

Abstract
White tail disease (WTD) of cultured Macrobrachium rosenbergii is caused by Macrobrachium rosenbergii nodavirus (MrNV) and extra small virus (XSV). Since both the viruses have small single strand RNA as genetic material with short generation time, they are more prone to mutations. Hence detection methods developed for one strain may be suboptimal for the detection of isolates from the different geographical locations. In the present study two new genomic based methods (RT-PCR and dot-blot hybridization) along with one immunological method (polyclonal antibodies based detection) were developed for the detection of Indian isolates of MrNV and XSV. Among genomic based methods, RT-PCR assay developed was most sensitive. Sensitivity of detection of RT-PCR was 1 fg (both MrNV and XSV) of total RNA extracted from purified viral inoculum preparation. In case of WTD positive whole tissue total RNA, the limit of detection was 10 fg for both MrNV and XSV. Dot-blot hybridization had a detection limit of 10 pg and 0.1 ng for MrNV and XSV respectively when RNA extracted from viral inoculum preparation was used; 0.1 ng and 1 ng when WTD positive whole tissue total RNA was used. Polyclonal antibodies against recombinant proteins (MrNV and XSV capsid) were synthesised. Western blotting and indirect ELISA revealed that the antibodies produced to be specific and highly sensitive. Recombinant protein (antigen) of MrNV and XSV capsid were detected at the dilution of 1:8000. However in case of infected prawn tissue sample, MrNV and XSV were detected at the dilution of 1:32,000 and 1:64,000 respectively. All methods developed are field applicable.
Keywords: MrNV, XSV, Molecular detection, Indian isolate
Introduction
Giant freshwater prawn, Macrobrachium rosenbergii is an important cultured palaemonid, and its culture represents economically important activity in many of the developing Southeast Asian countries. Global production of freshwater prawn has got tremendous scope for the growth due to market demand; however, production could be seriously affected by the occurrence of white tail disease (WTD). WTD affects post larvae (PL) and juveniles of M. rosenbergii in hatcheries and farms, causing mortalities up to 100% in 2–3 days after infection [32]. For the first time WTD was reported from French West Indies [1], subsequently from Taiwan [44], China [25], India [32], Thailand [47], Australia [22], Malaysia [31] and in 2016 it was reported from Indonesia [18].
Macrobrachium rosenbergii nodavirus (MrNV) along with extra small virus (XSV) are the causative agents of WTD [25]. MrNV is a small, icosahedral, non-enveloped positive-sense virus with a size of 26–27 nm in diameter and belongs to the family Nodaviridae and proposed genus Gammanodavirus [4, 19, 20]. Smaller virus among the two, XSV is also non-enveloped and has a diameter of 14–16 nm with a single-stranded positive-sense RNA genome of approximately 0.8 kilobases [25, 39]. Clinical signs of the disease include lethargy, loss of appetite and whitish colouration of the muscle at the abdominal region in the earlier stage that spreads throughout the body including cephalothorax in the final stage [2, 9, 34].
The methods available for the detection of causative agents of WTD include electron microscopy, RT-PCR, RT-LAMP, in situ hybridization, dot-blot hybridization and antibody-based methods includes S-ELISA, TAS-ELISA [12, 24–26, 30, 38, 40]. The study conducted by Sahul Hameed et al. [32] showed the presence of only XSV in the diseased animals. Hence it became important to develop methods for the detection of both the viruses,’ i.e. MrNV and XSV causing WTD.
As viral borne infections are difficult to control, only preventive measures are important, which includes early diagnosis can help in disease management. It is also important for daily monitoring of prawn health to help in restrain outbreaks. Generally, viruses mutate at a faster rate due to short generation time and high mutation rates; viruses with smaller genome tend to mutate even faster and among viruses, RNA viruses are comparatively more prone to mutation due to lack of proofreading activity of RNA dependent RNA polymerases (RdRp) involved in their genome replication. The mutation rate of single-stranded RNA virus is higher when compared with dsRNA viruses even though the reasons behind this phenomenon are not known. As a result, new strains of viruses, particularly RNA viruses, constantly evolve [6, 23, 35, 36]. MrNV and XSV both being small, single-stranded RNA viruses tend to undergo rapid mutations, and this is evident by the studies on nucleotide and amino acid sequences of different geographical isolates [19, 46]. Hence already established detection methods developed for different geographical strains may be suboptimal for the detection of the Indian strains. Reports on sequences of RNA1 and RNA2 (GenBank accession no. JQ418295 and JQ418298) of MrNV, Indian isolate [19] and availability of partial sequence of XSV of Indian isolate (GenBank accession no. JQ418299) helped in addressing the problem. In this manuscript, we describe two newly developed genomic-based detection techniques (RT-PCR and dot-blot hybridization) and one antibody-based technique specific for the Indian isolates of MrNV and XSV. The methods were also tested successfully for the field samples.
Materials and methods
Sample collection and virus purification
Suspected samples showing clinical signs of WTD (whitish colouration of the muscle) were collected from two hatcheries situated along East coast of India (Andhra Pradesh) during December month of 2012. Two separate samples were collected from both the hatcheries. Collected samples were washed in DEPC-treated water, transferred to RNase free sterile tubes and transported quickly (within 12 h) to the laboratory using an ice-chilled box. Upon, arrival samples were stored at − 80 °C until used. Suspected samples were tested for WTD by RT-PCR using primers, UniShNodaF (5′ATGCAGTGGACGAACGTCAA 3′) and UniShNodaR (5′ TTACCACGTTATGAGGTCGC3′) designed by Naveen Kumar et al. [19]. Positive samples were homogenized in a sterile homogenizer using TN buffer (20 mM Tris–HCl and 0.4 M NaCl, pH 7.4) and a 10% (w/v) homogenate suspension was prepared. The suspended homogenate was centrifuged at 5000 g for 20 min at 4 °C and the collected supernatant was again centrifuged. The supernatant was filtered through 0.45 μm pore membrane (Merck-Millipore, USA) followed by a subsequent filtration through 0.22 μm membrane (Merck-Millipore, USA). This viral inoculum filtrate was further confirmed for the presence of WTD causing agents by RT-PCR. The filtrate was stored at − 80 °C until used.
Experimental infection
Healthy PL of M. rosenbergii were purchased from hatcheries in Orissa and Maharashtra, India which was confirmed negative for WTD by RT-PCR upon arrival. PL was reconciled in the live fish laboratory of the department with suitable aeration and feeding. Experimental infection was carried out by immersion challenge for 12 h. Healthy juveniles (MrNV and XSV PCR negative) of M. rosenbergii weighing approximately 0.1 g were placed in an aquarium containing freshwater maintained at the temperature range 27 to 30 °C with continuous aeration. Viral inoculum prepared, as described earlier, was added to the rearing medium at the rate of 0.1% of the total rearing medium [3, 28, 45]. Test animals were observed regularly for clinical signs of WTD. Dead animals showing whitish discolouration at different time intervals were grouped into ten groups, viz., group 1 till group 10 consisting of 20 juveniles each and were treated as separate samples. All ten samples were examined by RT-PCR, dot-blot hybridization and immunological assays developed in the present study.
Total RNA extraction
Total RNA was extracted using TRIzol reagent (Invitrogen, USA) following the manufacturer’s instructions with some modifications and stored at − 80 °C until used. Isopropyl alcohol was used for precipitating out RNA. The obtained RNA pellet was washed with 75% ethanol and then dissolved in 50 μl DEPC treated ultrapure water (Millipore, USA). The concentration and purity of RNA were determined using NanoDrop ND 1000 spectrophotometer (Thermo Scientific, USA).
RT-PCR
Primer sets were designed for the conservative region of both MrNV and XSV (Table 1) of Indian isolates using primer3 software and were custom synthesised by Bioserve Biotechnologies (Hyderabad, India). GenBank accession numbers of the Indian isolates used for the primer design are JQ418298; AM114036; HQ637179; GU300102 in case of MrNV. In case of XSV we used the following sequences: JQ418299; AM114037; NC_043494; HQ637180. Primers showed 100 percent match with all Indian isolates in both the cases (MrNV and XSV). cDNA was reverse transcribed from total RNA using 20 pmol of specific reverse primer in a 20 μl reaction also consisting of 4 µl of 5 × buffer (250mMTris-HCl, pH 8.3; 250 mM KCl, 20 mM MgCl2),2 µl of 0.1 M dithiothreitol (DTT), (Fermentas, Canada), 200 µM of each of the four deoxyribonucleotide triphosphate (dNTPs), 20 U RNase inhibitor (Ambion, USA) and100 U of Revert Aid H minus M-mul V RT enzyme (Fermentas, Canada). The final volume was made up using DEPC treated water. Reverse transcription was carried out in a thermocycler at 42 °C for 1 h. 2 µl of the reverse-transcribed cDNA was used in PCR. Post standardization, PCR for both MrNV and XSV detection was carried out in a 30 µl reaction mixture containing 1 × assay buffer with 1.25 mM MgCl2, 200 µM of each of the 4 dNTPs, 1U of Taq polymerase (Bangalore GeNei, Bengaluru) and 10 pmol of each primer. PCR was performed in a thermocycler (BioRad, USA) with initial denaturation of 95 °C for 3 min and thirty cycles of denaturation at 95 °C for 40 s, annealing at 50 °C (both MrNV and XSV) for 30 s and extension at 72 °C for 30 s with a final delay of 5 min at 72 °C. The amplified PCR products were resolved by electrophoresis on 2% agarose gel, stained with ethidium bromide (0.5 µg/ml) and photographed using a gel documentation system (BioRad, USA). Sequencing of PCR products using both forward and reverse primers with an automated ABI 3100 Genetic analyser using fluorescent label dye terminators (SciGenom, India) was carried out for result confirmation. Amplification of β-actin gene of M. rosenbergii using primers (Table 1) developed by Liu et al. [13] was used as an internal control to monitor the RNA decay. The sensitivity of the primer sets was determined by ten-fold successive dilution of total RNA. To check the specificity of the primer sets cDNA synthesised to the total RNA extract of the tissue samples of healthy M. rosenbergii and P. monodon and also DNA extracted from WSSV and IHHNV infected P. monodon samples were used. No template controls were also included.
Table 1.
Sequences of the primers used in the present study. The length of amplified DNA fragments is given in parentheses
| Primers | Sequence (5′-3′) | Purpose |
|---|---|---|
| MrNVForward (151 bp) | CAAAGTCCGAGGTTCTAACA | Detection |
| MrNVReverse | ATATCTGACTGCAGCCTTGT | Detection |
| XSVForward (152 bp) | TTGGGTCATACCGTAATAGG | Detection |
| XSVReverse | ATAAGAGCCTTCATCAACGA | Detection |
| β-actin-Forward (193 bp) | TATGCACTTCCTCATGCCATC | Internal control |
| β-actin-Reverse | AGGAGGCGGCAGTGGTCAT | Internal control |
| MrNV DBHF (564 bp) | ATGGCTAGAGGTAAACAAAATTC | DBH |
| MrNV DBHR | TCATTGATCATCACGCCTGACA | DBH |
| XSV DBHF (525 bp) | ATGAATAAGCGCATTAATAAT | DBH |
| XSV DBHR | TTACTGTTCGGAGTCCCAATA | DBH |
| MrNV PEF (564 bp) | GGGCCGGATCCATGGCTAGAGGTAAACAAAATTC | PE |
| MrNV PER | GGCCAAGCTTTCATTGATCATCACGCCTGACA | PE |
| XSV PEF (525 bp) | GGGCCGGATCCATGAATAAGCGCATTAATAAT | PE |
| XSV PER | GGCCAAGCTTTTACTGTTCGGAGTCCCAATA | PE |
Underlined nucleotide sequences are Bam I and Hind III restriction sites at forward and reverse primers respectively
DBH, primer used for probe development for dot-blot hybridization method; PE, primers used for amplification of gene used for protein expression
Dot-blot hybridization
Digoxigenin (DIG)-labelled probes were prepared using PCR products of the recombinant vectors of capsid genes of MrNV and XSV (used for recombinant protein expression). The primers used were MrNV DBF and MrNV DBR for MrNV capsid gene amplification and XSV DBF and XSV DBR for XSV capsid gene amplification (Table 1) (amplification conditions were same as given in the next section). The probes were labelled with DIG using 3′-end labelling kit (Roche Diagnostics, Germany). Dot blot using DIG-labelled probes was performed as per the manufacturer’s instruction with minor modifications to confirm the presence of MrNV and XSV in the tested sample. To perform dot blot, total RNA was denatured using MOPS buffer and was spotted onto a nylon membrane (Pall Corporation, BioTrace™ NT, USA). The membrane was dried at 50 °C for 1 h and cross-linked using a UV light for 3 min. After UV cross-linking, pre-hybridization was carried out for 60 min at 42 °C in 50% formamide, 2 × saline sodium citrate (SSC), 50 mM sodium phosphate, 2% blocking reagent, 0.1% sodium sarkosyl and 7% sodium dodecyl sulphate (SDS). Pre-hybridization solution was removed from the hybridization bag and replaced with 1 ml hybridization solution (Roche Diagnostics, Germany) per 10 cm2 membrane and incubated along with 25 ng/ml denatured probes for overnight hybridization at 42 °C with gentle agitation. The membrane was removed from the bag and washed with the 2 × SSC for 5 min at 25 °C under constant agitation in a clean glass petri plate. The membrane was then washed twice with 0.5 × SSC, 0.1% SDS solution (pre-warm to wash temperature) at 68 °C under constant agitation for 15 min each followed by washing with maleic acid buffer(100 mM maleic acid, 150 mM NaCl, pH 7.5, 0.3% Tween 20) for 5 min at room temperature. After hybridization and stringent washes, the membrane was blocked using blocking solution (100 mM maleic acid, 150 mM NaCl, pH 7.5, 1% blocking reagent) and finally washed once more with wash buffer (100 mM Tris–HCL, pH 9.5, 100 mM NaCl, 50 mM MgCl2) for 5 min. Then the membrane paper was incubated with antibody solution (Roche Life Science, Germany) for 30 min. Finally, the colour was developed by adding freshly prepared colour substrate solution (according to the manufacturer’s protocol). After incubation in the dark for a few minutes to 1 h the reaction was stopped by keeping the membrane in distilled water. Hybridization was performed using total RNA extracted from tissue sample and purified viral filtrate. The sensitivity of the probes was determined by successive tenfold dilution of total RNA. Specificity was tested by using the same controls used for RT-PCR. Total RNA from healthy prawns were used as negative control, and recombinant plasmids of MrNV and XSV capsid genes were used as positive controls.
Antibody-based detection method
Protein expression (MrNV and XSV capsid genes) and purification of expressed proteins for polyclonal antibody production
Complementary DNA was synthesized and PCR amplification was carried out for capsid genes of MrNV and XSV by designing primers based on the nucleotide sequence data generated for Indian isolates in our previous study (GenBank accession no. JQ418295, JQ418298 and JQ418299) [19]. PCR was carried out in a thermocycler (BioRad, USA) with initial denaturation of 95 °C for 5 min and thirty cycles of denaturation at 95 °C for 1 min, annealing at 58 and 45 °C (MrNV and XSV capsid genes respectively) for 1 min and extension at 72 °C for 1 min with a final delay of 10 min at 72 °C. PCR products of capsid genes of MrNV and XSV was generated using primers with BamHI and HindIII restriction sites at sense and anti-sense primers, respectively (Table 1). Amplified capsid genes of MrNV and XSV were ligated to pET-32a (+) (Novagen, USA) and pQE-30 UA vectors (Qiagen, Germany) respectively after restriction digestion. Recombinant pET-32a (+) and pQE-30 UA vectors were transformed into competent Escherichia coli DH5α cells (Invitrogen, USA) and E. coli SG 13009 cells (Qiagen, Germany) respectively. The recombinant transformants were selected using blue/white screening and ampicillin (100 μg/ml) and kanamysin (25 μg/ml) selection on Luria Bertani (LB) agar (HiMedia, India). The presences of the gene insert in the colonies containing recombinant plasmid were confirmed by PCR. For further, confirmation randomly selected PCR positive colonies containing a recombinant vector of the gene inserts were sequenced (SciGenom, India). The recombinant plasmids of the capsid gene of MrNV in DH5α cells were purified and transformed to expression competent E. coli BL21 (DE3)pLysS cells (Invitrogen, USA). The transformants were selected using ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) on LB agar. Positive clones were further confirmed by PCR. For protein expression overnight grown cultures of PCR positive recombinant clones (recombinant plasmid of capsid gene of MrNV in BL21 (DE3)pLysS cells and capsid gene of XSV in SG 13009) were inoculated in to LB broth containing ampicillin (100 μg/ml) and kanamysin (25 μg/ml) or chloramphenicol (34 μg/ml) in case of BL21 cells. Cultures grown to a density of 0.6 to 0.7 at OD600 were induced with a final concentration of 1 mM isopropyl β-D-thiogalactoside (IPTG). Post induction, cultures were grown for 4 h with vigorous shaking and expression of the recombinant proteins were examined by separation on 12% SDS-PAGE [11]. The N-terminal 6 × His-tagged recombinant proteins were purified in nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (Qiagen, Germany) using guanidine hydrochloride denaturation method according to manufacturer’s instruction. The eluted proteins were dialyzed against 10 mMTris, pH 8.0, 0.1% Triton X-100 overnight at 4 °C to remove guanidine hydrochloride. Concentrations of purified proteins were estimated using bicinchoninic acid (BCA) method [37] following kit manufacturer’s instruction (Merck, India).
Polyclonal antibody production against recombinant proteins
Polyclonal antibodies were produced against purified recombinant proteins (capsid proteins of MrNV and XSV) by immunizing white rabbits (2.5 to 3 kg) with 200 μg recombinant proteins by intramuscular injections at 7-day intervals for 4 consecutive weeks. Freund’s complete adjuvant was used for the first injection, and subsequently, Freund’s incomplete adjuvant was used. Animals were bled, a week after the last dose by cardiac puncture and sera were separated and stored at − 20 °C in aliquots until used.
Western blot analysis
Prawn samples for Western blotting were prepared by the method described by Magbanua et al. [17] with minor modifications. Samples were homogenized using TN buffer (20mMTris-HCl and 0.4 M NaCl, pH 7.4) and 10% (w/v) suspension was prepared. The homogenate was centrifuged at 5000 g for 20 min at 4 °C and the collected supernatant was recentrifuged at 10,000 g for another 20 min at 4 °C. The final supernatant was mixed with equal volume of 2 × loading buffer (100 mM Tris–Cl, pH 6.8; 4% (w/v) SDS; 20% (v/v) glycerol; 0.1% (w/v) bromophenol blue; 200 mM β-mercaptoethanol) and heated at 95 °C for 10 min. The samples were clarified further by centrifuging again and 20 μl of this was used for loading. Similarly, bacterial cells expressing recombinant capsid proteins as well as purified recombinant capsid proteins were also prepared. Samples were loaded into the wells of a 12% SDS-PAGE gel and electrophoresed (BioRad, USA). Western blotting was performed as per the procedure of Towbin, Staehelin and Gordon [42] using the serum of immunized rabbit. After SDS-PAGE, proteins were transferred to a nitrocellulose membrane using protein electrophoresis unit (BioRad, USA). Then the membrane was blocked by soaking with 3% (w/v) BSA (HiMedia, Mumbai) at 4 °C for overnight. After washing with PBS, membrane was incubated with respective primary antibodies produced against recombinant proteins for 1 h at room temperature on a rotor shaker. Visualization of the band was achieved by incubation of the membrane with secondary antibodies (goat anti-rabbit antibodies) conjugated with horseradish peroxidase (HRP) at room temperature for 1 h followed by development using diaminobenzine-hydrogen peroxide (DAB-H2O2). Tissue samples of virus-free M. rosenbergii and P. monodon, as well as WSSV and IHHNV, infected P. monodon tissues were used to check the cross-reactivity of the primary antibodies produced. Experimentally infected prawns were also tested using the antibodies produced.
Indirect ELISA
Indirect ELISA was used to check the application of the primary antibodies produced in the detection of MrNV and XSV [7]. Wells of polystyrene ELISA plates were coated separately with 10 µg/ml of antigen in carbonate bicarbonate buffer and incubated overnight at 4 °C. After washing with phosphate-buffered saline (PBS), non-specific sites were blocked by 3% BSA in PBS and further incubated overnight at 4 °C. After washing thrice with phosphate-buffered saline Tween-20 (PBST), plates were incubated for 1 h at 37 °C with primary antibodies in PBS (produced against recombinant capsid MrNV and XSV) (1:8000 dilutions). Post incubation plates were washed thrice with PBST and incubated with 100 µl/well of horseradish peroxidise-conjugated goat anti-rabbit immunoglobulins (Sigma Aldrich, USA) at 1:2000 dilutions in PBS for 1 h at 37 °C. After three times washing with PBST, enzyme activity was determined by adding 50 µl of freshly prepared solution of tetramethylbenzidine hydrogen peroxide (TMB/H2O2) into each well. Incubation was carried out for 5 min at room temperature in the dark and 100 µl of 2.5 N sulphuric acid was added into wells to stop the reaction. Enzyme activity was read at 450 nm in ELISA reader (ELX 800, Bio-Teck instrument, USA). Sensitivity of the antibodies was determined by serial dilution of the primary antibodies in PBS. Purified recombinant proteins, as well as WTD positive tissue samples, were used to check the application of the antibodies produced. Experimentally infected prawns were also checked for the reaction. Controls used in Western blotting to check cross-reactivity were rechecked for the reactions.
Results
RT-PCR for the detection of MrNV and XSV
Primers for RT-PCR developed were based on the sequence data generated for Indian isolates of MrNV and XSV. Post standardization of primer annealing temperature, primers and salt concentration for the detection, a single expected band of size ≈ 150 bp (MrNV is 151 bp and XSV is 152 bp) was visualized by agarose gel electrophoresis (Fig. 1a, b). Sensitivity of the presently developed methods was tested using successive tenfold serially diluted total RNA extracted from purified viral filtrate and whole tissue sample (WTD positive). Total RNA ranging from 1 µg to 0.1 fg were tested. The lowest detection limit of the presently developed RT-PCR assays for both MrNV and XSV was 1 fg (Fig. 2a, b) when total RNA extracted from purified viral inoculum was used. However, when RNA extracted from whole tissue sample was used presently developed assays showed sensitivity up to 10 fg of total RNA for both the viruses. Experimentally infected dead prawns grouped in to ten separate samples (group 1 to group 10) were used for the validation of the present assay developed for the detection of both MrNV and XSV. All ten samples which showed clinical signs of WTD showed positive for both MrNV and XSV (total RNA concentration of all samples were more than 200 ng). Sequence of the amplicons of both MrNV and XSV revealed 100% match with the Indian isolates. Negative control (total RNA of healthy prawns) did not show any amplification while the internal control (β-actin of M. rosenbergii) (193 bp) did show amplification. Non MrNV and XSV controls used to test specificity of the primers did not show any amplification proving the primers to be specific.
Fig. 1.
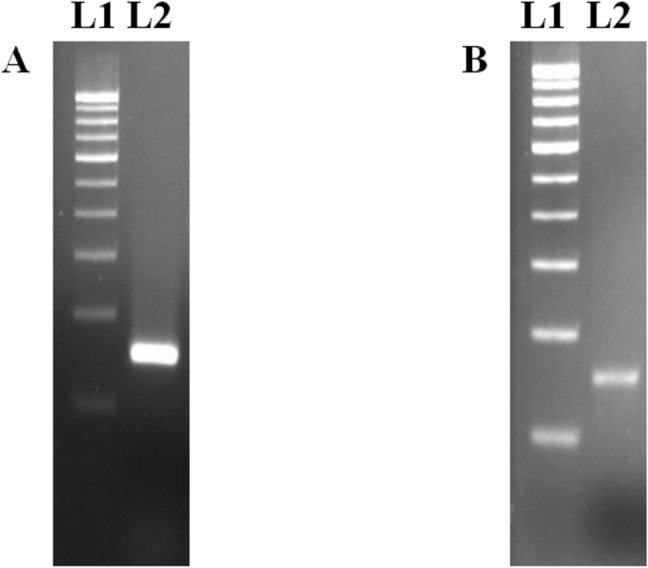
Detection of Macrobrachium rosenbergii nodavirus (MrNV) and extra small virus (XSV) by RT-PCR. a Lane L1: 100 bp ladder; Lane L2: MrNV (151 bp). b Lane L1: 100 bp ladder; Lane L2: XSV (152 bp)
Fig. 2.

Evaluation of sensitivity of the RT-PCR assay developed in the present study using total RNA extracted from the viral filtrate. a MrNV; b XSV. Lane M: 100 bp ladder marker; Lanes 1–11: RT-PCR assay results for the ten-fold dilution of total RNA ranging from 1 µg to 0.1 fg RNA
Detection of MrNV and XSV by dot-blot hybridization
Dot-blot hybridization for MrNV and XSV was performed using DIG-labelled PCR products of cloned MrNV and XSV capsid genes. MrNV and XSV were successfully detected by the dot-blot hybridization method developed in this study. To check sensitivity; same serially diluted RNA used for RT-PCR was used for dot-blot hybridization also. The lowest detection limit for MrNV was 10 pg of RNA when total RNA extracted from viral inoculum was used (Fig. 3a). However, when whole tissue total RNA was used, presently developed assay showed reaction up to 0.1 ng. In case of XSV the sensitivity of the presently developed method for purified viral filtrate and whole tissue sample were 0.1 ng (Fig. 3b) and 1 ng of total RNA, respectively. Experimentally infected samples which showed clinical signs of WTD and which were RT-PCR positive for both MrNV and XSV also showed positive reaction for dot-blot hybridization (Fig. 3c, d). Negative control (total RNA of healthy prawns) did not show any reaction while the positive controls (recombinant vectors of MrNV and XSV capsid genes) showed reactions. Same non MrNV and XSV controls used in RT-PCR were used to test specificity of the dot-blot assay developed and the probes did not show any reactions showing that the probes developed are specific.
Fig. 3.

Detection of MrNV and XSV by dot-blot hybridization assay developed in the present study. a Evaluation of detection threshold of the MrNV detection probes using total RNA extracted from viral filtrate (1:100 ng; 2:10 ng; 3:1 ng; 4:100 pg; 5:10 pg; 6:1 pg); b Evaluation of detection threshold of the XSV detection probes using total RNA extracted from viral filtrate (1:100 ng; 2:10 ng; 3:1 ng; 4:100 pg; 5:10 pg; 6:1 pg); c, d Dot hybridization using DIG-labelled probes for the experimentally infected samples; c MrNV, d XSV. *P: Positive; N: Negative
Cloning, expression and purification of recombinant proteins
PCR products of capsid genes of MrNV (564 bp) and XSV (525 bp) (Fig. 4a, b) were ligated to pET-32a (+) and pQE-30 UA expression vectors respectively after restriction digestion. Sequencing of the insert revealed both the genes had a putative methionine start site in the frame with the coding sequence of the vector and 100% match was revealed when the sequence were compared with those of Indian isolates. Expression of recombinant fusion proteins were carried out by inducing with 1 mM IPTG using BL21 (DE3)pLysS cells (MrNV capsid gene) and SG 13009 cells (XSV capsid gene). As expected, expressed fusion proteins of MrNV and XSV capsid showed an approximate molecular weight of 38.6 and 22 kDa respectively when run on 12% SDS-PAGE (Fig. 5). Recombinant proteins were purified using Ni-NTA affinity column chromatography. Concentration of purified recombinant proteins were 450 (MrNV capsid) and 600 (XSV capsid) µg/mL.
Fig. 4.
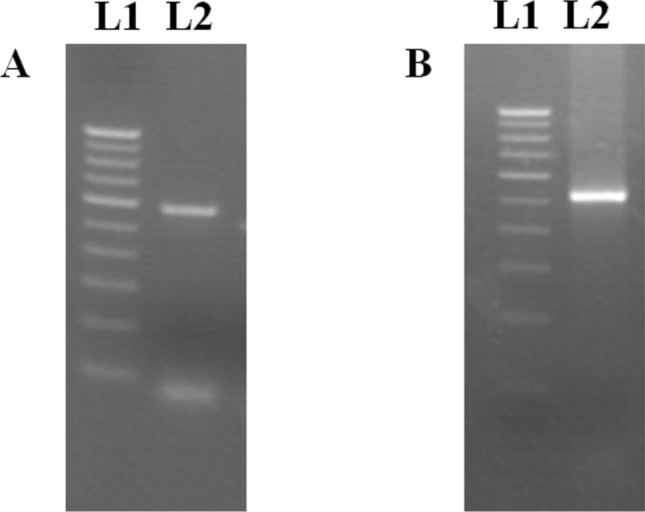
RT-PCR amplicon of capsid genes of MrNV and XSV. a Lane L1: Marker (100 bp); Lane L2: MrNV capsid (564 bp). b Lane L1: Marker (100 bp); Lane 2: XSV capsid (525 bp)
Fig. 5.

Coomassie brilliant blue-stained SDS-PAGE showing expressed recombinant proteins. Lane M: Protein marker; Lane 1: Uninduced recombinant (MrNV capsid) E. coli BL21 (DE3)pLysS cells; Lane 2: IPTG induced recombinant (MrNV capsid) E. coli BL21(DE3)pLysS cells; Lane 3: Uninduced recombinant (XSV capsid) SG13009 E. coli cells; Lane 4: IPTG induced recombinant (XSV capsid) SG13009 E. coli cells; Lanes 5 and 6: Western blot analysis using rabbit polyclonal antibody raised against purified capsid proteins of MrNV and XSV; Lane 5: Positive reactions for purified MrNV capsid protein; Lane 6: Positive reactions for purified XSV capsid protein
Specificity and sensitivity of polyclonal antibodies in detection of MrNV and XSV antigens
Antiserum against MrNV and XSV capsid proteins were generated separately using purified recombinant proteins by injecting the white rabbits. Western blotting was carried out using polyclonal antibodies obtained after third booster. The antibodies were highly specific and showed bands of expected size corresponding to 38.6 and 22 kDa for MrNV and XSV capsid proteins respectively (Fig. 5) when E. coli cells expressing proteins were Western blotted with the help of SDS-PAGE. Similarly, purified proteins as well as experimentally infected RT-PCR positive prawn samples also showed positive reactions with antibodies produced. Tissue samples of virus free M. rosenbergii and P. monodon as well as WSSV and IHHNV infected P. monodon tissues did not show any reactions confirming the polyclonal antibodies produced to be specific to MrNV and XSV only.
Indirect ELISA that was carried out using the polyclonal antibodies produced after the third booster showed that the antibodies produced were highly sensitive. Polyclonal antibodies showed reaction at 1:8000 dilutions in case of both MrNV and XSV recombinant protein antigens.
However when RT-PCR positive tissue samples were used the titre for MrNV and XSV obtained were 1:32,000 and 1:64,000 respectively. Negative controls used in Western blotting to check specificity were also used in indirect ELISA and similar kinds of results were obtained. RT-PCR positive tissue samples when used did not show any deviations from the previous results.
Discussion
WTD has the potential to crash the M. rosenbergii aquaculture industry in the years to come. Prawn production in India alone reduced to 2000 tonnes in 2009 from 40,000 tonnes in 2003, which may be attributed to outbreaks of WTD and other factors [8, 29, 33]. Since MrNV and XSV are highly prone for mutations, presently developed methods help in optimal detection of Indian strains. The role of individual virus (MrNV and XSV) in the pathogenesis of WTD is not clearly understood. Even though there are some evidence to show MrNV plays a key role in the pathogenesis of WTD [10] the role of XSV cannot be neglected [11, 16]. Studies from Zhang et al. [48] have shown XSV to be a satellite virus and several studies have shown that satellite virus acts as pathogenicity factor and also interfere and suppress RNAi mechanisms [27]. Presence of only XSV in the diseased animal was also reported in one of the studies [32]. Hence it became important to develop methods for the detection of both the viruses causing WTD.
RT-PCR is one of the most sensitive diagnostic methods when compared to other methods in detection of any pathogen and this method can also be applied for routine health monitoring and screening of carriers and broodstocks [38]. Several studies have shown nested or semi-nested PCR to be more sensitive than conventional single-step PCR [5, 14, 15, 41]. However, major drawbacks of nested PCR are high risk of cross-contamination and it is also cumbersome and more time consuming compared to conventional single-step PCR. A sensitive single-step PCR is sufficient enough for the diagnostics and regular screening hence the present study focused on developing a single-step PCR by designing primers for the nucleotide sequences of Indian isolates of MrNV and XSV. Total RNA extracted from the purified viral filtrate was used to establish the specificity and sensitivity of RT-PCR initially. Then the standardized methods were applied for the tissue samples and challenge study samples. For all tissue samples, RNA was extracted from the muscle tissue cells of the abdomen region, which is one of the target cells of both the viruses. To improve specificity of the method developed, reverse transcription was carried out using the specific reverse primers of the respective virus. Parameters such as primer concentration, annealing temperature and salt concentration were standardized for the best possible results and are given in the material and methods section. As the annealing temperatures for the detection of both the viruses are the same (50 °C), multiplex RT-PCR for simultaneous detection of both MrNV and XSV can also be standardized. Since multiplex PCR compromises with the sensitivity of the test, it was not further developed. Shorter amplicon size (~ 150 bp) in case of both MrNV and XSV detection in combination with quick denaturation and annealing steps (30 s each), the method developed has faster detection when compared to other assays developed with larger amplicon size [32, 41, 43]. The detection limit of the present RT-PCR method developed was 1 fg when total RNA extracted from the purified viral filtrate was used for both the viruses. However, sensitivity reduced for both the viruses to 10 fg when RNA from whole tissue sample was used. The reduction of sensitivity of total RNA from the whole tissue can be attributed to the presence of tissue RNA which increases the total RNA concentration and might be inhibitory for the RT-PCR. Similar kind of inhibition was also noted when higher RNA concentration was used by Sri Widada et al. [38] and Sahul Hameed et al. [32]. All the challenge study samples were positive for both MrNV and XSV by RT-PCR assay developed in the study providing evidence for the assay to be field applicable. In this study, all positive samples tested were positive for both MrNV and XSV, unlike in some studies where some samples were only positive for either XSV [32] or MrNV [39].
Dot-blot hybridization was performed using DIG-labelled PCR products of cloned MrNV and XSV capsid gene. Same serially diluted RNA used for RT-PCR was used for dot-blot hybridization and 10 pg of RNA was sufficient for successive dot-blot hybridization (MrNV) when RNA extracted from the viral filtrate was used. When the whole tissue sample was used, the reaction was seen for 0.1 ng of RNA. In the case of XSV, the sensitivity for virus filtrate and whole tissue were 0.1 ng and 1 ng respectively. Experimentally infected samples which were RT-PCR positive for both MrNV and XSV also showed positive reaction for dot-blot hybridization. However, since dot-blot is not an amplification method the sensitivity is lower than RT-PCR assay developed. The sensitivity of detection of MrNV was more when compared to XSV in both filtrate and tissue samples, possibly due to the presence of high MrNV copy numbers in the samples used. Similar observations were also made by Sri Widada and Bonami [39]. Dot-blot hybridization techniques developed for the detection of MrNV and XSV were also very specific similar to RT-PCR. Even though the RNA of both MrNV and XSV are single-stranded and are more prone for degradation, we observed no RNA degradation during the process of both RT-PCR and dot-blot hybridization which was monitored using β-actin gene of M. rosenbergii. However, suitable precautionary steps were taken while handling the total RNA. In comparison with RT-PCR, dot-blot hybridization method is cumbersome and requires a high degree of technical skill.
Available antibody-based diagnostic methods for MrNV includes S-ELISA [30] and TAS-ELISA [26] and for XSV, it is polyclonal antibodies raised against the whole virus [33] and recombinant viral protein [21]. In the majority of cases, antibodies were produced against the whole virus. Lack of susceptible cell lines for the propagation of prawn viruses and requirement of high skill and cost are few of the limitations in producing antibodies for the whole viruses. Purification of viruses from infected tissues is cumbersome and it is usually not possible to purify a single virus in the case of WTD even through ultracentrifugation [48]. In some cases, viruses were purified by slicing SDS-PAGE, which is time-consuming and yield limited quantity of protein which is contaminated with other tissue protein. Hyperimmune sera produced against such contaminated virus preparation are less suitable for diagnostic use. In the present study, detection of MrNV and XSV was carried out using antibodies produced against recombinant capsid proteins of MrNV and XSV. Since both MrNV and XSV do not pose any envelope, targeting capsid proteins for the immunological detection was found to be suitable. The polyclonal serums were specific and were highly sensitive against MrNV and XSV antigens with high titer values and details of the same are given in the results section. Earlier studies also suggest both ELISA and Western blot are as sensitive as RT-PCR and were able to detect MrNV and XSV post 24 h of infection [33].
It can be concluded that the presently developed genomic and immunological methods for the detection of Indian isolates of MrNV and XSV are sensitive and specific and can be used for WTD diagnosis and disease control in India. Among all the methods developed, we propose the use RT-PCR regularly for diagnosis and screening since it is less cumbersome comparatively and generally more sensitive.
Acknowledgements
The financial support received from the COE-Programme support to aquaculture and marine biotechnology, Department of Biotechnology, Government of India, towards this study is gratefully acknowledged.
Compliance with ethical standards
Conflict of interest
There is no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Arcier JM, Herman F, Lightner DV, Redman R, Mari J, Bonami JR. A viral disease associated with mortalities in hatchery-reared postlarvae of the giant freshwater prawn Macrobrachium rosenbergii. Dis Aquat Org. 1999;38:177–181. [Google Scholar]
- 2.Bonami JR, Sri WJ. Viral diseases of the giant fresh water prawns Macrobrachium rosenbergii: a review. J Invertebr Pathol. 2011;106:131–142. doi: 10.1016/j.jip.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Chen LL, Lo CF, Chiu YL, Chang CF, Kou GH. Natural and experimental infection of white spot syndrome virus WSSV in benthic larvae of mud crab Scylla serrata. Dis Aquat Org. 2000;40:157–161. doi: 10.3354/dao040157. [DOI] [PubMed] [Google Scholar]
- 4.Chong LC, Ganesan H, Yong CY, Tan WS, Ho KL. Expression, purification and characterization of the dimeric protruding domain of Macrobrachium rosenbergii nodavirus capsid protein expressed in Escherichia coli. PLoS ONE. 2019;14(2):e0211740. doi: 10.1371/journal.pone.0211740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalla Valle L, Zanella L, Patarnello P, Paolucci L, Belvedere P, Colombo L. Development of a sensitive diagnostic assay for fish nervous necrosis virus based on RT-PCR plus nested PCR. J Fish Dis. 2000;23:321–327. [Google Scholar]
- 6.Elena SF, Sanjuan R. Adaptive value of high mutation rates of RNA viruses: separating causes from consequences. J Virol. 2005;79:11555–11558. doi: 10.1128/JVI.79.18.11555-11558.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engvall E, Perlman P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8(9):871–874. doi: 10.1016/0019-2791(71)90454-x. [DOI] [PubMed] [Google Scholar]
- 8.Farook MA, Madan N, Taju G, Abdul Majeed S, Nambi KSN, Sundar Raj N, et al. Production of recombinant capsid protein of Macrobrachium rosenbergii nodavirus (r-MCP43) of giant freshwater prawn, M rosenbergii (de Man) for immunological diagnostic methods. J Fish Dis. 2014;37:703–710. doi: 10.1111/jfd.12156. [DOI] [PubMed] [Google Scholar]
- 9.Hsieh CY, Wu ZB, Tung MC, Tu C, Lo SP, Chang TC, et al. In situ hybridization and RT-PCR detection of Macrobrachium rosenbergii nodavirus in giant freshwater prawn, Macrobrachium rosenbergii (de Man), in Taiwan. J Fish Dis. 2006;29:665–671. doi: 10.1111/j.1365-2761.2006.00762.x. [DOI] [PubMed] [Google Scholar]
- 10.Jariyaponga P, Pudgerd A, Weerachatyanukul W, Hirono I, Senapinf S, Dhar AK, et al. Construction of an infectious Macrobrachium rosenbergii nodavirus from cDNA clones in Sf9 cells and improved recovery of viral RNA with AZT treatment. Aquaculture. 2018;483:111–119. [Google Scholar]
- 11.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 12.Lin F, Liu L, Hao GJ, Sheng PC, Cao Z, Zhou Y, Lv P, Xu T, Shen J, Chen K. The development and application of a duplex reverse transcription loop-mediated isothermal amplification assay combined with a lateral flow dipstick method for Macrobrachium rosenbergii nodavirus and extra small virus isolated in China. Mol Cell Probes. 2018;40:1–7. doi: 10.1016/j.mcp.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Yang WJ, Zhu XJ, Karouna-Renier NK, Rao RK. Molecular cloning and expression of two HSP70 genes in the prawn, Macrobrachium rosenbergii. Cell Stress Chaperones. 2004;9:313–323. doi: 10.1379/CSC-40R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo CF, Leu JH, Ho CH, Chen CH, Peng SE, Chen YT, et al. Detection of baculovirus associated with white spot syndrome (WSBV) in penaeid shrimps using polymerase chain reaction. Dis Aquat Org. 1996;25:133–141. [Google Scholar]
- 15.Lo CF, Ho CH, Peng SE, Chen CH, Hsu HC, Chiu YL, et al. Virus-associated white spot syndrome of shrimp in Taiwan: a review. Fish Pathol. 1996;33:365–371. [Google Scholar]
- 16.Low C-F, MdYusoff MR, Kuppusamy G, Ahmad Nadzri NF. Molecular biology of Macrobrachium rosenbergii nodavirus infection in giant freshwater prawn. J Fish Dis. 2018;41(12):1771–1781. doi: 10.1111/jfd.12895. [DOI] [PubMed] [Google Scholar]
- 17.Magbanua FO, Natividad KT, Migo VP, Alfafara CG, de la Pena FO, Miranda RO, et al. White Spot Syndrome virus (WSSV) in cultured Penaeus monodon in the Philippines. Dis Aquat Org. 2000;42:77–82. doi: 10.3354/dao042077. [DOI] [PubMed] [Google Scholar]
- 18.Murwantoko M, Bimantara A, Roosmanto R, Kawaichi M. Macrobrachium rosenbergii nodavirus infection in a giant freshwater prawn hatchery in Indonesia. SpringerPlus. 2016;5(1):1729. doi: 10.1186/s40064-016-3127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naveen Kumar S, Shekar M, Karunasagar I, Karunasagar I. Genetic analysis of RNA1 and RNA2 of Macrobrachium rosenbergii nodavirus (MrNV) isolated from India. Virus Res. 2013;173:377–385. doi: 10.1016/j.virusres.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Naveen Kumar S, Hassan MA, Mahmoud MA, Al-Ansari A, Al-Shwared WK. Betanodavirus infection in reared marine fishes along the Arabian Gulf. Aquacult Int. 2017;25(4):1543–1554. [Google Scholar]
- 21.Neethi V, Sivakumar N, Kumar K, Rajendran KV, Makesh M. Production and application of polyclonal antibodies against recombinant capsid protein of extra small virus of Macrobrachium rosenbergii. Indian J Virol. 2012;23(3):374–378. doi: 10.1007/s13337-012-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owens L, La Fauce K, Juntunen K, Hayakijkosol O, Zeng C. Macrobrachium rosenbergii nodavirus disease (white tail disease) in Australia. Dis Aquat Org. 2009;85:175–180. doi: 10.3354/dao02086. [DOI] [PubMed] [Google Scholar]
- 23.Peck KM, Lauringa AS. Complexities of viral mutation rates. J Virol. 2018;92(14):e01031–e1117. doi: 10.1128/JVI.01031-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pillai D, Bonami JR, Sri WJ. Rapid detection of Macrobrachium rosenbergii nodavirus (MrNV) and extra small virus (XSV), the pathogenic agents of white tail disease of Macrobrachium rosenbergii (De Man), by loop-mediated isothermal amplification. J Fish Dis. 2006;29:275–283. doi: 10.1111/j.1365-2761.2006.00718.x. [DOI] [PubMed] [Google Scholar]
- 25.Qian D, Shi Z, Zhang S, Cao Z, Liu W, Li L, et al. Extra small virus-like particles (XSV) and nodavirus associated with whitish muscle disease in the giant freshwater prawn, Macrobrachium rosenbergii. J Fish Dis. 2003;26:521–527. doi: 10.1046/j.1365-2761.2003.00486.x. [DOI] [PubMed] [Google Scholar]
- 26.Qian D, Liu W, Jianxiang W, Yu L. Preparation of monoclonal antibody against Macrobrachium rosenbergii nodavirus and applications of TAS–ELISA for virus diagnosis in post-larvae hatcheries in East China during 2000–2004. Aquaculture. 2006;261:1144–1150. [Google Scholar]
- 27.Qiu W, Scholthof KB. Satellite panicum mosaic virus capsid protein elicits symptoms on a non-host plant and interferes with a suppressor of virus-induced gene silencing. Mol Plant-Microbe Interact. 2004;17:263–271. doi: 10.1094/MPMI.2004.17.3.263. [DOI] [PubMed] [Google Scholar]
- 28.Ravi M, Nazeer Basha A, Sarathi M, RosaIdalia HH, SriWidada J, Bonami JR, et al. Studies on the occurrence of white tail disease (WTD) caused by MrNV and XSV in hatchery-reared post-larvae of Penaeus indicus and P. monodon. Aquaculture. 2009;292:117–120. [Google Scholar]
- 29.Ravi M, Sahul Hameed AS. Experimental transmission of Macrobrachium rosenbergii nodavirus (MrNV) and extra small virus (XSV) in Macrobrachium malcolmsonii and Macrobrachium rude. Aquacult Int. 2014;23(1):195–201. [Google Scholar]
- 30.Romestand B, Bonami JR. A sandwich enzyme linked immunosorbent assay (S-ELISA) for detection of MrNV in the giant freshwater prawn, Macrobarchium rosenbergii (de Man) J Fish Dis. 2003;26:71–75. doi: 10.1046/j.1365-2761.2003.00432.x. [DOI] [PubMed] [Google Scholar]
- 31.Saedi TA, Hassan M, Wen TS, Khatijah Y, Hassan DM, Kua CB, et al. Detection and phylogenetic profiling of nodavirus associated with white tail disease in Malaysian Macrobrachium rosenbergii (de Man) Mol Biol Rep. 2012;39:5785–5790. doi: 10.1007/s11033-011-1389-7. [DOI] [PubMed] [Google Scholar]
- 32.Sahul Hameed AS, Yoganandhan K, Sri Widada J, Bonami JR. Studies on the occurrence and RT-PCR detection of Macrobrachium rosenbergii nodavirus and extra small virus-like particles associated with white tail disease of Macrobrachium rosenbergii in India. Aquaculture. 2004;238:127–133. [Google Scholar]
- 33.Sahul Hameed AS, Ravi M, Farook MA, Taju G, Hernandez-Herrera RI, Bonami JR. Screening the post-larvae of Macrobrachium rosenbergii for early detection of Macrobrachium rosenbergii nodavirus (MrNV) and extra small virus (XSV) by RT–PCR and immunological techniques. Aquaculture. 2011;317:42–47. [Google Scholar]
- 34.Sahul Hameed AS, Bonami JR. White tail disease of freshwater prawn, Macrobrachium rosenbergii. Indian. J Virol. 2012;23(2):134–140. doi: 10.1007/s13337-012-0087-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanjuan R, Nebot MR, Chirico N, Mansky LM, Belshaw R. Viral mutation rates. J Virol. 2010;84:9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanjuan R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73:4433–4448. doi: 10.1007/s00018-016-2299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith DK, Krohn RL, Hermanson GT, Mallia AK, Gartne FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 38.Sri Widada J, Durand S, Cambournac I, Qian D, Shi Z, Dejonghe E, et al. Genome-based detection methods of Macrobrachium rosenbergii nodavirus, a pathogen of the giant freshwater prawn, Macrobrachium rosenbergii: dot-blot, in situ hybridization and RT-PCR. J Fish Dis. 2003;26:583–590. doi: 10.1046/j.1365-2761.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 39.Sri Widada J, Bonami JR. Characteristics of the monocistronic genome of extra small virus, a virus like particle associated with Macrobrachium rosenbergii nodavirus: possible candidate for a new species of satellite virus. J Gen Virol. 2004;85:643–646. doi: 10.1099/vir.0.79777-0. [DOI] [PubMed] [Google Scholar]
- 40.Sri Widada J, Richard V, Shi Z, Qian D, Bonami JR. Dot-blot hybridization and RT-PCR detection of extra small virus (XSV) associated with white tail disease of prawn Macrobrachium rosenbergii. Dis Aquat Org. 2004;58:83–87. doi: 10.3354/dao058083. [DOI] [PubMed] [Google Scholar]
- 41.Sudhakaran R, Ishaq Ahmed VP, Haribabu P, Mukherjee SC, Sri Widada J, Bonami JR, et al. Experimental vertical transmission of Macrobrachium rosenbergii nodavirus (MrNV) and extra small virus (XSV) from brooders to progeny in Macrobrachium rosenbergii and Artemia. J Fish Dis. 2007;30:27–35. doi: 10.1111/j.1365-2761.2007.00774.x. [DOI] [PubMed] [Google Scholar]
- 42.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tripathy S, Sahoo PK, Kumari J, Mishra BK, Sarangi N, Ayyappan S. Multiplex RT-PCR detection and sequence comparison of viruses MrNV and XSV associated with white tail disease in Macrobrachium rosenbergii. Aquaculture. 2006;258:134–139. [Google Scholar]
- 44.Tung CW, Wang CS, Chen SN. Histological and electron microscopy study on Macrobrachium muscle virus (MMV) infection in the giant freshwater prawn, Macrobrachium rosenbergii (de Man), cultured in Taiwan. J Fish Dis. 1999;22:1–5. [Google Scholar]
- 45.Venegas CA, Nonaka L, Mushiake K, Nishizawa T, Muroga K. Pathogenicity of penaeid rod-shaped DNA virus PRDV to kuruma prawn at different development stages. Fish Pathol. 1999;170:179–194. [Google Scholar]
- 46.Wang CS, Chang JS, Shih HH, Chen SN. RT-PCR amplification and sequence analysis of extra small virus associated with white tail disease of Macrobrachium rosenbergii (de Man) cultured in Taiwan. J Fish Dis. 2007;30:127–132. doi: 10.1111/j.1365-2761.2007.00793.x. [DOI] [PubMed] [Google Scholar]
- 47.Yoganandhan K, Leartvibhas M, Sriwongpuk S, Limsuwan C. White tail disease of the giant freshwater prawn Macrobrachium rosenbergii in Thailand. Dis Aquat Org. 2006;69:255–258. doi: 10.3354/dao069255. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Wang J, Yuan J, Li L, Zhang Z, Bonami JR, et al. Quantitative relationship of two viruses (MrNV and XSV) in white-tail disease of Macrobrachium rosenbergii. Dis Aquat Org. 2006;71:11–17. doi: 10.3354/dao071011. [DOI] [PubMed] [Google Scholar]