

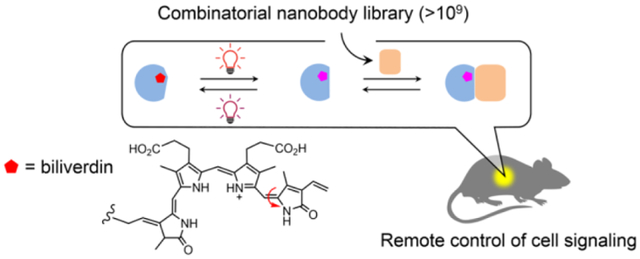
Abstract
Protein dimerization systems controlled by red light with increased tissue penetration depth are a highly needed tool for clinical applications such as cell and gene therapies. However, mammalian applications of existing red light-induced dimerization systems are hampered by limitations of their two components: a photosensory protein (or photoreceptor) which often requires a mammalian exogenous chromophore and a naturally occurring photoreceptor binding protein typically having a complex structure and non-ideal binding properties. Here, we introduce an efficient, generalizable method (COMBINES-LID) for creating highly specific, reversible light-induced heterodimerization systems independent of any existing binders to a photoreceptor. It involves a two-step binder screen (phage display and yeast two-hybrid) of a combinatorial nanobody library to obtain binders that selectively engage a light-activated form of a photoswitchable protein or domain not the dark form. Proof-of-principle was provided by engineering nanobody-based, red light-induced dimerization (nanoReD) systems comprising a truncated bacterial phytochrome sensory module using a mammalian endogenous chromophore, biliverdin, and light-form specific nanobodies. Selected nanoReD systems were biochemically characterized, exhibiting low dark activity and high induction specificity, and further demonstrated for the reversible control of protein translocation and activation of gene expression in mice. Overall, COMBINES-LID opens new opportunities for creating genetically encoded actuators for the optical manipulation of biological processes.
Keywords: Light-induced dimerization, nanobody, combinatorial antibody library, bacterial phytochrome, phage display, optogenetics
Graphical Abstract
Since the invention of using light to control gene expression by Quail and coworkers in 2002,1 light-induced protein dimerization (LID) systems have been increasingly used to manipulate biological processes in living organisms.2–3 Similar to chemically induced dimerization (CID),4–5 LID systems can be genetically encoded to provide noninvasive control of molecular proximity, which is a fundamental regulator of gene regulation, cell signaling and metabolism, among other processes.6 For in vivo applications, LID offers spatiotemporal resolution unmatched by CID7 and, unlike chemical approaches, is not limited by toxicity, unintended effects of chemical inducers, or difficulties associated with drug delivery.
Different from single-component actuator systems such as microbial opsins,8 LID comprises two separate proteins or domains which serve as a sensor and an effector. The sensory function is initiated by i) light-induced chromophore isomerization or chromophore-protein bond formation triggering a conformational change of a chromophore-bound photosensory protein (hereafter named ‘conformation switcher’), or ii) photolytic release of a caged ligand or isomerization of a photoswitchable ligand that serves as a dimerization inducer.9 Naturally occurring conformation switchers widely exist in all kingdoms of life and many have been identified and characterized in the past three decades (Table S1). They have diverse structural and optical properties, offering flexible choices for in vivo applications. Many use metabolites widely shared from bacteria to humans as chromophores; for example, riboflavin-5’-phosphate bound to light-oxygen-voltage (LOV) sensing domains10 and biliverdin, a heme-derived linear tetrapyrrole found in bacterial phytochromes (BphPs).11
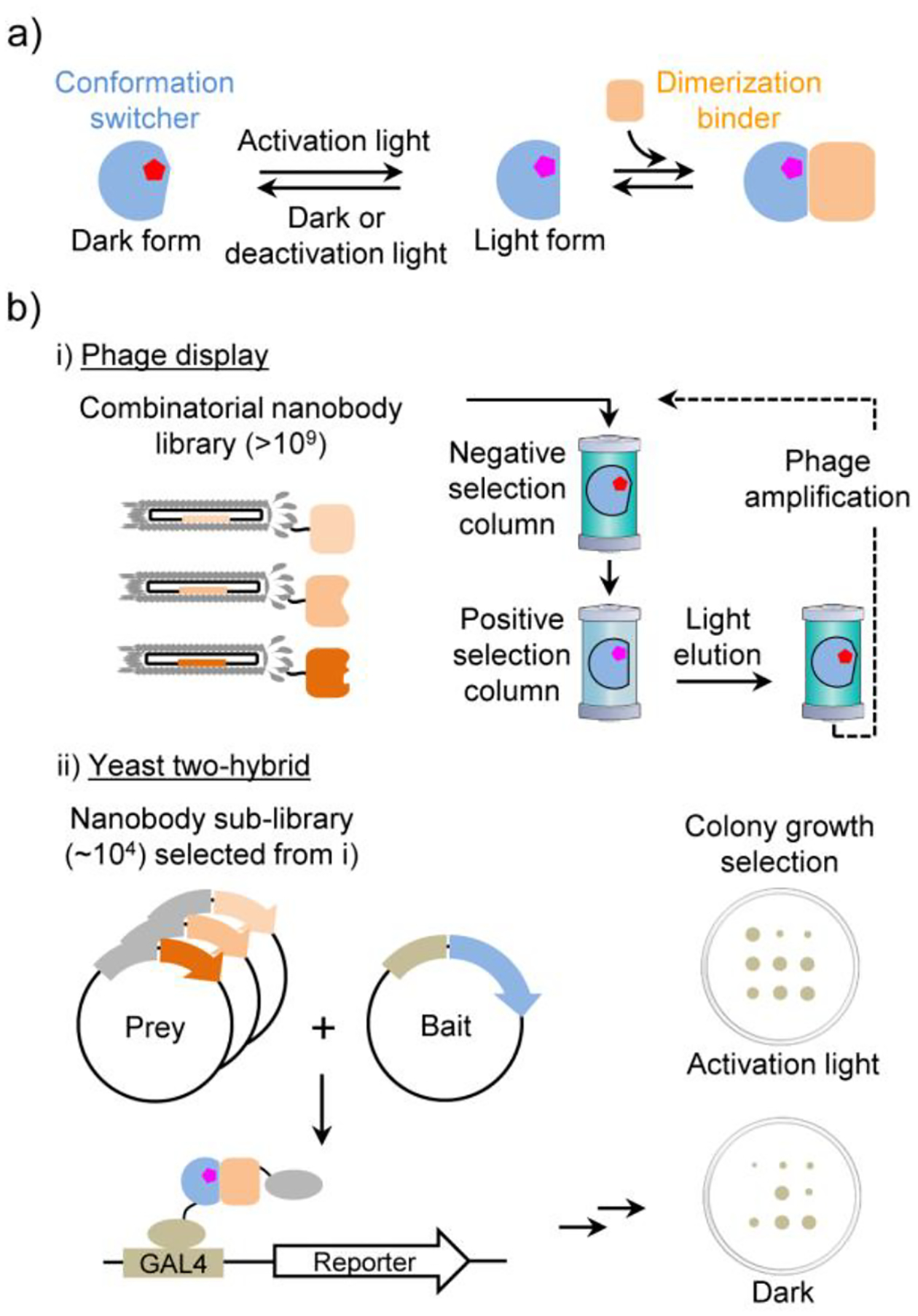
The effector function of LID is executed by a ‘dimerization binder’ which specifically binds to the conformation switcher in its light form—the state after a light-induced conformational change occurring to its thermally stable state in the dark, or the dark form (Figure 1a). Many natural conformation switchers do not modulate protein-protein interaction and thus are associated with any dimerization binder; for example, in phytochromes, conformation switcher domains (known as a photosensory module) are typically fused with an enzymatic domain or module to allosterically regulate catalytic activity.12 Although a few natural dimerization binders have been identified (Table S1), the in vivo performance such as the basal binding in the dark (or dark activity) is yet to be improved. It is yet difficult to design new dimerization binders with suitable specificity, sensitivity, and kinetics.
Figure 1.
a) LID mechanism. b) Principle of the COMBINES-LID method.
For deep-tissue applications in animals, LID is required to sense an optical input in the 650‒900 nm region, known as a tissue transparency window,13 because tissue absorbance, autofluorescence, and light scattering are minimized in this region.14–15 Phytochromes including plant and cyanobacterial phytochromes, BphPs, and cyanobacteriochromes use a class of linear tetrapyrroles, bilins, as chromophores that can sense low-energy optical signals in the far-red and near-infrared (NIR) range.16 Plant and cyanobacterial phytochromes use non-mammalian chromophores such as phycocyanobilin and phytochromobilin (Table S1), which imposes challenges for in vivo mammalian applications. In contrast, BphPs use biliverdin, a ubiquitous endogenous bilin in mammalian cells,11 thus avoiding exogenous administration of the chromophore. However, so far only a natural dimerization binder, PpsR2, a ~50 kilodalton (kDa) transcriptional regulatory protein, was identified to bind to Rhodopseudomonas palustris BphP1 (RpBphP1), a ~160 kDa homodimeric protein.17–18 Its close homolog, RsPpsR from Rhodobacter sphaeroides was found to interact with DNA and self-assemble as a dimer or higher oligomers,19 which might result in unwanted cellular outputs. Thus, a truncated form of PpsR2, Q-PAS1, was generated to eliminate DNA binding while maintain binding to RpBphP1.20–21 However, both PpsR2 and Q-PAS1 LIDs exhibited a significant dark activity,18, 20, 22 likely due to the nonspecific binding to the dark form of RpBphP1.
Here we aimed to create reversible LID systems with decreased protein sizes and minimized dark activity by de novo engineering of small dimerization binders to a minimal photoactive module excised from a full-length BphP. So far, light form-specific binders were mainly obtained by in vitro selection due to the feasibility to manipulate light sensitive proteins in a screening assay. Initial successes have been reported using phage display to screen computationally designed binders for a LOV2 domain23 and a random surface mutation library of an albumin-binding domain targeting the LOV2 and a photoactive yellow protein.24 Inspired by these works, we sought to establish a robust, generalizable method by coupling in vitro and cell-based screening to create new LID systems suitable for mammalian applications. To facilitate implementation by other labs, we chose to screen one of the mostly used small binders, single-domain antibody (or nanobody), a 12–15 kDa functional antibody domain with a universal scaffold and three variable complementarity-determining regions (CDRs).25
RESULTS AND DISCUSSION
Screening of Dimerization Binders for DrBphP.
We devised a two-step screening method, combinatorial binders-enabled selection of LID (COMBINES-LID), which involves phage display to enrich binders that only bind to the BphP light form and then yeast two-hybrid (Y2H) screening of the enriched sub-library to select for in vivo activity (Figure 1b). A high-quality synthetic combinatorial nanobody library generated in our previous work26 was used. These nanobodies have an optimized scaffold27 and rationally randomized CDRs with an estimated sequence diversity of 1.23 to 7.14 × 109.
The photosensory module truncated from Deinococcus radiodurans phytochrome (DrBphP) was chosen as the conformation switcher. Its light and dark forms can be stably generated for a screening assay by activating far-red (e.g., 654 nm) and deactivating NIR (e.g., 775 nm) illuminations, respectively. The photoswitching efficiency is close to that of the full-length DrBphP.28–29 By contrast, the similarly excised module of RpBphP1 showed impaired photoconversion.17 This tridomain module (Figure 2a; hereafter named DrBphP for simplicity) comprising a Per-ARNT-Sim (PAS), a cGMP phosphodiesterase-adenylate cyclase-FhlA (GAF), and a phytochrome-specific (PHY) domains was expressed as a ~60 kDa fusion bearing a C-terminal AviTag and HisTag, incubated with biliverdin, purified, and biotinylated (Figure S1) to serve as a bait for phage display. The photoconversion of the purified protein was confirmed by measuring the spectra of the light and dark forms (Figure S2).
Figure 2.
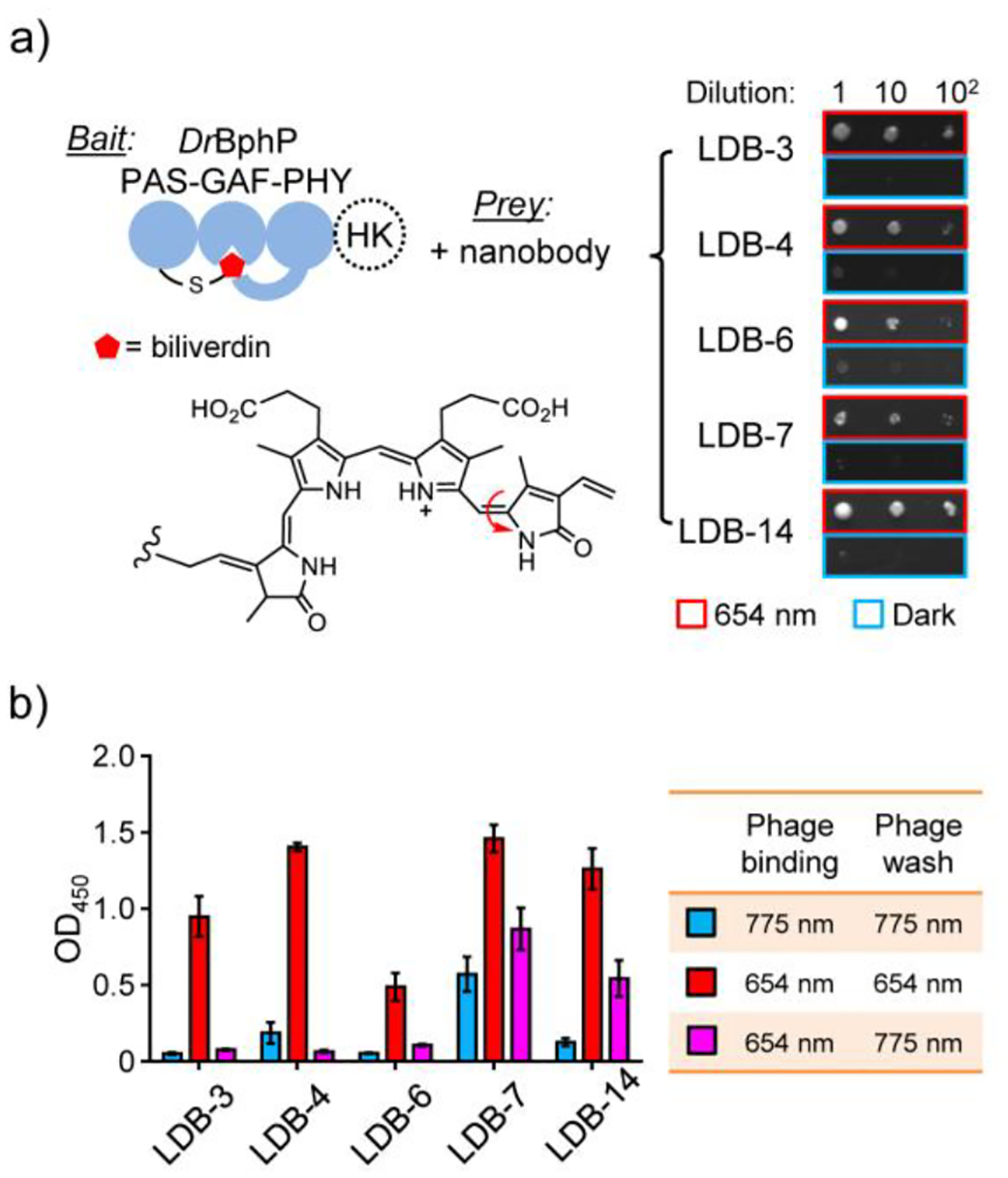
Y2H and single phage ELISA analyses of dimerization binder candidates. a) Y2H assay with the biliverdin-bound DrBphP photosensory module as a bait and nanobodies as preys. The tridomain module was obtained by removing a histidine kinase (HK) domain from the full-length DrBphP. A serial dilution of Y2HGold cells resuspended in 0.9% NaCl were spotted on SD/-Ade/-His/-Leu/-Trp plates (no biliverdin added) and grown under the 654-nm illumination (0.03 mW/cm2) or in the dark. A representative result from three independent experiments is shown on the right. b) ELISA analysis of nanobody binding specificity and reversibility. Phage-displayed nanobodies were bound to DrBphP immobilized in microtiter plates, which were illuminated with the 654-nm (0.3 mW/cm2) or 775-nm (0.2 mW/cm2) lights during the binding and wash steps. Data represent mean values of 3 measurements; error bars, standard deviation.
We hypothesized that specific and reversible dimerization binders are critical for the in vivo performance of LID, such as a low dark activity. To enhance selection efficiency, we used column chromatography to continuously separate phage-displayed nanobodies between the stationary and mobile phases as they passed through a column. Binding specificity was selected by loading the library onto two connected transparent columns, the first (negative selection) preloaded with biotinylated DrBphP photoconverted to the dark form and the second (positive selection) to the light form (Figure S3a). Thus, nanobodies captured in the second column should have zero or very low affinity to the dark form. Next, reversible binders were collected by eluting only dissociated nanobodies from the second column after switching the light to dark form by the 775-nm illumination. After four-round phage binding and elution with gradually decreased illumination times (Figure S3b), the final eluent was estimated to contain ~90% light-eluted clones (Table S2) with the sequence diversity of up to ~104.
In vitro selected nanobodies were subcloned into a Y2H sub-library for the cell-based screening of cytoplasmic expression and binding specificity. Although some nanobodies can be functionally expressed as intracellular binders,27 many are unstable leading to loss of function due to the inability of disulfide bond formation in the reducing cytoplasmic environment. Y2H was selected for the sub-library screening due to its suitable throughput and cost-effectiveness. Y2HGold cells were co-transformed with plasmids carrying genes of DrBphP and nanobodies and selected on SD/-Ade/-His/-Leu/-Trp agar plates under the 654-nm illumination. ~2,000 fully grown colonies were picked, inoculated into 1-mL SD/-Leu/-Trp medium, and replica spotted onto the agar plates to compare colony growth under the illumination and in the dark (Figure S4). Five candidates growing only under the illumination (Figure 2a) and with diverse CDR sequences (Table S3) were selected for further characterization.
To confirm the binding specificity and reversibility, we assayed selected nanobodies by single phage enzyme-linked immunosorbent assay (ELISA). Phage displayed-nanobodies were first bound to the dark and light forms of biotinylated DrBphP immobilized in streptavidin-coated microtiter plates. To maintain the dark or light form, or to convert the light to dark form, the plates were kept under 654- and/or 775-nm illumination during the phage binding and wash steps. As expected, all candidates showed light-form binding specificity with non-detectable (LDB-3 and LDB-6) to relatively low (LDB-4, LDB-7, and LDB-14) binding to the dark form (Figure 2b). Bound nanobodies were almost completely (LDB-3, LDB-4, and LDB-6) or partially (LDB-7 and LDB-14) washed off after converting the light to dark form.
Specificity Validation in Mammalian Cells.
To determine whether nanobody candidates are suitable for mammalian applications, we first tested their expression in human embryonic kidney 293T (HEK293T) cells. It is known that the same protein-protein interaction (PPI) found in yeast might not be detected in mammalian cells due to protein expression or stability issues; for example, a recent comparison of PPI assays in different hosts found that only half of human PPIs detected in yeast were also seen in HEK293T, and vice versa.30 Thus, we were interested to know the success rate of nanobodies selected by COMBINES-LID that can be functionally expressed in mammalian cells.
To compare in vivo activity, we assayed proteins by mammalian two-hybrid (M2H)31 under a standardized condition. Specifically, DrBphP was fused with an N-terminal GAL4 DNA binding domain (BD) and nanobodies with a C-terminal p65 transcriptional activation domain (AD) to control the expression of a firefly luciferase reporter (Figure 3a). After transient co-transfection with the BD and AD, and reporter plasmids, cells were cultured for 24 hours in the dark to express the DrBphP and nanobodies, and then maintained in the dark or under 654-nm illumination for another 24 hours to compare the luciferase expression. Dark activity and specificity were analyzed by comparing the dark expression with a negative control (i.e., only DrBphP was expressed) and the light-induced expression with the dark expression, respectively. All candidates showed low dark activities, and the dark expression levels were close to the control (Figure 3b). LDB-3 and LDB-14 showed the high specificity; their light-induced expression levels were increased by ~19.5 and ~19.1 folds, respectively. We also tested the expression under 775-nm illumination, which was close to the dark levels (Figure S5). However, the other three candidates did not show obvious light activation. To understand their loss of the expected activity, we investigated protein expression in HEK293T. Specifically, all nanobodies bearing a C-terminal SNAP-tag were expressed, fluorescently labelled, and analyzed by sodium dodecyl sulfate gel electrophoresis (SDS-PAGE). All nanobodies were found with detectable expression; however, compared with LDB-3 and LDB-14, the other three nanobodies showed drastically decreased levels of full-length proteins with degraded fragments, suggesting that they might not be stable in the host cells (Figure S6).
Figure 3.
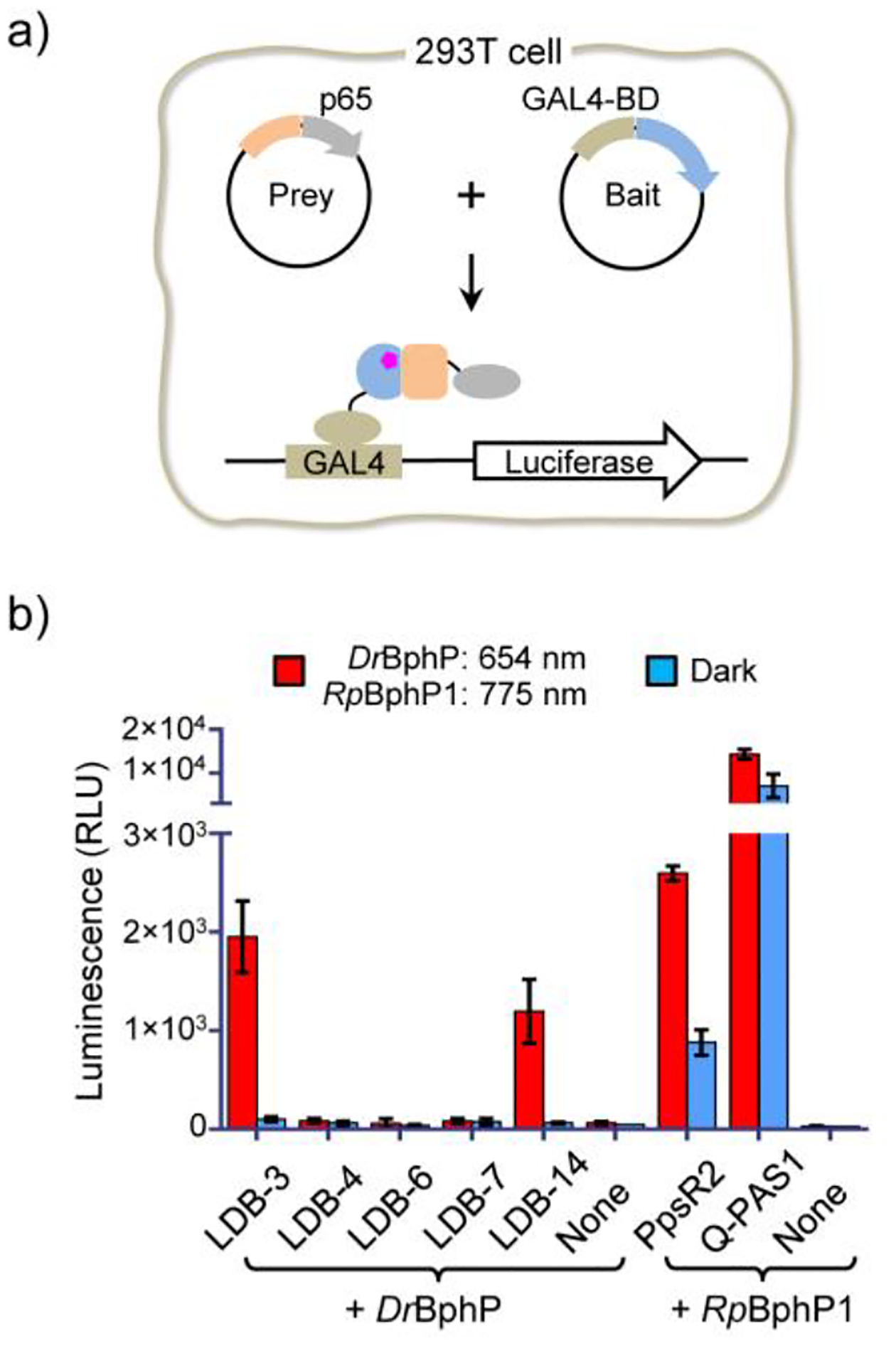
Nanobody specificity validation in mammalian cells. a) Schematic of the M2H assay. DrBphP and nanobody genes were inserted into the bait and prey plasmids, respectively. b) Specificity comparison of LID systems. HEK293T cells were transiently co-transfected with the bait, prey, and GAL4UAS-luciferase reporter plasmids (~0.25 μg each) in a 0.5 mL culture (no biliverdin added). None, the negative control transfected with only the bait and the luciferase reporter plasmids. Cells were maintained under the illumination (654 nm (0.2 mW/cm2) or 775 nm (0.2 mW/cm2)) or in the dark for 24 hours before measuring luciferase levels. Different from DrBphP, RpBphP1 is required to be converted to the light form by a NIR (e.g., 775-nm) light. Data represent mean values of 3 measurements; error bars, standard deviation.
We compared the specificity of the nanobody-based LIDs with the RpBphP1-PpsR2 and RpBphP1-Q-PAS1 systems by M2H. The RpBphP1-based LIDs showed light-enhanced expression (Figure 3b). However, due to the high dark activity, light-induced expression was only increased by ~2.95 and ~2.02 folds. Our observed specificity of the RpBphP1 systems is much lower than originally reported,19,20 but is close to that recently achieved in Escherichia coli.32 To further assess the specificity, we investigated impacts of extra supply of biliverdin and decreased cellular levels of LID proteins. Of note, supplementing culture media with biliverdin enhanced the light induction and further decreased the dark activity of the nanobody/DrBphP LIDs (Figure S7a), suggesting that the in vivo performance can be further improved by increasing biliverdin binding to DrBphP. Although the addition of biliverdin also lowered the dark activity of the RpBphP1 LIDs, the dark levels were still high (Figure S7a). In HeLa cells with lower levels of LID proteins than those in HEK293T cells, although the light induction levels were reduced, the relative specificity of the nanobody/DrBphP- and RpBphP1 systems was similar in both cell lines (Figure S7b). As suggested by a mathematical model of LID,33 the specificity is determined not only by relative binding affinities of dimerization binders to dark and light forms of a conformation switcher, but also by their effective cellular concentrations, which unfortunately are difficult to measure in our experiment. Nevertheless, our comparison with the same transfection and culture condition suggests that LDB-3 and LDB-14 offer significantly enhanced dimerization specificity even without addition of the exogenous chromophore.
Biochemical Characterization of LDB-3 and LDB-14.
We biochemically assessed selected nanobodies to understand their binding mechanisms. We first sought to detect light-induced DrBphP-nanobody complexes using analytical size-exclusion chromatography (SEC). LDB-3 and LDB-14 were bacterially expressed and purified with yields of ~2–3 milligrams per liter of culture. SEC data showed that LDB-3 was a mixture of the monomer and dimer and LDB-14 mainly the monomer and both nanobodies dimerized at increased concentrations (Figure S8a). Consistent with a previous report,29 both the light and dark forms of DrBphP were eluted mainly as homodimers (Figure S8b). After DrBphP was illuminated and then incubated with LDB-3 or LDB-14 in the dark, split SEC peaks of DrBphP were observed only for the light form (Figure 4a), implying complex formation. Complexes were confirmed by SDS-PAGE detection of coeluted DrBphP and nanobodies (Figure 4b). Interestingly, gel analysis revealed that LDB-3 was coeluted with DrBphP later than LDB-14, suggesting that they might have different binding configurations or affinities. Consistent with the single phage ELISA result (Figure 2b), LDB-14 appeared to weakly interact with the dark form since a faint nanobody band was detected in the dark-form DrBphP factions. We next sought to determine which domain(s) of DrBphP bind to LDB-3 and LDB-14 by splitting DrBphP into two soluble expressed portions, PAS-GAF and PHY, and then detecting their binding by ELISA. Interestingly, both portions showed binding to the nanobodies (Figure S9), implying that both might be engaged in the heterodimerization.
Figure 4.
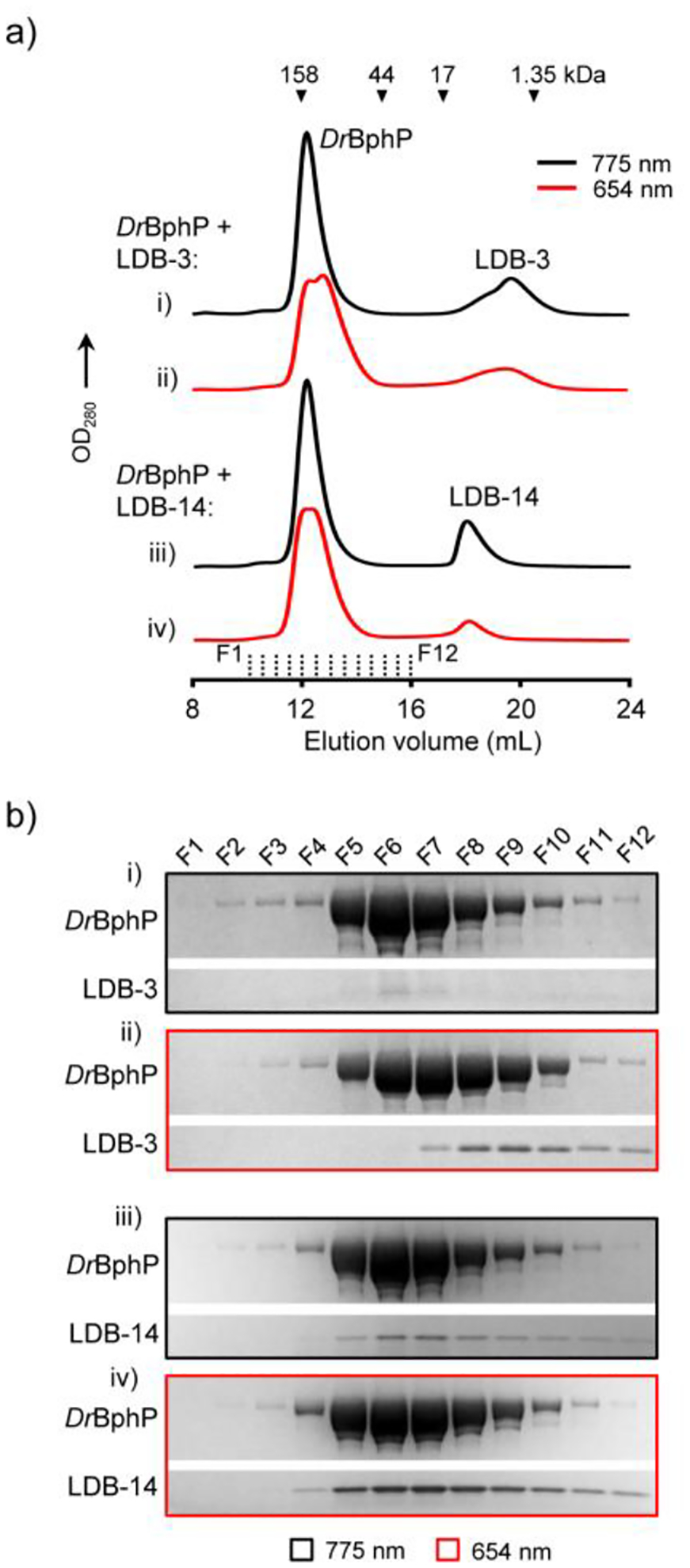
Detection of light-induced DrBphP-nanobody complexes. a) Analytical SEC. ~6 μM (final concentration) DrBphP after the 654-nm (0.2 mW/cm2) or 775-nm (0.8 mW/cm2) illumination for 5 min were incubated with ~5 μM (final concentration) LDB-3 or LDB-14 in the dark. 500 μL mixtures were loaded onto a Superdex 200 Increase 10/300 GL column pre-equilibrated with 1× PBS buffer and eluted at 0.75 mL/min at 4°C. Elution volumes of protein standards are marked by triangles. b) SDS-PAGE analysis of the fractions (500 μL each) collected in the SEC (marked by dash lines in a)) and concentrated by trichloroacetic acid precipitation. Only gel regions showing DrBphP and nanobody bands are shown.
We studied the thermodynamics of nanobody binding by isothermal titration calorimetry (ITC). Binding data obtained by titrating LDB-3 or LDB-14 into a photoconverted light-form sample were fitted using a one-site model (R2 = ~0.99) to give apparent dissociation constants (KDapps) of 1.01 and 0.47 μM, respectively (Figure 5 and Table S4). The binding site number of DrBphP was calculated to be ~0.6, consistent with a previous small-angle X-ray scattering result that only 64% of the protein molecules in the photoconverted sample in equilibrium were in the light-state conformation.29 Unexpectedly, the thermograph of LDB-3 titration into the dark form showed significant heat exchange: a clear transition from heat release to absorption when more LDB-3 was added, which was not observed for LDB-3 titration into the light form (Figure 5a). This transition suggests that the exothermic binding of LDB-3 to the dark form might be coupled to an endothermic process, which is likely to be the LDB-3 dimer dissociation (Figure S10). In other words, LDB-3 binding to the dark form could be inhibited by LDB-3 dimerization, thus providing a mechanism to explain the observed low dark activity of LDB-3. Thermodynamic simulation of the competitive binding processes suggests that the KD of heterodimerization of LDB-3 and the dark form is likely above 100 μM (refer to Supplementary Note). LDB-14 was found to only bind to a small fraction (~15%) of DrBphP in the dark-form sample but did not generate measurable heat of binding with the major population (Figure 5b). The minor fraction was likely the protein switched to the light form during our sample preparation. Thus, although the binding to the dark form is too weak to be determined by ITC, we estimate that the specificity of both nanobodies towards the light form is above 100 folds.
Figure 5.
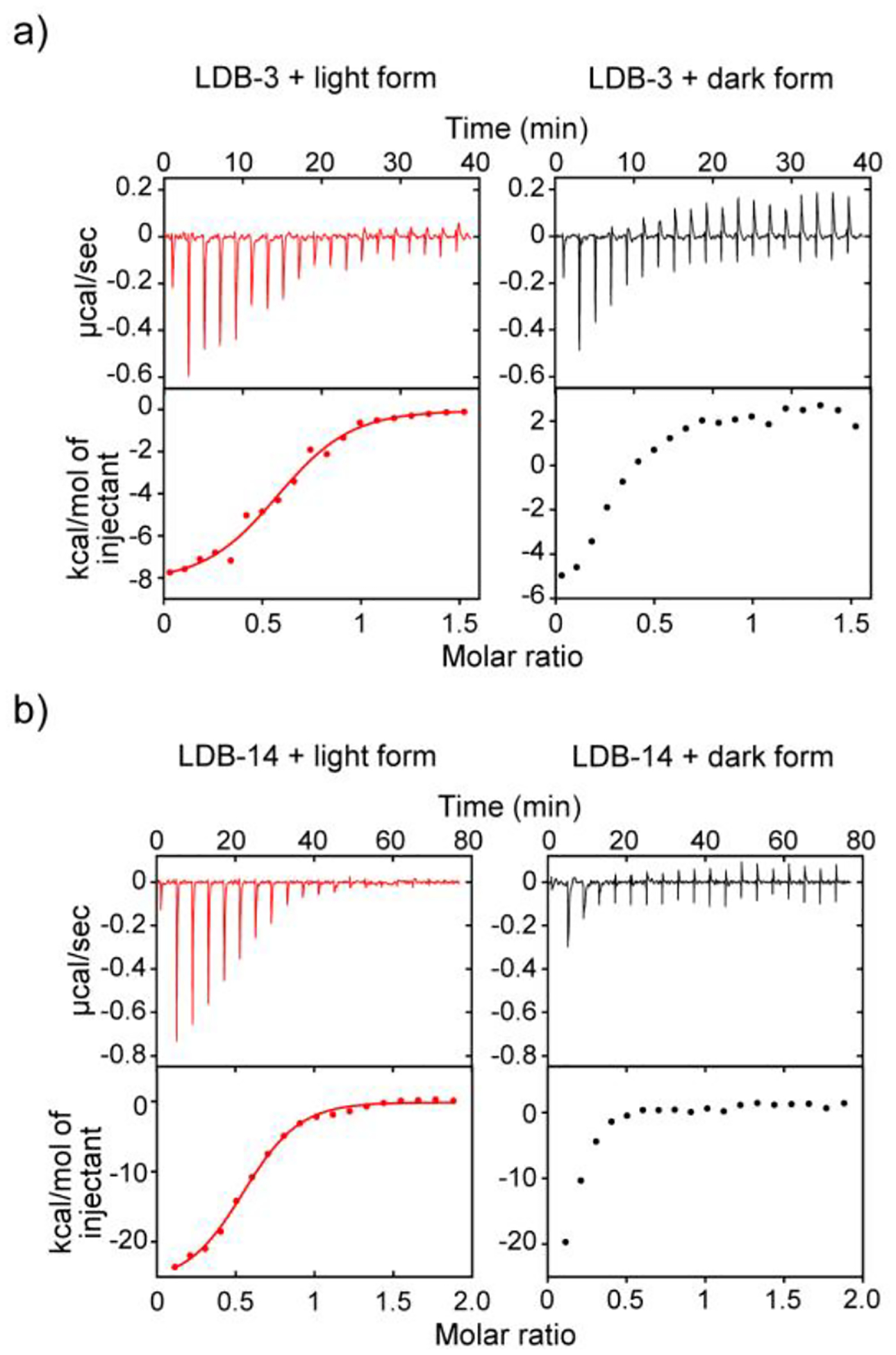
ITC thermographs of the nanobody binding. a) 80 μM LDB-3 and b) 50 μM LDB-14 were titrated into 10 μM and 5 μM DrBphP, respectively. The light and dark forms were converted by the 654-nm (0.2 mW/cm2) and 775-nm (0.2 mW/cm2) lights, respectively. The raw data (top) and the integration of heats (bottom) for each titration are shown.
The binding kinetics of LDB-3 and LDB-14 were measured by Bio-Layer Interferometry (BLI). The assay was performed by incubating the light or dark form with nanobodies immobilized on streptavidin biosensors. The result revealed that, compared with LDB-14, LDB-3 has a weaker binding affinity to the light form mainly due to a ~4.9-fold faster dissociation from the DrBphP (koff = ~18.5 × 10−2s−1) (Figure S11 and Table S5). Theoretically, the fast dissociation could offer higher temporal resolution for the reversible control of LID.33 The BLI analysis of the dark form binding was not straightforward because the white light signal applied to BLI biosensors might partially convert the biosensor-bound DrBphP to the light form.
Since DrBphP photoconversion and nanobody binding might reciprocally affect each other, we asked whether the nanobody binding can slow the photoconversion to the dark form. To test this, we illuminated DrBphP with different exposure times and light intensities and measured the percentage of the dark form in the protein by the ratio of absorption at 750 nm (A750) to 700 nm (A700) (Figure S12a). Compared with unbound DrBphP, the nanobody-bound DrBphP has a slower photoconversion (Figure S12b), implying that the nanobody binding can potentially stabilize the light form. At a relatively low light intensity, the LDB-3-bound DrBphP had a slightly faster photoconversion to the dark state than the LDB-14-bound DrBphP, likely due to the faster dissociation of the DrBphP-LDB-3 complex. Together, our biochemical data support the high specificity and reversibility of the nanobody-based LID systems.
Red Light-Activated Gene Expression.
To develop in vivo applications, we focused on light-induced gene expression. We first determined the time-course response of light-induced activation of luciferase expression in HEK293T cells. Under the same culture and transfection condition, luciferase levels with or without 654-nm illumination were measured at seven time points up to 72 hours. For both LDB-3 and LDB-14, luciferase levels after illumination reached half maximum and maximum at 12 and 24 hours, respectively (Figure S13). The half-maximum and maximum luciferase levels in cells expressing LDB-3 were ~2.5-fold higher than those of cells expressing LDB-14, which seems to be correlated with in vivo nanobody stability (Figure S6).
The luciferase assay required releasing the protein by cell lysis, so we also directly measured green fluorescent protein (GFP) expression by fluorescence imaging. Specifically, HEK293T cells were transiently co-transfected with LID genes to control the transcription of a chromosomally integrated GFP gene. Imaging analysis showed extremely low GFP expression in cells kept in the dark (Figure 6a). In LDB-3- and LDB-14-expressing cells, light-induced GFP levels were similar and, compared with the dark levels, increased by ~44 and ~39 folds, respectively (Figure 6b).
Figure 6.
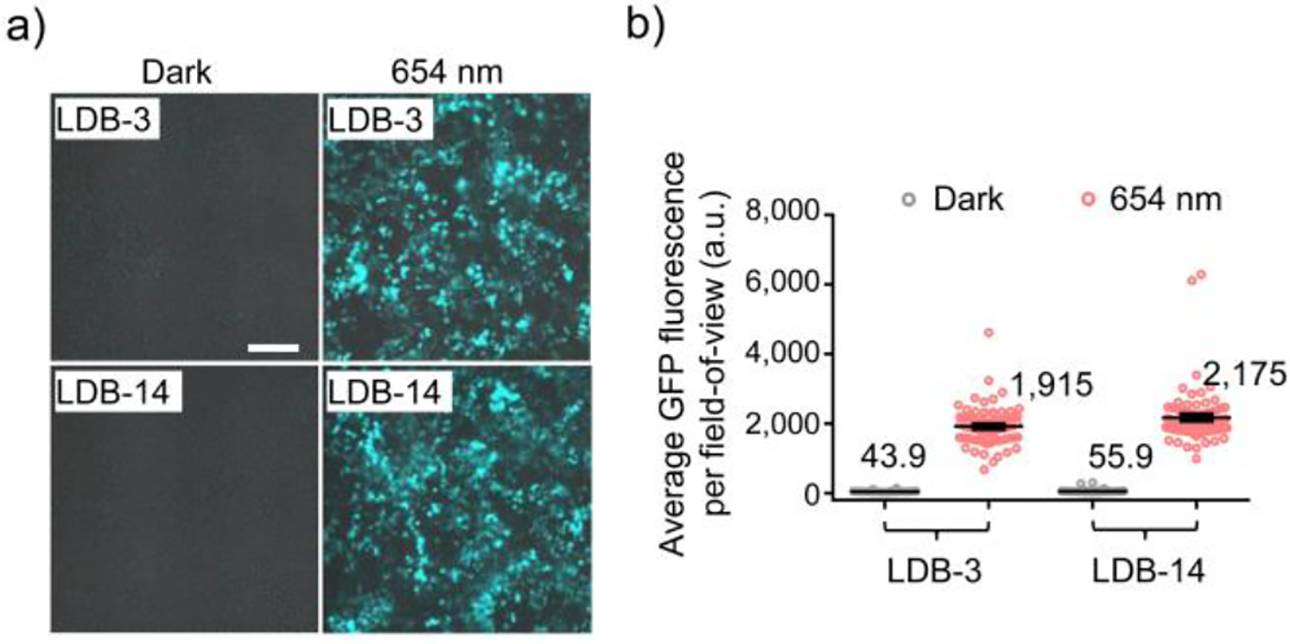
Red light-induced GFP expression. a) Representative overlaid brightfield and fluorescence images of GFP-expressing HEK293T cells. Cells were transduced with a lentiviral GFP expression vector and then co-transfected with the LID plasmids after 654-nm (0.2 mW/cm2) illumination or in the dark for 48 hours. No biliverdin was supplemented in the medium. Scale bar, 200 μm. b) Comparison of GFP fluorescence intensities in fields-of-view (FOVs). Data represent mean values of 78 FOVs; error bars, standard error of the mean (SEM).
Reversible Control of Protein Translocation.
To directly visualize LID complex formation in cells, we coexpressed LDB-3 and DrBphP bearing C-terminal mCherry and monomeric GFP (AcGFP), respectively, in HEK293T cells. To facilitate the complex detection, DrBphP-AcGFP was anchored to plasma membrane via a C-terminal membrane-targeting CAAX motif (Figure 7a). After 24-hour coexpression of the two proteins, cells were imaged immediately after three sequential illumination by 775, 654 and 775-nm lights to detect the translocation of LDB-3-mCherry. Obvious colocalization of GFP and mCherry signals adjacent to plasma membrane was only observed for the 654-nm illumination, not for the 775-nm illumination (Figure 7b). Intensity profiling analysis of the colocalization region showed that the mCherry signal increased by 1.31-fold and after the third-step 775-nm illumination for 10 min dropped to 1.11-fold (Figure 7c). Thus, our result reveals that significant amounts of LDB-3 and DrBphP can be engaged in the light-switchable complex formation and dissociation.
Figure 7.
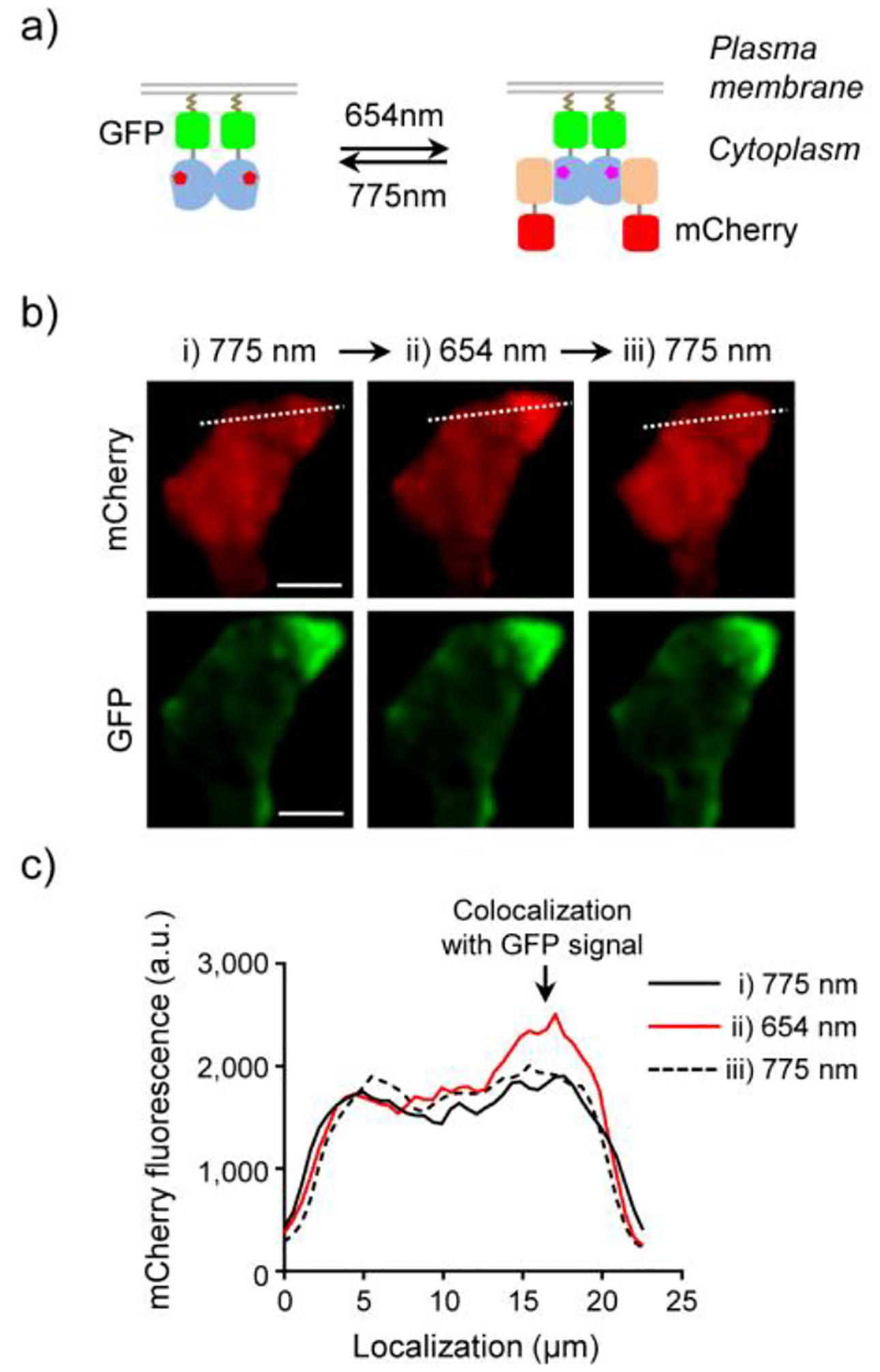
Light-switchable protein translocation. a) Schematic of the light-induced reversible binding of cytoplasmic LDB-3-mCherry to membrane-bound DrBphP-AcGFP. b) Fluorescence images of a HEK293T cell coexpressing LDB-3-mCherry (red) and DrBphP-AcGFP (green) for 24 h and then subjected to three illumination steps: i) 775 nm (0.2 mW/cm2, 10 min; left), ii) 654 nm (0.2 mW/cm2, 2 min; middle), and iii) 775 nm (0.2 mW/cm2, 10 min; right). Bars, 10 μm. c) Intensity profile of mCherry fluorescence intensities along white dashed lines in b). 5 μM biliverdin was supplemented in the medium.
In Vivo Transcription Activation in Mice.
Finally, to demonstrate the gene activation in living animals, we subcutaneously injected the HEK293T cells with LDB-3-controlled luciferase expression into nude mice, remotely activated the transcription by the 654-nm light, and quantified luciferase expression by in vivo bioluminescence imaging. As expected, mice kept in the dark were detected with low bioluminescence signals and those received the 24-hour illumination had a ~25-fold average increase of bioluminescence (Figure 8a–b), which is consistent with the data obtained with the cultured cells (Figure S13). To demonstrate the light activation in a deeper tissue, the HEK293T cells were transplanted into the mouse liver. The light induction was performed similarly as above; a ~31-fold increase of bioluminescence was observed (Figure 8c–d). Considering cells had been exposed to light during the subcutaneous injection or liver transplantation, the dark activity is expected to be even lower if light exposure can be completely avoided.
Figure 8.
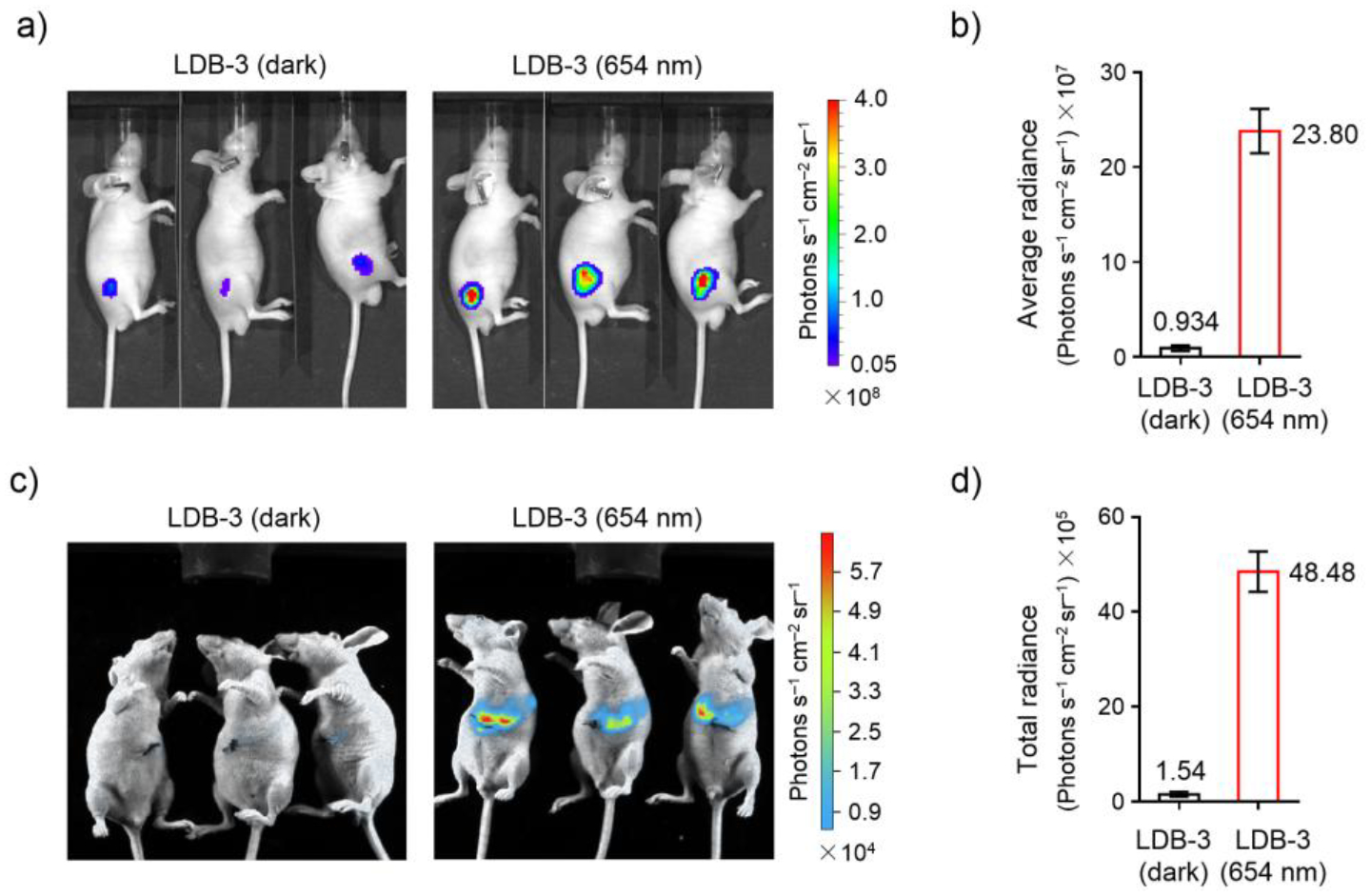
In vivo transcription activation in mice. a) Bioluminescence imaging of nude mice after subcutaneous injection with HEK293T cells co-transfected with the LID and the luciferase reporter plasmids and then kept under 645-nm illumination (0.03 mW/cm2) or in the dark for 24 hours. b) Comparison of average bioluminescence intensities of imaged mice in a). Data represent the mean ± SEM (n = 3 mice per group). c) Bioluminescence imaging of nude mice after transplantation of the co-transfected HEK293T cells into the liver and then treated similarly as in a). d) Comparison of total bioluminescence intensities of imaged mice in c). Data represent the mean ± SEM (n = 3 mice per group). Different detection sensitivities in a) and c) were due to different in vivo imaging systems used.
CONCLUSIONS
Our work demonstrated that COMBINES-LID is efficient for creating highly specific LID systems. This method screened a generic combinatorial nanobody library using fast, cost-effective phage display and Y2H techniques to obtain high-quality, mammalian-applicable binders without need for further engineering of binding affinity and specificity, thus offering a short turnaround time (Figure S14). It relies on using protein targets with photo-switchable conformational states to select binder specificity, and thus can potentially be applied to a large array of photoswitchable proteins (Table S1) to create orthogonal LID systems with diverse optical and structural properties. Applicable dimerization binders can also include those with other scaffolds such as non-immunoglobulin34 and computationally designed scaffolds.35
The LDB-3 and LDB-14-DrBphP LID systems, now named ‘nanoReD1’ and ‘nanoReD2’, respectively, have simplified structures and improved in vivo performance, overcoming the intrinsic limitations of naturally occurring BphP LID and derivatives. Although these systems have only been tested for controlling gene expression and protein translocation, they should be useful for controlling many other cellular processes, for example, the spatiotemporal activation of chimeric antigen receptor T (CAR-T) cells.36–37 Their use of the mammalian endogenous metabolite as chromophore and the compatibility of deep tissue penetration offer the unique potential to address clinical challenges such as CAR-T therapy targeting solid tumors.
METHODS
Plasmid construction.
Primers and protein coding sequence (CDSs) for plasmid construction were synthesized by Integrated DNA Technologies (IDT) or amplified from other plasmids. Primers are listed in Tables S6. CDSs, noncommercial plasmid sequences, and subcloning insertion sites are listed in Table S7. The subcloning was performed using a Gibson assembly protocol recently described,38 except for the PAS-GAF-Avi-His and PHY-Avi-His which were constructed by Q5 Site-Directed Mutagenesis Kit (New England Biolabs, E0554S).
Protein expression and purification.
DrBphP-Avi-His and DrBphP-His were expressed in Escherichia coli and purified by Ni-affinity and size-exclusion chromatography. In brief, Escherichia coli C41(DE3) cells (Lucigen) were transformed with a DrBphP-Avi-His or DrBphP-His expression construct and grown in 2×YT medium at 37°C to an OD600 of ~0.6 before induction with 0.1% arabinose at 25°C for overnight. Harvested cell pellets from 1-liter cultures were resuspended in 40 mL ice-cold lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 10 mM imidazole, 10% glycerol) for sonication. The supernatant after centrifugation at 15,000×g, 4°C for 30 min was loaded onto a 5 mL HisTrap column (GE Healthcare) pre-equilibrated with the lysis buffer. The column was washed with a washing buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 20 mM imidazole, 10% glycerol) and then His-tagged DrBphP was eluted with an elution buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 500 mM imidazole, 10% glycerol). Eluates were desalted with a HiPrep 26/10 column (GE Healthcare) pre-equilibrated with a storage buffer (1×PBS, 5% glycerol). Fractions were pooled and incubated with biliverdin (Frontier Scientific) with molar ratio of 1:20 at 4°C overnight and then loaded onto a HiPrep 26/10 desalting column (GE Healthcare) pre-equilibrated with a storage buffer (1×PBS, 5% glycerol). Eluates were concentrated with Amicon Ultra-15 centrifugal filter units (30 kDa cutoff; Millipore). Concentrated proteins were loaded onto a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) pre-equilibrated with a storage buffer (1×PBS, 5% glycerol). Eluted proteins were concentrated, examined by SDS-PAGE, and quantified by a Bradford assay (BioRad), then flash frozen by liquid N2 and stored at −80°C. PAS-GAF-Avi-His, PHY-Avi-His and PHY-SNAP-Avi-His were expressed and purified as the same as described above.
His-tagged or Avi-Tagged nanobodies were expressed in Escherichia coli strain WK6 and purified by Ni-affinity and size-exclusion chromatography as we previously reported.27
Protein biotinylation.
DrBphP-Avi-His was biotinylated by BirA using a BirA-500 kit (Avidity). Typically, 200 μL BiomixA (10× concentration: 0.5 M bicine buffer, pH 8.3), 200 μL BiomixB (10× concentration: 100 mM ATP, 100 mM Mg(OAc)2, 500 μM d-biotin), 200 μL BIO200 (10× concentration: 500 μM d-biotin), 20 μL 1 mg/mL BirA, and DrBphP-Avi-His (final concentration at ~2.4 mg/mL) were mixed with H2O to a final volume of 2 mL. The biotinylation mixture was incubated at 37°C for 1 h and then loaded onto a HiPrep 26/10 desalting column (GE Healthcare) pre-equilibrated with a storage buffer (1×PBS, 5% glycerol). Eluted proteins were concentrated, examined by SDS-PAGE, and quantified by the Bradford assay, flash frozen by liquid N2, and stored at −80°C. LDB-3-Avi-His, LDB-14-Avi-His, PAS-GAF-Avi-His, PHY-Avi-His and PHY-SNAP-Avi-His were biotinylated similarly as DrBphP.
Phage display selection.
The combinatorial nanobody phage library was prepared as previously described.27 Dimerization binders were selected using 775-nm and 654-nm illuminations for the negative and positive selections, respectively. Briefly, 1.2 mL 20 μM biotinylated DrBphP-Avi-His was bound to 600 μL streptavidin agarose resin (Thermo Scientific) and blocked with 1% casein and 1% BSA in 1×PBS pH 7.4 for 30 min at 4°C in the dark. The resins were divided by a 2:1 ratio to pack the negative and positive selection columns (HR 5/5, GE Healthcare), respectively. As shown in Figure S3, both columns were connected to AKTA FPLC system and equilibrated with ~10 mL PBS buffer at 0.5 mL/min. The negative and positive selection columns were illuminated with 775 ± 14 nm light (FC-LED-780M, Prizmatix) at 0.8 mW/cm22 for 10 min and 654 ± 11 nm light (FC-LED-655A, Prizmatix) at 0.3 mW/cm2 for 10 min and then wrapped with aluminum foil. The light intensity was measured with an optical power meter (PM100D, Thorlabs) connected to a S130C probe (Thorlabs). In each round, phage-displayed nanobodies were loaded onto the columns equilibrated with 1× PBS at a flow rate of 0.04 mL/min. Next, the negative selection column was removed and the positive selection column washed with ~30 mL 0.05 % PBST (1×PBS with 0.05% v/v Tween 20) at a flow rate at 0.5 mL/min until the UV 280 nm baseline became stable (i.e., non-bound phages were washed out). Prior to the illumination, 2 mL flow through was collected as a “pre-elution fraction” at 0.5 mL/min immediately. The flow rate was decreased to 0 and then the positive selection column was illuminated with the 775-nm light (0.8 mW/cm2) for a given time (refer to Figure S3). A 2-mL fraction was collected as a “light-elution fraction” at 0.5 mL/min immediately after the illumination. The percentage of phages specifically eluted by the light was estimated by comparing phage counts in the pre-elution and light-elution fractions. The light eluted phages were amplified and used as an input for next round biopanning.
Y2H screening.
CDSs of the enriched nanobody library after four rounds of the biopanning were subcloned into pGADT7 to create a sub-library as preys. DrBphP was inserted to pGBKT7 as the bait. Y2HGold cells were co-transformed with bait and prey plasmids, plated onto SD/-Ade/-His/-Leu/-Trp plates under the 654-nm illumination (0.03 mW/cm2), and incubated at 30°C for 4–5 days. ~2,000 well-grown clones were picked and grew in 1-mL SD/-Leu/-Trp medium in deep 96- well plates under the 654-nm illumination (0.03 mW/cm2) for 24 h. 1-μL cells of each clone were replica spotted to SD/–Ade/–His/–Leu/–Trp plates and incubated under the 654-nm illumination (0.03 mW/cm2) or in the dark for 2–3 days. Clones showing significantly faster growth under the illumination were picked for further analysis. Because clones picked from the plates were often contaminated with a small amount of other clones, plasmids were purified from yeast, transformed into an E. coli DH5α strain to select clones carrying pGADT7 on LB Agar plates with Ampicillin (100 μg/mL) and then identify those carrying correct nanobody genes by Sanger sequencing. To further confirm the gene activation specificity, sequenced preys and the bait were again co-transformed into Y2HGold cells; non-diluted and diluted (1/10 and 1/100) cells were spotted onto SD/-Ade/-His/-Leu/-Trp plates to compare colony growth under the illumination and in the dark. Sequence- and specificity-validated clones were chosen for further analyses.
Phage ELISA.
E. coli electrocompetent TG1 cells were transformed with pADL-23c inserted with selected nanobody candidates. Colonies were inoculated into 250 μL media (2×TY, 2% glucose, 100 μg/mL ampicillin) in deep 96-well plates and grown at 37°C for overnight. 10-μL cultures were inoculated into 500 μL fresh media and cells were grown to OD600 = ~0.5 and infected by CM13 helper phage with the multiplicity of infection (MOI) of ~18. Cells were shaken at 37°C for 45 min, added with kanamycin (50 μg/mL, the final concentration), and grown at 25°C for overnight. Plates were centrifuged for 30 min at 3,000×g and phage-containing supernatants were transferred to fresh plates for an ELISA assay. Specifically, ELISA plates (Nunc MaxiSorp, Thermo Fisher Scientific) were coated with 100 μL 5 μg/mL streptavidin in a coating buffer (100 mM carbonate buffer, pH 8.6) at 4°C for overnight. After washing five times with 0.05% PBST (1×PBS with 0.05% v/v Tween 20), each well was added with 100 μL 2 μM biotinylated DrBphP-Avi-His, PAS-GAF-Avi-His, PHY-Avi-His, or PHY-SNAP-Avi-His and incubated at room temperature (r.t.) for 1 h in the dark. Wells were washed five times with 0.05% PBST, blocked with 1% casein in 1× PBS, and then illuminated by the 654-nm (0.3 mW/cm2) or 775-nm (0.2 mW/cm2) light for 10 min. 100 μL phage supernatants were added and incubated at r.t. for 1h in dark. Wells were washed 10 times with 0.05% PBST and then illuminated with corresponding light (654 nm at 0.3 mW/cm2 or 775 nm at 0.2 mW/cm2) for 10 min before washing five times with 0.05% PBST. Wells were added with 100 μL HRP-M13 major coat protein antibody (RL-ph1, Santa Cruz Biotechnology; 1:10,000 dilution with 1× PBS, 1% casein) and incubated at r.t. for 1 h in the dark. A colorimetric detection was performed using a 1-Step Ultra TMB ELISA substrate solution (Thermo Fisher Scientific); OD450 was measured with a SpectraMax Plus 384 microplate reader (Molecular Devices).
Mammalian two-hybrid assay.
HEK293T (ATCC, CRL-3216) and HeLa cells (ATCC-CCL-2) were grown in a Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and with or without biliverdin (Frontier Scientific) in a humidified incubator (Forma Scientific) under 5% CO2 at 37°C. For a firefly luciferase assay, cells were grown in 24-well plates (Greiner Bio-One) to ~60% confluence and transiently co-transfected with the DrBphP bait and nanobody preys, and a luciferase reporter plasmid (Addgene, #64125) in a 1:1:1 ratio (0.25 μg each into a ~500 μL medium). After the transfection, culture medium was changed in 6 h and then cells were kept in the darkness for another 18 h prior to the transcription activation. The activation was performed by continuously illuminating cells with the 654-nm light at 0.2 mW/cm2 for 24 h; cells were kept in the dark as the control. The time-course luciferase assay was performed as described above with different transcription induction times.
Luciferase levels were measured with a firefly luciferase glow assay kit (Pierce) following the manual. Briefly, after the transcription activation, cells were washed with 1× PBS, added with 150 μl of 1× cell lysis buffer, and incubated at 4°C for 30 min. 20 μl cell lysate from each well was transferred into a black 96-well plate (CELLSTAR, Greiner Bio-One, Cat # 655079) and mixed with 50 μl of a Working Solution. Bioluminescence signals were measured with a SpectraMax i3 plate reader (Molecular Devices) after incubation in the dark at r.t. for 10 min.
The luciferase assay of RpBphP1-based systems was performed under the same condition, except that the transcription activation was performed with the 775-nm (0.2 mW/cm2) illumination, because different from DrBphP, RpBphP1 is converted to the light form by the NIR illumination.
Analysis of nanobody stability in mammalian cells.
~2×105 HEK293T cells were seeded in 6-well plates (Thermo Fisher Scientific, catalog # 140675) in DMEM supplemented with 10% FBS, and incubated under 5% CO2 at 37°C for overnight. Cells in a 1.5-mL medium were transiently transfected with plasmids (2.5 μg each) encoding nanobody–SNAP-tag fusions using lipofectamine 2000 (Thermo Fisher Scientific). After 36-h incubation, the medium was removed and cells were washed with 1× PBS twice, dissociated from the plate by digestion with a 1× Trypsin-EDTA Solution (Thermo Fisher Scientific, catalog # R001100), and collected in 15-mL conical tubes. Cells were washed with 1 mL 1× PBS and re-suspended in 250 μL ice cold 1× PBS for sonication. After centrifugation at 20,000g for 10 min, ~50 μL supernatants were incubated with 1 μM (final concentration) SNAP-Surface 649 (New England Biolabs, catalog # S9159S) for 1 h at r.t. to label SNAP-tagged proteins. Labelled samples were boiled for 10 min at 95°C in an SDS sample loading buffer before loaded onto an SDS-PAGE gel. The gel was scanned by an Odyssey CLx imaging system (Li-cor Biosciences).
Analytical SEC.
Interactions of DrBphP with LDB-3 and LDB-14 were analyzed by analytical SEC. Samples were loaded onto a Superdex 200 Increase 10/300 GL column (GE Healthcare) pre-equilibrated with 1× PBS and eluted at 0.75 mL/min at 4°C. The column was calibrated with molecular weight standards (Bio-Rad, catalog # 1511901). Light-sensitive samples were prepared in a dark room and the column and sample syringes were all covered by aluminum foil to avoid light exposure.
To detect the complex formation, DrBphP-His was photoconverted to the dark and light forms by the 775-nm (0.8 mW/cm2, 10 min) and 654-nm (0.2 mW/cm2, 5 min) illumination, respectively. ~6 μM (final concentration) DrBphP-His was added with ~5 μM (final concentration) LDB-3-His or LDB-14-His and incubated at r.t. for 30 min in the dark before loading a 500 μL mixture onto the column. 500-μL fractions with an elution volume between 8 and 16 mL were collected and proteins in each fraction were precipitated by trichloroacetic acid (TCA) for SDS-PAGE analysis. Briefly, 55 μL 100% TCA was mixed with each fraction and incubated at −20°C for 30 min. After centrifugation at 20,000×g, 4°C for 15 min, supernatants were removed, and pellets were washed with 600 μL ice-cold acetone three times and then dried in air. Pellets were resuspended and boiled in the SDS loading buffer and analyzed by SDS-PAGE.
Isothermal titration calorimetry.
Binding affinities and thermodynamics of LDB-3 and LDB-14 to DrBphP were measured by a MicroCal PEAQ-ITC device (Malvern) at 25°C. Specifically, ~ 210 μL of 10 or 5 μM DrBphP-His was loaded to a sample cell and then illuminated by the 654-nm (0.2 mW/cm2, 5 min) or 775-nm (0.8 mW/cm2, 15 min) light. ~38 μL 80 μM LDB-3-His and 50 μM LDB-14-His were titrated into 10 and 5 μM DrBphP-His in the cell, respectively, by 19 injections (2 μL each) from a syringe. Background heat transfer caused by the nanobody dilution was measured by conducting a titration of LDB-3-His (80 μM) or LDB-14-His (50 μM) into a 1× PBS buffer alone. Titration of 1× PBS buffer into DrBphP-His (10 μM) was also conducted as the control.
Raw ITC data were analyzed by NITPIC version 1.2.7.39 To find a suitable range for each injection, cut-off differentials for the injection end was changed to 0.1. The fitting equation for a one-site model is , where y represents the heat of injection, x represents the molar ratio, and b, k, L, x0 are related parameters. The integrated data of LDB-3 and LDB-14 titrated to the light form were fitted with the above equation by using a “curve_fit” function in the Python-SciPy package, which generated KDapp and other thermodynamic parameters in Table S4.
Bio-layer interferometry.
LDB-3 and LDB-14 binding kinetics were analyzed using an Octet RED96 system (ForteBio) and Streptavidin (SA) biosensors. Briefly, 20 μg/mL biotinylated LDB-3-Avi-His or LDB-14-Avi-His was immobilized on SA biosensors in 1× PBS buffer (pH 7.4). A duplicate set of sensors was incubated in the buffer without any protein to measure background binding. All sensors were blocked with a buffer (1× PBS, pH 7.4, 0.05% Tween-20, 0.2% BSA, and 10 ng/mL biocytin) before the binding assay. Serial dilutions of DrBphP-His in an assay buffer (1× PBS, pH 7.4, 0.05% Tween-20, and 0.2% BSA) were illuminated with the 654-nm (0.3 mW cm−2, 5 min) or 775-nm (0.2 mW cm−2, 10 min) light before binding to the nanobodies. The assay was performed in black 96-well plates with a total working volume of 0.2 mL per well at r.t. Raw data were analyzed by an Octet data analysis software V9.0 (ForteBio) using a double-reference-subtraction protocol to subtract signals related to nonspecific binding, background, and signal drift caused by sensor variability.
Apparent dissociation constants (KDapps) were calculated by the steady-state analysis and the fitting with a global 1:1 model. The fitting of apparent dissociation rate constant (koffapp) was found to be more reliable (or less DrBphP-His concentration dependent) than the fitting of apparent binding rate constant (konapp), so only koffapp was calculated by fitting with the equation, , where C represents the level of binding, C0 the binding at the start of dissociation, A an asymptote, and t time. koffapp for each binding was calculated using the “curve_fit” function in the Python-SciPy package. After obtaining KDapp and koffapp, konapp was calculated by . Of note, compared with the fitting result, the dissociation curves were slightly tailed (Figure S11), likely due to the contribution from DrBphP dimer dissociation.
DrBphP photoconversion analysis.
The DrBphP photoconversion rate was analyzed by absorption spectroscopy. Absorption spectra (500–900 nm) of DrBphP samples were obtained using a SpectraMax Plus 384 microplate reader (Molecular Devices). DrBphP-His was added in a quartz micro cuvette (Yixing Purshee Optical Elements) and then converted to the light or dark form by the 654-nm (0.5 mW/cm2, 2 min) or 775-nm (0.3 mW/cm2, 10 min) illumination before collecting spectra. To monitor the real-time photoconversion to the dark form, ~400 μL 5 μM (final concentration) DrBphP-His samples added with or without 5 μM (final concentration) LDB-3-His or LDB-14-His in the cuvette were first converted to the light form by the 654-nm (0.5 mW/cm2) illumination for 2 min and then immediately converted by the 775-nm (0.3 or 0.05 mW/cm2) illumination with different exposure times before collecting spectra. The ratio of A750/A700 was normalized to the range (0–1) to monitor the photoconversion process.
GFP imaging.
HEK293T cells were seeded in 10 cm Nunclon Delta Surface culture dishes (Thermo Scientific) in DMEM supplemented with 10% FBS in the humidified incubator under 5% CO2 at 37°C. They were co-transfected with 10 μg a pGreenFire1-Gal4 lentivector (System Biosciences, catalog # TR017PA-1) and lentivirus-packing plasmids (5μg each) including PMDL, REV and VSVG by a calcium phosphate transfection method. The medium was changed in 6 h after the transfection and the virus was harvested after incubation for another 72 h. To separate the virus from the medium, the medium was centrifuged at 500×g for 5 min and the supernatant was passing through a Millex-HV filter (0.45 μm, Merck Millipore). 2.5 out of 10 mL filtered virus was used to infect HEK293T cells cultured in another 10 cm dish under 50% confluence, with 10 μg/mL polybrene (Merck Millipore), for 24 h.
Lentivirus-transduced HEK293T cells were seeded in 35 mm glass bottom microwell dishes coated with poly-D-lysine (MatTek, catalog # P35GC-0–10-C) at a density of 1×105 cells per dish. On the second day, cells were transiently co-transfected with the GAL4-BD–DrBphP and nanobody-p65 plasmids (1.25 μg each) using lipofectamine 2000 (Thermo Fisher Scientific) and incubated for overnight. For each nanobody candidate, two dishes were needed for the illumination and the dark control; after the transfection, dishes were immediately covered by aluminum foil to avoid light exposure. On the third day, cells were under the 654-nm (0.2 mW/cm2) illumination or maintained in the dark for another 48 h. Prior to fluorescence imaging, cells were fixed by 4% paraformaldehyde for 10 min and washed with 1× PBS.
GFP images were acquired using a Nikon Ti-E automated inverted microscope equipped with a Perfect Focus System, a Nikon 20×/0.75-NA Plan Apo Lambda objective, a linear encoded motorized stage (Nikon Ti-S-ER), and an Andor iXon Ultra 888 EMCCD camera (16-bit dynamic range, 1,024×1,024 array with 13-μm pixels). Cells were illuminated by a SPECTRA X LED illuminator (Lumencor) coupled with an excitation filter (448 ± 19 nm) and a filter cube mounted with a dichroic mirror (506 nm) and an emission filter (510 ± 20 nm) (Chroma). Culture dishes were scanned under the GFP and a brightfield channels. Acquired GFP images (dark and light condition) were analyzed by MATLAB for quantifying the fluorescence intensity. Specifically, fluorescence signals in all pixels were subtracted by an average background value (i.e., the median of the pixel intensity distribution in each field-of-view (FOV)) and integrated for each FOV. For each condition, 78 FOVs were sampled for statistical analysis.
Protein Translocation.
HEK293T cells were seeded in the 24 well plates (Thermo Scientific) in DMDM supplemented with 10% FBS and cultured at 37°C with 5% CO2 provided. A total ~0.5 μg of DrBphP-AcGFP-CAAX and LDB-3-mCherry plasmids (1:1 ratio) were co-transfected into the cells using lipofectamine 3000 (Thermo Fisher) when 70% confluence was reached. 6 hour later, the medium was replaced with the fresh medium supplemented with 5 μM biliverdin and then plates were covered by aluminum foil and maintained in the dark for another 18 h before imaging.
Cell images were acquired using a Nikon spinning disk confocal microscope equipped with a Ti2-E inverted frame with full automation, a Perfect Focus System, a Nikon 20×/0.50-NA Plan Fluor objective, and a Photometrics Prime 95B camera. Cells were imaged under GFP and mCherry channels illuminated by 488 and 561 nm lasers, respectively, and the emission filters were 523 ± 18 nm and 605 ± 26 (Chroma), respectively. Standard flat bottom 24 well plates were sequentially illuminated by the Prizmatix LEDs from the top of wells and then imaged at both channels. Acquired images were analyzed by ImageJ.
Transcription activation in mice.
Animal data were collected in Third Institute of Oceanography (TIO) of Ministry of Natural Resources following a protocol approved by TIO’s Animal Care and Use Committee. 6 to 8-week-old male BALB/c nude mice of ~20 g body weight were used for the in vivo transcription activation. Like the M2H assay, HEK293T cells were co-transfected with the plasmids encoding LDB-3-p65, GAL4-DB–DrBphP, and the luciferase reporter. 6 h after the transfection, cells were used for in vivo assays.
For subcutaneous injection, each mouse was injected with ~5×106 cells supplemented with 100 μL DMEM medium, and kept in a conventional cage in the dark for 24 h. In another 24 h, mice in a light treatment group (n = 3) were placed in a transparent cage illuminated by a LED array at 0.03 mW/cm2 and those in the control group (n = 3) remained in the dark. Mice were subjected to in vivo imaging using an IVIS Spectrum instrument (PerkinElmer) in luminescence mode with an open emission filter. Throughout the imaging process, animals were maintained under anesthesia with 1.5% vaporized isoflurane. Prior to imaging, 200 μL of 15 mg/mL D-Luciferin sodium salt solution (US Everbright) was intravenously injected through a tail vein. Data were analyzed using a Living Image 3.0 software (Perkin Elmer). For liver transplantation, ~2×106 cells supplemented with 20 μL DMEM medium were transplanted into the subcapsular region of the liver. Then, the mice were treated as describe above. After treatment, the mice were subjected to bioluminescence imaging using a Newton7.0 instrument (Vilber).
Supplementary Material
ACKNOWLEDGMENTS
We thank N. Zheng and T. Hinds for the help on ITC assay, A. Merz for confocal imaging, Y. Wu, E. J. Quitevis and M. Moussa for binder screening, and T. Lin, M. Dinh and X. Kuang for protein production. This work, except the collection of in vivo animal data, was supported by a grant from the U.S. National Institutes of Health (R35GM128918 to L.G.), a Safeway Pilot Award (to L.G.), and a startup fund of the University of Washington (to L.G.). The collection of animal data was supported by a grant from the Chinese Ministry of Natural Resources (12110600000018003927 to Z.L). K.W. was supported by an Institute for Protein Design summer fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1101/2020.07.09.174003
Details of primers and plasmid constructs, phage display biopanning and Y2H screening, protein SDS-PAGE and absorption spectra, SEC, ITC, BLI, and ELISA assays, the time-course analysis of light-induced luciferase expression, a flowchart and timeline of COMBINES-LID, a summary of photoswitchable proteins or domains that can potentially be used as conformational switchers in LID systems, and the thermodynamic modeling of competitive hetero- and homo-dimerization. (PDF)
A provisional patent related to this work has been filed by the University of Washington.
Contributor Information
Zhimin Huang, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Zengpeng Li, Key Laboratory of Marine Genetic Resources, State Key Laboratory Breeding Base of Marine Genetic Resources, Fujian Key Laboratory of Marine Genetic Resources, Fujian Collaborative Innovation Centre for Exploitation and Utilization of Marine Biological Resources, Third Institute of Oceanography Ministry of Natural Resources, Xiamen, 361005, P.R. China; Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Xiao Zhang, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Shoukai Kang, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Runze Dong, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Li Sun, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Xiaonan Fu, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
David Vaisar, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Kurumi Watanabe, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
Liangcai Gu, Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, United States.
REFERENCES
- (1).Shimizu-Sato S; Huq E; Tepperman JM; Quail PH (2002) A light-switchable gene promoter system. Nat. Biotechnol 20, 1041–1044. [DOI] [PubMed] [Google Scholar]
- (2).Kolar K; Weber W (2017) Synthetic biological approaches to optogenetically control cell signaling. Curr. Opin. Biotechnol 47, 112–119. [DOI] [PubMed] [Google Scholar]
- (3).Klewer L; Wu YW (2019) Light-induced dimerization approaches to control cellular processes. Chem.: Eur. J 25, 12452–12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Spencer DM; Wandless TJ; Schreiber SL; Crabtree GR (1993) Controlling signal transduction with synthetic ligands. Science 262, 1019–1024. [DOI] [PubMed] [Google Scholar]
- (5).Stanton BZ; Chory EJ; Crabtree GR (2018) Chemically induced proximity in biology and medicine. Science 359, eaao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Klemm JD; Schreiber SL; Crabtree GR (1998) Dimerization as a regulatory mechanism in signal transduction. Annu. Rev. Immunol 16, 569–592. [DOI] [PubMed] [Google Scholar]
- (7).Kennedy MJ; Hughes RM; Peteya LA; Schwartz JW; Ehlers MD; Tucker CL (2010) Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang F; Vierock J; Yizhar O; Fenno LE; Tsunoda S; Kianianmomeni A; Prigge M; Berndt A; Cushman J; Polle J; Magnuson J; Hegemann P; Deisseroth K (2011) The Microbial Opsin Family of Optogenetic Tools. Cell 147, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mayer G; Heckel A (2006) Biologically active molecules with a “light switch”. Angew. Chem. Int. Ed. Engl 45, 4900–4921. [DOI] [PubMed] [Google Scholar]
- (10).Christie JM; Salomon M; Nozue K; Wada M; Briggs WR (1999) LOV (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (nph1): Binding sites for the chromophore flavin mononucleotide. Proc. Natl. Acad. Sci. U. S. A 96, 8779–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bhoo SH; Davis SJ; Walker J; Karniol B; Vierstra RD (2001) Bacteriophytochromes are photochromic histidine kinases using a biliverdin chromophore. Nature 414, 776–779. [DOI] [PubMed] [Google Scholar]
- (12).Rockwell NC; Su YS; Lagarias JC (2006) Phytochrome structure and signaling mechanisms. Annu. Rev. Plant Biol 57, 837–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Weissleder R (2001) A clearer vision for in vivo imaging. Nat. Biotechnol 19, 316–317. [DOI] [PubMed] [Google Scholar]
- (14).Weissleder R; Ntziachristos V (2003) Shedding light onto live molecular targets. Nat. Med 9, 123–128. [DOI] [PubMed] [Google Scholar]
- (15).Jobsis FF (1977) Noninvasive, infrared monitoring of cerebral and myocardial oxygen sufficiency and circulatory parameters. Science 198, 1264–1267. [DOI] [PubMed] [Google Scholar]
- (16).Chernov KG; Redchuk TA; Omelina ES; Verkhushaa VV (2017) Near-infrared fluorescent proteins, biosensors, and optogenetic tools engineered from phytochromes. Chem. Rev 117, 6423–6446. [DOI] [PubMed] [Google Scholar]
- (17).Bellini D; Papiz MZ (2012) Structure of a bacteriophytochrome and light-stimulated protomer swapping with a gene repressor. Structure 20, 1436–1446. [DOI] [PubMed] [Google Scholar]
- (18).Kaberniuk AA; Shemetov AA; Verkhusha VV (2016) A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nat. Methods 13, 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Winkler A; Heintz U; Lindner R; Reinstein J; Shoeman RL; Schlichting I (2013) A ternary AppA-PpsR-DNA complex mediates light regulation of photosynthesis-related gene expression. Nat. Struct. Mol. Biol 20, 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Redchuk TA; Omelina ES; Chernov KG; Verkhusha VV (2017) Near-infrared optogenetic pair for protein regulation and spectral multiplexing. Nat. Chem. Biol 13, 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Redchuk TA; Karasev MM; Verkhusha PV; Donnelly SK; Hulsemann M; Virtanen J; Moore HM; Vartiainen MK; Hodgson L; Verkhusha VV (2020) Optogenetic regulation of endogenous proteins. Nat. Commun 11, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kojadinovic M; Laugraud A; Vuillet L; Fardoux J; Hannibal L; Adriano JM; Bouyer P; Giraud E; Vermeglio A (2008) Dual role for a bacteriophytochrome in the bioenergetic control of Rhodopsdeudomonas palustris: Enhancement of photosystem synthesis and limitation of respiration. Biochim. Biophys. Acta 1777, 163–172. [DOI] [PubMed] [Google Scholar]
- (23).Guntas G; Hallett RA; Zimmerman SP; Williams T; Yumerefendi H; Bear JE; Kuhlman B (2015) Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. U. S. A 112, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Reis JM; Xu XL; McDonald S; Woloschuk RM; Jaikaran ASI; Vizeacoumar FS; Woolley GA (2018) Uppalapati, M., Discovering selective binders for photoswitchable proteins using phage display. ACS Synth. Biol 7, 2355–2364. [DOI] [PubMed] [Google Scholar]
- (25).Muyldermans S, Nanobodies: natural single-domain antibodies (2013) Annu. Rev. Biochem 82, 775–797. [DOI] [PubMed] [Google Scholar]
- (26).Kang S; Davidsen K; Gomez-Castillo L; Jiang H; Fu X; Li Z; Liang Y; Jahn M; Moussa M; DiMaio F; Gu L (2019) COMBINES-CID: An efficient method for de novo engineering of highly specific chemically induced protein dimerization systems. J. Am. Chem. Soc 141, 10948–10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Moutel S; Bery N; Bernard V; Keller L; Lemesre E; de Marco A; Ligat L; Rain JC; Favre G; Olichon A; Perez F (2016) NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife 5, e16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wagner JR; Zhang JR; von Stetten D; Guenther M; Murgida DH; Mroginski MA; Walker JM; Forest KT; Hildebrandt P; Vierstra RD (2008) Mutational analysis of Deinococcus radiodurans bacteriophytochrome reveals key amino acids necessary for the photochromicity and proton exchange cycle of phytochromes. J. Biol. Chem 283, 12212–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Takala H; Bjorling A; Berntsson O; Lehtivuori H; Niebling S; Hoernke M; Kosheleva I; Henning R; Menzel A; Ihalainen JA; Westenhoff S (2014) Signal amplification and transduction in phytochrome photosensors. Nature 509, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Choi SG; Olivet J; Cassonnet P; Vidalain PO; Lucks K; Lambourne L; Spirohn K; Lemmens I; Dos Santos M; Demeret C; Jones L; Rangarajan S; Bian W; Coutant EP; Janin YL; van Der Werf S; Trepte P; Wanke EE; De Las Rivas J; Tavernier J; Twizere JC; Hao T; Hill DE; Vidal M; Calderwood MA; Jacob Y (2019) Maximizing binary interactome mapping with a minimal number of assays. Nat. Commun 10, 3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lievens S; Lemmens I; Tavernier J (2009) Mammalian two-hybrids come of age. Trends Biochem. Sci 34, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ong NT; Olson EJ; Tabor JJ (2018) Engineering an E. coli near-infrared light sensor. ACS Synth. Biol 7, 240–248. [DOI] [PubMed] [Google Scholar]
- (33).Niu J; Ben Johny M; Dick IE; Inoue T (2016) Following optogenetic dimerizers and quantitative prospects. Biophys. J 111, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Skrlec K; Strukelj B; Berlec A (2015) Non-immunoglobulin scaffolds: a focus on their targets. Trends Biotechnol. 33, 408–418. [DOI] [PubMed] [Google Scholar]
- (35).Chevalier A; Silva DA; Rocklin GJ; Hicks DR; Vergara R; Murapa P; Bernard SM; Zhang L; Lam KH; Yao G; Bahl CD; Miyashita SI; Goreshnik I; Fuller JT; Koday MT; Jenkins CM; Colvin T; Carter L; Bohn A; Bryan CM; Fernández-Velasco DA; Stewart L; Dong M; Huang X; Jin R; Wilson IA; Fuller DH; Baker D (2017) Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wu CY; Roybal KT; Puchner EM; Onuffer J; Lim WA (2015) Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Huang ZL; Wu YQ; Allen ME; Pan YJ; Kyriakakis P; Lu SY; Chang YJ; Wang X; Chien S; Wang YX (2020) Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv 6, eaay9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Xia YZ; Li K; Li JJ; Wang TQ; Gu LC; Xun LY (2019) T5 exonuclease-dependent assembly offers a low-cost method for efficient cloning and site-directed mutagenesis. Nucleic Acids Res. 47, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Scheuermann TH; Brautigam CA (2015) High-precision, automated integration of multiple isothermal titration calorimetric thermograms: New features of NITPIC. Methods 76, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.