

Abstract
The accumulation of neurotoxic amyloid-beta (Aβ) in the brain is a characteristic hallmark of Alzheimer’s disease (AD). The blood-brain barrier (BBB) provides a large surface area and has been shown to be an important mediator for removal of brain Aβ. Both, the ABC transporter P-glycoprotein (ABCB1/P-gp) and the receptor low-density lipoprotein receptor-related protein 1 (LRP1) have been implicated to play crucial roles in Aβ efflux from brain. Here, with immunoprecipitation experiments, co-immunostainings and dual inhibition of ABCB1/P-gp and LRP1, we show that both proteins are functionally linked, mediating a concerted transcytosis of Aβ through endothelial cells. Late-onset AD risk factor Phosphatidylinositol binding clathrin assembly protein (PICALM) is associated with both ABCB1/P-gp and LRP1 representing a functional link and guiding both proteins through the brain endothelium. Together, our results give more mechanistic insight on Aβ transport across the BBB and show that the functional interplay of different clearance proteins is needed for the rapid removal of Aβ from the brain.
Keywords: Low-density lipoprotein receptor-related protein 1 (LRP1), ABC transporter B1/P-glycoprotein (ABCB1/P-gp), Phosphatidylinositol-binding clathrin assembly protein (PICALM), Amyloid-beta (Aβ), Blood-brain barrier (BBB), Transcytosis, Alzheimer’s disease (AD), Endothelial cell, Endothelium, Clearance
1. Introduction
The accumulation of neurotoxic peptide beta-amyloid (Aβ) is a characteristic hallmark of Alzheimer’s disease (AD) (Selkoe, 2001; Hardy, 2006; Aleksis et al., 2017). In the sporadic form of AD, the production of Aβ is not altered from healthy individuals. However, failures in clearance mechanisms lead to an time-dependent accumulation of Aβ peptides (Bateman et al., 2006; Mawuenyega et al., 2010). In the healthy brain, several highly efficient clearance mechanisms normally exceed the removal of Aβ over its production (Tarasoff-Conway et al., 2015; Storck and Pietrzik, 2017). The blood-brain barrier (BBB)-forming capillaries in the central nervous system (CNS) contribute to a large part (up to 70%) to the rapid removal of excessive brain Aβ (Bell et al., 2009; Krohn et al., 2011; Tarasoff-Conway et al., 2015; Storck et al., 2016). To date, several receptors (Deane et al., 2005; Bell et al., 2007; Storck et al., 2016) and transporters (Kuhnke et al., 2007; Do et al., 2013; Elali and Rivest, 2013; Pahnke et al., 2014; Dodacki et al., 2017) have been described that facilitate the efflux across the BBB into the periphery, where Aβ is then degraded by peripheral organs (Shibata et al., 2000).
One of the most prominent receptors that transports Aβ across the endothelium is low-density lipoprotein receptor-related protein 1 (LRP1) (Shibata et al., 2000; Storck et al., 2016). It is highly expressed in all functional elements of the so called neurovascular unit that constitutes the BBB (Deane et al., 2009; Kanekiyo et al., 2012; Storck et al., 2016; Liu et al., 2017). LRP1 has been described to rapidly remove soluble Aβ from the interstitial fluid (ISF) by both endocytosis and lysosomal degradation or in the BBB, transcytosis across cells. Due to its fast endocytosis rate compared to other members of the low-density receptor (LDLR) family (Li et al., 2001), LRP1 is important for the rapid removal of Aβ from brain. LRP1 is synthesized as a 600 kDa protein and cleaved by furin into two subunits: 1) an extracellular N-terminal ligand-binding subunit (alpha-chain) which binds more than 40 structurally diverse ligands; 2) the intracellular subunit which contains two NPxY motifs and is important for adaptor protein binding, signaling and endocytosis (Roebroek et al., 2006; Reekmans et al., 2010). The distal NPxYxxL motif plays distinct roles in the functionality of LRP1. It has been shown that cells from mice carrying a knock-in mutation in this sequence exhibit reduced internalization rates for LRP1 (Roebroek et al., 2006; Pflanzner et al., 2010). Moreover, the NPxYxxL motif is crucial for basolateral sorting of LRP1 in polarized epithelial cells (Marzolo et al., 2003). In human capillaries LRP1 has been shown to be primarily expressed at the abluminal, brain-facing side of polarized endothelial cells (Zhao et al., 2015). A recent study has found that LRP1 in endothelial cells is associated with phosphatidylinositol binding clathrin assembly (PICALM) protein (Zhao et al., 2015). Several genome-wide association studies have identified PICALM as a consistent link to AD (Harold et al., 2009; Jun et al., 2010; Kauwe et al., 2011; Lambert et al., 2011; Lee et al., 2011; Piaceri et al., 2011; Ferrari et al., 2012; Kamboh et al., 2012; Schnetz-Boutaud et al., 2012; Parikh et al., 2014; Xu et al., 2015). PICALM influences clathrin–dependent endocytosis of the Aβ–LRP1 complex and therefore, affects Aβ clearance across the BBB by association with LRP1 after Aβ internalization, directing LRP1 to the early endosome and sorting endosome, leading to Aβ transcytosis. It has been shown that a reduction of PICALM expression aggravates Aβ pathology in a mouse model by reducing brain clearance of Aβ (Zhao et al., 2015).
Another protein that mediates efflux of Aβ across the BBB is the ABC transporter ABCB1 also known as P-glycoprotein (ABCB1/P-gp). ABCB1/P-pg is expressed at the luminal, blood-facing side of the endothelium and normally prevents brain entry of xenobiotics from the blood stream by its proposed flippase mechanism (Miller et al., 2008; Hartz et al., 2010; Hartz et al., et al., 2016). The mechanism of ABCB1/P-gp in efflux of brain-derived Aβ is not fully understood. One reason is its luminal expression in the endothelium. Hence, ABCB1/P-gp has no direct access to soluble Aβ in the ISF. Another reason is the size and charge distribution of Aβ as most other ABCB1/P-gp substrates are much smaller molecules. Therefore, the exact mechanism of action by which ABCB1/P-gp mediates the efflux of brain derived Aβ remains elusive. All proteins, LRP1, ABCB1/P-gp and PICLAM have been linked to AD. PICALM reductions in brain endothelium in AD correlate with Aβ and AD neuropathology and cognitive impairment, LRP1 and ABCB1/P-gp are both reduced in AD patients and loss of either reduces Aβ transport across the BBB and exacerbates cognitive impairments in mouse models (Kang et al., 2000; Shibata et al., 2000; Cirrito et al., 2005; Zhao et al., 2015; Storck et al., 2016). Here, we show that in brain endothelial cells these three proteins are functionally linked and that, in the brain-to-blood transcytosis machinery, the functional interplay of all three proteins is needed for rapid Aβ transport across the brain endothelium.
2. Materials and methods
2.1. Animals
All animal studies were conducted in compliance with European and German guidelines for the care and use of laboratory animals and were approved by the Central Animal Facility of the University of Mainz and the ethical committee on animal care and use of Rhineland-Palatinate, Germany or approved by the Institutional Animal Care and Use Committee of the University of Kentucky (protocol#: 2014–1233, PI: Hartz). They were carried out in strict accordance with AAALAC regulations, the US Department of Agriculture Animal Welfare Act, and the Guide for the Care and Use of Laboratory Animals of the NIH. Male, 10–12 week old CD-1 mice were purchased from Charles River Laboratories (Portage, MA, USA). Animals were group-housed and kept under controlled environmental conditions (21 °C; 51–62% relative humidity; 12-hour light/dark cycle) using an EcoFlo Allentown ventilation system (Allentown Inc., Allentown, NJ, USA). Animals were monitored at least once a day and had free access to tap water and Harlan Teklad Chow 2918 rodent feed (Harlan Laboratories Inc., Indianapolis, IN, USA). After shipping, animals were allowed to acclimate to their new environment for at least 7 days prior to experiments. Inducible, brain endothelial–specific LRP1 knockout (Lrp1BE−/−) and littermate control (Lrp1BEfl/fl) mice were used as described before (Storck et al., 2016). Generation of mice with inactive LRP1 (LRP1 NPxYxxL knock-in) has been described in detail before (Roebroek et al., 2006; Reekmans et al., 2010; Pflanzner et al., 2011).
2.2. Reagents and antibodies
[125I]-Aβ1–42 was purchased from Phoenix Peptide, and [14C]-inulin was purchased from PerkinElmer. PSC833 (Valspodar, SML0572), Cyclosporin A (30024), rabbit anti-Rab7 (R4779, WB: 3 μg/mL), rabbit anti-PICALM (HPA019053, WB: 1:250, IP: 3μL/sample, ICC: 1:500), rabbit anti-β-actin (A2066, WB: 1:1000), HRP-conjugated goat anti-mouse (A9169; WB: 1:5000), HRP-conjugated goat anti-rabbit (A5278, WB: 1:10,000) and HRP-conjugated rabbit anti-goat (A5420, WB: 1:2000) were purchased from Sigma-Aldrich. 11E2 mouse anti-LRP1 was purified from hybridoma cell culture as described before (Storck et al., 2016). Rabbit anti-LRP1 for immunohistochemistry (IHC) in isolated capillaries was purchased from Abcam (ab92544; Cambridge, MA, USA). Polyclonal 1704 rabbit anti-LRP1 (WB: 1: 10,000, IP: 3 μL/sample, ICC: 1:2000) was generated as described before (Pietrzik et al., 2002). H-241 rabbit anti-Mdr (sc-8313, WB: 1:1000, IP: 3 μL/sample – detects MDR1&MDR3 mouse/rat/human), D11 mouse anti-Mdr1 (sc55510, ICC: 1:100), goat anti-Rab11 (sc-6565, WB: 1:200) were purchased from Santa Cruz Biotechnology. Polyclonal rabbit anti RAB11A/B (15903-1-AP, IP: 3μL/sample, ICC: 1:500) was purchased from Proteintech. Alexa-Fluor 546-conjugated goat anti-rabbit (A11010) and Alexa-Fluor 488-conjugated goat anti-mouse (A11001) for ICC were purchased from Thermo Fisher Scientific. Alexa-Fluor 488-conjugated secondary IgG used for IHC was obtained from Invitrogen (Carlsbad, CA, USA).
2.3. Isolation and culture of primary brain capillary endothelial cells
2.3.1. Mouse brain endothelial cells
Primary mouse brain capillary endothelial cells were isolated from 10- to 12-week-old mice according to a standard protocol as described previously (Pflanzner et al., 2011; Pflanzner et al., 2012; Storck et al., 2016). Cells were plated on 24-well Transwell filters (pore size, 0.4 μm; surface area, 33.6 mm2; Greiner Bio-One) coated with collagen IV/fibronectin (Sigma-Aldrich). Cultures were maintained in DMEM supplemented with 20% plasma-derived bovine serum (First Link), 100 U/ml penicillin (Gibco), 100 μg/ml streptomycin (Gibco), 2 mM l-glutamine (Gibco), 4 μg/ml puromycin (Alexis), and 30 μg/ml endothelial cell growth supplement (Sigma-Aldrich) at 37 °C and 5% CO2. Cells were cultured in the cellZscope device, in which transendothelial electrical resistance (TEER) and capacitance were monitored over time. Puromycin was withdrawn after 4 days in culture. When cells reached confluency and the capacitance was around 1 μF/cm2, culture medium was removed and serum-free DMEM/Ham’s F12 (Gibco) medium containing 1 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin was added. 550 nM hydrocortisone (Sigma-Aldrich) was supplemented to induce high TEER (≥80 Ohms/cm2). The following day transport studies were performed.
2.3.2. Porcine brain endothelial cells
Primary porcine brain endothelial cells were isolated as described previously (Mahringer et al., 2009). Cells were plated in Earlés medium 199 (FG0615, Biochrom) supplemented with 0.7 mM l-glutamine, 10 mg/ml penicillin/streptomycin and 10% FCS on 6 well (pore size, 0.4 μm; surface area, 425.4 mm2; Greiner Bio-One) and 24-well Transwell filters (pore size, 0.4 μm; surface area, 33.6 mm2; Greiner Bio-One) coated with collagen IV/fibronectin (Sigma-Aldrich). Cells on 24-well Transwell filters were cultured in the cellZscope device, in which TEER and capacitance were monitored over time. Puromycin was withdrawn after 2 days in culture. When cells reached confluency and the capacitance was around 1 μF/cm2, culture medium was removed and serum-free DMEM/Ham’s F12 (Gibco) medium containing 0.7 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin was added. 550 nM hydrocortisone (Sigma-Aldrich) was supplemented to induce high TEER (≥300 Ohms/cm2). The following day the hydrocortisone-supplemented, serum-free media was changed; transport studies were performed 48 h after serum starvation. To some cells 50 μM chloroquine was added 12 h prior to transport studies to raise the intracellular pH and inhibit receptor-mediated transport function.
2.4. Brain capillary isolation
For immunoblot analysis, capillaries were isolated using dextran gradient centrifugation followed by sequential cell-strainer filtration as previously described (Storck et al., 2016). First, mouse brains were harvested, forebrains were pooled, meninges were removed, and the tissue was mechanically dissociated in ice-cold phosphate-buffered saline (PBS) containing 2% fetal bovine serum (FBS) using a stainless steel razor blade followed by vigorously pipetting. Dextran (70 kDa, Carl Roth) was subsequently added to yield a final concentration of 16% and the samples were thoroughly mixed on ice. The samples were then centrifuged at 6000g for 15 min. The microvessel-depleted brain (brain tissue minus capillaries) remained on top of the dextran gradient. The capillary pellet located at the bottom of the tubes was collected and sequentially filtered through a 100 μm and 45 μm cell strainer (BD Falcon). The capillaries remaining on top of the 45 μm cell strainer were collected in PBS and lysed for immunoblot analysis.
For IHC, Aβ transport studies and ABCB1/P-gp transport assays brain capillaries were isolated as described previously (Hartz et al., 2008). In brief, mice were euthanized by CO2 inhalation and then decapitated. Brains were removed, dissected, and homogenized in ice-cold PBS buffer (2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4, 0.9 mM CaCl2, and 0.5 mM MgCl2 supplemented with 5 mM d-glucose, 1 mM sodium pyruvate, pH 7.4). Ficoll was added to a final concentration of 15%, and the homogenate was centrifuged at 5800g for 15 min at 4 °C. After resuspending the pellet in 1% bovine serum albumin (BSA) in PBS, the capillary suspension was passed over a glass bead column. Capillaries adhering to the glass beads were collected by gentle agitation in 1% BSA/PBS. Capillaries were washed with BSA-free PBS and used for experiments.
2.5. Transport assay in isolated capillaries
2.5.1. ABCB1/P-gp transport activity
To determine ABCB1/P-gp transport activity, freshly isolated brain capillaries were incubated for 1 h at room temperature with the fluorescent ABCB1/P-gp-specific substrate NBD-cyclosporin A (NBD-CSA, 2 μM in PBS buffer) (Hartz et al., 2008). As before, specific, luminal NBD-CSA fluorescence was taken as the difference between total luminal fluorescence and fluorescence in the presence of the ABCB1/P-gp-specific inhibitor PSC833 (5 μM) (Hartz et al., 2008) or 11E2 anti-LRP1 antibody (15 μg/mL) (Storck et al., 2016).
2.5.2. ABCB1/P-gp-mediated Aβ transport
For Aβ-transport studies, isolated capillaries were incubated for 1 h at room temperature with 5 μM HiLyte™-hAβ1–42 (AnaSpec, Fremont, CA, USA) in PBS buffer in the presence or absence of 5 μM PSC833 or 10 μM Cyclosporine A (CSA) and/or 15 μg/mL 11E2 anti-LRP1 antibody. For each treatment, images of 7–10 capillaries were acquired by confocal microscopy Leica TCS SP5 confocal microscope with a 63 × 1.2 NA water immersion objective (Leica Instruments, Wetzlar, Germany). Luminal fluorescence intensity was quantitated using Image J v.1.48v (Wayne Rasband, NIH, USA).
2.6. In vitro transcytosis of [125I] Aβ1–42
Aβ transcytosis across endothelial monolayers was studied in a standard transport model as described before (Pflanzner et al., 2010; Pflanzner et al., 2011; Pflanzner et al., 2012; Storck et al., 2016). [125I] Aβ1−42 (0.2 nM) and 1 μCi/ml [14C]-inulin, a marker for paracellular diffusion, were added to serum-free media supplemented with 550 nM hydrocortisone and 40 mM HEPES and incubated at 37 °C. For pharmacological ABCB1/P-gp inhibition 5 μM PSC833 or 10 μM CSA was added to the luminal compartment. For inhibition of LRP1 15 μg/mL 11E2 anti-LRP1 antibody was added to the abluminal compartment. To study blood-to-brain transport, samples were taken from the luminal compartment after 60 min. To investigate the amount of intact [125I] Aβ1−42 transported to the luminal side, TCA was added to a final concentration of 8% and incubated for 10 min at 4 °C. Samples were then centrifuged at 10,000g for 10 min. Pellets (representing intact [125I] Aβ1−42) were counted for [125I]. Transport of intact [125I] Aβ1−42 across the monolayer was calculated as Aβ1−42 transcytosis quotient (TQ) using the following formula: Aβ1−42 TQ = ([125I] − Aβ1−42 luminal/[125I] – Aβ1−42 input)/([14C] − inulin acceptor/[14C] − inulin input). Probes were counted on a Wallac Wizard2 2470 automatic γ-counter (PerkinElmer) for [125I] or on a Tri-Carb 2800 TR Liquid Scintillation Analyser (PerkinElmer) for [14C].
2.7. LRP1 immunostaining of mouse brain capillaries
Isolated brain capillaries were fixed with 3% paraformaldehyde/0.25% glutaraldehyde for 30 min at room temperature. After washing with PBS, capillaries were permeabilized with 0.5% Triton X-100 for 30 min and washed with PBS. Capillaries were blocked with 1% BSA for 60 min and incubated overnight at 4 °C with a 1:500 dilution of monoclonal antibody against LRP1 (ab92544; Abcam, Cambridge, MA, USA). After primary antibody incubation, capillaries were washed and incubated with Alexa-Fluor 488-conjugated secondary IgG (1:750, Invitrogen, Carlsbad, CA, USA) for 1 h at 37 °C. Nuclei were counter-stained with 2 μg/ml DAPI. Negative controls for each treatment were processed without primary antibody and showed negligible background fluorescence (data not shown). Immunofluorescence was visualized by confocal microscopy (Zeiss LSM 710 inverted confocal microscope, 40× 1.2NA water immersion objective, 488 nm line of argon laser, Carl Zeiss Inc., Thornwood, NY, USA).
2.8. Uptake of [125I]-Aβ1−42
Internalization of [125I]-Aβ1−42 was performed as previously described for LRP1 ligand alpha-2 macroglobulin (Pflanzner et al., 2011). Briefly, confluent cells were blocked for 30 min with medium containing 4 μg/mL BSA and incubated for 1 h at 37 °C in medium (supplemented with 1 mM Ca2+, 20 mM 4-(2-hydroxyethyl)-1-piper-azineethanesulfonic acid (HEPES), 4 μg/mL BSA) plus 0.1 nM [125I]-Aβ1−42 in the presence of either 15 μg/ml 11E2 anti-LRP1 or 15 μg/ml unspecific mouse IgG. Subsequently, cells were washed 5 times with ice-cold PBS. Remaining surface bound [125I]-Aβ1–42 was removed by two washes with acidic PBS/HCl (pH 2) before cells were lysed in 0.2 N NaOH. TCA was added to a final concentration of 50% and incubated for 10 min at 4 °C. Samples were then centrifuged at 10,000g for 10 min. Pellets (representing intact intracellular [125I-Aβ1–42) were counted on a Wallac Wizard2 2470 automatic γ-counter (PerkinElmer) for [125I].
2.9. Cell culture of immortalized cell lines
bEnd.3 cells (ATCC, Manassas, VA, USA) and wildtype (wt) or mouse embryonic fibroblasts (MEF) deficient for LRP1 (LRP1−/−) were described previously (Kang et al., 2000; Pietrzik et al., 2002; Pflanzner et al., 2011; Meister et al., 2015) and cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) high glucose medium containing 10% fetal bovine serum and 100 U/ml penicillin/streptomycin (Gibco). For Aβ stimulation experiments, bend.3 cells were washed twice with PBS and stimulated with 1 nM Aβ1–40 in serum-free media for 5 min at 37 °C. The same procedure was conducted for co-immunocytochemistry of ABCB1/P-gp together with other proteins involved in Aβ transport.
2.10. Immunoprecipitation, cell lysis and immunoblot analysis
bEnd.3 cells were grown on 150-mm dishes. For immunoprecipitation 3–4 confluent dishes were lysed in 400μL CHAPS lysis buffer (50 mM Tris–Cl pH 7.5, 150 mM NaCl, 0.02% NaN3, 0.5% CHAPS) supplemented with a cocktail proteinase inhibitors [PhosStop, Complete, Roche Applied Science]). Homogenates were centrifuged for 20 min at 15,000g, and the supernatant was collected. 3 μL of rabbit anti-PICALM (HPA019053), 3 μL of 1704 anti-LRP1 or 3 μL of rabbit anti-Mdr (sc-8313) antibody was added to the samples and rotated at 4 °C overnight. 30 μL of equilibrated ProteinA-beads (17-0618-01, GE Healthcare) were added and rotated for 4 h at 4 °C. Beads were washed three times with CHAPS lysis buffer and proteins were eluted with 15 μL 2× Roti-Load (Carl-Roth) for 5 min at 95 °C. Co-immunoprecipitated proteins were subjected to SDS-gel electrophoresis using tris-glycine gels (BioRad) and transferred onto nitrocellulose membranes (Millipore) and then blocked in 5% (w/v) skim milk in TBST (20 mM Tris, 137 mM NaCl, 0.1% [v/v] Tween-20). Proteins were detected with antibodies described under 2.2. Chemiluminescence signal detection was carried out using HRP-conjugated secondary antibodies and ELC assay solutions (Millipore and Thermo Scientific Pierce) and LAS-3000 mini (Fujifilm). Western blots signals were quantified using NIH ImageJ (version 1.44).
For immunoblot analysis of capillaries, isolated capillary fragments were solubilized in lysis buffer (50 mM TrisOH, 150 mM NaCl, 0.02% [w/v] NaN3, 1% [v/v] Nonidet P-40 supplemented with a cocktail of phosphatase and proteinase inhibitors [PhosStop, Complete, Roche Applied Science]). Homogenates were centrifuged for 20 min at 15,000g, and the supernatant was collected. 10 μg of capillary lysate was separated on 4–12% Bis-Tris gels (NuPAGE™, Invitrogen) gels by SDS-PAGE, transferred onto nitrocellulose membranes (Millipore)
2.11. Endocytosis assay of FITC-labeled human alpha-2-macroglobulin
The endocytosis assay was adapted from the procedure performed previously (Reekmans et al., 2010). Briefly, subconfluent cells on cover slips were washed with PBS and pre-incubated with fresh serum-free medium containing 0.2% BSA to block unspecific binding. After 30 min the cells were then incubated for 1 h at 37 °C in fresh serum-free medium containing 50 μg/ml FITC-labeled human alpha-2-macro-globulin (α2m, Biomac) in the presence of 100 μM chloroquine (Sigma-Aldrich) to inhibit lysosomal activity and either 15 μg/mL unspecific mouse IgG, 15 μg/mL 11E2 anti-LRP1, 5 μM PCS833, or 10 μM CSA. Cells were next washed three times with ice-cold PBS and the remaining surface bound FITC-α 2 M was removed by incubating the cells for 2 min with PBS containing 10 U/ml heparin on ice. Samples were embedded in Prolong Gold Antifade Reagent (Invitrogen), dried overnight at room temperature, and analyzed with the confocal laser scanning microscope LSM710 microscope (Zeiss) equipped with ZEN 2011 SP2 software.
2.12. Immunocytochemistry
bEnd.3 cells were fixed in ice-cold methanol for 30 min. Unspecific binding sites were blocked using a solution of 10% (v/v) goat serum and 1% (w/v) BSA in PBS and incubated in primary antibody solution at 37 °C for 1 h, washed, and incubated at room temperature with secondary antibodies for 90 min. Samples were embedded in Prolong Gold Antifade Reagent (Invitrogen), dried overnight at room temperature, and analyzed with the confocal laser scanning microscop LSM710 microscope (Zeiss) equipped with ZEN 2011 SP2 software.
3. Results
3.1. Lrp1 knockout endothelial cells show decreased levels of ABCB1/P-glycoprotein
Recently, we developed mice that allow for tamoxifen-inducible deletion of Lrp1 in brain endothelial cells (Lrp1BE−/−) (Storck et al., 2016). In various in vivo and in vitro experiments we have shown that deletion of Lrp1 in the endothelium decreased the BBB-mediated efflux of Aβ about 50 percent as compared to LRP1-expressing wildtype endothelium. A detailed analysis of transporter expression ex vivo in isolated capillaries revealed that Lrp1 knockout endothelial cells showed about 60 percent reduction in ABCB1/P-gp expression (Fig. 1A and B) compared to control (Lrp1BEfl/fl) endothelial cells. Vice versa, Cirrito and colleagues have shown previously that LRP1 expression is reduced in isolated capillaries of global ABCB1/P-gp knockout mice (Cirrito et al., 2005). Therefore, our results and studies from other groups suggest that the LRP1 receptor and the ABC transporter ABCB1/P-gp are functionally linked and that knockout of either gene results in a reduction of gene expression of the other.
Fig. 1.
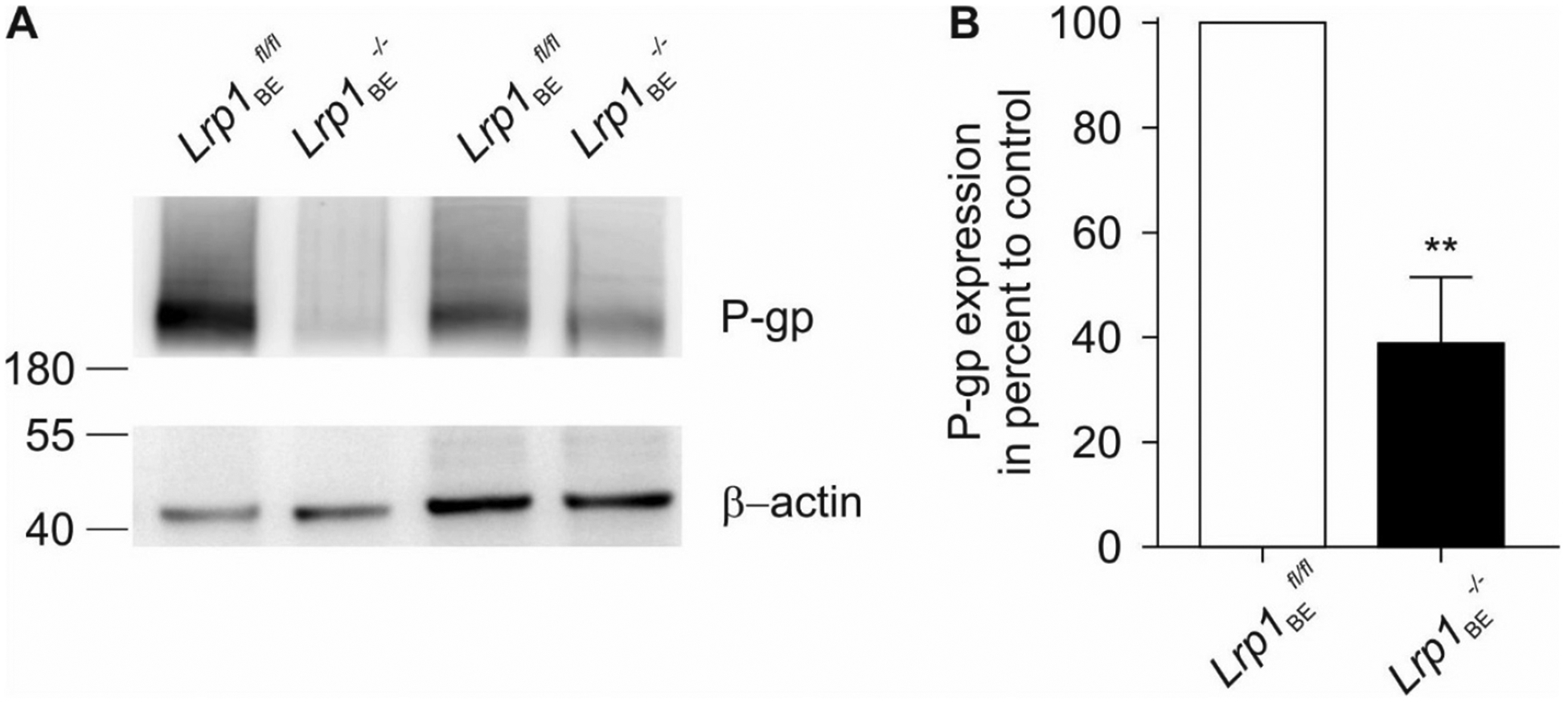
ABCB1/P-gp expression is decreased in Lrp1 knockout capillaries. (A) Representative immunoblots of ABCB1/P-gp in ex vivo capillaries isolated from control (Lrp1BEfl/fl) and brain endothelial-specific Lrp1 knockout (Lrp1BE−/−) mice. An anti-β-actin immunoblot is shown as a loading control. (B) Quantification of relative abundance of ABCB1/P-gp expression of n = 5 independent experiments. For each isolation ≥3 mice per group were used (mean ± SEM). For statistical analyses, unpaired t test was used. **P < 0.01.
3.2. Specific ABCB1/P-gp inhibition has no effect on Aβ transport when LRP1 is absent or inactive
Since LRP1 and ABCB1/P-gp are both implicated in the efflux of Aβ from brain, we aimed to discriminate their individual roles in Aβ transport across the brain endothelium. Therefore, we conducted in vitro transport studies with radiolabeled Aβ across primary endothelial cells isolated from Lrp1BE−/− and control mice. Confirming our previous findings, Aβ transport was reduced in Lrp1 knockout endothelial cells (Fig. 2A). Interestingly, pharmacological ABCB1/P-gp inhibition by PSC833, a specific ABCB1/P-gp inhibitor, reduced Aβ transport across control (Lrp1BEfl/fl) endothelial cells but had no additional effect on Aβ transport across an Lrp1 knockout endothelial monolayer (Fig. 2A). Pharmacological inhibition of ABCB1/P-gp by another ABCB1/P-gp substrate CSA showed the same effect as PSC833 and inhibited Aβ transport across control (Lrp1BEfl/fl) endothelial cells but had no additional effect on Aβ transport on Lrp1 knockout endothelial cells (Fig. 2B).
Fig. 2.
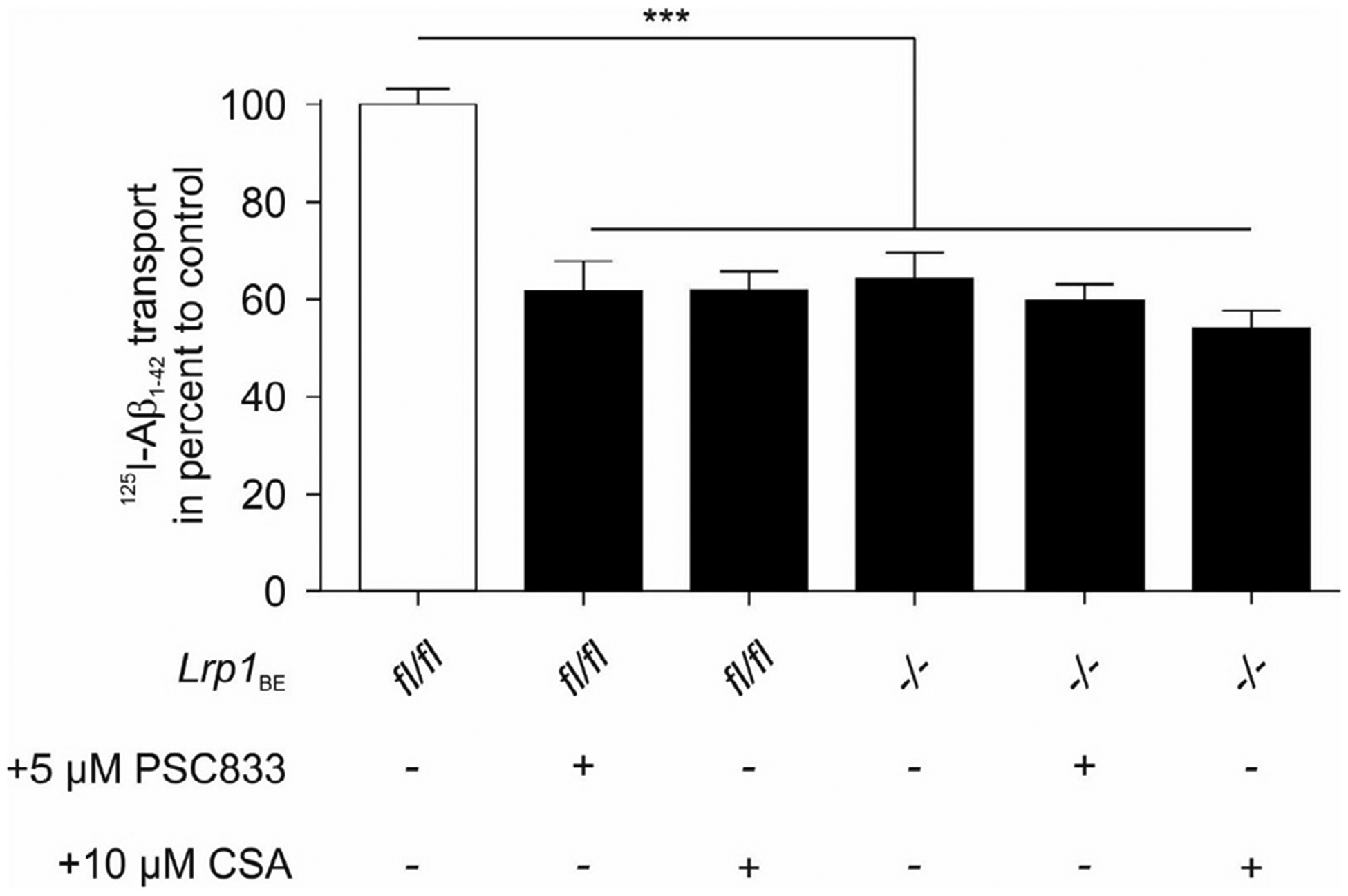
No effect of ABCB1/P-gp inhibition on [125I] Aβ1−42 transcytosis across Lrp1 knockout endothelial cells. [125I]-Aβ1−42 transport across LRP1 knockout (Lrp1BE−/−) and control (Lrp1BE−fl/fl) primary mouse brain capillary endothelial cell monolayer was studied in the presence of 1 μCi/ml [14C]-inulin to normalize for passive diffusion. Transcytosis was analyzed in the brain-to-blood direction (abluminal to luminal) by measuring the dpm for [14C]-inulin and the cpm for the TCA-precipitable [125I]-radioactivity. Transport rates were normalized to transport rates of control Lrp1BEfl/fl brain endothelial cells in the absence of any inhibitors. Transport was studied at a physiological concentration of 0.1 nM [125I]-Aβ1−42 and 5 μM PSC833 or 10 μM Cyclosporine A (CSA). Data represent mean ± SEM of n = 18; n = 21, n = n = 27, n = 17, n = 11 from left to right of at least 2 independent experiments. For statistical analyses, repeated-measures ANOVA followed by Bonferroni multiple comparisons was used. ***P < 0.001.
In order to understand whether the effect on reduced Aβ transport in Lrp1 knockout endothelial cells is due to Lrp1 deletion or a reduction in ABCB1/P-gp expression, we repeated the Aβ transport studies with endothelial cells isolated from mice carrying a knock-in mutation in the NPxYxxL amino acid sequence of Lrp1 (Roebroek et al., 2006; Reekmans et al., 2010). This knock-in mutation inactivates receptor endocytosis and therefore, inhibits LRP1 transport function since the NPxYxxL sequence is important for LRP1 internalization. Analysis of ABCB1/P-gp expression in capillaries isolated from LRP1 NPxYxxL knock-in mice showed enhanced expression of ABCB1/P-gp compared to capillaries isolated from control animals. Despite enhanced ABCB1/P-gp expression (Fig. 3A), LRP1 NPxYxxL knock-in endothelial cells showed about 50 percent reduction in transport of [125I]-Aβ1–42 across an endothelial monolayer (Fig. 3B) similar to previous transport studies using the less aggregation-prone isoform Aβ1–40 (Pflanzner et al., 2011). Also in this setup, pharmacological inhibition of ABCB1/P-gp showed about 50 percent reduction in Aβ transport across endothelial cells isolated from control mice but had no significant effect in LRP1 NPxYxxL knock-in endothelial cells (Fig. 3).
Fig. 3.
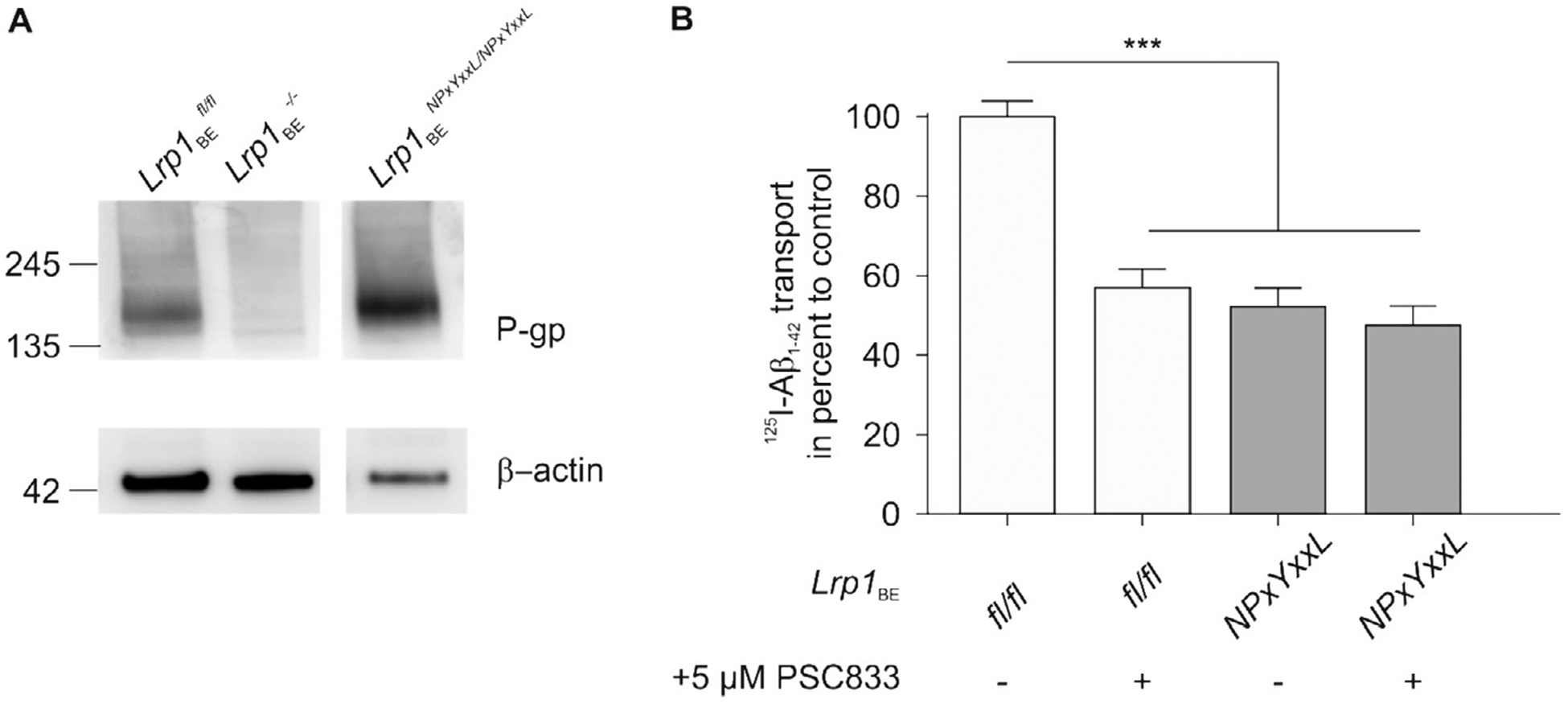
Despite enhanced ABCB1/P-gp expression in LRP1 NPxYxxL knock-in endothelial cells, ABCB1/P-gp inhibition has no effect on [125I] Aβ1−42 transcytosis. (A) Immunoblot analysis of ABCB1/P-gp in ex vivo capillaries isolated from control (Lrp1BEfl/fl), endothelial-specific Lrp1 knockout (Lrp1BE−/−) and LRP1 NPxYxxL knock-in (Lrp1BENPxYxxL/NPxYxxL) mice. Representative result of n = 2 independent isolations with n ≥ 3 mice per group. Lysates were analyzed on the same Western blot but rearranged for clearer presentation(B) [125I]-Aβ1−42 transport across the primary mouse brain capillary endothelial cell monolayer was studied in the presence of 1 μCi/ml [14C]-inulin to normalize for passive diffusion. Transcytosis was analyzed in the brain-to-blood direction (abluminal to luminal) by measuring the dpm for [14C]-inulin and the cpm for the TCA-precipitable [125I] radioactivity. Transport rates were normalized to transport rates of Lrp1BEfl/fl brain endothelial cells in the absence of PSC833. Transport was studied at a physiological concentration of 0.1 nM [125I]-Aβ1−42. Data represent mean ± SEM of n = 12; n = 20, n = 19, n = 14 from left to right of 3 independent experiments. For statistical analyses, repeated-measures ANOVA followed by Bonferroni multiple comparisons was used. ***P < 0.001.
3.3. The anti-LRP1 antibody 11E2, is a potent inhibitor for LRP1-mediated Aβ transport that does not interfere with ABCB1/P-gp function
As we realized that deciphering the individual roles of ABCB1/P-gp and LRP1 in Aβ transport using genetic LRP1 models is limited due to the changes in ABCB1/P-gp expression upon LRP1 modulation, we aimed to inhibit LRP1 pharmacologically. Receptor-associated protein (RAP) has widely been used to inhibit LRP1 function but its specificity has been debated due to its binding capacity to other members of the LDLR family (Storck and Pietrzik, 2017). Recently, we developed the monoclonal LRP1-antibody 11E2 that binds to LRP1 N-terminus (Storck et al., 2016). Uptake studies of radiolabeled Aβ in wild-type mouse embryonic fibroblast (MEF) cells expressing LRP1 revealed that 11E2 monoclonal anti-LRP1 antibody significantly decreased the uptake of Aβ (Fig. 4A) but had no significant effect of Aβ uptake in MEF cells deficient for Lrp1. (Fig. 4B). Next, we determined ABCB1/P-gp transport activity in isolated brain capillaries by using an assay we previously described (Hartz et al., 2010; Hartz et al., 2016; Hartz et al., 2017). In this assay, isolated brain capillaries are incubated with the fluorescent ABCB1/P-gp substrate NBD-cyclosporin A (NBD-CSA, 2 μM) for 1 h to steady state. Capillaries are then imaged with a confocal microscope followed by quantitative image analysis of NBD-CSA fluorescence in the capillary lumen. In brain capillaries with low ABCB1/P-gp transport activity, less NBD-CSA is transported into the capillary lumen compared to control capillaries, resulting in lower luminal NBD-CSA fluorescence (Fig. 4C). Thus, luminal NBD-CSA fluorescence levels is a measure for ABCB1/P-gp transport activity. Despite its effect of blocking LRP1-mediated uptake of Aβ into wt MEF cells (Fig. 4A), and high LRP1 expression in isolated capillaries from CD-31 mice (Fig. 4D + E) the 11E2 anti-LRP1 antibody did not have any effect on the general function of ABCB1/P-gp activity as determined by an assay using fluorescently labeled ABCB1/P-gp substrate NBD-CSA (Fig. 4C). These data indicate that the monoclonal 11E2 anti-LRP1 antibody effectively blocked LRP1-mediated uptake of Aβ but did not interfere with ABCB1/P-gp transport activity.
Fig. 4.
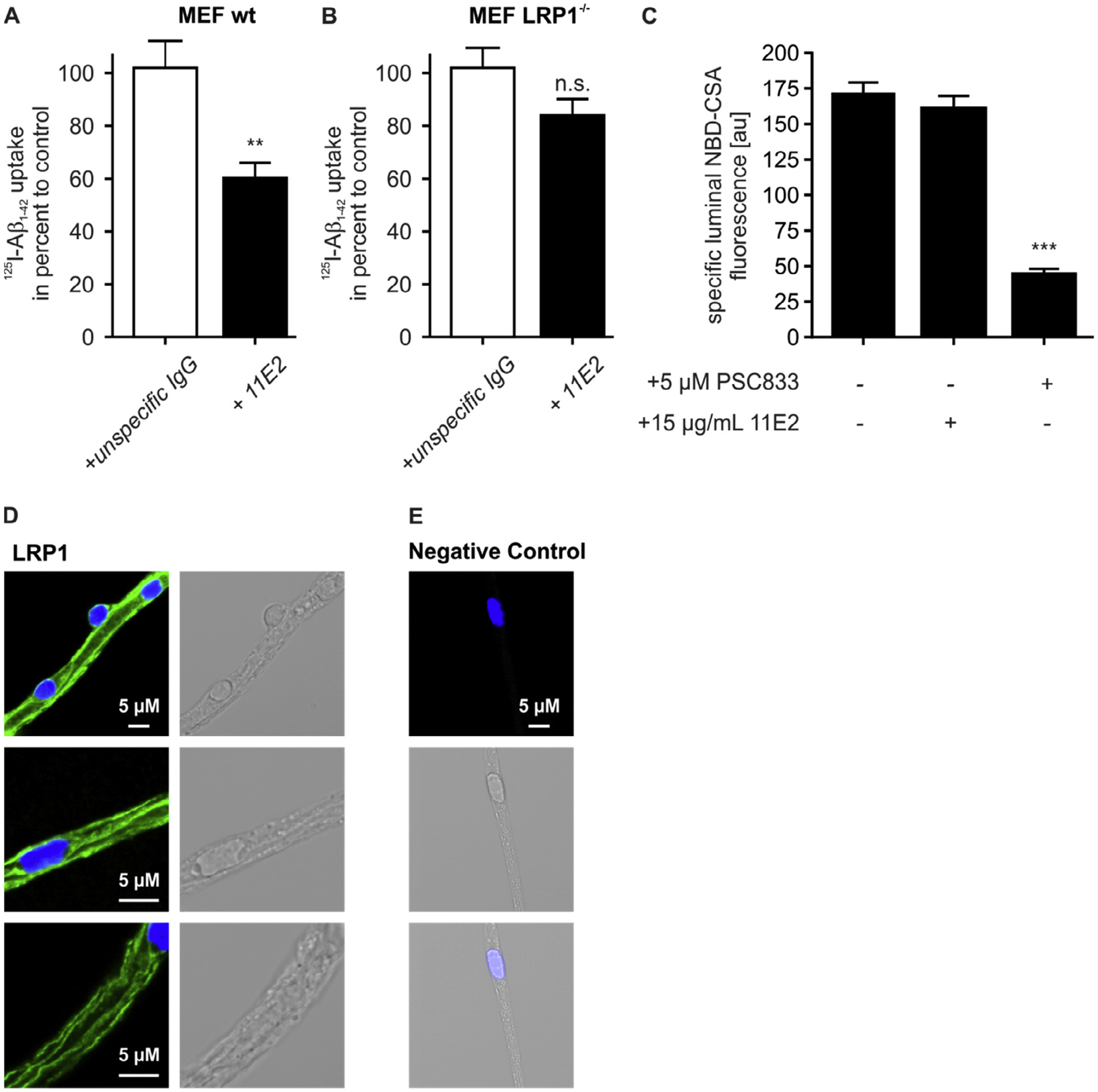
11E2 anti-LRP1 antibody is a potent LRP1 inhibitor that does not interfere with ABCB1/P-gp function. (A) 11E2 anti-LRP1 antibody reduces the uptake of 0.1 nM [125I] Aβ1−42 in wt mouse embryonic fobroblasts (MEF wt) but (B) has no effect on the uptake of 0.1 nM [125I] Aβ1−42 in Lrp1-defecient mouse embryonic fobroblasts (MEF LRP1−/−) results of (A) and (B) represent the mean and SEM of n ≥ 9 of one experiment. (C) 11E2 anti-LRP1 antibody has no effect on ABCB1/P-gp function. For luminal NBD-CSA fluorescence, each data point represents the mean value of 7 capillaries from a single preparation (pooled tissue from 30 CD-1 mice); variability is given by SEM bars. Units are arbitrary fluorescence units (scale 0–255). (D) Immunofluorescent staining for LRP1 in isolated brain capillaries. Scale bar: 5 μm. For statistical analyses, repeated-measures ANOVA followed by Bonferroni multiple comparisons was used. **P < 0.01, ***P < 0.001.
3.4. Dual inhibition of ABCB1/P-gp and LRP1 does not have an additive effect on Aβ transport
To exclude species-specific differences in the regulation of ABCB1/P-gp and LRP1 in mice we additionally measured Aβ transport across a porcine endothelial cell monolayer and in isolated wildtype ex vivo mouse capillaries in the presence of LRP1 and/or ABCB1/P-gp inhibitors. First, however, we verified that both ABCB1/P-gp PSC833 and CSA had no effect on LRP1 function. We performed uptake studies of FITC-alpha-2-macroglobulin (α2m), a specific LRP1 ligand, in the presence or absence of the inhibitors. Whereas 11E2 anti-LRP1 antibody almost completely blocked uptake of FITC-α2m into the cell (Fig. 5B), neither PCS833 (Fig. 5C) nor CSA (Fig. 5D) showed any effect. This demonstrates that the ABCB1/P-gp inhibitors do not affect the general function of LRP1.
Fig. 5.
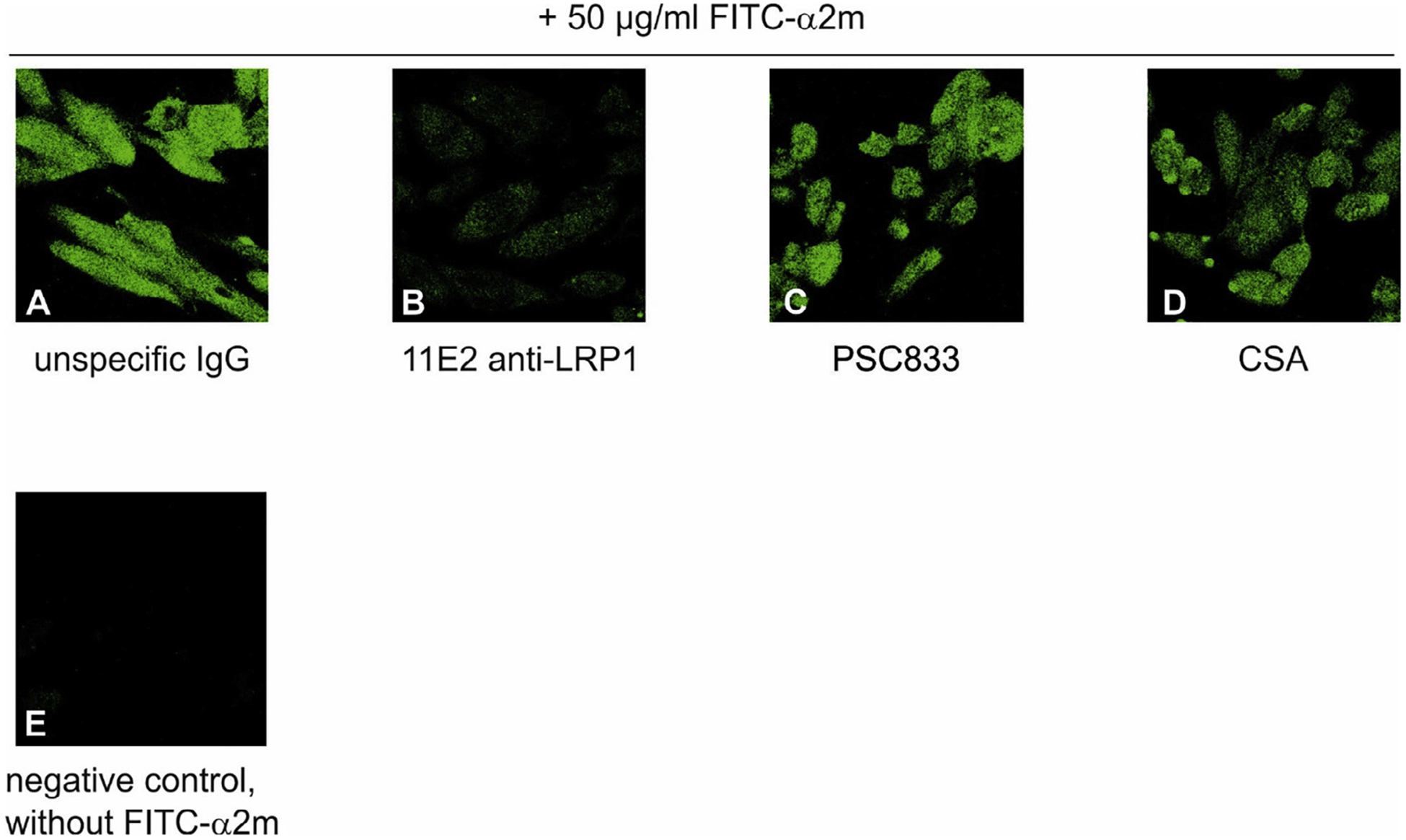
ABCB1/P-gp inhibitors PSC833 and CSA do not affect LRP1 function Uptake of FITC-labeled alpha-macroglobulin (FITC-α2m) into LRP1-expressing CHO K1 cells. Cells were incubated in the presence of 50 μg/ml FITC-α2m and 15 μg/mL unspecific IgG (A), 15 μg/mL 11E2 anti-LRP1 (B), 5 μM PSC833 (C) or 10 μM Cyclosporine A (CSA) for 60 min, washed, fixed and subjected to fluorescence detection. Negative control without FITC-α2m (D).
Interestingly, in the Aβ transport studies across a porcine endothelial cells and in isolated mouse capillaries pharmacological inhibition of LRP1 using the 11E2 antibody showed the same effect as ABCB1/P-gp inhibition using ABCB1/P-gp inhibitors PSC833 or CSA (Fig. 6). Moreover, simultaneous inhibition of ABCB1/P-gp and LRP1 did not have any additive effect on Aβ transport (Fig. 6). One explanation for the lack of an additive effect of both inhibitors could be the interaction of both receptors in intracellular endocytosis pathways. To identify a common pathway, we treated cells with 50 μM chloroquine, which raises the intracellular pH and prevents the disassociation of ligand-receptor complexes in intracellular vesicles. This treatment indeed inhibited transport of Aβ as expected (Fig. 6A) suggesting that receptor-mediated transport processes are involved in transcytosis. Since chloroquine treatment and inhibition of either ABCB1/P-gp or LRP1 had the same effect as simultaneous inhibition of both proteins, we speculated that LRP1 and ABCB1/P-gp mediate a concerted transport of Aβ across murine and porcine brain endothelia.
Fig. 6.
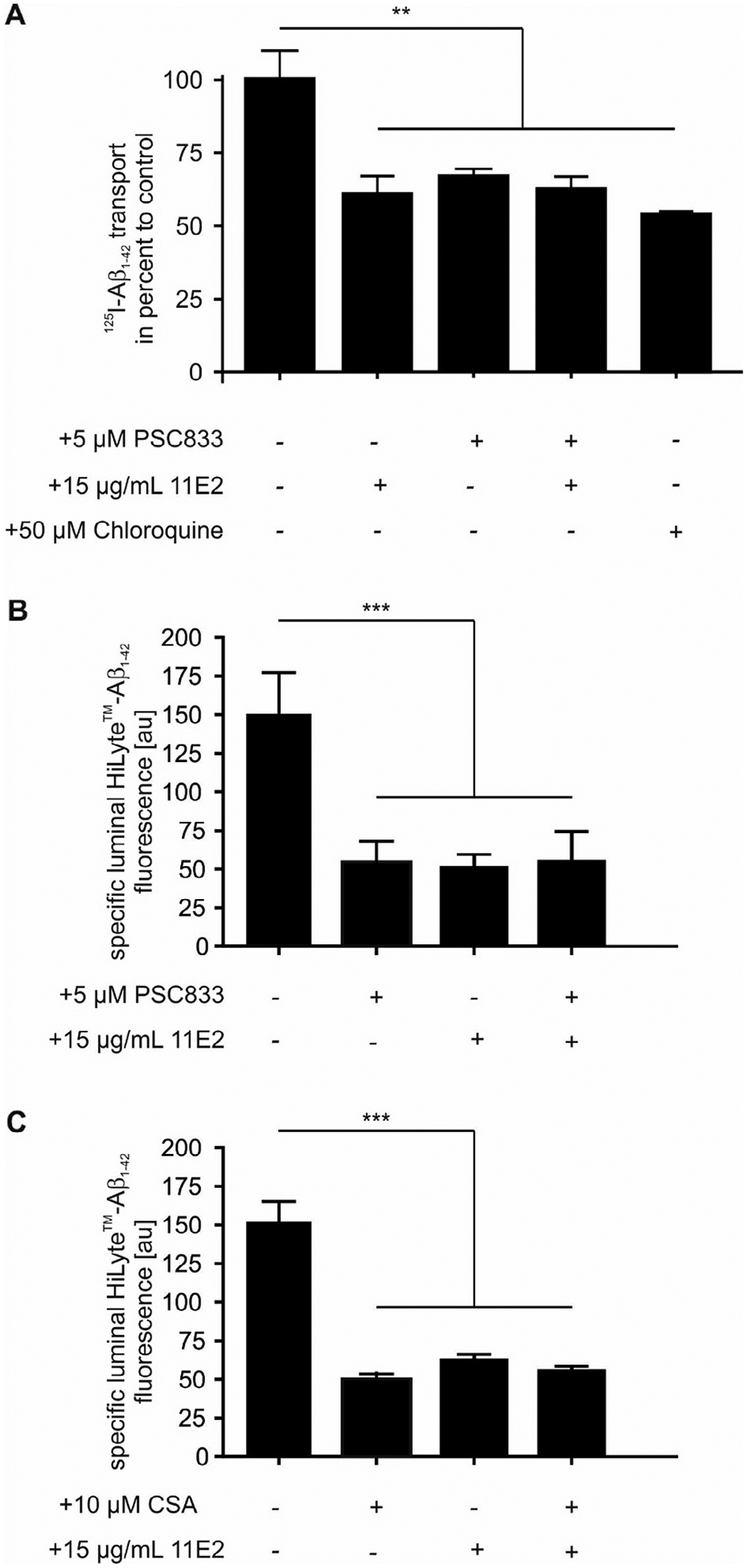
Simultaneous inhibition of LRP1 and ABCB1/P-gp has no additional effect on Aβ1−42 transcytosis. (A) Aβ1−42 transport across a primary porcine brain endothelial monolayer [125I]-Aβ1−42 transport was studied in the presence of 1 μCi/ml [14C]-inulin to normalize for passive diffusion. Transcytosis was analyzed in the brain-to-blood direction (abluminal to luminal) by measuring the dpm for [14C]-inulin and the cpm for the TCA-precipitable [125I] radioactivity. Transport rates were normalized to transport rates of untreated brain endothelial cells. Transport was studied at a physiological concentration of 0.2 nM [125I]-Aβ1−42. For ABCB1/P-gp Inhibition 5 μM PSC833 and for LRP1 inhibition 20 μg/mL 11E2 anti-LRP1 was used. Data represent mean ± SEM of n = 12; n = 20, n = 19, n = 14 from left to right of two independent experiment. The chloroquine group represents data of n = 4 of one experiment. (B + C) Aβ1−42 transport in isolated capillaries. For luminal HiLyte™-Aβ1–42 fluorescence, each data point represents the mean value of 10 capillaries from a single preparation (pooled tissue from 30 CD-1 mice); variability is given by SEM bars. For ABCB1/P-gp Inhibition 5 μM PSC833 (B) or 10 μM Cyclosporine A (CSA) (C) and for LRP1 inhibition 15 μg/mL 11E2 anti-LRP1 was used. Units are arbitrary fluorescence units (scale 0–255). For statistical analyses, repeated-measures ANOVA followed by Bonferroni multiple comparisons was used. **P < 0.01; ***P < 0.001.
3.5. PICALM links LRP1- and ABCB1/P-gp-mediated transport in endothelial cells
If LRP1 and ABCB1/P-gp mediate a concerted efflux of Aβ across brain endothelial cells, the proteins have to interact or be present in a common intracellular vesicle. In order to study the connection between ABCB1/P-gp and LRP1, we investigated whether we can co-immunoprecipitate LRP1 and ABCB1/P-gp in immortalized brain endothelial cells bEnd.3 and isolated brain capillaries. Using 1704, an anti-LRP1 antibody (Pietrzik et al., 2002), we could immunoprecipitate LRP1. Also, ABCB1/P-gp was co-immunoprecipitated together with LRP1 from bEnd.3 lysates (Fig. 7A + B) and isolated capillaries (Fig. 8). Vice versa, we were able to co-immunoprecipitate LRP1 with an anti-ABCB1/P-gp antibody (Figs. 7B, 8) implicating that both proteins either interact directly or exist in a common cellular vesicle. Previously, it has PICALM has been shown to regulate internalization of Aβ bound to LRP1. PICALM knockdown mice show reduced Aβ transport across the BBB and knockdown of PICALM in human primary endothelial cells inhibits LRP1 internalization. Aβ exposure triggered the accumulation of LRP1 and PICALM in Rab11-positive vesicles. The authors claim, that by facilitating LRP1 internalization, PICALM guides Aβ-trafficking to Rab5- and Rab11-positive vesicles leading to Aβ endothelial transcytosis (Zhao et al., 2015). The effects of PICALM on LRP1 internalization are well characterized but our data suggested an interplay of LRP1 and ABCB1/P-gp in Aβ efflux through the endothelium. Previously, we have shown that Aβ triggers the internalization of ABCB1/P-gp from the cell membrane into the cell (Hartz et al., 2016). We analyzed whether PICALM is also associated with ABCB1/P-gp and might be involved in the trafficking of ABCB1/P-gp. Immunoprecipitation of PICALM in the endothelial cell line bEnd.3 as well as in isolated capillaries from mice revealed that PICALM is associated with LRP1 and Rab11 (Figs. 7C + 8A). Consistent with previous findings, we found that PICALM is not associated with Rab7, a marker for the late endosome that directs its cargo for lysosomal degradation (Fig. 6C) (Zhao et al., 2015). Interestingly, we found that also the ABC transporter ABCB1/P-gp can be immunoprecipitated together with PICALM in bENd.3 lysates (Fig. 7) and isolated capillaries (Fig. 8B). This implicates that PICALM which mediates LRP1 endocytosis into the cell and which is present in the cell membrane, in early endosomes and in sorting endosomes, at some point, is also associated with ABCB1/P-gp. Sorting endosome marker Rab11 was co-immunoprecipitated with both, LRP1 and P-pg (Figs. 7B + 8A) suggesting that sorting endosomes may represent a vesicle where both LRP1 and ABCB1/P-gp are present and where a potential transfer of Aβ could take place.
Fig. 7.

Co-immunoprecipitation of LRP1, ABCB1/P-gp and PICALM. (A and B) Co-immunoprecipitation of LRP1 and ABCB1/P-gp in brain endothelial cell line bEnd.3 using different antibodies. Rab11 can be co-immunoprecipitated with both ABCB1/P-gp and LRP1. (C) Co-immunoprecipitation of PICALM and LRP1, ABCB1/P-gp, Rab11 but not Rab7. Representative results of experiments done twice (A + B), Data shown from the ABCB1/P-gp-IP (B) is the result of one experiment.
Fig. 8.
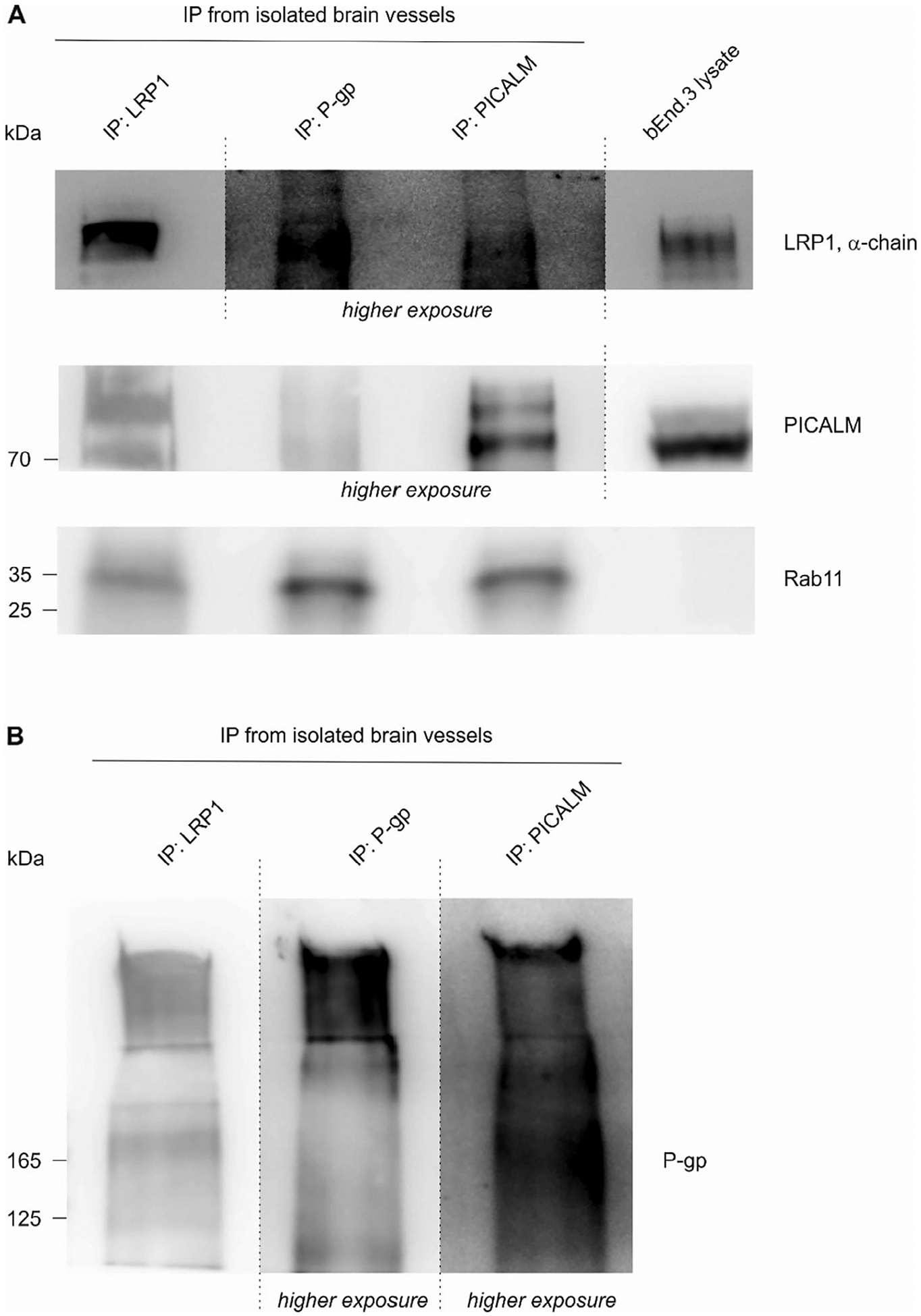
Co-immunoprecipitation of LRP1, ABCB1/P-gp, PICALM and Rab11 from isolated capillaries. (A + B) The three proteins LRP1, ABCB1/P-gp are associated with each other and are present in Rab11-positive vesicles in isolated capillaries, Data shown is the result of one experiment from capillaries isolated from 10 mice.
Previously, we have shown that ABCB1/P-gp is internalized upon Aβ exposure (Hartz et al., 2016). In oder to verify our immunoprecipitation data and support our idea that LRP1 and ABCB1/P-gp meet in the sorting endosome for Aβ transfer, we co-stained ABCB1/P-gp together with LRP1, PICALM or Rab11 in cultivated bEnd.3 cells (Fig. 9) after treatment with Aβ for 5 min. We found that ABCB1/P-gp was highly expressed at the cell membrane but also co-localized with LRP1 inside the cell. PICALM staining showed a punctual staining pattern within bEnd.3 cells that could represent endocytoic vesicels. Moreover, PICALM staining showed co-localization with ABCB1/P-gp staining inside the cell. Rab11 could be detected in structures that looked like small vesicles also co-localizing with ABCB1/P-gp inside the cell. These data verified our immunoprecipitation data, showing that ABCB1/P-gp interacts with LRP1, PICALM and Rab11 in brain endothelial cells.
Fig. 9.
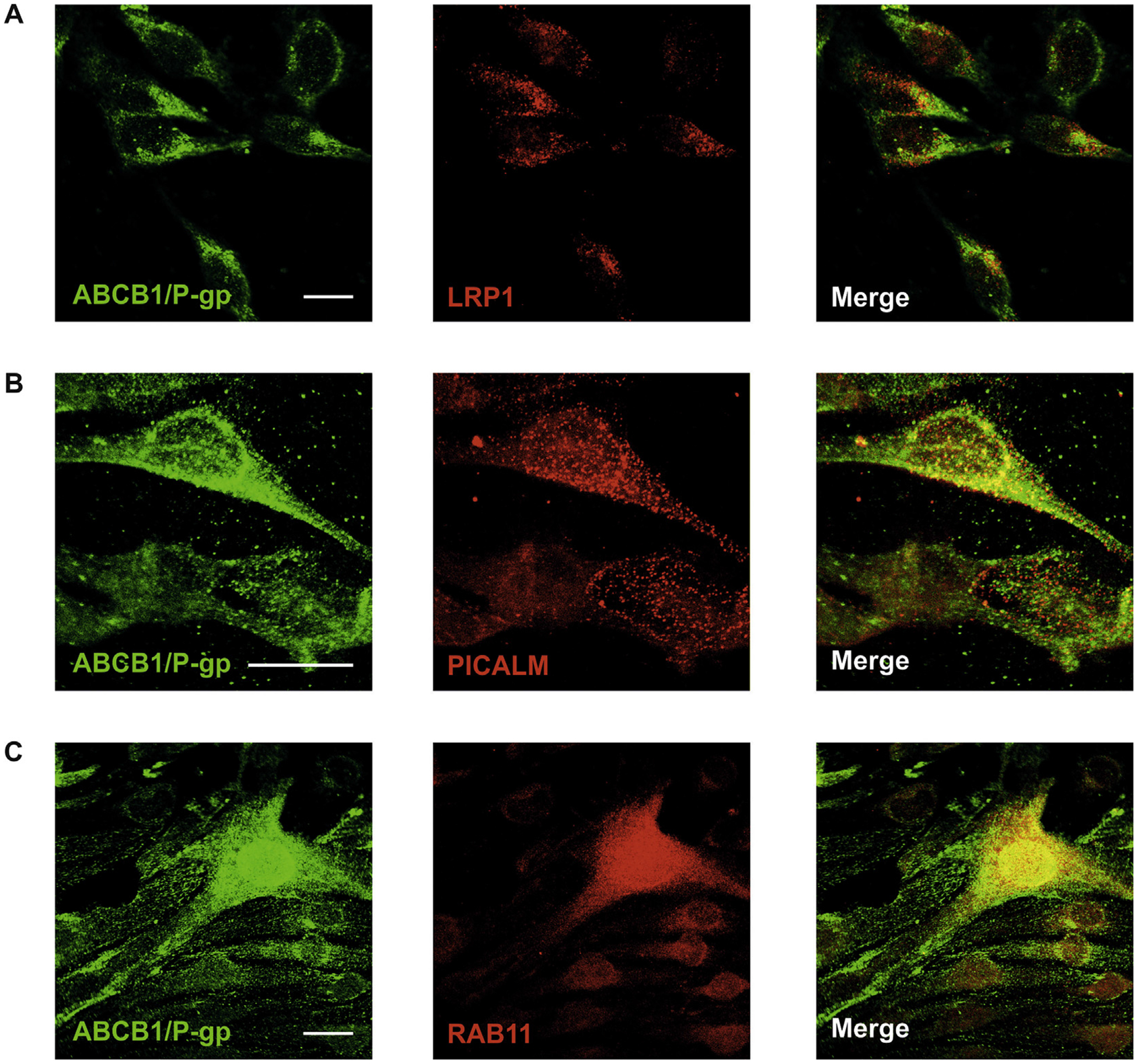
Colocalization of ABCB1/P-gp with other proteins involved in Aβ transport. ABCB1/P-gp co-localizes with LRP1 (A), PICALM (B) and Rab11 (C) in bEnd.3 cells. Scale bar: 20 μm.
As the study by Zhao and colleages showed an enhanced recruitment of LRP1 and PICALM to Rab11-positive vesicles upon Aβ exposure to endothelial cells, we investigated if this was also the case for ABCB1/P-gp. Confirming previous results by Zhao and colleages, we found that in endothelial cells, higher amounts of PICALM and LRP1 could be co-immunoprecipitated with Rab11 upon exposure to 1 nM Aβ for 5 min (Fig. 10). Moreover, also higher amounts of ABCB1/P-gp could be co-immunoprecipitated with Rab11 upon Aβ exposure. As it has been shown that ACB transporters including ABCB1/P-gp in intracellular vesicles remain active in their transport function (Eytan et al., 1997) our results suggest that ABCB1/P-gp, along with PICALM and LRP1, is recruited to Rab11-positive vesicles for the rapid clearance of Aβ out of the cell. Conclusively, our results, for the first time, show that LRP1 and ABCB1/P-gp are associated with each other inside the cell and mediate a concerted transcytosis of Aβ through brain endothelial cells. Moreover, our data show that for the rapid transcytosis of Aβ through the cells the function of both LRP1 and ABCB1/P-gp is needed.
Fig. 10.
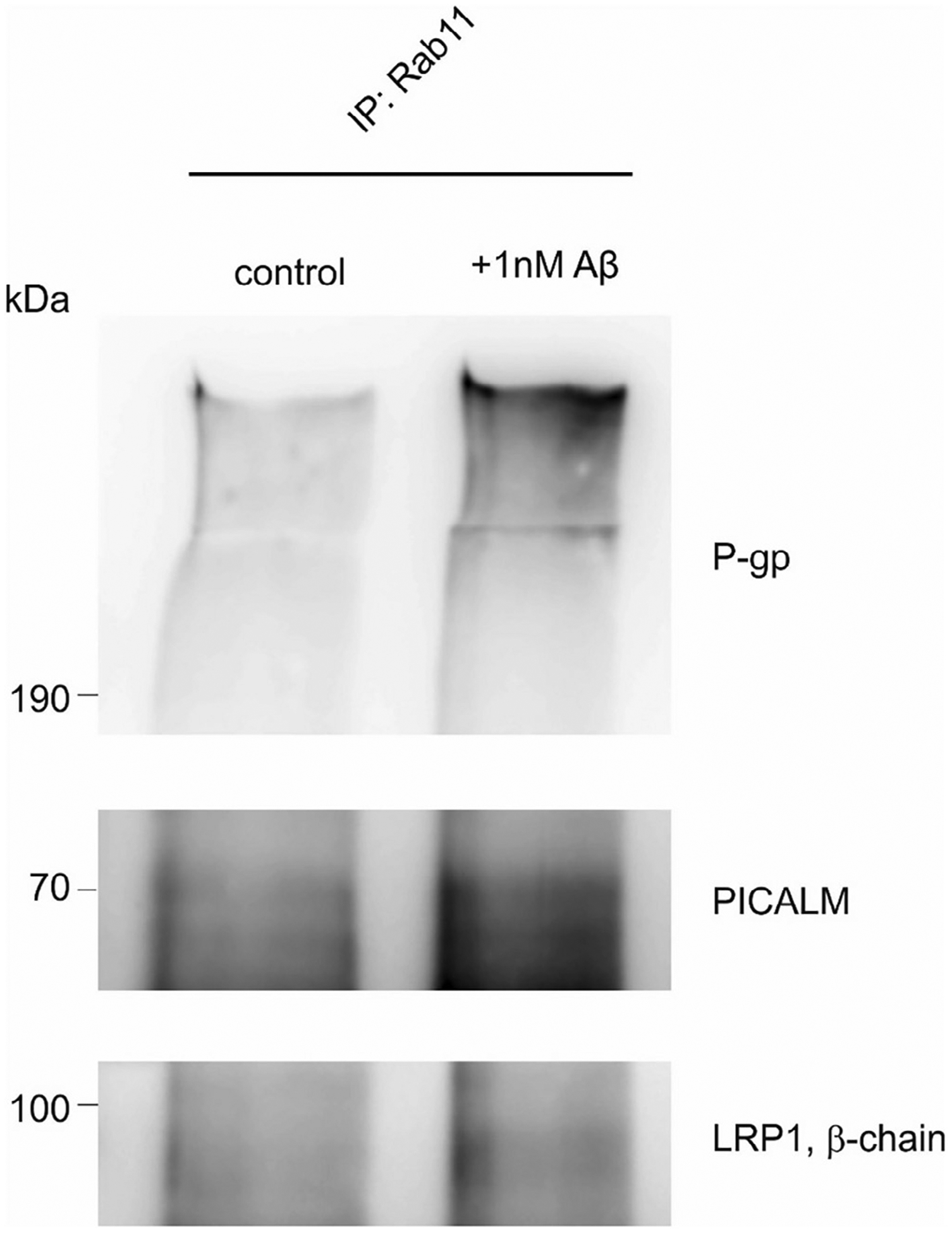
Co-immunoprecipitation of LRP1, ABCB1/P-gp and PICALM with Rab11 after Aβ stimulation. bEnd3 were stimulated with 1 nM Aβ1–40 for 5 min.
4. Discussion
Numerous studies have shown that transport across the BBB is an important elimination route for the rapid removal of brain Aβ (Shibata et al., 2000; Vogelgesang et al., 2004; Cirrito et al., 2005; Hartz et al., 2010; Krohn et al., 2011; Storck et al., 2016). However, the exact molecular mechanisms of Aβ transcytosis have not been fully understood. Both, ABCB1/P-gp and LRP1 have been implicated to play a crucial role in the efflux of Aβ across the brain endothelium (Shibata et al., 2000; Vogelgesang et al., 2002; Vogelgesang et al., 2004; Cirrito et al., 2005; Hartz et al., 2010; Storck et al., 2016). Several studies have shown a downregulation of ABCB1/P-gp and LRP1 in brains and capillaries during aging and in patients and mouse models of AD suggesting that both proteins are somehow linked to AD pathology (Vogelgesang et al., 2002; Vogelgesang et al., 2004; Cirrito et al., 2005; Hartz et al., 2010; Hartz et al., 2016; Storck et al., 2016). As ABCB1/P-gp is located at the luminal, blood-facing side of the endothelium, it has always been questioned how the ABC transporter gets access to brain-derived Aβ. Here, we show that LRP1 is a partner of ABCB1/P-pg providing abluminally-endocytosed Aβ, handing it over to ABCB1/P-gp inside the cell for luminal efflux into blood. Our data furthermore indicate that the LRP1 receptor and the ABC transporter ABCB1/P-gp cannot mediate Aβ efflux on their own but need a mandatory molecular partner for Aβ transcytosis. Both proteins are present in Rab11-positive sorting endosomes where the potential transfer of Aβ may take place (Figs. 7 + 8). The exact intracellular molecular release of Aβ by LRP1 and mechanistic transfer to ABCB1/P-gp is still elusive. Moreover, our data suggest that in brain endothelial cells, next to a rapid LRP1-ABCB1/P-gp efflux machinery, other clearance mechanisms involving other transporters and receptors do exist. Our studies show, that upon inhibition of both LRP1 and ABCB1/P-gp, transport from the abluminal to the luminal compartment across an primary brain endothelial monolayer in vitro is inhibited but not completely abolished. Also, previous studies from our groups have shown that in mice where LRP1 was deleted from brain endothelial cells, which according to our new set of data is sufficient to block the LRP1-ABCB1/P-gp efflux machinery, Aβ peptides can be detected in the blood stream suggesting additional clearance mechanisms from brain. The existence other clearance mechanisms at the BBB has already been suggested by Bell et al. (2007) who in their studies elegantly show that Aβ bound to Apolipoprotein J (APOJ) can be cleared via low-density receptor-related protein-2 (LRP2) that is expressed in brain endothelial cells. If this LRP2/APOJ-dependent clearance also needs a efflux transporter at the luminal side of the brain endothelium (e. g. ABCB1/P-gp or other ABC transporters) remains to be studied in further detail. Also, other ABC transporter and SLC transporters have been shown to play a role in Aβ clearance from brain (Krohn et al., 2011; Do et al., 2012; Dodacki et al., 2017) and/or uptake of Aβ from the periphery (Do et al., 2013). In future studies it will be interesting to see whether these membrane transporters are also connected with each other or even have a common functional partner in brain endothelial cells. An additional important regulator for Aβ efflux seems to be the cytosolic protein PICALM that, in the case of LRP1/P-pg-mediated clearance, appears to direct both LRP1- and ABCB1/P-gp-trafficking inside the cell and is an intracellular molecular link between both proteins (Figs. 7–9). It has shown before that PICALM takes over a central role in Aβ blood-brain barrier transcytosis and clearance: Picalm deficiency in mice diminished Aβ clearance across the BBB and accelerated Aβ pathology in a manner that was reversible by endothelial PICALM re-expression. Using primary human brain endothelial monolayers, Zhao et al., elegantly showed that PICALM regulated PICALM/clathrin-dependent internalization of Aβ bound to LRP1 inside the cells and guided Aβ trafficking to Rab5 and Rab11. Whereas PICALM’s role in LRP1 trafficking through the endothelial cell has been shown before (Zhao et al., 2015), this is the first study to our knowledge that shows that PICALM is associated with ABCB1/P-gp. Like LRP1 and P-pg, PICALM is highly expressed in brain microvessels (Zhao et al., 2015) (more then 2-fold higher than in capillary-depleted brain homogenates) suggesting an important role in protein trafficking in endothelial cells. Several case-controlled genome wide association (GWAS) studies have repeatedly shown the association between various PICALM loci and late onset AD (Harold et al., 2009; Jun et al., 2010; Kauwe et al., et al., 2011; Lambert et al., 2011; Lee et al., 2011; Piaceri et al., 2011; Ferrari et al., 2012; Kamboh et al., 2012; Schnetz-Boutaud et al., 2012; Parikh et al., 2014; Xu et al., 2015). To date, several molecular pathways have been identified how PICALM modulates AD pathology implicating that PICALM has different roles in Aβ metabolism in different cell types of the brain (Kanatsu et al., 2014; Xu et al., 2015; Zhao et al., 2015). Collectively, our findings suggest that in brain endothelial cells PICALM seems to be important for the rapid LRP1/ABCB1/P-gp-mediated Aβ efflux. As each of the three proteins seem to play their own crucial role in rapid Aβ clearance from the brain, the development of future therapeutic strategies that target the removal of excessive brain Aβ should, therefore, take into consideration that the functional interplay of different cellular components may be necessary for the treatment or prevention of AD pathology.
Funding
The project was funded by collaborative grants to Jens Pahnke and Claus Pietrzik of the Deutsche Forschungsgemeinschaft (PA930/12, PI 379/8-1) and JPND joint EU grant (PROP-AD: BMBF #01ED1605 - Germany, NFR #260786 - Norway) within Horizon 2020/European Union (#643417 - JPco-fuND agreement) and the intramural funding program of the University Medical Center of the Johannes-Gutenberg University Mainz to Steffen Storck.
This project was also supported by grant number 2R01AG039621 from the National Institute on Aging (to A.M.S.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health.
The work of J.P. was also financed by the following grants: Deutsche Forschungsgemeinschaft/Germany (DFG PA930/9); Leibniz Society/Germany (SAW-2015-IPB-2); HelseSØ/Norway (2016062); VIAA/Latvia (NFI/R/2014/023); Norsk forskningsrådet/Norway (247179 NeuroGeM, 251290 FRIMEDIO).
NeuroGeM is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organisations under the aegis of JPND - www.jpnd.eu (CIHR - Canada, BMBF - Germany, NRF #247179 - Norway, ZonMW - The Netherlands).
PROP-AD is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organisations under the aegis of JPND - www.jpnd.eu (AKA #301228 - Finland, BMBF #01ED1605 - Germany, CSO-MOH #30000-12631 - Israel, NFR #260786 - Norway, SRC #2015-06795 - Sweden).
References
- Aleksis R, Oleskovs F, Jaudzems K, Pahnke J, Biverstal H, 2017. Structural studies of amyloid-beta peptides: unlocking the mechanism of aggregation and the associated toxicity. Biochimie 140, 176–192. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM, 2006. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med 12 (7), 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, Streb JW, Guo H, Rubio A, Van Nostrand W, Miano JM, Zlokovic BV, 2009. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat. Cell Biol 11 (2), 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV, 2007. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab 27 (5), 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM, 2005. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Invest 115 (11), 3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Bell RD, Sagare A, Zlokovic BV, 2009. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 8 (1), 16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, Wu Z, Holtzman DM, Zlokovic BV, 2005. IgG-assisted age-dependent clearance of Alzheimer’s amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J. Neurosci 25 (50), 11495–11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do TM, Bedussi B, Chasseigneaux S, Dodacki A, Yapo C, Chacun H, Scherrmann JM, Farinotti R, Bourasset F, 2013. Oatp1a4 and an L-thyroxine-sensitive transporter mediate the mouse blood-brain barrier transport of amyloid-beta peptide. J. Alzheimers Dis 36 (3), 555–561. [DOI] [PubMed] [Google Scholar]
- Do TM, Noel-Hudson MS, Ribes S, Besengez C, Smirnova M, Cisternino S, Buyse M, Calon F, Chimini G, Chacun H, Scherrmann JM, Farinotti R, Bourasset F, 2012. ABCG2- and ABCG4-mediated efflux of amyloid-beta peptide 1–40 at the mouse blood-brain barrier. J. Alzheimers Dis 30 (1), 155–166. [DOI] [PubMed] [Google Scholar]
- Dodacki A, Wortman M, Saubamea B, Chasseigneaux S, Nicolic S, Prince N, Lochus M, Raveu AL, Decleves X, Scherrmann JM, Patel SB, Bourasset F, 2017. Expression and function of Abcg4 in the mouse blood-brain barrier: role in restricting the brain entry of amyloid-beta peptide. Sci. Rep 7 (1), 13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elali A, Rivest S, 2013. The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer’s disease. Front. Physiol 4, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eytan GD, Regev R, Oren G, Hurwitz CD, Assaraf YG, 1997. Efficiency of P-glycoprotein-mediated exclusion of rhodamine dyes from multidrug-resistant cells is determined by their passive transmembrane movement rate. Eur. J. Biochem 248 (1), 104–112. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Moreno JH, Minhajuddin AT, O’Bryant SE, Reisch JS, Barber RC, Momeni P, 2012. Implication of common and disease specific variants in CLU, CR1, and PICALM. Neurobiol. Aging 33 (8) 1846 e1847–1818. [DOI] [PubMed] [Google Scholar]
- Hardy J, 2006. Alzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis 9 (3 Suppl), 151–153. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, Heun R, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J, 2009. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet 41 (10), 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Block ML, Hong JS, Miller DS, 2008. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 22 (8), 2723–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Miller DS, Bauer B, 2010. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol 77 (5), 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Pekcec A, Soldner EL, Zhong Y, Schlichtiger J, Bauer B, 2017. P-gp protein expression and transport activity in rodent seizure models and human epilepsy. Mol. Pharm 14 (4), 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz AM, Zhong Y, Wolf A, LeVine H 3rd, Miller DS, Bauer B, 2016. Abeta40 reduces P-glycoprotein at the blood-brain barrier through the ubiquitin-proteasome pathway. J. Neurosci 36 (6), 1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Naj AC, Beecham GW, Wang LS, Buros J, Gallins PJ, Buxbaum JD, Ertekin-Taner N, Fallin MD, Friedland R, Inzelberg R, Kramer P, Rogaeva E, St George-Hyslop P, Alzheimer’s Disease Genetics C, Cantwell LB, Dombroski BA, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Lunetta KL, Martin ER, Montine TJ, Goate AM, Blacker D, Tsuang DW, Beekly D, Cupples LA, Hakonarson H, Kukull W, Foroud TM, Haines J, Mayeux R, Farrer LA, Pericak-Vance MA, Schellenberg GD, 2010. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol 67 (12), 1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboh MI, Minster RL, Demirci FY, Ganguli M, Dekosky ST, Lopez OL, Barmada MM, 2012. Association of CLU and PICALM variants with Alzheimer’s disease. Neurobiol. Aging 33 (3), 518–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu K, Morohashi Y, Suzuki M, Kuroda H, Watanabe T, Tomita T, Iwatsubo T, 2014. Decreased CALM expression reduces Abeta42 to total Abeta ratio through clathrin-mediated endocytosis of gamma-secretase. Nat. Commun 5, 3386. [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G, 2012. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J. Neurosci 32 (46), 16458–16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH, 2000. Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Invest 106 (9), 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauwe JS, Cruchaga C, Karch CM, Sadler B, Lee M, Mayo K, Latu W, Su’a M, Fagan AM, Holtzman DM, Morris JC, Alzheimer’s Disease Neuroimaging I, Goate AM, 2011. Fine mapping of genetic variants in BIN1, CLU, CR1 and PICALM for association with cerebrospinal fluid biomarkers for Alzheimer’s disease. PLoS One 6 (2), e15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krohn M, Lange C, Hofrichter J, Scheffler K, Stenzel J, Steffen J, Schumacher T, Bruning T, Plath AS, Alfen F, Schmidt A, Winter F, Rateitschak K, Wree A, Gsponer J, Walker LC, Pahnke J, 2011. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Invest 121 (10), 3924–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I, Cascorbi I, Walker LC, Kroemer HK, Warzok RW, Vogelgesang S, 2007. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides–implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 17 (4), 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Zelenika D, Hiltunen M, Chouraki V, Combarros O, Bullido MJ, Tognoni G, Fievet N, Boland A, Arosio B, Coto E, Del Zompo M, Mateo I, Frank-Garcia A, Helisalmi S, Porcellini E, Pilotto A, Forti P, Ferri R, Delepine M, Scarpini E, Siciliano G, Solfrizzi V, Sorbi S, Spalletta G, Ravaglia G, Valdivieso F, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Licastro F, Lathrop M, Soininen H, Amouyel P, 2011. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol. Aging 32 (4) 756 e711–755. [DOI] [PubMed] [Google Scholar]
- Lee JH, Cheng R, Barral S, Reitz C, Medrano M, Lantigua R, Jimenez-Velazquez IZ, Rogaeva E, St George-Hyslop PH, Mayeux R, 2011. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch. Neurol 68 (3), 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu W, Marzolo MP, Bu G, 2001. Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J. Biol. Chem 276 (21), 18000–18006. [DOI] [PubMed] [Google Scholar]
- Liu CC, Hu J, Zhao N, Wang J, Wang N, Cirrito JR, Kanekiyo T, Holtzman DM, Bu G, 2017. Astrocytic LRP1 mediates brain abeta clearance and impacts amyloid deposition. J. Neurosci 37 (15), 4023–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahringer A, Delzer J, Fricker G, 2009. A fluorescence-based in vitro assay for drug interactions with breast cancer resistance protein (BCRP, ABCG2). Eur. J. Pharm. Biopharm 72 (3), 605–613. [DOI] [PubMed] [Google Scholar]
- Marzolo MP, Yuseff MI, Retamal C, Donoso M, Ezquer F, Farfan P, Li Y, Bu G, 2003. Differential distribution of low-density lipoprotein-receptor-related protein (LRP) and megalin in polarized epithelial cells is determined by their cytoplasmic domains. Traffic 4 (4), 273–288. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ, 2010. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330 (6012), 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister S, Storck SE, Hameister E, Behl C, Weggen S, Clement AM, Pietrzik CU, 2015. Expression of the ALS-causing variant hSOD1(G93A) leads to an impaired integrity and altered regulation of claudin-5 expression in an in vitro blood-spinal cord barrier model. J. Cereb. Blood Flow Metab 35 (7), 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DS, Bauer B, Hartz AM, 2008. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev 60 (2), 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahnke J, Frohlich C, Paarmann K, Krohn M, Bogdanovic N, Arsland D, Winblad B, 2014. Cerebral ABC transporter-common mechanisms may modulate neurode-generative diseases and depression in elderly subjects. Arch. Med. Res 45 (8), 738–743. [DOI] [PubMed] [Google Scholar]
- Parikh I, Fardo DW, Estus S, 2014. Genetics of PICALM expression and Alzheimer’s disease. PLoS One 9 (3), e91242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflanzner T, Janko MC, Andre-Dohmen B, Reuss S, Weggen S, Roebroek AJ, Kuhlmann CR, Pietrzik CU, 2011. LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol. Aging 32 (12) 2323 e2321–2311. [DOI] [PubMed] [Google Scholar]
- Pflanzner T, Kuhlmann CR, Pietrzik CU, 2010. Blood-brain-barrier models for the investigation of transporter- and receptor-mediated amyloid-beta clearance in Alzheimer’s disease. Curr. Alzheimer Res 7 (7), 578–590. [DOI] [PubMed] [Google Scholar]
- Pflanzner T, Petsch B, Andre-Dohmen B, Muller-Schiffmann A, Tschickardt S, Weggen S, Stitz L, Korth C, Pietrzik CU, 2012. Cellular prion protein participates in amyloid-beta transcytosis across the blood-brain barrier. J. Cereb. Blood Flow Metab 32 (4), 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaceri I, Bagnoli S, Lucenteforte E, Mancuso M, Tedde A, Siciliano G, Piacentini S, Bracco L, Sorbi S, Nacmias B, 2011. Implication of a genetic variant at PICALM in Alzheimer’s disease patients and centenarians. J. Alzheimers Dis 24 (3), 409–413. [DOI] [PubMed] [Google Scholar]
- Pietrzik CU, Busse T, Merriam DE, Weggen S, Koo EH, 2002. The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J. 21 (21), 5691–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reekmans SM, Pflanzner T, Gordts PL, Isbert S, Zimmermann P, Annaert W, Weggen S, Roebroek AJ, Pietrzik CU, 2010. Inactivation of the proximal NPXY motif impairs early steps in LRP1 biosynthesis. Cell Mol. Life Sci 67 (1), 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roebroek AJ, Reekmans S, Lauwers A, Feyaerts N, Smeijers L, Hartmann D, 2006. Mutant Lrp1 knock-in mice generated by recombinase-mediated cassette exchange reveal differential importance of the NPXY motifs in the intracellular domain of LRP1 for normal fetal development. Mol. Cell Biol 26 (2), 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnetz-Boutaud NC, Hoffman J, Coe JE, Murdock DG, Pericak-Vance MA, Haines JL, 2012. Identification and confirmation of an exonic splicing enhancer variation in exon 5 of the Alzheimer disease associated PICALM gene. Ann. Hum. Genet 76 (6), 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, 2001. Clearing the brain’s amyloid cobwebs. Neuron 32 (2), 177–180. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV, 2000. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest 106 (12), 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storck SE, Meister S, Nahrath J, Meissner JN, Schubert N, Di Spiezio A, Baches S, Vandenbroucke RE, Bouter Y, Prikulis I, Korth C, Weggen S, Heimann A, Schwaninger M, Bayer TA, Pietrzik CU, 2016. Endothelial LRP1 transports amyloid-beta1–42 across the blood-brain barrier. J. Clin. Invest 126 (1), 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storck SE, Pietrzik CU, 2017. Endothelial LRP1 – a potential target for the treatment of Alzheimer’s disease: theme: drug discovery, development and delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res 34 (12), 2637–2651. [DOI] [PubMed] [Google Scholar]
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Menard J, Zetterberg H, Wisniewski T, de Leon MJ, 2015. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol 11 (8), 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW, 2002. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 12 (7), 535–541. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, Siegmund W, Walker LC, Pahnke J, 2004. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res 1 (2), 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Tan L, Yu JT, 2015. The role of PICALM in Alzheimer’s disease. Mol. Neurobiol 52 (1), 399–413. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, Owens NC, Rege SV, Si G, Ahuja A, Zhu D, Miller CA, Schneider JA, Maeda M, Maeda T, Sugawara T, Ichida JK, Zlokovic BV, 2015. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci 18 (7), 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]