Impact of Chromatographic Hydrophobicity Measurements and Aromaticity. Drug Discovery Today 2011, 16, 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Thomas M. G.; De Rycker M.; Ajakane M.; Albrecht S.; Alvarez-Pedraglio A. I.; Boesche M.; Brand S.; Campbell L.; Cantizani-Perez J.; Cleghorn L. A. T.; Copley R. C. B.; Crouch S. D.; Daugan A.; Drewes G.; Ferrer S.; Ghidelli-Disse S.; Gonzalez S.; Gresham S. L.; Hill A. P.; Hindley S. J.; Lowe R. M.; MacKenzie C. J.; MacLean L.; Manthri S.; Martin F.; Miguel-Siles J.; Nguyen V. L.; Norval S.; Osuna-Cabello M.; Woodland A.; Patterson S.; Pena I.; Quesada-Campos M. T.; Reid I. H.; Revill C.; Riley J.; Ruiz-Gomez J. R.; Shishikura Y.; Simeons F. R. C.; Smith A.; Smith V. C.; Spinks D.; Stojanovski L.; Thomas J.; Thompson S.; Underwood T.; Gray D. W.; Fiandor J. M.; Gilbert I. H.; Wyatt P. G.; Read K. D.; Miles T. J. Identification of GSK3186899/DDD853651 as a Preclinical Development Candidate for the Treatment of Visceral Leishmaniasis. J. Med. Chem. 2019, 63, 1180–1202. 10.1021/acs.jmedchem.8b01218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egbertson M. S.; Moritz H. M.; Melamed J. Y.; Han W.; Perlow D. S.; Kuo M. S.; Embrey M.; Vacca J. P.; Zrada M. M.; Cortes A. R.; Wallace A.; Leonard Y.; Hazuda D. J.; Miller M. D.; Felock P. J.; Stillmock K. A.; Witmer M. V.; Schleif W.; Gabryelski L. J.; Moyer G.; Ellis J. D.; Jin L.; Xu W.; Braun M. P.; Kassahun K.; Tsou N. N.; Young S. D. A Potent and Orally Active HIV-1 Integrase Inhibitor. Bioorg. Med. Chem. Lett. 2007, 17, 1392–1398. 10.1016/j.bmcl.2006.11.080. [DOI] [PubMed] [Google Scholar]
- King C. D.; Rios G. R.; Green M. D.; Tephly T. R. UPD-Glucuronosyltransferases. Curr. Drug Metab. 2000, 1, 143–161. 10.2174/1389200003339171. [DOI] [PubMed] [Google Scholar]
- Scott J. S.; deSchoolmeester J.; Kilgour E.; Mayers R. M.; Packer M. J.; Hargreaves D.; Gerhardt S.; Ogg D. J.; Rees A.; Selmi N.; Stocker A.; Swales J. G.; Whittamore P. R. Novel Acidic 11β-Hydroxysteroid Dehydrogenase Type 1 (11β-HSD1) Inhibitor with Reduced Acyl Glucuronide Liability: The Discovery of 4-[4-(2-Adamantylcarbamoyl)-5-tert-butyl-pyrazol-1-yl]benzoic Acid (AZD8329). J. Med. Chem. 2012, 55, 10136–10147. 10.1021/jm301252n. [DOI] [PubMed] [Google Scholar]
- Croxtall J. D.; Keam S. J. Raltegravir. Drugs 2009, 69, 1059–1075. 10.2165/00003495-200969080-00007. [DOI] [PubMed] [Google Scholar]
- a Hazuda D. J.; Anthony N. J.; Gomez R. P.; Jolly S. M.; Wai J. S.; Zhuang L.; Fisher T. E.; Embrey M.; Guare J. P. Jr.; Egbertson M. S.; Vacca J. P.; Huff J. R.; Felock P. J.; Witmer M. V.; Stillmock K. A.; Danovich R.; Grobler J.; Miller M. D.; Espeseth A. S.; Jin L.; Chen I. W.; Lin J. H.; Kassahun K.; Ellis J. D.; Wong B. K.; Xu W.; Pearson P. G.; Schleif W. A.; Cortese R.; Emini E.; Summa V.; Holloway M. K.; Young S. D. A Naphthyridine Carboxamide Provides Evidence for Discordant Resistance Between Mechanistically Identical Inhibitors of HIV-1 Integrase. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11233–11238. 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Little S.; Drusano G.; Schooley R. In Antiviral Effect of L-000870810, a Novel HIV-1 Integrase Inhibitor, in HIV-1 Infected Patients, 12th Conference on Retroviruses and Opportunistic Infections, Boston, MA, Abstract 161, Feb 22–25, 2005.
- Bowes J.; Brown A. J.; Hamon J.; Jarolimek W.; Sridhar A.; Waldron G.; Whitebread S. Reducing Safety-Related Drug Attrition: The Use of in Vitro Pharmacological Profiling. Nat. Rev. Drug Discovery 2012, 11, 909–922. 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- Wall R. J.; Moniz S.; Thomas M. G.; Norval S.; Ko E. J.; Marco M.; Miles T. J.; Gilbert I. H.; Horn D.; Fairlamb A. H.; Wyllie S. Antitrypanosomal 8-Hydroxy-Naphthyridines are Chelators of Divalent Transition Metals. Antimicrob. Agents Chemother. 2018, 62, e00235-18 10.1128/AAC.00235-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns B. A.; Weatherhead J. G.; Allen S. H.; Thompson J. B.; Garvey E. P.; Foster S. A.; Jeffrey J. L.; Miller W. H. The Use of Oxadiazole and Triazole Substituted Naphthyridines as HIV-1 Integrase Inhibitors. Part 1: Establishing the Pharmacophore. Bioorg. Med. Chem. Lett. 2009, 19, 1802–1806. 10.1016/j.bmcl.2009.01.090. [DOI] [PubMed] [Google Scholar]
- Crescenzi B.; Gardelli C.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Petrocchi A.; Poma M.; Rowley M.; Scarpelli R.; Summa V.. N-Substituted Hydroxypyrimidinone Carboxamide Inhibitors of HIV Integrase. WO2003035077, 2003.
- Zeng L.-F.; Wang Y.; Kazemi R.; Xu S.; Xu Z.-L.; Sanchez T. W.; Yang L.-M.; Debnath B.; Odde S.; Xie H.; Zheng Y.-T.; Ding J.; Neamati N.; Long Y.-Q. Repositioning HIV-1 Integrase Inhibitors for Cancer Therapeutics: 1,6-Naphthyridine-7-carboxamide as a Promising Scaffold with Drug-like Properties. J. Med. Chem. 2012, 55, 9492–9509. 10.1021/jm300667v. [DOI] [PubMed] [Google Scholar]
- Brand S.; Ko E. J.; Viayna E.; Thompson S.; Spinks D.; Thomas M.; Sandberg L.; Francisco A. F.; Jayawardhana S.; Smith V. C.; Jansen C.; De Rycker M.; Thomas J.; MacLean L.; Osuna-Cabello M.; Riley J.; Scullion P.; Stojanovski L.; Simeons F. R. C.; Epemolu O.; Shishikura Y.; Crouch S. D.; Bakshi T. S.; Nixon C. J.; Reid I. H.; Hill A. P.; Underwood T. Z.; Hindley S. J.; Robinson S. A.; Kelly J. M.; Fiandor J. M.; Wyatt P. G.; Marco M.; Miles T. J.; Read K. D.; Gilbert I. H. Discovery and Optimization of 5-Amino1,2,3-triazole-4-carboxamide Series against Trypanosoma cruzi. J. Med. Chem. 2017, 60, 7284–7299. 10.1021/acs.jmedchem.7b00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.