

Abstract
Cell fate determination requires faithful execution of gene expression programs, which are increasingly recognized to respond to metabolic inputs. In particular, the family of α-ketoglutarate (αKG)-dependent dioxygenases, which include several chromatin-modifying enzymes, are emerging as key mediators of metabolic control of cell fate. αKG-dependent dioxygenases consume the metabolite αKG (also known as 2-oxoglutarate) as an obligate cosubstrate and are inhibited by succinate, fumarate, and 2-hydroxyglutarate. Here, we review the role of these metabolites in the control of dioxygenase activity and cell fate programs. We discuss the biochemical and transcriptional mechanisms enabling these metabolites to control cell fate and review evidence that nutrient availability shapes tissue-specific fate programs via αKG-dependent dioxygenases.
Cell Fate Determination Responds to Metabolic Inputs
Development and homeostasis of multicellular organisms depends on cells acquiring and maintaining the correct fate at the right place and time. Cell fate determination (see Glossary), wherein less differentiated cells progressively acquire specific fates and functions, is essential for proper embryogenesis and maintenance of postnatal tissue homeostasis by stem cells. In both the embryo and postnatal tissues, cell fate determination depends both on inductive signals from the environment and the competence of cells to respond appropriately to these signals [1,2]. Accordingly, dysregulation of either extracellular cues or their downstream intracellular responses compromise cell fate programs and results in diseases ranging from birth defects to cancer [1,2]. Consequently, dissecting the molecular regulation of cell fate decisions is critically important for understanding the mechanistic basis of both normal physiology and disease states.
Increasingly, metabolites are recognized as important modulators of the regulatory programs that control cell fate. In particular, chemical modifications on DNA and histones provide a critical avenue for cells to control activation of gene expression programs that specify cell identity [3]. These chemical modifications are derived from intermediates of cellular metabolism, most notably S-adenosylmethionine and acetyl-CoA, which serve as the donors for methylation and acetylation modifications, respectively. Enzymes that remove these modifications often also require metabolites as critical cosubstrates. Accordingly, fluctuations in the availability of key metabolites that modulate activity of chromatin-modifying enzymes are postulated to contribute to transcriptional regulation by shaping the chromatin landscape [4].
Intracellular metabolite levels are responsive to both cell-intrinsic metabolic pathway activity as well as extrinsic cues from the microenvironment, including growth factors and nutrient availability. Many inputs, including tissue lineage, proliferative status, and nutrient availability, collectively determine the metabolic demands of individual cells [5]. In turn, cell type-specific metabolic demands influence cellular proliferation and fate in response to microenvironmental changes, including changes in key nutrients [6,7]. Increasing evidence additionally suggests that tissues experience distinct nutrient microenvironments and that heterogeneous availability of extracellular nutrients can modulate cell fate by controlling the availability of metabolites that regulate the chromatin landscape [7–11]. In this manner, intracellular metabolites are emerging as critical components of cell fate determination programs, capable of integrating extracellular nutrient status and intracellular biochemical demands to influence transcriptional networks and cell fate decisions.
Here, we review the proposed mechanisms by which select extracellular nutrients and intracellular metabolites shape pathways determining lineage-specific cell fates. We focus on metabolic intermediates of the tricarboxylic acid (TCA) cycle (Figure 1A). Multiple nutrients, most notably glucose and glutamine, feed into the TCA cycle and several TCA cycle intermediates, including α-ketoglutarate (αKG, also known as 2-oxoglutarate/2OG), succinate, and fumarate have been shown to regulate gene expression programs in various contexts [4,5]. We discuss evidence that regulation of gene expression and cell fate by TCA cycle metabolites is context-dependent, with metabolites acting both upstream and downstream of lineage-specific signaling and specification programs to shape cell fate. Finally, we discuss how nutrient availability creates permissive environments for cell fate outcomes by regulating intracellular metabolic pathways.
Figure 1. Metabolic Regulation of α-Ketoglutarate-Dependent Dioxygenases.
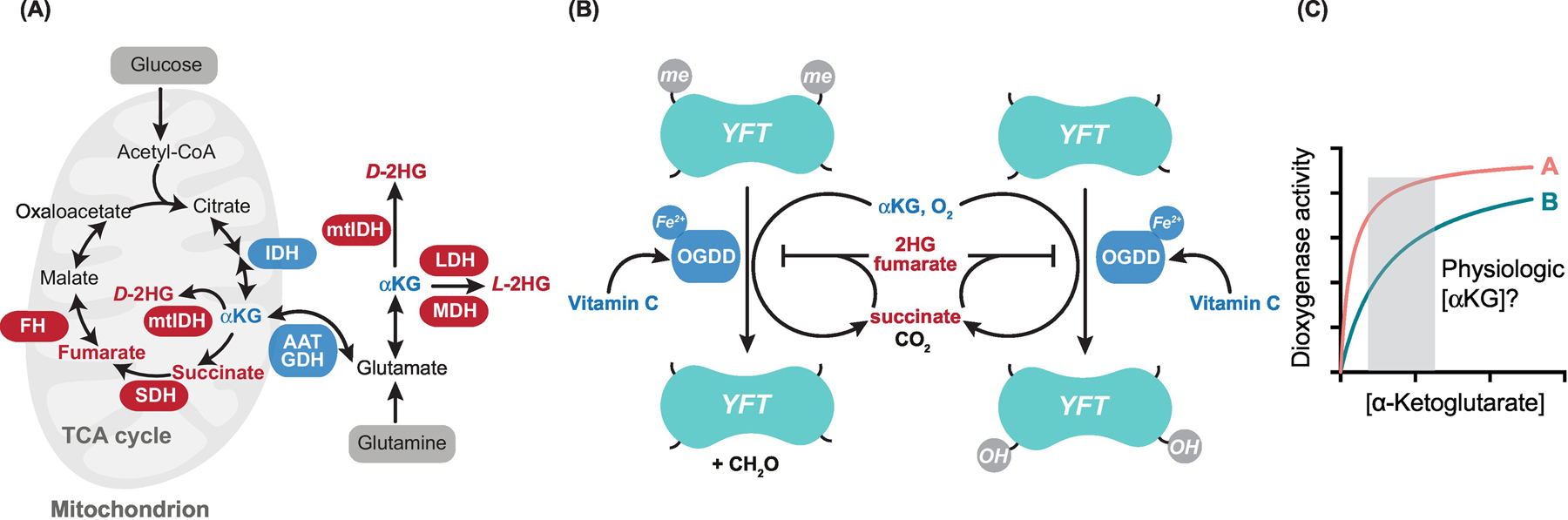
(A) Schematic of key pathways involved in synthesis and break down of α-ketoglutarate (αKG), 2-hydroxyglutarate (2HG), fumarate, and succinate. Enzymes directly involved in αKG metabolism are shown in blue, those involved in 2HG, fumarate, and succinate metabolism are shown in red. (B) Generalized schematic of αKG-dependent dioxygenase (also known as 2-oxoglutarate dependent dioxygenase or OGDD) action on your favorite target (YFT). Dioxygenases catalyze net demethylation or hydroxylation reactions using αKG and molecular oxygen as cosubstrates and producing succinate as a by-product. Vitamin C, oxygen, and αKG can promote dioxygenase activity, whereas succinate, fumarate, and 2HG have been shown to suppress their activity. (C) Enzymatic assays provide potential insights into metabolic regulation of dioxygenase catalytic activity. In this example, dioxygenase B is expected to be sensitive to physiological fluctuations in αKG concentrations, whereas dioxygenase A will be less sensitive. However, it remains unclear what true physiological αKG concentrations are, given that dioxygenases may be sensitive to compartmentalized metabolite pools. Abbreviations: FH, Fumarate hydratase; IDH, isocitrate dehydrogenase; mtIDH, IDH mutations; SDH, succinate dehydrogenase; TCA, tricarboxylic acid.
αKG-Dependent Dioxygenases as Metabolic Mediators of Cell Fate Control
Mechanistically, changes in TCA cycle metabolites are postulated to affect cell fate by regulating activity of α-ketoglutarate-dependent dioxygenases [12,13]. The family of αKG-dependent dioxygenases includes Jumonji C-domain lysine demethylases (JmjC-KDMs), ten-eleven translocation (TET) DNA cytosine-oxidizing enzymes, and prolyl hydroxylases (PHDs). TCA cycle intermediates serve both as critical cosubstrates and competitive inhibitors of αKG-dependent dioxygenases: the enzymes consume αKG and molecular oxygen as part of their reaction cycle, yielding succinate and carbon dioxide, as well as formaldehyde in the case of net demethylation reactions [4]. αKG-dependent dioxygenases are additionally activated by ascorbate (Vitamin C) and inhibited by TCA cycle metabolites, including succinate and fumarate, as well as the related metabolite 2-hydroxyglutarate (2HG), which can exist in cells as both a D and L enantiomer (Figure 1B). Mutations in several of the enzymes that are involved in the production and breakdown of these metabolites, including isocitrate dehydrogenase (IDH1 and IDH2), succinate dehydrogenase (SDH), and fumarate hydratase (FH) have been shown to facilitate cancer progression by disrupting normal cell fate decisions [5] (Box 1, Figure 1A). Even absent of these mutations, however, intracellular abundances of αKG, ascorbate, succinate, fumarate, and 2HG have been shown to regulate cell fate, including in stem cells, immune cells, and cancer cells [4,5].
Box 1. Cancer-Associated Mutations Hijack Metabolic Control of Cell Fate.
The powerful ability of metabolites to regulate cell fate programs is underscored by the pathological impairments in normal differentiation that arise as a result of recurrent oncogenic mutations in metabolic enzymes, including IDH1/2, SDH, and FH, leading to accumulation of D-2HG, succinate, and fumarate, respectively [20,75,76]. These metabolites competitively inhibit αKG-dependent dioxygenases and are often suggested to exert similar functions in blocking stem cell differentiation [19,20,22,23,42]. In vitro differentiation of mesenchymal, hepatic, hematopoietic, and neural progenitors are all suppressed by expression of mtIDH1/2 or 2HG treatment alone [12,19,25,41]. In vivo, expression of mtIDH in the brain, bone marrow, and liver all lead to expansion of resident progenitor populations and suppression of terminal differentiation [25,77,78]. SDH-deficient paragangliomas arise in the oxygen-sensing carotid bodies, a neural crest-derived organ, and Sdhd loss is associated with carotid body hypertrophy and reduced differentiation [79,80]. Together, these studies suggest that inhibitory metabolites act by suppressing the competence of stem cells to respond to appropriate differentiation stimuli, such that stem cell division is skewed towards self-renewal.
Uncontrolled self-renewal of stem cells at the expense of differentiation facilitates tumor initiation and progression; therefore, understanding the degree to which intracellular metabolites regulate stem cell fate will provide critical insight into mechanisms of tumor initiation [7]. In classic models of cell fate induction, cellular competence to respond to inductive signals for fate changes can be suppressed by direct inhibitors of that signaling pathway. By analogy, the pervasive observation that oncometabolites block differentiation across multiple lineages raises the possibility that αKG could be an inductive signal for adult stem cell differentiation that is undermined by oncogenic metabolic adaptations. In support of this model, ascorbate, which promotes αKG-dependent dioxygenase function, drives HSC differentiation and suppresses leukemogenesis [81,82]. Exogenous αKG also promotes cytokine-induced HSC differentiation [83] and αKG is necessary and sufficient for effector T cell differentiation [48,64]. Notably, the effects of αKG extend beyond tissues that are prone to transformation by oncometabolites. In both intestinal and epidermal stem cells, αKG is sufficient for stem cell differentiation and can suppress tumor initiation and progression [7,26]. The effects of αKG extend to transformed cells: in pancreatic cancer cells, p53 restoration drives αKG-dependent differentiation, and αKG alone is sufficient to recapitulate the effects of p53 [84]. When tested in these settings, succinate showed little effect except under conditions of αKG accumulation [7,84], consistent with αKG acting as an inductive signal and its antagonist establishing repressive thresholds for cell fate changes. In this manner, mutations in metabolic enzymes that lead to accumulation of 2HG, succinate, and fumarate may be akin to mutations in key tissue-specific signaling pathways that normally act to restrain stem cell self-renewal.
The observation that TCA cycle metabolites can control αKG-dependent dioxygenases and cell fate raises major questions, including how a nonspecific signal such as TCA cycle intermediate abundance can lead to a specific outcome in cell fate. One possibility is that the effect of a change in metabolite abundance is read out by a cell based on the relative affinity of dioxygenases for that metabolite (Figure 1C). For example, in vitro studies suggest that L- but not D-2HG inhibits prolyl hydroxylase function [12,14] and even closely related dioxygenases can exhibit vastly different affinity for the same metabolite [15,16]. However, this biochemical logic for metabolic control of specific dioxygenase activity relies largely on in vitro assessment of enzyme–metabolite affinities, which have several important limitations to understanding metabolic control of cell fate. First, kinetic constants determined in vitro may not reflect enzyme behavior in a complex environment in vivo. Second, current approaches to measure metabolite concentrations provide information on whole cell or tissue abundances, while chromatin modifying αKG-dependent dioxygenases may be sensitive only to nucleo-cytosolic levels of metabolites, where substrates may be more (or less) limiting [17]. For example, cancer-associated IDH mutations (mtIDH), which convert αKG to 2HG, occur in both cytosolic IDH1 and mitochondrial IDH2, but mtIDH1 traditionally drives lower production of 2HG than mtIDH2. However, 2HG production by mtIDH1 is increased upon ectopic targeting to the mitochondria, potentially reflecting limited cytosolic substrate availability [17]. Whether or not nucleo-cytosolic dioxygenases compete for a limited pool of αKG remains to be explored.
An additional challenge in understanding how changes in select TCA cycle metabolites may coordinately influence cell fate is the fact that multiple dioxygenase-sensitive marks are often affected by metabolic changes [18–21]. For example, succinate and fumarate accumulation in cancer cells is linked to HIF stabilization and hypermethylation of DNA as well as multiple histone lysine residues [20,22,23]. The ability of a single metabolite to influence multiple relevant pathways highlights the importance of these molecules but poses a challenge in understanding which changes represent biologically relevant, primary responses. For example, fumarate accumulation in FH-deficient cells was thought to drive tumorigenesis in the kidney via inhibition of PHDs and subsequent stabilization of HIFs. However, double knockout of HIF1α and HIF2α did not prevent FH loss-induced renal cyst formation; rather, fumarate covalently modifies reactive cysteines on KEAP1, a major negative regulator of NRF2, thereby driving cyst formation independent of αKG-dependent dioxygenase inhibition [24]. Indeed, many studies provide evidence that metabolites regulate cell fate decisions and dioxygenase-sensitive marks but remain correlative with regards to the precise mechanisms by which these effects are mediated.
The most rigorous approach to studying metabolic control of cell fate will involve both cell-free biochemical studies and genetic experiments in relevant in vivo systems. This combined approach recently revealed a critical role for molecular oxygen in muscle differentiation. αKG-dependent dioxygenases consume oxygen as part of their reaction cycle and therefore may directly respond to hypoxia. The authors found that hypoxia impaired myogenic differentiation by inhibiting the αKG-dependent H3K27me3 demethylase UTX, which has a low oxygen affinity in vitro. Notably, the closely related H3K27me3 demethylase, JMJD3, has relatively high oxygen affinity and was not sensitive to hypoxia. Mutagenesis of key residues in UTX’s catalytic domain to resemble that of JMJD3 was sufficient to increase oxygen affinity and restore differentiation in hypoxia [16]. Analogous experiments manipulating dioxygenase sensitivity to TCA cycle metabolites will be necessary to identify the enzymes that directly mediate the effects of metabolites on cell fate decisions.
The ability of metabolites to exert specific effects on cell fate is perhaps best illustrated by studies highlighting the context-specific effects of oncometabolites (Box 2). The distinct linage bias of different oncogenic metabolic mutations suggests that metabolites likely regulate cell fate decisions in a tissue-specific manner. Thus, it is critical to understand how metabolites act in the context of the particular cohort of lineage-specific signals and factors experienced by individual cells. In subsequent sections we review potential mechanisms by which lineage factors and environmental inputs might shape the cellular response to individual metabolites. Collectively, these studies raise two related but separate points: specific metabolites may be required for particular chromatin changes and cell fate decisions, and different lineages may exhibit distinct responses and sensitivities to metabolites.
Box 2. Cancer-Associated Mutations Suggest Lineage-Specific Effects of Metabolites.
Despite evidence that 2HG, succinate, and fumarate act via similar biochemical mechanisms, mutations that drive accumulation of these oncometabolites occur in distinct lineages. For example, acute myeloid leukemia (AML) and gliomas harbor IDH but not SDH and FH mutations, whereas the opposite is true for renal cell carcinoma and neuroendocrine tumors [20,85,86]. The context-specific effect of these oncogenic mutations has been best studied in hematopoiesis, which is fueled by HSCs. Consistent with an oncogenic function for 2HG, expression of mtIDH1/2 or treatment with D-2HG increases proliferation and self-renewal of HSCs and hematopoietic progenitors in vitro and in vivo [13,42,77]. Accordingly, mtIDH2 cooperates with additional oncogenic hits to drive leukemogenesis in mice [34]. In contrast, both SDH and FH loss lead to impaired HSC maintenance and function and FH loss suppresses leukemic transformation [87,88].
One possible explanation for disparate effects of IDH, SDH, and FH mutations is that SDH and FH are components of the core oxidative TCA cycle, whereas IDH1 and IDH2 are not. Thus, it may be that HSCs are unable to cope with TCA cycle truncation. Alternatively, D-2HG may exert distinct effects from succinate and fumarate on αKG-dependent dioxygenases: whereas mtIDH1/2 inhibits both DNA and histone demethylation, FH loss in HSCs only inhibits histone demethylation [42,77,88]. Fumarate dioxygenase-independent functions [24,89]. Altogether, these studies support distinct effects of succinate, fumarate, and 2HG accumulation on HSC behavior.
Multiple mutations in IDH1 and IDH2 are sufficient to induce D-2HG and block HSC differentiation [13,42,77]. In AML, IDH1/2 mutations are mutually exclusive with TET2 mutations, and TET2 loss of function phenocopies mtIDH expression, supporting the hypothesis that 2HG accumulation plays a major role promoting leukemic transformation by antagonizing TET2 function [13,42]. Even within the hematopoietic lineage, however, IDH mutations may exert distinct effects. For example, IDH mutations occur only at R172 of IDH2 in angioimmunoblastic T cell lymphoma, where they co-occur with TET2 mutations. IDH2 R172 mutants produce significantly more 2HG than other mutants and only IDH2 R172 mutant expression disrupts T cell differentiation in mouse models [90]. These data suggest that metabolite dosage may have cell type-specific effects and exert distinct effects along a developmental trajectory. Thus, gaining an understanding of how metabolites cooperate with lineage-specific transcription factors and signaling pathways may provide insight into why metabolites exhibit cell type-specific effects.
Metabolic Interaction with Lineage-Specific Transcription Factors
Given the critical role transcription factors (TFs) and cell type-specific enhancer landscapes play in establishing and maintaining cell identity, it is likely that effects of metabolites will vary according to their ability to interact with lineage-specific transcriptional machinery. In this section we discuss two potential mechanisms by which metabolites may regulate cell fate changes: first, we review evidence that metabolites can directly control TF abundance and expression; and second, we review data suggesting that metabolites regulate TF activity by globally modulating coactivator function. Since different cell fate changes are likely to rely on distinct TF and coactivator cohorts, these data may provide an explanation for lineage-specific effects of metabolites.
In some cases, metabolites may act upstream of lineage TFs. In the liver, mutant IDH1 expression blunts progenitor differentiation into hepatocytes by silencing the master TF HNF4α [25]. In intestinal tumor organoids, αKG is sufficient to suppress activation of the master intestinal stem cell regulator β-catenin and induce differentiation [26]. Despite correlations with changes in DNA and histone methylation at TF promoters and target gene loci, the exact mechanisms by which metabolites affect expression and activity of these specific TFs are not well understood. Appealing mechanisms by which metabolites may act upstream of master lineage regulators are via post-transcriptional and post-translational regulation of TF expression. The RNA demethylases FTO and ALKBH5, which demethylate N6-methyladenosine (m6A), are αKG-dependent dioxygenases. m6A destabilizes transcripts and can control expression of key TFs in hematopoietic stem cells (HSCs) and human embryonic stem cells (ESCs) [27,28]. 2HG suppresses FTO activity in leukemia cells, leading to decreased expression of the lineage TF CCAAT enhancer binding protein α (C/EBPα) that enforces normal HSC quiescence and myeloid differentiation [29,30]. In this manner, direct metabolic regulation of key TF expression may enable specific cell fate outcomes.
Metabolites also exert post-translational control over TFs, as exemplified by the dominant role of αKG-dependent prolyl hydroxylation in the control of HIF1α stability [14,22]. Studies in macrophages recently uncovered the ability of PHDs to regulate other TFs during cell fate decisions: αKG is necessary for anti-inflammatory M2 polarization and sufficient to suppress proinflammatory M1 polarization. While the effects of αKG on M2 polarization required the H3K27me3-demethylase JMJD3, αKG suppressed M1 polarization by inhibiting the master inflammatory TF NFκB via prolyl-hydroxylase-dependent modification of its activator IKKβ. Accordingly, a hydroxylation-dead IKKβ mutant rendered M1 polarization resistant to αKG [31]. Thus, exploring post-transcriptional and post-translational regulation of TFs by αKG-dependent enzymes may identify new layers of regulation by which metabolites can induce cell fate changes (Figure 2A).
Figure 2. Mechanisms of Metabolic Control of Cell Fate.
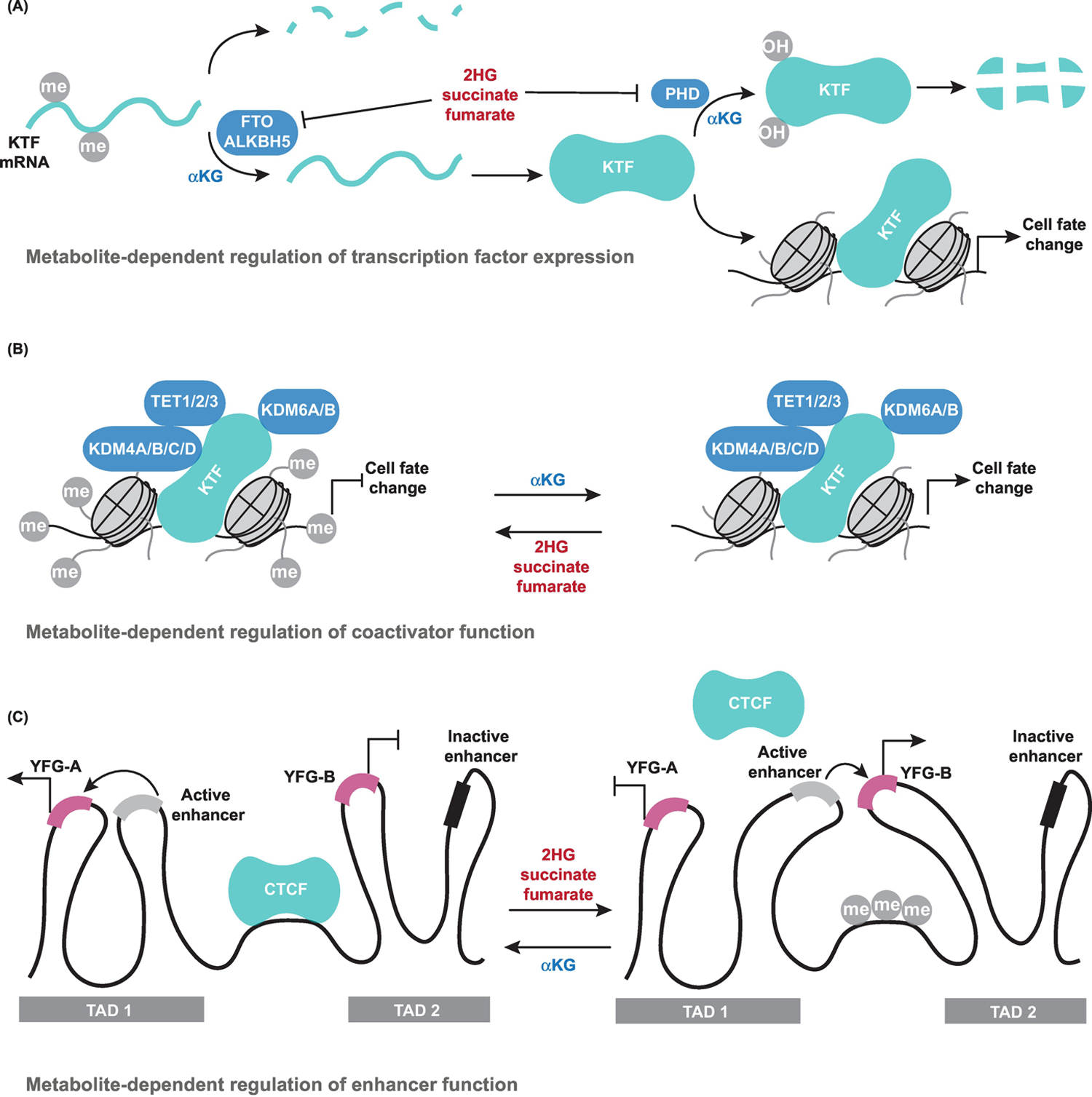
(A) α-Ketoglutarate (αKG)-dependent dioxygenases can directly impact expression of key transcription factors (KTFs) by regulating either mRNA methylation via FTO and ALKBH5 or protein hydroxylation by prolyl hydroxylases (PHDs), each of which trigger target degradation. (B) Metabolites can regulate transcription factor (TF) function by influencing function of transcriptional coactivators, a subset of which are αKG-dependent dioxygenases. Transcription factors recruit coactivators such as ten-eleven translocation (TET) DNA cytosine oxidizing enzymes and Jumonji C-domain lysine demethylases (KDMs) to target loci in order to locally remodel chromatin. (C) Metabolites can affect enhancer function and long-range chromatin interactions by controlling CCCTC-binding factor (CTCF) binding to DNA, which is suppressed by DNA methylation. αKG enforces topologically associated domains (TAD) architecture in cells by facilitating CTCF binding, whereas 2-hydroxyglutarate (2HG), succinate, and fumarate disrupt TAD architecture. An example scenario illustrates how, in the presence of CTCF, a cell type-specific active enhancer drives expression of your favorite gene-A (YFG-A), whereas YFG-B is suppressed. Upon loss of CTCF binding, however, TAD boundaries are disrupted and the active enhancer drives expression of YFG-B.
TFs regulate gene expression by recruiting coactivators and/or corepressors to target loci [3]. Many coactivators, including TETs and H3K27me3 demethylases JMJD3/UTX, require αKG as a cosubstrate, raising the possibility that metabolites function in cell fate decisions by potentiating or suppressing coactivator activity (Figure 2B). This model has been best studied in mouse ESCs. ESCs can be maintained in vitro in a heterogeneous state of metastable pluripotency or as a homogeneous population of cells in the naïve ground state of pluripotency via culture in specific media formulations. The naïve ground state of pluripotency is intrinsically associated with an accumulation of αKG at the expense of succinate, resulting in an increased αKG/succinate ratio, due to reduced αKG catabolism in the TCA cycle [6,18]. Notably, supplementing metastable ESCs with αKG is sufficient to facilitate DNA and histone demethylation and increase pluripotent self-renewal [18].
The ground state of pluripotency is characterized by a robust TF network, the action of which is facilitated in part by recruitment of TET1/2 to key pluripotency loci [32]. This robust transcriptional network enables naive, but not metastable, ESCs to maintain growth and pluripotency upon inhibition of the coactivator BRD4, which binds acetylated histones to activate transcription and is usually essential for maintenance of gene expression programs [33]. The ability of pluripotency TFs to confer BRD4 independence and sustain self-renewal is sensitive to metabolic perturbations: glutamine starvation, which depletes αKG, blunts expression of key pluripotency genes and ascorbate enhances self-renewal in the presence of BRD4 inhibitors [33]. Intriguingly, mtIDH acute myeloid leukemia (AML) is highly sensitive to BRD4 inhibition [34]. Whether or not this sensitivity is due to decreased ability of αKG-dependent dioxygenases to function downstream of master hematopoietic TFs upon 2HG accumulation remains to be explored but would be consistent with the data in ESCs.
Core pluripotency TFs are known to drive their own expression, creating a positive feedback loop, and can remain bound to target genes even upon induction of differentiation stimuli [35,36]. This enables ESCs to revert to pluripotency upon withdrawal of differentiation stimuli, so long as these key TFs remain chromatin bound [36]. Thus, if αKG acts by promoting pluripotency TF function, loss of these TFs from chromatin late in differentiation may blunt the effects of αKG. Consistently, αKG can only sustain ESC pluripotency when provided early after a differentiation stimulus, when expression of pluripotency TFs remains high [37]. Differentiation is associated with a decrease in αKG that coincides with the loss of pluripotency TFs; conversely, overexpression of key pluripotency factors such as NANOG or activated STAT3 are sufficient to increase αKG, suggesting that αKG accumulation may be a component of the feedforward loop of the pluripotency network [6,37].
Notably, αKG does not always enhance ESC self-renewal. In more committed pluripotent cells, such as human ESCs and mouse postimplantation epiblast ESCs, αKG facilitates differentiation, which is antagonized by succinate [38,39]. The differential response to αKG may reflect either the more committed state of human and mouse epiblast ESCs or the particular differentiation stimulus used in each condition. Alternatively, αKG may exert different outcomes as a result of distinct requirements for self-renewal versus differentiation in different cell states, either due to different cohorts of TFs or different required changes in chromatin to achieve a cell state change. Notably, naïve and more committed, primed states of pluripotency have dramatically different levels of DNA methylation [32] and thus may have different sensitivity to metabolic perturbations that favor demethylation. Given that αKG can influence both TF expression directly and function indirectly, metabolites are likely to play numerous roles in particular cell fate decisions depending on the specific requirements of that cell state.
Recent evidence suggests that oncometabolites also act by modulating coactivator action at lineage-specific genes. For example, during muscle differentiation, the master TF MYOD recruits H3K9me2/3 demethylases to target loci to promote expression [40]. Whereas normal myogenic differentiation is accompanied by selective hypomethylation of H3K9 at myogenic targets, mtIDH1 expression drives global, apparently nonselective increases in H3K9me3. Accordingly, mtIDH1 expression suppresses MYOD-mediated differentiation, which is rescued by genetic inhibition of H3K9 methyltransferases. These results support a model wherein 2HG accumulation in myoblasts establishes a global chromatin landscape that is incompatible with differentiation, whereas locus specificity during differentiation is achieved by TF recruitment of coactivators [40]. Consistently, work in neural, mesenchymal, and hematopoietic cells shows that IDH mutations render stem cells unable to appropriately respond to differentiation stimuli, although the interaction between master TFs, coactivators, and metabolites remain to be explored in these settings [13,19,41,42]. Thus, the ability of metabolites to regulate specific cell fate changes is likely due to the combinatorial effects on lineage-specific factor expression and function and gaining a detailed understanding of context-specific effects of metabolites will likely require integration of these multiple regulatory nodes.
Metabolic Control of Lineage-Specific Enhancers
A key component of cell fate decisions involves TF-mediated establishment and binding of cell type-specific enhancers [3]. Active enhancers are marked by acetylation of H3K27 and can be suppressed by H3K27me3 and DNA methylation [3]. Accordingly, metabolic regulation of αKG-dependent dioxygenases can directly regulate enhancers. For example, FH-deficient renal cells exhibit increased DNA and H3K27 methylation at a putative enhancer for the microRNA cluster miR-200. Fumarate-mediated enhancer methylation suppressed the antimetastatic miR-200 cluster, activating the epithelial–mesenchymal transition (EMT). Exogenous αKG reversed fumarate-induced EMT, suggesting that ongoing αKG-dependent dioxygenase activity may be required for maintenance of the renal epithelial phenotype [43].
Recent studies profiling three-dimensional chromatin landscapes and the ubiquitously expressed TF CCCTC-binding factor (CTCF) have provided additional insights into the interactions between metabolites and enhancers. The genome is organized into discrete regulatory units called topologically associated domains (TADs), the boundaries of which are established by CTCF binding [44]. Enhancers preferentially drive expression of genes within their own TADs; accordingly, CTCF loss increases interboundary interactions at the expense of intraboundary interactions, thereby modifying enhancer–promoter contacts and altering gene expression [44]. DNA methylation is emerging as a critical barrier to CTCF binding and TAD regulation that is susceptible to metabolic perturbation [44]. In both mtIDH gliomas and SDH-deficient gastrointestinal stromal tumors, hypermethylation of CTCF binding sites is associated with increased interactions between key receptor tyrosine kinase (RTK) genes and constitutive lineage-specific enhancers normally outside of their domains, resulting in elevated RTK expression [45,46]. Similarly, experiments performed in a human ESC model of glioma demonstrated that mtIDH suppressed differentiation by disrupting CTCF binding, leading to reduced interaction of the SOX2 locus with an active enhancer [47]. Collectively, these studies demonstrate the potential of oncometabolites to influence enhancer activity towards target genes (Figure 2C).
Intriguingly, CTCF is ubiquitously expressed, and many of its binding sites are shared between different cell types [44]. Therefore, metabolic regulation of CTCF binding per se is not sufficient to result in tissue-specific effects; rather, the particular enhancer landscape of a cell will play a key role in determining the transcriptional outcomes of altered CTCF binding. For example, αKG increases CTCF binding at overlapping loci in ESCs and T cells, but induces cell type-specific changes in gene expression based on the active lineage-specific enhancers in the vicinity of CTCF binding sites [48]. Altogether, these data support a model wherein αKG and related metabolites contribute to cell fate regulation by controlling three-dimensional chromatin architecture, but specific cell fate outcomes are driven by the enhancer landscape of a cell. Most likely, specific transcriptional programs reflect the integration of multiple inputs, including TF expression, chromatin architecture, and metabolite abundance, all of which can vary in response to developmental or environmental cues.
Nutrients Regulate Cell Fate Determination Programs
In vivo cell fate changes do not occur in isolation, but rather in a spatiotemporally defined manner that is under the control of a cell’s microenvironmental milieu, or niche [2]. Many niche-derived signals can induce changes in intracellular metabolism. For example, growth factor signaling acutely increases nutrient uptake and stromal cells can provide metabolic support to stem cells by directly providing nutrients [49,50]. Stem cells of multiple tissues, including those of the blood, brain, and skin, are in close proximity to the vasculature, suggesting that their microenvironment may be closely regulated by continuous blood supply [51–53]. Given recent evidence that the extracellular nutrient milieu may vary in a tissue-specific manner [8,9], we discuss the possibility that nutrient availability within stem cell niches could contribute to cell fate decisions through αKG-dependent dioxygenases (Figure 3).
Figure 3. Niche Regulation of α-Ketoglutarate (αKG)-Dependent Dioxygenases.
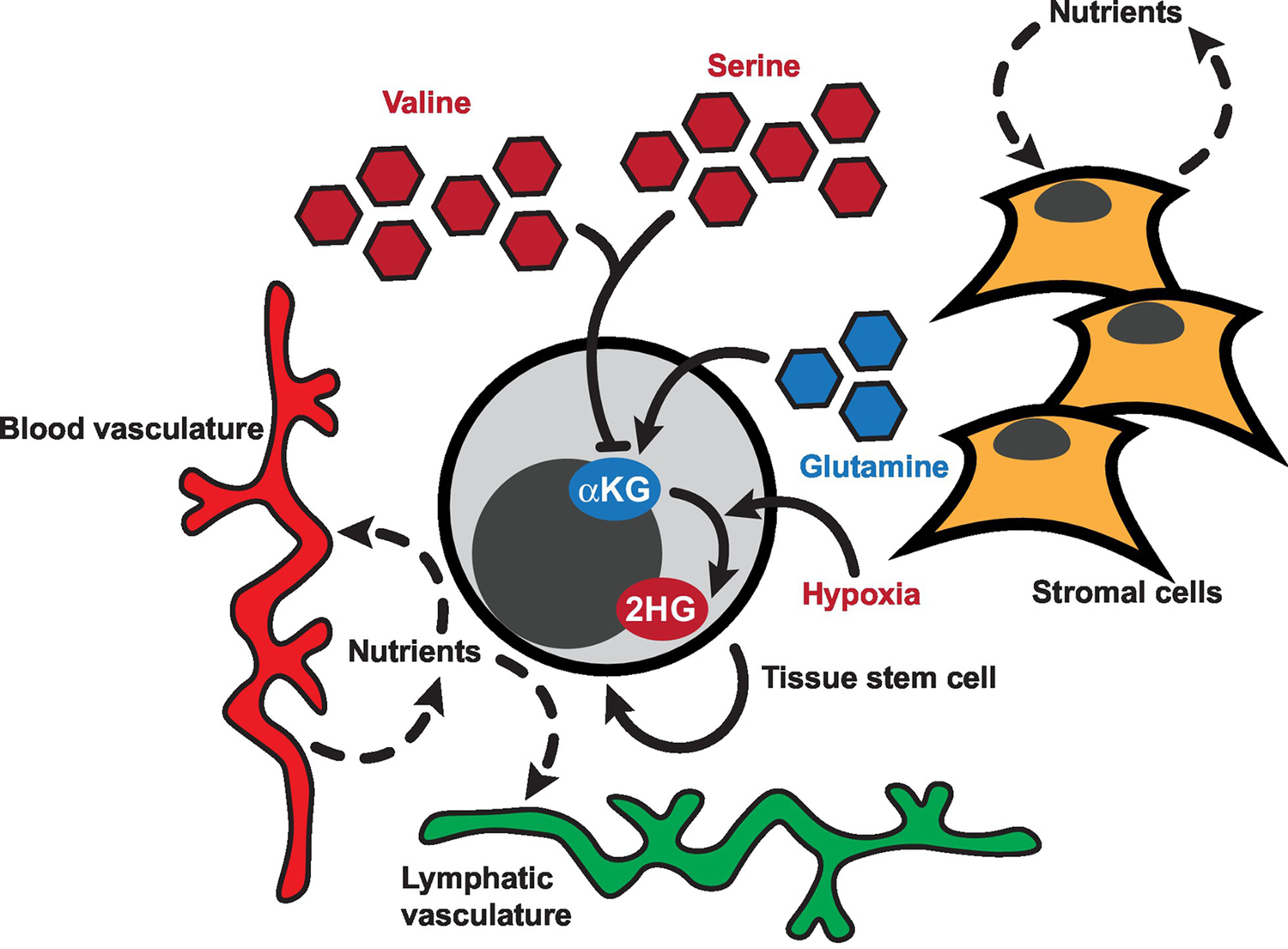
Summary of potential regulators of αKG-dependent dioxygenase activity within the stem cell niche. αKG is produced from glutamine-derived glutamate via transamination reactions, which can be suppressed or reversed by extracellular amino acids such as valine and serine. Hypoxia directly antagonizes dioxygenase activity and facilitates production of 2-hydroxyglutarate (2HG) from αKG. The role of niche cells in regulation of αKG-dependent dioxygenase activity has yet to be explored. Niche cells may add another layer of metabolic regulation of dioxygenase function, as many stem cells reside in proximity to the vasculature and stromal cells may provide nutrients to stem cells and/or compete with stem cells for nutrients within the niche.
Oxygen
How cells adapt to hypoxia has been thoroughly reviewed elsewhere [54] and here we highlight how αKG-dependent dioxygenases contribute to the cellular response to changes in oxygen availability. While all αKG-dependent dioxygenases consume oxygen, only a subset have sufficiently low affinity for oxygen to be limited by physiological levels of hypoxia [16]. However, hypoxic conditions, which induce intracellular acidification and high intracellular NADH/NAD+, favor promiscuous reduction of αKG to L-2HG by lactate dehydrogenase and malate dehydrogenase [55,56]. This rise in L-2HG is necessary and sufficient for hypoxic hypermethylation of histones in certain contexts [57]. Collectively, these findings suggest that hypoxia may block αKG-dependent dioxygenase activity through multiple mechanisms.
Intriguingly, many stem cells, including neural stem cells and HSCs, reside in hypoxic niches [58–60]. Notably, malignant ependymomas, derived from hypoxic fetal neural precursors, require hypoxia to proliferate due to metabolic requirements to maintain histone methylation and acetylation [61]. Whether or not metabolic responses to hypoxia also contribute to normal stem cell behavior remains an open question. Hypoxia-induced 2HG could create a nonpermissive environment for differentiation, thereby coupling hypoxic niches to homeostatic stem cell self-renewal. While the majority of HSCs reside in hypoxic niches, some studies report a small percentage of progenitors in well-oxygenated regions [62,63]. Whether these HSCs are functionally distinct in their ability to differentiate remains to be seen, but an intriguing possibility is that reduced 2HG levels in better-perfused niches facilitates differentiation. In this scenario, IDH1/2 mutations would effectively un-couple HSC differentiation from niche regulation via constitutive 2HG production.
Amino Acids
In most proliferating cells in vitro, glutamine is the major contributor to the carbon backbone of the TCA cycle. Glutamine is first deamidated to glutamate, which is then deaminated to αKG via transaminase reactions or glutamate dehydrogenase. Consequently, manipulating extracellular glutamine levels modulates TCA cycle metabolites, cell fate, and proliferation [5]. As discussed earlier, reduced catabolism of glutamine-derived αKG in the TCA cycle enables naïve mouse ESCs to maintain an elevated αKG/succinate ratio and to proliferate in the absence of glutamine [6,18]. Accordingly, glutamine has two distinct roles in regulating ESC fate. Transient glutamine withdrawal selects for ESCs with enhanced self-renewal due to death of more committed cells. However, because glutamine is the major source of αKG, prolonged glutamine starvation decreases ESC self-renewal. Resupplementation of glutamine after a transient withdrawal therefore enables naïve ESCs to recover expression of key pluripotency markers [6].
Glutamine is also a key regulator of αKG abundance and cell fate in adult tissues. In macrophages, glutamine starvation blunts M2 polarization and favors a proinflammatory phenotype, which can be reversed by αKG supplementation [31]. Similarly, glutamine starvation in T cells prevents effector differentiation, which can be restored via αKG treatment [48,64]. Glutamine starved or L-2HG treated T cells adopt a memory T cell fate, a subset of which may have stem cell properties [48,65]. Similarly, glutamine starvation enhances stemness of intestinal organoids harboring premalignant oncogenic mutations and increases tumor initiating capacity [26]. While the above studies manipulated glutamine levels in vitro, heterogeneity in glutamine availability may also be an important determinant of cell fate in vivo. For example, glutamine deficiency in melanoma cores is sufficient to deplete αKG and blunt cancer cell differentiation and increasing dietary glutamine increases αKG levels and impairs tumorigenesis [10,66]. Altogether, these data are consistent with glutamine availability creating a permissive environment for cell fate determination by enabling αKG accumulation. Thus, anticancer therapies aimed at suppressing glutamine uptake and catabolism may inadvertently select for the most aggressive stem cells in a tumor.
In proliferating cells, conversion of glutamate to αKG is largely accomplished by transaminase enzymes, which reversibly donate the amine nitrogen of glutamate to a ketoacid in order to synthesize a non-essential amino acid (NEAA) and αKG. The particular transaminase that predominantly contributes to αKG may vary in a tissue-specific manner. In IDH wild type AML, catabolism of branched chain amino acids (BCAA) valine, leucine, and isoleucine by branched chain aminotransferase 1 (BCAT1) restricts intracellular αKG levels, leading to DNA hypermethylation and HIF1α stabilization. Notably, BCAT1 loss blunts AML stem cell growth and induces myeloid differentiation [67]. While all cells require the essential BCAAs for protein synthesis, valine in particular is required for normal HSC maintenance for unknown reasons [68]. As BCAT activity may be cell type-specific [69], it is therefore intriguing to consider that valine supports HSC self-renewal by restricting intracellular αKG.
In other tissues, serine synthesis represents a notable source of nucleo-cytosolic αKG production via phosphoserine aminotransferase (PSAT1) activity. In breast cancer, hyperactivation of the serine synthesis pathway maintains hypoxic cancer cell survival and supports αKG production for TCA cycle anaplerosis [70,71]. In murine epidermal stem cells, in vivo restriction of serine and its immediate downstream metabolite glycine triggers de novo serine synthesis activation and accumulation of αKG, which is necessary and sufficient to drive stem cell differentiation, H3K27me3 demethylation, and tumor suppression [7]. Dietary serine and glycine restriction has proven effective in delaying tumorigenesis in several cancer models and it will be of interest to understand whether or not αKG-dependent stem cell differentiation plays a role in these systems [72,73]. Collectively, these studies demonstrate that dietary manipulation of extracellular nutrient availability is sufficient to regulate stem cell fate, in part by controlling intracellular αKG levels.
Concluding Remarks
Proper development and adult tissue function requires careful control over cell fate programs. The aforementioned studies suggest that metabolites act as both inductive signals and regulators of competence during cell fate determination; therefore, disruption of tissue-specific pathways that maintain TCA cycle homeostasis promote pathology, most notably by enforcing progenitor self-renewal and driving tumorigenesis. Metabolite abundances, determined by environmental nutrient availability and cell type-specific genetic programs, can collaborate closely with lineage-specific transcriptional programs to regulate cell fate. Although we focused on αKG-dependent dioxygenases, TCA cycle metabolites serve as substrates and inhibitors for many metabolic reactions that contribute to cellular proliferation and may accordingly influence tissue homeostasis [5,74]. Evidence across multiple tissues suggest a model wherein αKG acts as an inducer of cell fate changes, while succinate, fumarate, and 2HG largely restrict competence to appropriately execute cell fate programs. Accordingly, nutrients that fuel intracellular αKG pools may create permissive environments for cell fate decisions, while mutations that drive oncometabolite accumulation prevent αKG-mediated fate changes. The roles of specific oncometabolites in lineage-specific cancers warrants investigation into the role of αKG during development and differentiation of their normal tissue counterparts. More broadly, whether the tissue-specific profiles of cancer-associated mutations in chromatin modifying enzymes, such as TETs and DNA and histone methyltransferases, likewise predict metabolite-responsive nodes co-opted by tumors will be an important area of future investigation. Many questions remain (see Outstanding Questions), and improved technologies to enable measurement of extracellular, subcellular, and cell type-specific metabolite abundances in vivo, coupled with genetic experiments to modulate dioxygenase sensitivity to metabolites, will facilitate our ability to gain a deeper understanding of the physiological and pathological roles of metabolites in controlling cell fate.
Outstanding Questions.
What is the role of specific dioxygenases in mediating cell fate changes in response to metabolic fluctuations?
Do expression patterns of dioxygenases with varying affinities for metabolites provide a biochemical rationale for specific outcomes driven by metabolic perturbations?
Does differential regulation of subcellular metabolite pools contribute to metabolic control of αKG-dependent dioxygenases?
What is the role of metabolites within native tissue microenvironments in regulating cell fate? Do endogenous nutrients fluctuate sufficiently to modulate intracellular metabolite abundances to modify cell fate outcomes?
Dometabolites contribute tomaintenance of cell identity, or do they only facilitate/repress changes in fate? For example, do transcription factors require a basal level of αKG to sustain identity once a chromatin landscape is established, or do changes in αKG pools only enable dynamic reprogramming of gene expression?
Highlights.
The TCA cycle metabolites α-ketoglutarate (αKG), succinate, fumarate, and 2-hydroxyglutarate are emerging as regulators of cell fate decisions via control of αKG-dependent dioxygenases, which can facilitate demethylation of chromatin.
Metabolites display tissue-specific effects on cell fate, likely reflecting distinct lineage sensitivities to metabolites and unique transcriptional demands of cell state changes.
Metabolites are integrated with lineage-specific programs via post-transcriptional and post-translational control of transcription factors, control of coactivator function, and control of enhancer–promoter contacts.
Tissue-specific and systemic nutrient availability may modulate stem cell fate through αKG-dependent dioxygenases, highlighting the role of the TCA cycle as a central hub that integrates extracellular cues with cell fate decisions.
Acknowledgments
S.C.B. is a Ruth Kirschstein NIH Predoctoral fellow (F31CA236465). L.F. is a Searle Scholar. This work was additionally supported by grants to L.F. from The Starr Foundation (I11–0039), the Concern Foundation, the Anna Fuller Fund, The Edward Mallinckrodt, Jr. Foundation, the National Institutes of Health (R37 CA252305), and the Memorial Sloan Kettering Cancer Center Support Grant P30 CA008748. We thank S. Ellis for critical assessment of this review. We apologize to our colleagues whose work we could not cite due to space restrictions.
Glossary
- α-Ketoglutarate-dependent dioxygenases:
a family of >60 iron-containing enzymes that consume αKG and molecular oxygen as part of their reaction cycle and are inhibited by succinate, fumarate, and 2HG. α-Ketoglutarate-dependent dioxygenases are additionally activated by ascorbate (Vitamin C). In vitro enzymatic assays suggest that the Km of αKG-dependent dioxygenases for αKG are in the low (~1–50) micromolar range, whereas the Ki and IC50 of succinate, fumarate, and 2HG are in the upper micromolar to low millimolar range.
- Cell fate determination:
the process wherein cells progressively acquire specific identities. Specification relies on both cues from a cell’s environment and the competence of that cell to respond appropriately to these signals. For example, early in amphibian development, the Spemann-Mangold organizer can secrete factors to induce non-neural tissues to adopt a neural fate, but this plasticity is lost as development progresses. In postnatal life, stem cell fate determination maintains homeostasis and response to tissue injury.
- Jumonji C-domain lysine demethylases:
αKG-dependent dioxygenases that catalyze net demethylation of mono-, di-, and tri-methylated lysines, including on histone tails (e.g., H3K27me2/3, H3K9me2/3, H3K4me2/3).
- Oncometabolite:
metabolite [such as D-2HG, succinate, and fumarate] that accumulates in cancer as a result of mutations in metabolic enzymes (IDH1/2, SDH, and FH, respectively) and is thought to facilitate malignant development.
- Prolyl hydroxylases (PHDs):
αKG-dependent dioxygenases that catalyze hydroxylation of proline residues in target proteins. Proline hydroxylation stabilizes collagen helices and facilitates degradation of labile subunits of the hypoxia inducible factors (HIFs).
- Stem cells:
undifferentiated progenitors responsible for producing the cells of a tissue during embryogenesis and postnatal life. Stem cells balance two key cell fate decisions: self-renewal, to produce more stem cells, and differentiation, to produce the mature cells of a tissue. Stem cells include embryonic stem cells, which can organism, and tissue stem cells, which under homeostasis are restricted to production of one or more cell types within their cognate tissue.
- Ten-eleven translocation (TET) DNA cytosine-oxidizing enzymes:
αKG-dependent dioxygenases that catalyze iterative oxidation of methylated cytosines on DNA, thereby facilitating DNA demethylation.
- Transcription factors (TFs):
proteins that directly bind consensus sequences in DNA and recruit chromatin remodeling machinery to target loci, thereby activating or repressing gene expression. TFs can respond to exogenous stimuli (e.g., HIF in response to hypoxia) and often act in a coordinated manner to enforce a cell state (e.g., STAT3 and NANOG are part of a network that enforces pluripotency).
- Tricarboxylic acid (TCA) cycle:
a series of conserved chemical reactions by which carbohydrates, fatty acids, and/or amino acids are oxidized to generate reducing equivalents and precursors for macromolecule synthesis.
References
- 1.Bedzhov I et al. (2014) Developmental plasticity, cell fate specification and morphogenesis in the early mouse embryo. Philos. Trans. R. Soc. B Biol. Sci 369, 20130538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ge Y and Fuchs E (2018) Stretching the limits: from homeostasis to stem cell plasticity in wound healing and cancer. Nat. Rev. Genet 19, 311–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adam RC and Fuchs E (2016) The yin and yang of chromatin dynamics in stem cell fate selection. Trends Genet 32, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schvartzman JM et al. (2018) Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol 217, 2247–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Intlekofer AM and Finley LWS (2019) Metabolic signatures of cancer cells and stem cells. Nat. Metab 1, 177–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vardhana SA et al. (2019) Glutamine independence is a selectable feature of pluripotent stem cells. Nat. Metab 1, 676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baksh SC et al. (2020) Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat. Cell Biol 22, 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan MR et al. (2019) Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan MR et al. (2019) Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab 29, 1410–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan M et al. (2016) Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol 18, 1090–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elia I et al. (2019) Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koivunen P et al. (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Losman J-A et al. (2013) (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koivunen P et al. (2007) Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J. Biol. Chem 282, 4524–4532 [DOI] [PubMed] [Google Scholar]
- 15.Laukka T et al. (2018) Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J. Mol. Biol 430, 3081–3092 [DOI] [PubMed] [Google Scholar]
- 16.Chakraborty AA et al. (2019) Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward PS et al. (2013) The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem 288, 3804–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carey BW et al. (2014) Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu C et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao M et al. (2012) Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26, 1326–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chowdhury R et al. (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selak MA et al. (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7, 77–85 [DOI] [PubMed] [Google Scholar]
- 23.Letouzé E et al. (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23, 739–752 [DOI] [PubMed] [Google Scholar]
- 24.Adam J et al. (2011) Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saha SK et al. (2014) Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran TQ et al. (2020) α-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat. Cancer 1, 345–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weng H et al. (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell 22, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batista PJ et al. (2014) M6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasemann MS et al. (2014) C/EBPα is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet 10, e1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su R et al. (2018) R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell 172, 90–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu P-S et al. (2017) α-Ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol 18, 985–994 [DOI] [PubMed] [Google Scholar]
- 32.Martello G and Smith A (2014) The nature of embryonic stem cells. Annu. Rev. Cell Dev. Biol 30, 647–675 [DOI] [PubMed] [Google Scholar]
- 33.Finley LWS et al. (2018) Pluripotency transcription factors and Tet1/2 maintain Brd4-independent stem cell identity. Nat. Cell Biol 20, 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C et al. (2013) Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev 27, 1974–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X et al. (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 [DOI] [PubMed] [Google Scholar]
- 36.Hamilton WB et al. (2019) Dynamic lineage priming is driven via direct enhancer regulation by ERK. Nature 575, 355–360 [DOI] [PubMed] [Google Scholar]
- 37.Hwang I-Y et al. (2016) Psat1-dependent fluctuations in α-ketoglutarate affect the timing of ESC differentiation. Cell Metab 24, 494–501 [DOI] [PubMed] [Google Scholar]
- 38.TeSlaa T et al. (2016) α-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab 24, 485–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tischler J et al. (2019) Metabolic regulation of pluripotency and germ cell fate through α-ketoglutarate. EMBO J 38, e99518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schvartzman JM et al. (2019) 2-Hydroxyglutarate inhibits MyoD-mediated differentiation by preventing H3K9 demethylation. Proc. Natl. Acad. Sci. U. S. A 116, 12851–12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu C et al. (2013) Induction of sarcomas by mutant IDH2. Genes Dev 27, 1986–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Figueroa ME et al. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sciacovelli M et al. (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu M and Ren B (2017) The three-dimensional organization of mammalian genomes. Annu. Rev. Cell Dev. Biol 33, 265–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flavahan WA et al. (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flavahan WA et al. (2019) Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 575, 229–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Modrek AS et al. (2017) Low-grade astrocytoma mutations in IDH1, P53, and ATRX cooperate to block differentiation of human neural stem cells via repression of SOX2. Cell Rep 21, 1267–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chisolm DA et al. (2017) CCCTC-binding factor translates interleukin 2- and α-ketoglutarate-sensitive metabolic changes in T cells into context-dependent gene programs. Immunity 47, 251–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson CB and Bielska AA (2019) Growth factors stimulate anabolic metabolism by directing nutrient uptake. J. Biol. Chem 294, 17883–17888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodríguez-Colman MJ et al. (2017) Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 543, 424–427 [DOI] [PubMed] [Google Scholar]
- 51.Crane GM et al. (2017) Adult haematopoietic stem cell niches. Nat. Rev. Immunol 17, 573–590 [DOI] [PubMed] [Google Scholar]
- 52.Gur-Cohen S et al. (2019) Stem cell-driven lymphatic remodeling coordinates tissue regeneration. Science 366, 1218–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tavazoie M et al. (2008) A specialized vascular niche for adult neural stem cells. Cell Stem Cell 3, 279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee P et al. (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol 21, 268–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sergiy MN et al. (2016) Acidic pH is a metabolic switch for 2-hydroxyglutarate generation and signaling. J. Biol. Chem 291, 20188–20197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Intlekofer AM et al. (2017) L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol 13, 494–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Intlekofer AM et al. (2015) Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab 22, 304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mazumdar J et al. (2010) O2 regulates stem cells through Wnt/β-catenin signalling. Nat. Cell Biol 12, 1007–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simsek T et al. (2010) The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7, 380–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takubo K et al. (2013) Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michealraj KA et al. (2020) Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 181, 1329–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spencer JA et al. (2014) Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 508, 269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Acar M et al. (2015) Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526, 126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klysz D et al. (2015) Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal 8, ra97. [DOI] [PubMed] [Google Scholar]
- 65.Tyrakis PA et al. (2016) S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 540, 236–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ishak Gabra MB et al. (2020) Dietary glutamine supplementation suppresses epigenetically-activated oncogenic pathways to inhibit melanoma tumour growth. Nat. Commun 11, 3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raffel S et al. (2017) BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388 [DOI] [PubMed] [Google Scholar]
- 68.Taya Y et al. (2016) Depleting dietary valine permits non-myeloablative mouse hematopoietic stem cell transplantation. Science 354, 1152–1155 [DOI] [PubMed] [Google Scholar]
- 69.Mayers JR et al. (2016) Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Possemato R et al. (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Samanta D et al. (2016) PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res 76, 4430–4442 [DOI] [PubMed] [Google Scholar]
- 72.Maddocks ODK et al. (2017) Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376 [DOI] [PubMed] [Google Scholar]
- 73.Maddocks ODK et al. (2012) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McBrayer SK et al. (2018) Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175, 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ward PS et al. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dang L et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sasaki M et al. (2012) IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488, 656–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bardella C et al. (2016) Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell 30, 578–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Díaz-Castro B et al. (2015) Resistance of glia-like central and peripheral neural stem cells to genetically induced mitochondrial dysfunction—differential effects on neurogenesis. EMBO Rep 16, 1511–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Piruat JI et al. (2004) The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell. Biol 24, 10933–10940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Agathocleous M et al. (2017) Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cimmino L et al. (2017) Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ho TT et al. (2017) Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morris JP et al. (2019) α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mardis ER et al. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med 361, 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan H et al. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med 360, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bejarano-García JA et al. (2016) Sensitivity of hematopoietic stem cells to mitochondrial dysfunction by SdhD gene deletion. Cell Death Dis 7, e2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guitart AV et al. (2017) Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J. Exp. Med 214, 719–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kulkarni RA et al. (2019) A chemoproteomic portrait of the oncometabolite fumarate. Nat. Chem. Biol 15, 391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lemonnier F et al. (2016) The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc. Natl. Acad. Sci. U. S. A 113, 15084–15089 [DOI] [PMC free article] [PubMed] [Google Scholar]