

Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused over 1.4 million deaths worldwide. Repurposing existing drugs offers the fastest opportunity to identify new indications for existing drugs as a stable solution against coronavirus disease 2019 (COVID-19). The SARS-CoV-2 main protease (Mpro) is a critical target for designing potent antiviral agents against COVID-19. In this study, we identify potential inhibitors against COVID-19, using an amalgam of virtual screening, molecular dynamics (MD) simulations, and binding-free energy approaches from the Korea Chemical Bank drug repurposing (KCB-DR) database. The database screening of KCB-DR resulted in 149 binders. The dynamics of protein-drug complex formation for the seven top scoring drugs were investigated through MD simulations. Six drugs showed stable binding with active site of SARS-CoV-2 Mpro indicated by steady RMSD of protein backbone atoms and potential energy profiles. Furthermore, binding free energy calculations suggested the community-acquired bacterial pneumonia drug ceftaroline fosamil and the hepatitis C virus (HCV) protease inhibitor telaprevir are potent inhibitors against Mpro. Molecular dynamics and interaction analysis revealed that ceftaroline fosamil and telaprevir form hydrogen bonds with important active site residues such as Thr24, Thr25, His41, Thr45, Gly143, Ser144, Cys145, and Glu166 that is supported by crystallographic information of known inhibitors. Telaprevir has potential side effects, but its derivatives have good pharmacokinetic properties and are suggested to bind Mpro. We suggest the telaprevir derivatives and ceftaroline fosamil bind tightly with SARS-CoV-2 Mpro and should be validated through preclinical testing.
Keywords: COVID-19, SARS-CoV-2, Drug repurposing, Virtual screening, Molecular dynamics, Free energy calculations
Graphical abstract

1. Introduction
Coronavirus disease 2019 (COVID-19) is a pandemic viral pneumonia and a threat to global public health. COVID-19 has resulted in over 1.4 million deaths from more than 61.8 million cases worldwide as of Dec 1, 2020 [1]. COVID-19 is caused by a novel coronavirus (nCoV) termed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is a member of the larger coronavirus family including severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS CoV) [[2], [3], [4]]. The main protease (Mpro) of SARS-CoV-2 is a non-structural protein that is responsible for processing the polyprotein translated from viral RNA and is a promising therapeutic target for COVID-19 [[5], [6], [7]]. Early evidence in SARS-CoV suggested that Mpro inhibitors may abrogate the viral replication [8]. The atomic-level resolution of the SARS-CoV-2 protease structure must allow rational drug design against COVID-19 [4,9,10]. Drug repurposing (DR) provides the fastest possibilities to find existing drugs that may help treat COVID-19 [11]. Currently, the anti-HIV-1 (human immunodeficiency virus) drugs lopinavir and ritonavir have been repurposed for SARS-CoV-2 Mpro and tested in clinical trials in China [12]. Lopinavir and ritonavir act in combination to abrogate the function of an important enzyme ‘protease’ which is essential for the viral replication. The efficacy and safety of a combination of an anti-HIV-1 drug darunavir and cobicistat are under phase 3 trials for COVID-19 (NCT04252274). Another example of drug repurposing is the Ebola drug remdesivir that has been approved by the U.S. Food and Drug Administration (FDA) for treating COVID-19 patients [[13], [14], [15]]. Chloroquine and hydroxychloroquine have been used to treat malaria and certain inflammatory conditions showed encouraging results against COVID-19 in vitro [[16], [17], [18]]. Corticosteroid methylprednisolone (NCT04244591, NCT04273321), favipiravir (NCT04310228), abidol hydrochloride (NCT04254874, NCT04255017), oseltamivir (NCT04255017, NCT04261270), and danoprevir (NCT04291729) have been subjected to clinical studies in China. Several other antiviral treatments suggested for COVID-19 may include nucleoside analogs, neuraminidase inhibitors, tenofovir disoproxil, lamivudine, and umifenovir [19,20]. These studies reveal the potential of DR drugs in combating COVID-19. Here, we recruited an amalgam of docking-based virtual screening, molecular dynamics (MD) simulations, and binding-free energy approaches to identify suitable existing drugs for the treatment of COVID-19.
2. Materials and methods
2.1. Preparation of molecules
The experimentally determined X-ray crystal structure of SARS-CoV-2 Mpro (PDB: 6LU7) at a resolution of 2.16 Å was downloaded from RCSB Protein Data Bank (PDB) (https://www.rcsb.org/) [21]. The Mpro protein complexes with a peptide-like substrate inhibitor N3. Protein was prepared with the Clean Protein functionality of the Discovery Studio 2017 (DS) software. All water molecules were omitted, hydrogen atoms were added, and bond orders were corrected for the protein. The 2D structures of reference inhibitors lopinavir and ritonavir were downloaded from PubChem structures and subsequently energy-minimized using the CHARMm forcefield implemented under the Minimize Ligands module of DS. The resulting 3D structures ensured that they had the correct bond orders, bond lengths, and bond angles for all the ligands. Proteins structures of SARS-CoV (PDB: 2A5I) [22] and SARS-CoV-2 (PDB: 6LU7) [21] main proteases were superimposed using the Superimpose Proteins tool of the DS software. The sequence alignment between two proteins and root mean square deviation (RMSD) value of the protein structure superimposition were obtained.
2.2. High-throughput virtual screening
The Genetic Optimization for Ligand Docking (GOLD v5.2.2) program was used to find possible binders for SARS-CoV-2 Mpro [23]. The binding site of the SARS-CoV-2 Mpro was determined to be the residues around 10 Å of the bound cocrystal. The automatic genetic algorithm search option of GOLD considers a balance between speed and accuracy for large library screening procedures. The Virtual Screening module of GOLD was used with 30% search efficiency for screening of the Korea Chemical Bank drug repurposing (KCB-DR) database containing 1865 drugs. Further screening of compounds was performed based on the default scoring function Goldscore and a rescoring function Chemscore. Drugs with high Goldscore and low Chemscore binding energy values were selected in comparison with reference inhibitors. A total of 149 drugs binding with Mpro active site were screened. The screened compounds were further subjected to more exhaustive conformational searches using the genetic algorithm with 100% search efficiency. Twenty independent docking runs were performed for each molecule with GOLD. Furthermore, the seven top scoring drugs obtained by docking-based virtual screening were tested through MD simulations and binding free energy calculations.
2.3. Molecular dynamics simulation
MD simulations were performed to further understand the mechanism of protein-drug binding and to get dynamic information about the complex. The filtered hits from molecular docking studies along with reference inhibitors were subjected to MD simulations using the Groningen Machine for Chemical Simulations (GROMACS v5.1.4) package using CHARMM27 forcefield [24]. A separated simulation system was prepared for each selected molecule (Supplementary Table 1). The simulation parameters for all ligands were generated by SwisParam webserver program [25]. Simulations were carried out in dodecahedron boxes with the TIP3P water model, and systems were neutralized by adding counterions. The energy minimization step for each system was conducted using 50000 steps of the steepest descent algorithm by applying a maximum force of 1000 kJ/mol to avoid steric clashes. The minimized system was then equilibrated under NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature) independently. The NVT ensemble was applied for 500 ps at 300K using a V-rescale thermostat [26]. In the NPT ensemble, each system was equilibrated for 1 ns at 1 bar pressure controlled by Parrinnello-Rahman barostat [27]. The LINCS algorithm [28] and particle mesh Ewald (PME) were used to restrain the bonds of heavy atoms and electrostatic interactions, respectively [29]. All simulations were carried out under periodic boundary conditions to avoid edge effects. Finally, a production run of 50 ns for each equilibrated system was performed under NPT conditions. The results were analyzed with DS, GROMACS, and visual molecular dynamics (VMD) software. GROMACS program ‘gmx rms’ was used for calculating RMSD values of both protein and ligands. This program calculates RMSD values of atoms in a molecule with respect to a reference structure by least-square fitting the structure to the reference structure (t2 = 0) as given below
| (1) |
Here, , and ri(t) is the position of atom i at time t. The protein backbone atoms were selected for calculating protein RMSD values while all ligand atoms were selected for calculating ligand RMSD [30]. The root mean square fluctuation (RMSF) and radius of gyration (Rg) calculations were performed by ‘gmx rmsf’ and ‘gmx gyrate’ utilities of GROMACS program, respectively.
2.4. Binding free energy calculations
Prediction of the binding affinities of small-molecule inhibitors to their biological targets plays an important role in structure-based drug design [31]. In this study, we exploited a GROMACS compatible program ‘g_mmpbsa’ to predict the protein-ligand binding free energies [32]. For calculating binding free energy, 50 snapshots of protein-ligand complexes were selected evenly from 0 to 50ns of MD trajectories [33]. The protein-ligand binding free energy is calculated as:
| ΔGbinding = Gcomplex – (Gprotein + Gligand) | (2) |
| Gx = EMM + Gsolvation | (3) |
| EMM = Ebonded + Enon-bonded = Ebonded + (Evdw + Eelec) | (4) |
| Gsolvation = Gpolar + Gnon-polar | (5) |
| Gnon-polar = γSASA + b | (6) |
where x may indicate separated ligand or protein or a ligand-protein complex; EMM is the molecular mechanics potential energy in the vacuum; Gsolvation is the free energy of solvation; SASA is the solvent-accessible surface area; γ = 0.02267 kJ mol−1Å−2 or 0.0054 kcal mol−1Å−2 is a coefficient related to surface tension of the solvent; b = 3.849 kJ/mol or 0.916 kcal mol−1 is a fitting parameter. The binding interaction between protein and ligand was calculated in three terms such as solvation (ΔEsol), van der Waals (ΔEvdw), and the electrostatic (ΔEele) contribution. The final ΔGbind values for protein-ligand complexes were the average values from 0 to 50 ns of MD simulation trajectories.
3. Results
An exhaustive computational approach including high-throughput virtual screening, molecular docking, MD simulations, and free energy calculations was used to identify potential drugs against the SARS-CoV-2 main protease enzyme. A workflow of the current study is represented in Fig. 1 . Anti-HIV protease inhibitors lopinavir and ritonavir are recommended for the treatment of SARS-CoV and SARS-CoV-2 [4,34,35]. Therefore, we selected the bound cocrystal N3, lopinavir, and ritonavir as the reference inhibitors; these were used as standards for comparing the results obtained from our in silico study.
Fig. 1.

A workflow of the computational drug repurposing process for identifying potential drugs ceftaroline fosamil and telaprevir against the main protease of SARS-CoV-2.
3.1. Active site analysis of SARS-CoV-2 Mpro
SARS-CoV and SARS-CoV-2 main proteases are structurally similar and have a RMSD of 0.68 Å and highly conserved sequences with a striking sequence identity of 96.1% (Fig. 2 ) [36]. The close proximity of the inhibitor binding site is highly conserved and the only notable difference is an Ala46Ser mutation whose role is yet to be elucidated. The overall structure of the SARS-CoV-2 Mpro contains three domains: domain I (residues 8–101), domain II (residues 102–184), and domain III (residues 201–303). The active site of Mpro lies at the interface of domain I and domain II. The active site can further be divided into S1, S2, and S3 subsites [37]. The side chains of residues Phe140, His163, and Glu166, and main chain atoms of Leu141, Met165, Glu166, and His172 form the S1 subsite. The S1 subsite is a hydrophobic pocket that determines the substrate specificity in Mpro. The side chains of His41, Met49, Pro52, Met165, and Gln189 form a hydrophobic pocket at the S2 subsite that accommodates a leucine in the substrate. Further, the side chains of Pro168, Gln189, and Gln192 and main chain atoms of Glu166, Gln189, and Thr190 form the S3 subsite of Mpro. The residues His41 and Cys145 form the catalytic dyad required for proteolysis.
Fig. 2.

Sequence and structure comparison of SARS-CoV (PDB: 2A5I) and SARS-CoV-2 (PDB: 6LU7) main proteases. (A) Sequence alignment showing active site mutation of SARS-CoV-2 Mpro in the red box. (B) Alignment of SARS-CoV Mpro and SARS-CoV-2 Mpro structures shown in orange and cyan ribbons, respectively. (C) The active site of the SARS-CoV-2 Mpro bound with the N3 inhibitor shown in green sticks indicating different subsites as surface representation. (D) Superimposed structures of SARS-CoV Mpro and SARS-CoV-2 Mpro showing the location of Ala46Ser mutation near the active site. Fig. 2 is adopted from Fischer et al., 2020 [36].
3.2. High-throughput virtual screening
The experimentally determined three-dimensional structure of SARS-CoV-2 Mpro (PDB: 6LU7) was used for docking-based virtual screening of the KCB-DR database. A total of 149 drugs showed binding with Mpro active site (Supplementary Table 2). Of the 149 drug molecules identified in our study, we found that at least six drugs have antiviral effects against other viruses such as human immunodeficiency virus (HIV) and hepatitis C virus (HCV). Among these, fosamprenavir is a prodrug form of amprenavir and an established inhibitor of HIV-1 protease [38]. Moreover, several other HIV-1 protease inhibitors such as nelfinavir, tipranavir, and lopinavir and a HIV-1 integrase inhibitor elvitegravir were also identified in our study. Furthermore, an HCV NS3-4A protease inhibitor telaprevir was found to bind SARS-CoV-2 Mpro. Molecular conformations for all 149 drugs were obtained by more exhaustive use of the genetic algorithm search strategy. Docking studies of reference inhibitors N3, lopinavir, and ritonavir showed Goldscore values of 66.03, 60.86, and 48.53, respectively (Table 1 ). Likewise, Chemscore DG values of −26.64, −26.09, and −25.77 were obtained for N3, lopinavir, and ritonavir, respectively. Furthermore, a high Goldscore value of 66.03 and a low Chemscore value of −26.64 among reference inhibitors were used as the cutoff for candidate drug screening. Further screening was done on the basis of key molecular interactions exhibited by the drugs with the Mpro active site. The reference inhibitors formed hydrogen bonds with one or more residues such as Phe140, Gly143, His164, Glu166, Gln189, and Thr190 (Supplementary Table 3). Drugs having at least one hydrogen bond with the residues above were retained. Consequently, seven drugs were obtained and show good binding properties with Mpro. The screened drugs included ceftaroline fosamil, remikiren, everolimus, atorvastatin, sildenafil, clofazimine, and telaprevir. The drugs in complex with SARS-CoV-2 Mpro were subjected to MD simulations to study their dynamics of binding at the active site of the virus protein. MD simulations further suggested that only six drugs bind to the active site of the Mpro and two drugs show better binding energies than reference inhibitors. The details of MD simulations and binding free energy calculation results are discussed in sections 3.3, 3.4.
Table 1.
Docking scores and binding energies of SARS-CoV-2 Mpro in complex with reference inhibitors and potential drugs shown with respective standard deviations. The reference inhibitors are indicated in italics.
| No. | Name | Goldscore | Chemscore DG | MMPBSA Binding energy (kJ/mol) |
|---|---|---|---|---|
| 1 | Ceftaroline fosamil | 82.33 ± 11.72 | −33.62 ± 11.27 | −231.89 ± 36.36 |
| 2 | Telaprevir | 68.78 ± 10.04 | −29.32 ± 2.31 | −143.62 ± 16.69 |
| 3 | Ritonavir | 48.53 ± 12.07 | −25.77 ± 2.66 | −122.01 ± 17.88 |
| 4 | Clofazimine | 66.05 ± 1.17 | −33.41 ± 0.35 | −104.63 ± 11.65 |
| 5 | N3 | 66.03 ± 11.76 | −26.64 ± 4.86 | −104.39 ± 20.41 |
| 6 | Lopinavir | 60.86 ± 8.51 | −26.09 ± 4.46 | −101.17 ± 20.49 |
| 7 | Everolimus | 79.63 ± 13.78 | −28.02 ± 8.23 | −100.35 ± 45.38 |
| 8 | Sildenafil | 74.03 ± 7.79 | −28.85 ± 3.81 | −63.71 ± 32.34 |
| 9 | Atorvastatin | 76.06 ± 9.11 | −33.95 ± 5.71 | −59.72 ± 50.01 |
| 10 | Remikiren | 80.04 ± 11.09 | −26.76 ± 11.34 | 24.68 ± 14.97 |
3.3. Molecular dynamics simulations
MD simulations can simulate the dynamic behavior of molecular systems as a function of time. This approach provides an opportunity to understand the flexibility and dynamics of protein-drug binding. The drugs obtained from virtual screening and reference inhibitors were subjected to MD simulations for 50 ns in each system (Supplementary Table 1).
3.3.1. Stability of simulation systems
The RMSD of protein backbone atoms and potential energy profiles obtained from the simulations indicate the overall stability of the simulated systems (Supplementary Fig. S1) [39]. The RMSD values were between 0.15 nm and 0.36 nm for all the protein-drug complexes. Varying RMSD values at the initial few nanoseconds of the simulations indicate the initial adjustment of ligands at the active site of SARS-CoV-2 Mpro [[39], [40], [41]]. The analysis indicated that each system reached steady-state after convergence, and RMSD values remained within the acceptable value of <0.3 nm except the Mpro-Atorvastatin complex (0.36 nm) during the 50 ns simulation run [42].
The stability parameter potential energy (PE) was also analyzed (Supplementary Fig. S1). Our analysis found that each simulation system obtained constant average potential energy profiles and remained stable during the simulation period. The RMSF calculations showed comparatively high fluctuations in Glu47 and Tyr154 versus the other residues. These residues are located away from the active site at the periphery of the protein (Supplementary Fig. S2). The radius of gyration calculations also showed steady profiles for all complexes, which makes the system compact without any aberrant behavior during the simulations (Supplementary Fig. S3). These results suggest that all protein-drug complex systems are stable and reliable for further analysis of binding modes, important molecular interactions, and free energy calculations.
3.3.2. Dynamics of drug binding with SARS-CoV-2 Mpro
The substrate or inhibitor binding site of SARS-CoV-2 Mpro can be divided into several subsites such as S1, S1′, S2, and S3 (Fig. 2C) [43]. The binding events of reference inhibitors and drugs were assessed at these subsites. The reference inhibitor N3 occupied the S1, S1′, S2, and S3 subsites while lopinavir and ritonavir filled the S1′, S2, and S3 subsites during the simulations (Fig. 3 ). Telaprevir showed similar binding with SARS-CoV-2 Mpro as N3 inhibitor occupying the S1, S1′, S2, and S3 subsites (Fig. 4 A). Ceftaroline fosamil, atorvastatin, and clofazimine filled the S1′, S2, and S3 subsites (Fig. 4B–D). Sildenafil showed slightly different binding occupying S1, S1′, and S2, subsites while everolimus filled only the S1’ and S2 subsites (Fig. 4E and F). Interestingly, remikiren lost its binding with the SARS-CoV-2 Mpro active site and moved out of the binding pocket which is also evident through a high binding energy value discussed in the next section (Fig. 4G).
Fig. 3.

Binding events of reference inhibitors (A) N3, (B) lopinavir, and (C) ritonavir at S1, S1′, S2, and S3 pockets during 50 ns simulations.
Fig. 4.

Binding of potential drugs (A) telaprevir, (B) ceftaroline fosamil, (C) atorvastatin, (D) clofazimine, (E) sildenafil, (F) everolimus, and (G) remikiren with the SARS-CoV-2 Mpro active site during 50 ns simulations.
3.4. Binding free energy analysis
Structure-based drug design aims to generate small molecules with desirable pharmacological properties and high affinities to their specific protein target. Binding free energy calculations enable a better understanding of how a protein recognizes its biologically relevant ligand or small molecule inhibitor this ultimately leads to the design of potent therapeutics [44]. An ensemble of molecular conformations generated by MD simulations can be used for free energy calculations to show great promise in drug discovery [45]. Average binding energies of potential drugs against SARS-CoV-2 Mpro were calculated by taking multiple conformations at regular intervals from MD simulations [46]. The reference inhibitors showed binding energies of −104.39 kJ/mol, −101.17 kJ/mol, and −122.01 kJ/mol for N3, lopinavir, and ritonavir, respectively (Fig. 5 ). The favorable binding energies for potential drugs were in the range of −231.89 kJ/mol to −59.72 kJ/mol (Table 1). The reference inhibitors ritonavir, N3, and lopinavir showed binding energies of −122.01 kJ/mol, −104.39 kJ/mol, and −101.17 kJ/mol, respectively. The DR drugs ceftaroline fosamil, telaprevir, clofazimine, everolimus, sildenafil, atorvastatin, and remikiren showed binding energies of −231.89 kJ/mol, −143.62 kJ/mol, −104.63 kJ/mol, −100.35 kJ/mol, −63.71 kJ/mol, −59.72 kJ/mol, and 24.68 kJ/mol, respectively. Ceftaroline fosamil and telaprevir showed better binding energy scores than reference inhibitors. Only remikiren showed a positive binding energy (24.68 kJ/mol) justifying the loss of binding with SARS-CoV-2 Mpro active site; hence, it has little value as a potential drug against COVID-19. We inferred that ceftaroline fosamil and telaprevir can bind with SARS-CoV-2 Mpro more tightly than reference inhibitors.
Fig. 5.

Binding free energy profiles of SARS-CoV-2 Mpro in complex with reference inhibitors and potential drugs calculated by the MM-PBSA method.
3.5. Binding mode analysis of potential drugs
Binding modes of ceftaroline fosamil and telaprevir were assessed using average structures calculated from MD simulation trajectories. Superimposition of complexes of SARS-CoV-2 Mpro with reference inhibitors and drugs indicated that they bind at the same substrate/inhibitor binding cavity of the protein (Supplementary Fig. S4). Ceftaroline fosamil formed four hydrogen bond interactions with Thr24, Thr25, His41, and Thr45 (Fig. 6 ). Hydrogen bonding interaction of ceftaroline fosamil with Thr24 and Thr25 was further supported by recently published crystal structures of SARS-CoV-2 Mpro with inhibitors Z1348371854 (PDB: 5RF6), PCM-0102535 (PDB: 5RG0), and NCL-00025412 (PDB: 5RG3). Additionally, the binding of ceftaroline fosamil with SARS-CoV-2 Mpro was facilitated by hydrophobic interactions with residues such as Cys44, Ser46, Met49, Met165, Arg188, Gln189, Thr190, Ala191, and Gln192 (Table 2 ). van der Waals interactions with residues such as Thr24, Cys44, Thr45, Met49, His164, Met165, Glu166, Pro168, Val186, Asp187, Arg188, Gln189, Ala191, and Gln192 were also observed in the case of ceftaroline fosamil. Telaprevir formed five hydrogen bonds with residues such as Gly143, Ser144, Cys145, and Glu166. Binding site residues such as His41, Met49, Asn142, His163, His164, Met165, Arg188, Thr190, and Gln192 showed hydrophobic interactions with telaprevir. van der Waals interactions with residues such as Thr25, Thr26, leu27, His41, Met49, Phe140, Leu141, Asn142, His163, His164, Met165, Leu167, Pro168, His172, Arg188, Gln189, Thr190, and Gln192 were also identified.
Fig. 6.

Binding modes of top hit drugs (A) ceftaroline fosamil and (B) telaprevir in the active site of SARS-CoV-2 Mpro. The top panel indicates the 2D structures of the drugs. The middle panel represents the binding of drugs at subsites S1, S1′, S2, and S3 of SARS-CoV-2 Mpro. Ceftaroline fosamil and telaprevir are shown in stick form in green and orange, respectively. The lower panel demonstrates the 2D diagram of various molecular interactions exhibited by the drugs with SARS-CoV-2 Mpro. Ligands are represented as purple sticks while interacting amino acids are in orange sticks. Hydrogen bond interactions are indicated in dotted green lines with a distance measure between the hydrogen bond acceptor and donor atoms.
Table 2.
Molecular interactions exhibited by potential drugs with SARS-CoV-2 Mpro. The reference inhibitors are shown in italics.
| Compound | Hydrogen bond interactions |
Hydrophobic interactions | Van der Waals interactions | |||
|---|---|---|---|---|---|---|
| Ligand atom | Amino acid | Amino acid atom | Length (Å) | |||
| Ceftaroline fosamil | O2 | Thr24 | OG1 | 3.21 | Cys44, Ser46, Met49, Met165, Arg188, Gln189, Thr190, Ala191, Gln192 | Thr24, Cys44, Thr45, Met49, His164, Met165, Glu166, Pro168, Val186, Asp187, Arg188, Gln189, Ala191, Gln192 |
| N5 | Thr25 | OG1 | 2.92 | |||
| O18 | His41 | NE2 | 3.14 | |||
| O3 | Thr45 | OG1 | 3.24 | |||
| Telaprevir | O1 | Gly143 | N | 3.11 | His41, Met49, Asn142, His163, His164, Met165, Arg188, Thr190, Gln192 | Thr25, Thr26, leu27, His41, Met49, Phe140, Leu141, Asn142, His163,His164, Met165, Leu167, Pro168, His172, Arg188, Gln189, Thr190, Gln192 |
| O1 | Ser144 | N | 2.97 | |||
| O1 | Cys145 | N | 3.25 | |||
| O3 | Glu166 | O | 3.26 | |||
| O3 | Glu166 | N | 2.81 | |||
| N3 | O7 | Gly143 | N | 2.92 | Leu27, Met49, Cys145, Met165, Pro168, Ala191 | Thr25, Thr26, Leu27, His41, Val42, Tyr54, Leu141, Asn142, Cys145, His163, Met165, Leu167, Pro168, Thr190, Ala191, Gln192 |
| O7 | Ser144 | N | 3.21 | |||
| N5 | His164 | O | 3.61 | |||
| O8 | Glu166 | OE1 | 2.89 | |||
| O | Glu166 | N | 2.78 | |||
| N | Glu166 | O | 2.77 | |||
| N | Gln189 | O | 2.94 | |||
| N2 | Gln189 | O | 3.35 | |||
| Lopinavir | N3 | His41 | O | 2.78 | Met49, His164, Met165, Leu167, Pro168, Phe181, Val186 | Thr25, Thr26, Leu27, Val42, Ser46, Met49, Leu50, Cys85, Gly143, Cys145, Glu166, Leu167, Pro168, Val186, Asp187, Arg188, Thr190, Ala191, Gln192 |
| N4 | Gln189 | OE1 | 1.73 | |||
| Ritonavir | N6 | Ser46 | N | 2.41 | His41, Met49, Asn142, Gly143, Met165, Pro168 | His41, Thr45, Leu50, Asn142, Gly143, Met165, Leu167, Pro168, Asp187, Arg188, Gln189, Thr190, Ala191, Gln192 |
| O1 | Glu166 | N | 1.91 | |||
| N3 | Glu166 | O | 1.98 | |||
We further calculated the distance between hydrogen bond acceptor and donor atoms throughout the 50 ns simulations. The distances are represented as graphs which indicate that the average distance always remained less than the acceptable limit of 3.5 Å (Supplementary Fig. S5). The reference inhibitor N3 has extensive hydrogen bonding interactions with eight bonds with SARS-CoV-2 Mpro active site (Table 2). Lopinavir and ritonavir formed two and three hydrogen bonds with Mpro, respectively. Moreover, the inhibitors N3, lopinavir, and ritonavir formed hydrophobic interactions with residues such as Leu27, His41, Met49, Asn142, Gly143, Cys145, His164, Met165, Leu167, Pro168, Phe181, Val186, and Ala191 as well as van der Waals interactions with Thr25, Thr26, Leu27, His41, Val42, Ser46, Met49, Leu50, Tyr54, Cys85, Leu141, Asn142, Gly143, Cys145, His163, Met165, Glu166, Leu167, Pro168, Val186, Asp187, Arg188, Gln189, Thr190, Ala191, and Gln192. Ceftaroline fosamil and telaprevir exhibit several important molecular interactions with SARS-CoV-2 Mpro active site that are comparable with reference inhibitors. Our results demonstrate that ceftaroline fosamil and telaprevir bind tightly to SARS-CoV-2 Mpro and may inhibit its activity through several molecular interactions such as hydrogen bonds, hydrophobic interactions, and van der Waals interactions.
3.6. Substructure search
A substructure search in PubChem database was performed using telaprevir as an initial structure for identifying compounds having similar structures with different functional groups. We obtained 231 compounds that were further assessed for their good pharmacokinetic properties. The procedure of Kumar et al., 2015 was utilized for measuring the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the compounds using DS software [39]. A total of 14 compounds were obtained with desirable pharmacokinetic properties such as good aqueous solubility, non CYP2D6 binding, low hepatotoxicity, high intestinal absorption, and inability to cross Blood Brain Barrier (BBB). These compounds were further tested for their binding with SARS-CoV-2 Mpro active site through molecular docking. The compounds exhibited a range of docking Goldscores: 51.36 to 79.87; Chemscores were −36.75 to −22.24 (Supplementary Table 4). Finally, 11 compounds having better docking scores when compared to the parent compound telaprevir and reference inhibitors, N3, lopinavir, and ritonavir were retained. Besides the telaprevir forming hydrogen bonds with residues Gly143, Ser144, Cys145, and Glu166, its derivatives also showed hydrogen bonds with other important active site residues such as Asn142, His164, and Gln189 (2D interactions, Supplementary Table 4). These results indicate that telaprevir derivatives have better binding properties and safer pharmacokinetic profiles than telaprevir. The details of compounds with their 2D structures are shown in Table 3 .
Table 3.
Telaprevir derivatives identified by a substructure search in the PubChem database.
| No. | PubChem CID | IUPAC Name | 2D structure |
|---|---|---|---|
| 1 | 10211927 | (3S,3aS,6aR)-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-2-[(2R)-3,3-dimethyl-2-[[(2S)-2-phenyl-2-(pyrazine-2-carbonylamino)acetyl]amino]butanoyl]-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrole-3-carboxamide | 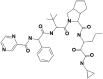 |
| 2 | 143545956 | (3S,3aS,6aS)-2-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-6-methylidene-1,3,3a,4,5,6a-hexahydrocyclopenta[c]pyrrole-3-carboxamide | 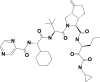 |
| 3 | 67566659 | (1R,2S,3S,6R,7S)-4-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-4-azatricyclo[5.2.1.02,6]dec-8-ene-3-carboxamide | 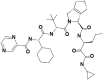 |
| 4 | 42617119 | (1S,7R)-4-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-4-azatricyclo[5.2.1.02,6]dec-8-ene-3-carboxamide | 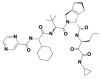 |
| 5 | 3011888 | (3S)–N-[1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-2-[(2S)-3,3-dimethyl-2-[[(2S)-2-phenyl-2-(pyrazine-2-carbonylamino)acetyl]amino]butanoyl]-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrole-3-carboxamide | 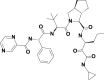 |
| 6 | 10283585 | (1S,3S,7R)-4-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-4-azatricyclo[5.2.1.02,6]dec-8-ene-3-carboxamide | 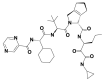 |
| 7 | 46241273 | (3S,3aS,6aR)-2-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-6-oxo-1,3,3a,4,5,6a-hexahydrocyclopenta[c]pyrrole-3-carboxamide | 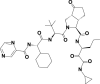 |
| 8 | 73356245 | (3S)–N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-2-[(2S)-3,3-dimethyl-2-[[(2S)-2-phenyl-2-(pyrazine-2-carbonylamino)acetyl]amino]butanoyl]-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrole-3-carboxamide | 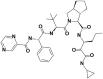 |
| 9 | 58907243 | (1S,2S,3S,6R,7R)-4-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-4-azatricyclo[5.2.1.02,6]dec-8-ene-3-carboxamide | 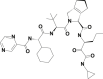 |
| 10 | 143044427 | (3aS,6aS)-2-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-6-methylidene-1,3,3a,4,5,6a-hexahydrocyclopenta[c]pyrrole-3-carboxamide | 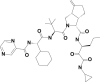 |
| 11 | 58907310 | (2S,3S,6R)-4-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-4-azatricyclo[5.2.1.02,6]dec-8-ene-3-carboxamide | 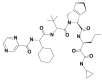 |
4. Discussion
The COVID-19 pandemic continues to spread chaos globally. The current situation compels researchers to find a fast and stable solution in terms of potent inhibitors against SARS-CoV-2. We used a computational drug repurposing approach to find existing drugs against COVID-19. Our docking-based virtual screening of KCB-DR database results suggested 7 drugs as potential binders. The screened compounds included approved drugs such as the cephalosporin antibacterial ceftaroline fosamil, remikiren (effective in the treatment of hypertension), antineoplastic drug everolimus, blood cholesterol-lowering drug atorvastatin, sildenafil effective in erectile dysfunction and pulmonary arterial hypertension, the anti-leprosy drug clofazimine, and the HCV NS3-4A protease inhibitor telaprevir.
Sildenafil has been under Phase 3 clinical trials in China (NCT04304313). Remikiren has been reported as a potential drug against COVID-19 by Nguyen et al. [47]. Telaprevir is predicted to bind effectively to SARS-COV-2 papain-like protease (PLpro) [48]. However, other potential drugs in our study have yet to be evaluated against COVID-19. The potential candidates were screened through exhaustive in silico approaches employing molecular docking, MD simulations, and free energy calculations. Our results suggested that ceftaroline fosamil and telaprevir have better binding energies than reference inhibitors. Ceftaroline fosamil occupies the S1′, S2, and S3 subsites while telaprevir filled the S1, S1′, S2, and S3 subsites of SARS-CoV-2 Mpro active site. Moreover, ceftaroline fosamil formed a close contact with Ser46 residue which is mutated (Ala46Ser) in SARS-CoV-2 Mpro versus SARS-CoV (Fig. 2D). SARS-CoV Mpro inhibitors may provide critical information about the molecular interactions necessary for targeting SARS-CoV-2 Mpro due to their high sequence and structure similarities (Table 4 ) [49]. This analysis suggested that complexes of SARS-CoV and bound inhibitors exhibit hydrogen bond formation with residues such as His41, Phe140, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Gln189, Thr190, and Gln192. Further, the SARS-CoV-2 Mpro binding site can accommodate a variety of inhibitors as evident from recent crystal structure complexes reported in the PDB database. The small size inhibitors such as carmofur (PDB: 7BUY) [50] and 2-Methyl-1-tetralone (PDB: 6YNQ) [51] may bind only at S1 and S2 subsites. Medium size inhibitors like boceprevir (PDB: 7BRP) [52], 7j (PDB: 6XMK) [53], and feline (PDB: 6WTJ) [54] may bind to S1′, S1, and S2 subsites. Some large inhibitors may bind to S1’, S1, S2, and may extend through S3 subsites, for example N3 (PDB: 6LU7) [21], alpha-ketoamide (PDB: 6Y2F) [43], and X77 (PDB: 6W63) [55]. These compounds have dissimilar structures and sizes, yet they bind with and inhibit the activity of SARS-CoV-2 Mpro. Therefore, a potential future perspective lies in the structure-activity relationship (SAR) studies which should provide greater insights into the selectivity of inhibitors of SARS-CoV-2 Mpro.
Table 4.
Hydrogen bond formation between SARS-CoV Mpro active site residues and bound inhibitors of the selected complexes.
| PDB Entry | Thr24 | Thr25 | Thr26 | His41 | Phe140 | Asn142 | Gly143 | Ser144 | Cys145 | His163 | His164 | Met165 | Glu166 | Gln189 | Thr190 | Gln192 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5RF6a | ✓ | ✓ | ||||||||||||||
| 5RG0a | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 5RG3a | ✓ | ✓ | ||||||||||||||
| 6LU7a | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 6Y2Fa | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 6Y2Ga | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| 1UK4 | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 1WOF | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 2A5I | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 2A5K | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| 2ACF | ||||||||||||||||
| 2ALV | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 2AMD | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 2AMQ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 2D2D | ✓ | ✓ | ||||||||||||||
| 2GTB | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||
| 2GX4 | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 2GZ7 | ✓ | ✓ | ||||||||||||||
| 2GZ8 | ✓ | ✓ | ||||||||||||||
| 2HOB | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||
| 2OP9 | ✓ | ✓ | ✓ | |||||||||||||
| 2Z3C | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 2Z3D | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 2Z3E | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| 2ZU4 | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 2ZU5 | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 3ATW | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 3AVZ | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 3AW0 | ✓ | ✓ | ||||||||||||||
| 3IWM | ✓ | ✓ | ||||||||||||||
| 3SN8 | ✓ | ✓ | ✓ | |||||||||||||
| 3SNA | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 3SNB | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 3SNC | ✓ | ✓ | ✓ | |||||||||||||
| 3SND | ✓ | ✓ | ||||||||||||||
| 3SNE | ✓ | ✓ | ||||||||||||||
| 3SZN | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 3TIT | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 3TIU | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 3TNS | ✓ | ✓ | ✓ | ✓ | ||||||||||||
| 3TNT | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||
| 3V3M | ✓ | ✓ | ||||||||||||||
| 3VB4 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 3VB5 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 3VB6 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| 3VB7 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| 4MDS | ✓ | ✓ | ||||||||||||||
| 6Y7M | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
Recently determined SARS-CoV-2 Mpro crystal structures.
Ceftaroline fosamil formed four hydrogen bond interactions including residues Thr24, Thr25, and His41 as observed in recently determined crystal structures of SARS-CoV-2 Mpro (Table 4). Interestingly, telaprevir formed hydrogen bond interactions with important residues such as Gly143, Ser144, Cys145, and Glu166 suggesting its relevance as a potential SARS-CoV-2 Mpro inhibitor. Telaprevir was previously used as a combination therapy against Hepatitis C virus (HCV) infection that was paired with ribavirin, peginterferon alfa-2a, and peginterferon alfa-2b [56]. Some side effects of telaprevir combination therapy were skin rashes and anemia in adverse events of treatment [57]. To avoid the side effects of telaprevir, analogs with better pharmacokinetic properties can be further synthesized and explored as Mpro inhibitors.
Thus, we performed a substructure search in the PubChem database to identify telaprevir derivative compounds. The 11 resulting compounds had desirable pharmacokinetic properties such as good aqueous solubility, non CYP2D6 binding, low hepatotoxicity, high intestinal absorption, and inability to cross the BBB besides good binding properties with SARS-CoV-2 Mpro active site. Therefore, we demonstrate that telaprevir derivative compounds identified by our in silico study have potential against COVID-19 and may bind to the SARS-CoV-2 main protease.
5. Conclusion
Our computational drug repurposing study identified ceftaroline fosamil and telaprevir as putative inhibitors against a critical COVID-19 drug target SARS-CoV-2 main protease. Our inhibitors formed hydrogen bonds with important residues such as Thr24, Thr25, His41, Gly143, Ser144, Cys145, and Glu166; these are consistent with the crystal structures of complexes of inhibitors bound with SARS-CoV and SARS-CoV-2 main proteases. A previously unexplored hydrogen bond interaction was identified in the case of ceftaroline fosamil with Thr45 located near the S1’ subsite of SARS-CoV-2 Mpro. Considering the side effects of the telaprevir, its derivative compounds with better pharmacokinetic properties are suggested as potential antiviral candidates against COVID-19. We recommend telaprevir analogs and ceftaroline fosamil identified in our study for in vitro testing and further validation through randomized clinical trials before being used in COVID-19 patients.
Author contributions
R.K., V.K., and K.W.L. conceived the idea of the project. R.K. and V.K. performed the experiments and compiled the manuscript. R.K. and K.W.L. analyzed the results. All the authors have read and approved the manuscript for submission.
Declaration of competing interest
The authors declare no conflicts of interest.
Acknowledgement
This work was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) and funded by the Korean government (MSIT) (No. NRF-2018M3A9A7057263).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2020.104186.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Cucinotta D., Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed. : Atenei Parmensis. 2020;91:157–160. doi: 10.23750/abm.v91i1.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W., China Novel Coronavirus I., Research T. A novel coronavirus from patients with pneumonia in China. N. Engl. J. Med. 2019;382:727–733. doi: 10.1056/NEJMoa2001017. (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu C., Zhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6:315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X., Wang X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genom.= Yi chuan xue bao. 2020;47:119–121. doi: 10.1016/j.jgg.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasan A., Paray B.A., Hussain A., Qadir F.A., Attar F., Aziz F.M., Sharifi M., Derakhshankhah H., Rasti B., Mehrabi M., Shahpasand K., Saboury A.A., Falahati M. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J. Biomol. Struct. Dyn. 2020:1–9. doi: 10.1080/07391102.2020.1754293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boopathi S., Poma A.B., Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1758788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J., Rabenau H., Doerr H.W., Hunsmann G., Otaka A., Tamamura H., Fujii N., Yamamoto N. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh A.K., Chapsal B.D., Weber I.T., Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Accounts Chem. Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 10.Ruigrok R.W., Crepin T., Hart D.J., Cusack S. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr. Opin. Struct. Biol. 2010;20:104–113. doi: 10.1016/j.sbi.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., Norris A., Sanseau P., Cavalla D., Pirmohamed M. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 12.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., Li X., Xia J., Chen N., Xiang J., Yu T., Bai T., Xie X., Zhang L., Li C., Yuan Y., Chen H., Li H., Huang H., Tu S., Gong F., Liu Y., Wei Y., Dong C., Zhou F., Gu X., Xu J., Liu Z., Zhang Y., Li H., Shang L., Wang K., Li K., Zhou X., Dong X., Qu Z., Lu S., Hu X., Ruan S., Luo S., Wu J., Peng L., Cheng F., Pan L., Zou J., Jia C., Wang J., Liu X., Wang S., Wu X., Ge Q., He J., Zhan H., Qiu F., Guo L., Huang C., Jaki T., Hayden F.G., Horby P.W., Zhang D., Wang C. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. N. Engl. J. Med. 2020;382(19):1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babadaei M.M.N., Hasan A., Vahdani Y., Bloukh S.H., Sharifi M., Kachooei E., Haghighat S., Falahati M. Development of remdesivir repositioning as a nucleotide analog against COVID-19 RNA dependent RNA polymerase. J. Biomol. Struct. Dyn. 2020:1–9. doi: 10.1080/07391102.2020.1767210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hendaus M.A. Remdesivir in the treatment of coronavirus disease 2019 (COVID-19): a simplified summary. J. Biomol. Struct. Dyn. 2020:1–6. doi: 10.1080/07391102.2020.1767691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colson P., Rolain J.M., Lagier J.C., Brouqui P., Raoult D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents. 2020:105932. doi: 10.1016/j.ijantimicag.2020.105932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao X., Ye F., Zhang M., Cui C., Huang B., Niu P., Liu X., Zhao L., Dong E., Song C., Zhan S., Lu R., Li H., Tan W., Liu D. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America; 2020. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Bioscience trends. 2020;14:72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 19.Lu H. Drug treatment options for the 2019-new coronavirus (2019-nCoV) Bioscience trends. 2020;14:69–71. doi: 10.5582/bst.2020.01020. [DOI] [PubMed] [Google Scholar]
- 20.Sk M.F., Roy R., Jonniya N.A., Poddar S., Kar P. Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1768149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 22.Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 24.Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D., Hess B., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32:2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 26.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126 doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 27.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 28.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 29.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 30.Kumar R., Lee Y.K., Jho Y.S. Martini coarse-grained model of hyaluronic acid for the structural change of its gel in the presence of monovalent and divalent salts. Int. J. Mol. Sci. 2020;21:4602. doi: 10.3390/ijms21134602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang C.Y., Sun H., Chen J., Nikolovska-Coleska Z., Wang S. Importance of ligand reorganization free energy in protein-ligand binding-affinity prediction. J. Am. Chem. Soc. 2009;131:13709–13721. doi: 10.1021/ja9039373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumari R., Kumar R., Open C. Source Drug Discovery, A. Lynn, g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 33.Kumar R., Parameswaran S., Bavi R., Baek A., Son M., Rampogu S., Park C., Lee G., Zeb A., Parate S., Rana R.M., Lee K.W. Investigation of novel chemical scaffolds targeting prolyl oligopeptidase for neurological therapeutics. J. Mol. Graph. Model. 2019;88:92–103. doi: 10.1016/j.jmgm.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X.W., Yap Y.L. Old drugs as lead compounds for a new disease? Binding analysis of SARS coronavirus main proteinase with HIV, psychotic and parasite drugs. Bioorg. Med. Chem. 2004;12:2517–2521. doi: 10.1016/j.bmc.2004.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong L., Hu S., Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19) Drug discoveries & therapeutics. 2020;14:58–60. doi: 10.5582/ddt.2020.01012. [DOI] [PubMed] [Google Scholar]
- 36.Fischer A., Sellner M., Neranjan S., Smieško M., Lill M.A. Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. Int. J. Mol. Sci. 2020;21(10):3626. doi: 10.3390/ijms21103626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chapman T.M., Plosker G.L., Perry C.M. Fosamprenavir: a review of its use in the management of antiretroviral therapy-naive patients with HIV infection. Drugs. 2004;64:2101–2124. doi: 10.2165/00003495-200464180-00014. [DOI] [PubMed] [Google Scholar]
- 39.Kumar R., Son M., Bavi R., Lee Y., Park C., Arulalapperumal V., Cao G.P., Kim H.H., Suh J.K., Kim Y.S., Kwon Y.J., Lee K.W. Novel chemical scaffolds of the tumor marker AKR1B10 inhibitors discovered by 3D QSAR pharmacophore modeling. Acta Pharmacol. Sin. 2015;36:998–1012. doi: 10.1038/aps.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu K., Kokubo H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations: a cross-docking study. J. Chem. Inf. Model. 2017;57:2514–2522. doi: 10.1021/acs.jcim.7b00412. [DOI] [PubMed] [Google Scholar]
- 41.Vettoretti G., Moroni E., Sattin S., Tao J., Agard D.A., Bernardi A., Colombo G. Molecular dynamics simulations reveal the mechanisms of allosteric activation of hsp90 by designed ligands. Sci. Rep. 2016;6:23830. doi: 10.1038/srep23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai H.H., Tsai C.J., Ma B., Nussinov R. In silico protein design by combinatorial assembly of protein building blocks. Protein Sci. : Publ.Protein.Soc. 2004;13:2753–2765. doi: 10.1110/ps.04774004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. 6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A., Cheatham T.E., 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Accounts Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 45.Klimovich P.V., Shirts M.R., Mobley D.L. Guidelines for the analysis of free energy calculations. J. Comput. Aided Mol. Des. 2015;29:397–411. doi: 10.1007/s10822-015-9840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spiliotopoulos D., Spitaleri A., Musco G. Exploring PHD fingers and H3K4me0 interactions with molecular dynamics simulations and binding free energy calculations: AIRE-PHD1, a comparative study. PloS One. 2012;7 doi: 10.1371/journal.pone.0046902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen D.D., Gao K., Chen J., Wang R., Wei G.-W. bioRxiv; 2020. Potentially Highly Potent Drugs for 2019-nCoV. 2020.2002.2005.936013. [Google Scholar]
- 48.Elzupir A.O. Inhibition of SARS-CoV-2 main protease 3CL(pro) by means of alpha-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J. Mol. Struct. 2020;1222:128878. doi: 10.1016/j.molstruc.2020.128878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li G., De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV), Nature reviews. Drug discovery. 2020;19:149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]
- 50.Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y., Zhu Y., Zhu C., Hu T., Du X., Duan Y., Yu J., Yang X., Yang X., Yang K., Liu X., Guddat L.W., Xiao G., Zhang L., Yang H., Rao Z. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- 51.Günther S., Reinke P.Y.A., Fernández-García Y., Lieske J., Lane T.J., Ginn H.M., Koua F.H.M., Ehrt C., Ewert W., Oberthuer D., Yefanov O., Meier S., Lorenzen K., Krichel B., Kopicki J.-D., Gelisio L., Brehm W., Dunkel I., Seychell B., Gieseler H., Norton-Baker B., Escudero-Pérez B., Domaracky M., Saouane S., Tolstikova A., White T.A., Hänle A., Groessler M., Fleckenstein H., Trost F., Galchenkova M., Gevorkov Y., Li C., Awel S., Peck A., Barthelmess M., Schlünzen F., Xavier P.L., Werner N., Andaleeb H., Ullah N., Falke S., Srinivasan V., Franca B.A., Schwinzer M., Brognaro H., Rogers C., Melo D., Zaitsev-Doyle J.J., Knoska J., Peña Murillo G.E., Mashhour A.R., Guicking F., Hennicke V., Fischer P., Hakanpää J., Meyer J., Gribbon P., Ellinger B., Kuzikov M., Wolf M., Beccari A.R., Bourenkov G., Stetten D.v., Pompidor G., Bento I., Panneerselvam S., Karpics I., Schneider T.R., Garcia Alai M.M., Niebling S., Günther C., Schmidt C., Schubert R., Han H., Boger J., Monteiro D.C.F., Zhang L., Sun X., Pletzer-Zelgert J., Wollenhaupt J., Feiler C.G., Weiss M.S., Schulz E.-C., Mehrabi P., Karničar K., Usenik A., Loboda J., Tidow H., Chari A., Hilgenfeld R., Uetrecht C., Cox R., Zaliani A., Beck T., Rarey M., Günther S., Turk D., Hinrichs W., Chapman H.N., Pearson A.R., Betzel C., Meents A. bioRxiv; 2020. Inhibition of SARS-CoV-2 Main Protease by Allosteric Drug-Binding. 2020.2011.2012.378422. [Google Scholar]
- 52.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y., Zhao C., Sun H., Huang B., Niu P., Song H., Shi Y., Li X., Tan W., Qi J., Gao G.F. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rathnayake A.D., Zheng J., Kim Y., Perera K.D., Mackin S., Meyerholz D.K., Kashipathy M.M., Battaile K.P., Lovell S., Perlman S., Groutas W.C., Chang K.O. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.abc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M.A., Young H.S., Tyrrell D.L., Vederas J.C., Lemieux M.J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mesecarr A.D. 2020. A Taxonomically-Driven Approach to Development of Potent, Broad-Spectrum Inhibitors of Coronavirus Main Protease Including SARS-CoV-2 (COVID-19) [DOI] [Google Scholar]
- 56.Sherman K.E., Flamm S.L., Afdhal N.H., Nelson D.R., Sulkowski M.S., Everson G.T., Fried M.W., Adler M., Reesink H.W., Martin M., Sankoh A.J., Adda N., Kauffman R.S., George S., Wright C.I., Poordad F., I.S. Team Response-guided telaprevir combination treatment for hepatitis C virus infection. N. Engl. J. Med. 2011;365:1014–1024. doi: 10.1056/NEJMoa1014463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muir A.J. Telaprevir for the treatment of chronic hepatitis C infection. Expert Rev. Anti-infect. Ther. 2011;9:1105–1114. doi: 10.1586/eri.11.133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.