

Abstract
Leigh syndrome is a genetically heterogeneous disorder resulting from deficient oxidative energy biogenesis. The syndrome is characterized by subacute episodic decompensations, transiently elevated lactate, and necrotizing brain lesions most often in the striatum and brainstem. Acute decompensation is often triggered by viral infections. Sequalae from repeated episodes leads to progressive neurological deterioration and death. The severity of Leigh syndrome varies widely, from a rapid demise in childhood to rare adult presentations. Although the causes of Leigh syndrome include genes affecting a variety of different pathways, more than 75 of them are nuclear or mitochondrial encoded genes involved in the assembly and catalytic activity of mitochondrial respiratory complex I.
Here we report the detailed clinical and molecular phenotype of two adults with mild presentations of NDUFS3 and NDUFAF6-related Leigh Syndrome. Mitochondrial assays revealed slightly reduced complex I activity in one proband and normal complex I activity in the other. The proband with NDUFS3-related Leigh syndrome was mildly affected and lived into adulthood with novel biallelic variants causing aberrant mRNA splicing (NM_004551.2:c.419G > A; p.Arg140Gln; NM_004551.2:c.381 + 6 T > C). The proband with NDUFAF6-related Leigh syndrome had biallelic variants that cause defects in mRNA splicing (NM_152416.3:c.371 T > C; p.Ile124Thr; NM_152416.3:c.420 + 2_420 + 3insTA). The mild phenotypes of these two individuals may be attributed to some residual production of normal NDUFS3 and NDUFAF6 proteins by NDUFS3 and NDUFAF6 mRNA isoforms alongside mutant transcripts. Taken together, these cases reported herein suggest that splice-regulatory variants to complex I proteins could result in milder phenotypes.
Keywords: Leigh syndrome, NDUFS3, NDUFAF6, Mitochondrial/rare disease, Splicing variants
1. Introduction
Leigh syndrome (LS), with an incidence of 1 per 40,000 live births, is one of the most common presentations of mitochondrial diseases [1]. A genetically heterogeneous disorder, LS can result from monogenic mutations in one of more than 75 nuclear or mitochondrial genes [2]. These mutations alter proteins that either form subunits of mitochondrial respiratory complexes or assist in their assembly and regulation, causing defects in the oxidative phosphorylation pathway and thereby inhibiting cellular ATP production [3]. NADH/ubiquinone oxidoreductase, the first mitochondrial respiration complex in the electron transport chain, transfers electrons from NADH to ubiquinone. NDUFS3, which denotes NADH/ubiquinone oxidoreductase core subunit 3, catalyzes the in vivo assembly of complex I in the mitochondrial matrix and provides it with the capacity to act as a hydrogenase [4]. NDUFAF6, or NADH/ubiquinone complex I assembly factor 6, also plays a role in the early assembly of complex I by regulating the biogenesis of mitochondrial subunit ND1 [5].
The clinical presentation of LS varies with each patient’s nuclear DNA or mitochondrial DNA (mtDNA) mutation(s). Chief diagnostic indicators of LS include bilateral striatal necrosis revealed by high T2 signal intensities cerebral MRI [1]. Symmetrical lesions in the brain-stem are also common. Additional supporting evidence for LS diagnosis includes decreased mitochondrial respiratory complex assembly and activity, as well as raised lactate concentration in the blood or cerebrospinal fluid. Patients can be diagnosed with Leigh-like syndrome if they also present with peripheral neurological symptoms or non-neurological manifestations, such as diabetes or abnormalities in the cardiovascular, renal, or gastrointestinal systems. Clinically, LS often presents in infancy with episodic decompensations, frequently triggered by acute viral infections, with symptoms such as poor sucking, loss of motor skills, dystonia, ataxia, abnormal eye movements, and peripheral neuropathy. As LS progresses, psychomotor regression and neurological deficits worsen and patients may succumb to respiratory or cardiac failure. Prognoses for LS patients are usually poor, and most patients do not live past mid-childhood or adolescence. Adult presentations of LS occur rarely [2].
A very small subset of LS patients present with relatively mild symptoms, manifesting some symptoms in childhood and survive into adulthood. Per the most recent review of long survival in complex IV-related LS, a total of eighteen patients with mutations in SURF1, an important gene in complex IV assembly, that presented in childhood have lived past age fourteen [6]. Recently, one adult-onset LS patient with a mutation in MT-ND3 (m.10197 G > A) recovered on treatment with coenzyme Q10 [7]. Long survival in childhood-onset LS has also been described in singular reports of patients with mutations to NDUFAF5 (33 years), MT-ND3 (20 years), and MT-ATP6 (58 years) [8,9]. Long survival in complex-I related LS is also poorly documented. Patients with mild symptoms are often prime candidates for missed diagnoses in the absence of strong radiological or biochemical findings. As a result, a recent review identified only two patients that have lived past the age of twenty following childhood-onset of complex-I related LS in a cohort of 130 [8].
Herein, we present the detailed phenotype of two adult LS probands with mild and monophasic/oligophasic presentations caused by splicing variants in NDUFS3 and NDUFAF6.
2. Materials and methods
The probands were admitted to the National Institutes of Health Clinical Center (NIH-CC) and enrolled in protocol 76-HG-0238, “Diagnosis and Treatment of Patients with Inborn Errors of Metabolism or Other Genetic Disorders,” approved by the National Human Genome Research Institute’s Institutional Review Board. After the parent gave written informed consent for proband A and proband B gave assent, the probands underwent a 5-day clinical evaluation.
2.1. Molecular analysis
DNA was extracted from peripheral whole blood samples and exome libraries were prepared using the Agilent SureSelect v5 PostCap or Nimblegen v3 protocols [10]. Exome sequencing was completed with the Illumina HiSeq2000 platform resulting in raw sequence FASTQs. Alignment to the human reference assembly hg19, variant calling, and joint genotyping were performed using the Illumina DRAGEN BioeIT Platform (v2.6). The VCF file was annotated using the Variant Effect Predictor (VEP v95) [6] and vcfanno (v0.2.9) [7], and Gemini (v0.20.1) [8] was used to query variants of interest. Each recessive variant was filtered for gnomAD population frequency < 1% and a gnomAD homozygote count fewer than 10. Variants in genes with known disease associations in Online Mendelian Inheritance in Man (OMIM) database (including NDUFAF6) were prioritized [9]. Sanger sequencing was performed to confirm the segregation of identified variants.
2.2. Cell culture
Primary dermal fibroblast cells were cultured from proband skin biopsies. Unaffected sex-matched primary dermal fibroblast cell lines GM23972 (Control A) and GM08398 (Control B) (Coriell Institute for Medical Research, Camden, NJ) were used as controls. Control A fibroblasts were submitted to a research study as a normal control with clinically normal brain MRI scans, neurological exam, and neuropsychological testing. Control B fibroblasts were sampled from the inguinal area and described as an “apparently healthy collection.” Fibroblasts were cultured at 37 °C with 5% CO2 in standard growth media: 90% DMEM – Dulbecco’s Modified Eagle Medium (11,995, Gibco/Thermofisher), 10% FBS – Fetal Bovine Serum (100–500, GeminiBio), and 1× PenStrep Glutamine (10,378,016, Gibco/Thermofisher). Fibroblasts were cultured until 70–80% confluency.
2.3. RNA extraction and reverse transcription
Adherent cells were collected from T75 flasks through trypsinization using TrypLE (A1285901, Gibco/Thermofisher) and homogenized through QIAshredder columns (Qiagen). Total RNA was extracted using the RNeasy Mini Kit (74,106, Qiagen) and residual genomic DNA was removed using the DNA-free™ DNA Removal Kit (AM1906, Thermofisher). Reverse transcription was conducted using High-Capacity RNA-to-cDNA™ Kit (4,387,406, ThermoFisher) for using 2 µg RNA per reaction for qPCR analysis.
2.4. qPCR
Quantitative real-time PCR was performed using Taqman specific primers for NDUFS3 (Hs00190028_m1, Thermofisher), NDUFAF6 (Hs00901868_m1, Thermofisher), and RPL13A (Hs04194366_g1). cDNA derived from proband and control fibroblasts was amplified using TaqMan universal PCR master mix and analyzed using CFX96 Touch Real-Time PCR Detection System (C1000, Bio-Rad) for gene expression analysis. Amplification conditions followed the Taqman standard curve protocol; results were normalized to RPL13A and analyzed using the ΔΔCt method.
2.5. PCR and gel electrophoresis
From proband-derived fibroblasts, mRNA was isolated using the RNeasy Mini Kit (74,106, Qiagen) and cDNA was reverse transcribed using the High-Capacity RNA-to-cDNA™ Kit (4,387,406, ThermoFisher). A PCR of the proband’s cDNA was run alongside two controls following the Platinum PCR Supermix protocol (11306–016, Invitrogen), using the following primer sequences: NDUFS3, NM_004551.2: 5’-ACTGTCAGACCACGGAATGA-3′ (forward) and 3’-GGATGTCCCTCGAAGCCATA-5′ (reverse); NDUFAF6, NM_152416.3: 5′- CTGGGGCACTGACCACTACT −3′ (forward) and 3’-TGCTTTAGGT GCAAGTGTGC-5′ (reverse). PCR product was electrophoresed on a 2% agarose gel using a Bio-Rad PowerPac (1,645,050, Bio-Rad). PCR products or gel-extracted bands were cloned using the TOPO kit (250,184, Invitrogen) following the manufacturer’s protocol, and sequenced using M13 primers.
2.6. Protein extraction
Adherent cells were washed twice with ice-cold PBS Buffer (RGF-3200, KD Medical) and scraped into RIPA Buffer (R0278, Sigma) supplemented with Halt Protease Inhibitor Cocktail Single Aliquots-Use (100×) (78,425, Thermofisher) for 30 min on ice to collect whole cell lysates. Protein lysates were quantified using a BCA assay (23,227, Thermofisher).
2.7. Western blot
Control and proband proteins were boiled with NuPAGE™ LDS Sample Buffer (4×) (NP0007, Invitrogen) and NuPAGE™ Sample Reducing Agent (10×) (NP0009, Invitrogen) for 5 min at 95 °C. The samples were resolved on 4–12% Bris-Tris Gels, 1.5 mm, 10-well (NP0335BOX, Invitrogen), transferred to a nitrocellulose (NC) membrane, and incubated overnight at 4 °C with the following antibodies: NDUFS3 (ab177471, Abcam) and β-actin (ab8226, Abcam). Bands were detected by incubation with IRDye® and VRDye™. Secondary antibodies were imaged on the Odyssey® CLx Infrared Imaging System. Densitometry analysis was performed using the Odyssey® imaging system. Experiments were performed in triplicate.
2.8. Respiratory chain complex activity
These activity assays were performed by a commercial clinical laboratory (Baylor Genetics). Electron transport chain enzymes in cells were assayed at 30 °C in duplicate using a temperature-controlled spectrophotometer. The activities of complex I (NADH:Ferricyanide dehydrogenase), complex II (succinate dehydrogenase), complex I + III (NADH:cytochrome c oxidoreductase), complex II + III (succinate:cytochrome c reductase), and complex IV (cytochrome c oxidase) were measured using different electron acceptors/donors. The increase or decrease of cytochrome c at 550 nm was measured for complex I + III, complex II + III, or complex IV. The activity of complex I was measured by following the oxidation of NADH at 340 nm. For complex II, the reduction of 2,6-dichloroindophenol (DCIP) at 600 nm was measured. Citrate synthesis was used as a marker for mitochondrial content. Enzyme activities were normalized against citrate synthase (CS) activity when CS activity was greater than one standard deviation above or below the control mean.
3. Results
3.1. Clinical cases
Proband A was born after an uneventful, full-term pregnancy to healthy unrelated African-American parents (Fig. 1A). At age five, he developed a weeklong episode of ataxia and motor deterioration and was admitted for a neurological and metabolic evaluation. Metabolic analyses were normal apart from elevated serum lactate of 4.1 mmol/L (normal, < 2.0), consistent with metabolic acidosis. Cerebral MRI showed abnormal bilateral T2 signal hyperintensities in the medulla and mild vermian atrophy. Genetic analyses for MELAS, MERRF, NARP, and mitochondrial deletion analysis were negative. The boy was diagnosed with subacute cerebellar ataxia and discharged, making a full recovery. He went on to acquire normal motor abilities but had learning difficulties, requiring an individualized education program at school. By the age of eighteen, he had spasmodic torticollis with a 6–8 Hz head tremor and neck pain leading to social withdraw. A neurological evaluation at age nineteen indicated torticollis, mild dystonic hand tremor, nystagmus, and mild bradykinesia. His sister also had developed episodic neurological deterioration related to infections in childhood and was reported to have an abnormal gait. The proband’s cranial MRI revealed a small cavitary lesion in the tail of the right caudate nucleus, as well as two punctate foci of cavitation and gliosis in the tail of the left caudate nucleus (Fig. 1B). He was referred to the Undiagnosed Diseases Program (UDP) at the National Institutes of Health (NIH) for further evaluation [11–13]. Comprehensive metabolic screening included electron transport chain (ETC) studies in fibroblasts (complex I activity = 66% of control mean) and a lactate and amino acid profile in the CSF, which was normal. Exome and mtDNA sequencing were performed. Fig. 1, Supplementary Fig. S1, Supplementary Fig. S2, Supplementary Fig. S3 and Supplementary Fig. S4.
Fig. 1.
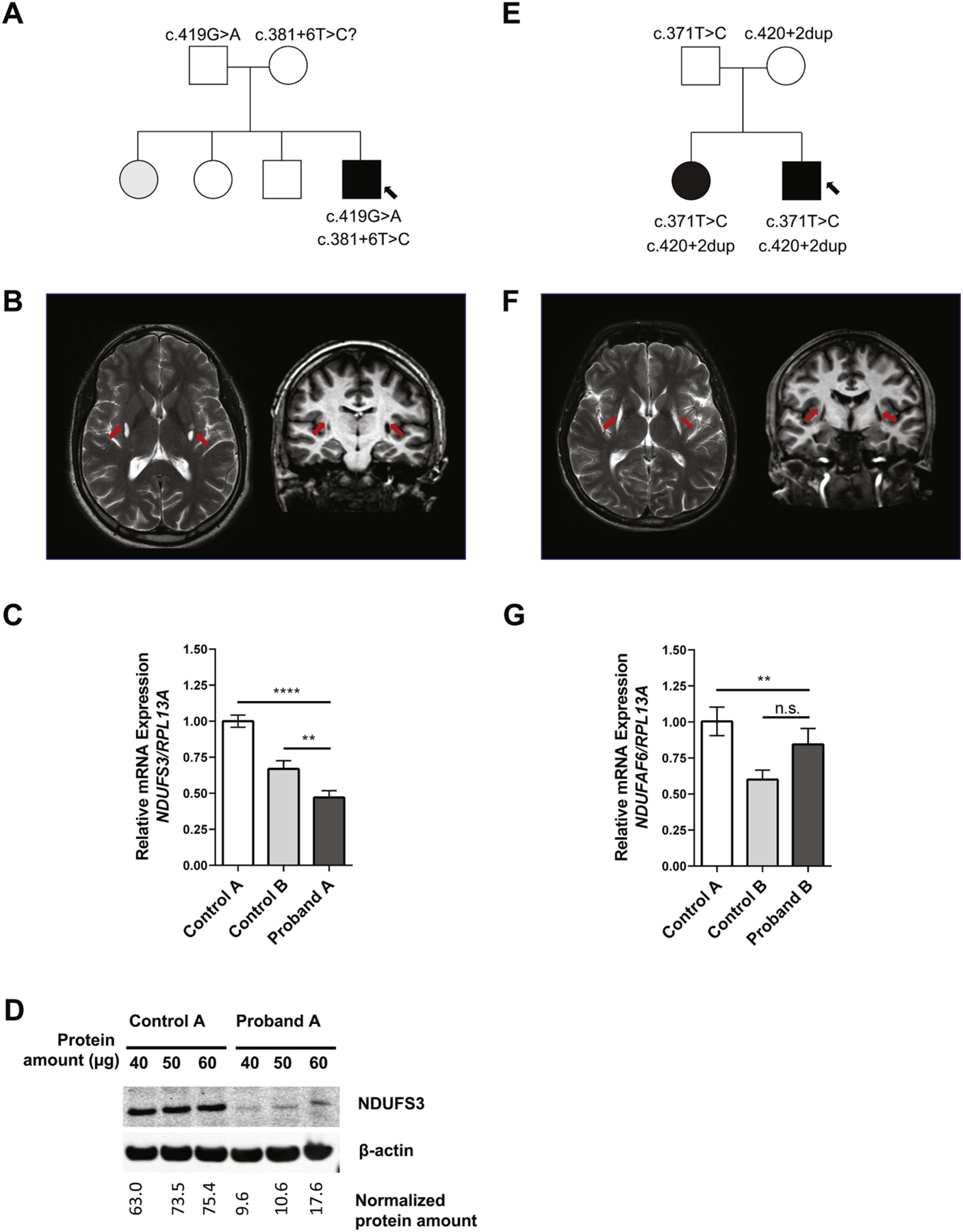
Clinical and Genetic Findings. (A) Pedigree for proband A’s family; affected members are shaded with black, and arrow points to proband A. (B) MRI images showing the characteristic T2 signal intensities in the basal ganglia of proband A (arrows). (C) mRNA expression analysis for NDUFS3 in proband A’s dermal fibroblasts as compared to unrelated control fibroblasts. Proband A displayed both a statistically significant reduction of mRNA compared to controls (****P < 0.001 in comparison to control A, **P < 0.01 in comparison to control B). Paired t-tests were conducted using GraphPad Prism8 software where asterisks indicate statistical significance. mRNA analysis was normalized to RPL13A. (D) Protein expression analysis show reduced amount of NDUFS3 (predicted to be at 25 kDa) in proband A as compared to control, normalized with (42 kDa). Three different protein amounts were loaded. (E) Pedigree for proband B’s family; affected members are shaded with black, and arrow points to proband B. (F) MRI images showing the characteristic T2 signal intensities in the basal ganglia of proband B (arrows). (G) mRNA expression analysis for NDUFAF6 in proband B’s dermal fibroblasts as compared to two unrelated control fibroblasts; mRNA analysis was normalized to RPL13A. Paired t-tests were conducted using GraphPad Prism8 software where asterisks indicate statistical significance (****P < 0.0001; **P < 0.01).
Proband B was born after an uneventful pregnancy to unaffected, non-consanguineous Hispanic parents (Fig. 1E). Prenatal and perinatal history was normal. At the age of 4, he and his one-year younger sister developed a high fever in the context of an acute viral illness. Both were admitted to the hospital for observation, had negative CSF examinations, and were discharged one week later. Within four weeks, both began to exhibit neurological symptoms including tiptoeing and increasingly severe focal hand dystonia which eventually became generalized. CT scan and MRI showed cavitary lesions of the putamina without mass effect or contrast enhancement. No white matter abnormalities were noted (Fig. 1F). Lactate and pyruvate levels were normal. Over the years, proband B underwent multiple investigations including a muscle biopsy that did not reveal abnormalities. He was treated empirically with riboflavin, ubiquinone, and carnitine to no symptomatic benefit. He has normal cognition, achieving a higher education degree, and is proficient in communicating in several languages but is profoundly impaired by symptoms of generalized dystonia. Bilateral deep brain stimulation (DBS) implants into internal globus pallidus (GPi) were of no symptomatic benefit. He was evaluated at age 27 by the NIH UDP. Other than his original episode at age four, he had no other episodes of acute neurological decline or metabolic decompensation. A comprehensive metabolic investigation including serum amino acids, pyruvate and lactate levels, and CSF lactate was normal. ETC studies were normal (complex I activity = 126% of the control mean). Mitochondrial DNA sequencing and WES were undertaken.
3.2. Bioinformatic and molecular data
Proband A.
Research analysis of exome sequencing data of proband A and his unaffected father confirmed compound heterozygous variants in NDUFS3 (NM_004551.2:c.419G > A; p.Arg140Gln; NM_004551.2:c.381 + 6 T > C) (Fig. S1A, B) in the proband. The c.419G > A variant in exon 5 (Fig. S2A) was identified in gnomAD with low frequency (0.000007952). The intronic 381 + 6 T > C variant was not identified in gnomAD, but a splice site mutation nearby was seen and was associated with LS in ClinVar (RCV000389911.1, RCV000332963.1). Neither variant is described as pathogenic in the literature. Sanger sequencing confirmed that the exonic c.419G > A variant was inherited from proband A’s father; it is pathogenic by in silico predictions: Complete Annotation Dependent Depletion (CADD) (see URLs) Phred-scaled score of 26.1; “Deleterious” (Score: 0.0) by SIFT (see URLs); and “Disease-Causing” (Probability: 1) by MutationTaster (see URLs). The arginine residue at position 140 of NDUFS3’s amino acid sequence is well conserved across multiple species (Fig. S1C). Proband A’s intronic variant (NM_004551.2:c.381 + 6 T > C) could be inherited from proband A’s mother or could be de novo. Neither the mother nor the affected sister could provide samples for sequencing analysis.
Alamut Visual’s splicing predictor, which synthesizes output from splice predictor bioinformatics software SpliceSiteFinder, MaxEntScan, NNSPLICE, and GeneSplicer, projected that the c.381 + 6 T > C could affect the nearest splice site, the donor site at the 3′ end of exon four. The exonic c.419G > A variant is predicted to cause the production of a new splicing acceptor site at the position of the exonic change, which is thirty-eight base pairs (bp) downstream from the start of exon five.
Proband B.
Exome sequencing of proband B, his parents, and an affected sister confirmed compound heterozygous variants in NDUFAF6 (NM_152416.3) (Fig. S3A, B) that were both identified in ClinVar as pathogenic: c.371 T > C, p.Ile124Thr and c.420 + 2_420 + 3insTA. The c.371 T > C in exon 3 (Fig. S4A) leads to a threonine change in a conserved amino acid (Fig. S3C) that has been described in a report detailing LS patients [19].
Alamut Visual’s splicing predictor projected that the intronic variant (NM_152416.3:c.420 + 2_420 + 3insTA) disrupts mRNA splicing by altering the splice donor site at the 3′ end of NDUFAF6’s exon 3.
3.3. Splicing variant analysis and expression studies
To test the in-silico splicing predictions, expression studies were conducted.
Proband A (NDUFS3).
On RT-PCR of cDNA derived from proband A’s fibroblasts visualized by gel electrophoresis, two distinct bands were seen, i.e., one shared with the controls, and one that was only present in the proband (Fig. S1D). Sanger sequencing of the gel-extracted PCR products revealed a 39 bp deletion beginning at the 5′ end of exon five (Fig. S2B) in 60% of analyzed transcripts (n = 25) that aligned with the in-silico predictions made regarding the c.419G > A variant. This results in an in-frame deletion of 13 amino acids (p.(Val129_Ile141del). Two other new transcript alterations were found related to the c.381 + 6 T > C variant (Fig. S2B): a 31 bp retention from intron four (c.381 + 5_381 + 6ins31); and a 19 bp deletion (c.215_233del) that spans 5′ end of three and 3′ end of exon four, which results to a frameshift variant (p.(Tyr72Cysfs*7)).
We then measured the expression of NDUFS3 mRNA transcripts by quantitative PCR (qPCR) and protein by Western blot. Proband-derived fibroblasts showed decreased expression of NDUFS3 mRNA and protein relative to two unaffected, healthy control samples (Fig. 1C, S1E). Since the NDUFS3 mRNA is reduced by ~50%, we predict that the alternate transcripts resulting from the c.381 + 6 T > C variant, which cause shifts in the reading frame, are targeted for nonsense-mediated decay (NMD). This is supported by our Western blot analysis using different amounts of protein loaded, which shows that proband A produces a reduced amount of total NDUFS3 protein (Fig. 1E). It is possible that the c.419G > A 39 bp deletion could result in a slightly shorter protein with no changes to the reading frame. Therefore, the barely-truncated mutant isoform is likely functional, not targeted for NMD, but less stable than the WT protein, which may help explain the proband’s residual complex I activity.
Proband B (NDUFAF6).
RT-PCR of cDNA derived from proband B’s fibroblasts visualized by gel electrophoresis showed several alternate mRNA transcripts (Fig. S3D). The presence of aberrant transcripts was confirmed by Sanger sequencing cloned cDNA. Mutant transcripts (Fig. S4B) included exon three skipping (33%), exon three skipping plus a 4 bp (GCTG) insertion, and a 4 bp insertion (Fig. S4B). NDUFAF6 mRNA expression, however, suggests that the production of both normal and aberrant transcripts remains unaffected by NMD (Fig. 1G). Future studies are required to confirm the role of NMD in transcript regulation in both probands.
4. Discussion
The prognosis for LS is usually very poor, with much of LS survival depending on which mitochondrial complex is affected and the extent to which the complex is deficient. For most complexes, individuals with partial deficiencies can be expected to live until their sixth or seventh year, while more complete deficiencies often result in infantile death. Rarely do patients live into their teenage years, and fewer still survive to adulthood. The probands described here are unusual given the relatively mild and oligosymptomatic nature of their decompensation episodes; they deviate significantly from the complex-I LS norm and are the only two known to live independently into their third decade of life despite presenting in childhood.
The probands’ mild clinical presentation is likely conferred by their normal (proband B) and near-normal (proband A) complex I activity resulting from their splicing variants, which are likely less deleterious than other types of variants reported in LS to date. Therefore, linking both probands’ neuroradiological and clinical phenotype to their respective splicing variants, in line with the diagnostic criteria for LS [1], was fundamental in preparing molecular diagnoses.
Although both of proband B’s variants had already been described and verified as pathogenic by other studies [19], our experiments showed that both of proband A’s variants can also be classified as pathogenic, per ACMG guidelines [14]. The intronic variant (NM_004551.2:c.381 + 6 T > C; pathogenic: PSV1, PS3, PM2, PM6) alters the canonical donor site at the 3′ end of exon four and results to two alternate transcripts, one of which leads to a frameshift variant (c.215_233del, p.(Tyr72Cysfs*7)). This variant might be de novo, although maternal inheritance cannot be excluded. The exonic variant (NM_004551.2:c.419G > A; p.Arg140Gln; pathogenic: PS3, PM1, PM2, PM4, PP2, PP3) activates an acceptor site in exon five, resulting in an in-frame deletion, and, presumably, shorter protein (c.384_422del, p.(Val129_Ile141del). Additionally, the variants combine to produce a damaging effect in functional studies as evidenced by functional studies showing reduced NDUFS3 protein production. Finally, multiple lines of computational evidence, including in-silico splicing predictors and conservation analyses (Fig. S1C, S3C) suggest a deleterious effect on the gene product. Nonetheless, our results could be bolstered by complementation assays, especially since both of our probands have normal and near-normal complex I activities in fibroblasts. It will also be important for future studies to evaluate complex I activities in muscle tissues to analyze potential differential expression.
Mutations in NDUFS3 have been identified as causing LS in two previous articles [15,16], both of which reported biallelic protein-coding non-synonymous missense mutations in patients who presented with severe symptoms (Table 1) and died at two and thirteen years of age, respectively. This contrasts with the presentation of proband A, who is now twenty-seven years old and who completed secondary education. However, the neuroimaging findings in proband A were consistent with the neuroradiological profiles of the other reported cases and the neuroimaging diagnostic criteria for LS in general [1,2]. A second cerebral MRI of the infantile NDUFS3-related LS patient, taken one year later, displayed severe disease progression [16] that included increased bilateral striatal necrosis and cerebellar atrophy, in contrast to the mild progression in proband A observed at a repeat MRI thirteen years after disease onset.
Table 1.
Summary of clinical findings in individuals with NDUFS3-related LS.
| Patient and source of study | Proband A, this study | MITO036–33463; [19] | Patient 1; [15] | Patient 2 (Fetus); [15] | Patient; [16] |
|---|---|---|---|---|---|
| Gender | Male | NR | Male | NR | Male |
| Age of onset | 5 years | NR | 9 years | NR | 7 months |
| Symptoms at onset | Ataxia and motor deterioration | Developmental delay, muscular hypotonia, lactic acidosis, rapid disease progression | Neck stiffness | NR | Torticollis |
| Developmental abnormality | +, learning difficulties | Developmental delay | Normal until 9 years; neuromotor regression thereafter | NR | NR |
| Neurological and cerebellar findings | Ataxia, spasmodic torticollis, head tremor, nystagmus, mild bradykinesia, dystonia | Muscular hypotonia, rapid disease progression | Neck stiffness, progressive axial dystonia with oral and pharyngeal motor dysfunction, dysphagia, and a tetraparetic syndrome | NR | Torticollis |
| Brain MRI | Abnormal bilateral T2 hyperintensities in the medulla and minor vermian atrophy (age five). Small cavitary lesions in the tails of the right caudate nucleus; two punctate foci of cavitation and gliosis in the tail of the left caudate nucleus (Age 19). | NR | Focal, symmetrical, necrotic lesions in the thalamus, brain stem and white matter along with optic atrophy. | NR | High T2 signal intensity in the white matter of hemispheres, basal ganglia, and brain stem. |
| State of life at last consultation, age | Alive, 22 years old | NR | Died of rapid multisystem deterioration at 13.5 years of age | NR | Died at 2 years of age |
| Complex I deficiency - muscle | – | Reduced, 28% | Reduced, 50 (normal, 243 ± 60) | Reduced in amniocytes, value NR | Reduced in lymphoblastoids, value NR |
| Complex I deficiency - fibroblasts | Reduced, 66% | Reduced, 36% | Reduced, 16 (normal, 21 to 39) | NR | NR |
| Pyruvate (plus malate) oxidation (Complex I) - muscle | Normal | NR | Reduced, 20 (normal, 94 ± 48) | Reduced in amniocytes, 6.9 (normal, 7.7 to 9.5) | NR |
| Pyruvate (plus malate) oxidation (Complex I) - fibroblast | Normal | NR | Reduced, 2.7 (normal, 3.3 to 6.8) | NR | |
| Serum lactate; normal range is 0.580–2.10 mmol/L | Elevated; 4.10 mmol/L | Elevated, value not reported | NR, CSF, 2.7a | NR | Elevated, value not reported |
| Serum pyruvate; normal range is 55–145 umol/L. | Normal | NR | NR | NR | Elevated, value not reported |
| Consanguinity | No | NR | No | No | No |
| Paternal NDUFS3b variant | c.419G > A (p.Arg140Gln) | c.532C > T (p.Arg199Trp) | c.595C > T (p.Arg199Trp) | c.595C > T (p.Arg199Trp) | c.418C > T (p.Arg140Trp) |
| Maternal NDUFS3 b variant | c.381 + 6 T > C? (produces abberant isoforms) | c.532C > T (p.Arg199Trp) | c.434C > T (p.Thr145Ile) | c.434C > T (p.Thr145Ile) | c.595C > T (p.Arg199Trp) |
“+” denotes the presence of a feature; “− “denotes the absence of a feature; “NR” represents not reported.
, CSF Lactate 2.7 mmol/L (normal < 2.0).
, Variant nomenclature in NDUFS3 based on NM_004551.2. “?” – denotes that the variant may be de novo, but also may be maternally inherited.
The compound heterozygous mutations to protein-coding regions presented by previous reports on NDUFS3-related LS produced a more severe phenotype and disease progression, potentially by rendering the mutant protein isoform nonfunctional, as evidenced by severely reduced complex I activity [15]. Indeed, the mutant NDUFS3 reported in [15] appeared more prone to aggregation than the WT isoform [4]. Proband A’s splicing variants may lead to the production of normal transcripts or semi-functional mutants that are still able to participate in complex I biogenesis, leaving a residual level of WT NDUFS3 protein or functional mutant protein that allows the probands’ mitochondria to retain near-normal oxidative function, at least in patient-derived fibroblasts. This may explain why the missense variants in previous studies [15,16] are more pathogenic than proband A’s splicing variants, but future studies will be required to support this hypothesis.
LS-causing mutations to NDUFAF6, in comparison with those to NDUFS3, are more common (Table 2), with some reports of splicing variants. For instance, two affected siblings with an intronic mutation (420 + 748C > T) and their resultant splicing defects have been described [18]. These patients, along with proband B, presented with prominent dysarthria but preserved cognitive functions. In contrast to proband B, they displayed complex I deficiency in fibroblasts and presented with more severe symptoms; no assertions can be made about survival or quality of life, as these siblings are five and seven years of age at the time of this report. Nonetheless, part of proband B’s presentation is consistent with that of other patients with NDUFAF6-related LS, including dystonia [17,18] and a neuroimaging finding of focal basal ganglia signal abnormalities.
Table 2.
Summary of clinical findings in individuals with NDUFAF6-related LS.
| Patient and source of study | Proband B (this study) | A1; [18] | A2; [18] | B; [17,18]a | C; [18] | P1; [20] | P2; [20] | P3; [20] | C8orf38 Patient P1; [21]; [22] |
C8orf38 Patient P2; [21] |
Pt512; [23] | Pt 598; [23] | Pt 101; [23] | Pt 330; [23] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gender | Male | Male | Female | Male | Male | Female | Male | Male | Female | Male | Female | Male | Male | Male |
| Age of onset | 4 years | 21 months | 12 months | 3.5 years | 5 years | 30 months | 30 months | 17 months | 10 months | 7 months | Newborn | 2 months | 6 years | 17 months |
| Symptoms at onset | Febrile episode, and months later toe walking and focal hand dystonia | Febrile episode and temporary loss of autonomous gait | Insidious, limb dysmetria and trunk titubation | Insidious, gross motor and language difficulties, dystonic movements and decreased fine motor ability. | Insidious, gait unsteadiness and motor coordination problems | Insidious, toe walking and speech difficulties | Insidious, toe walking and speech difficulties | Insidious, toe walking and speech difficulties | Focal right hand seizures, ataxia, rigidity, decreased movement and strength. | Focal right hand seizures, ataxia, rigidity, decreased movement and strength. | NR | NR | NR | NR |
| Developmental abnormality | Motor regression | Psychomotor regression | Psychomotor delay | Motor regression. Difficulty in verbal fluency but verbal comprehension preserved. | Psychomotor regression | Neuromotor regression | Neuromotor regression | Neuromotor regression | Neuromotor regression. | Neuromotor regression. | Neurological regression | Neurological regression | Neurological regression | Muscle atrophy |
| Neurological and cerebellar findings | Dysarthria, ataxic gait, dystonia | Ataxic gait, fine tremor, dysmetria, drooling, hypertonia, dystonia | Ataxic gait, limb dysmetria, trunk titubation, dystonia | Dysarthria, dystonic movements, decreased fine motor abilities, dystonia | Drooling, dysarthria, oromandibular dystonia, extrapyramidal hypertonia, mild camptocormic gait, dystonia | Dysarthria, stuttering, dysphagia, dystonia | Dysarthria, stuttering, dysphagia, dystonia | Dysarthria, stuttering, dysphagia, dystonia | Focal right hand seizures, ataxia, rigidity, decreased movement and strength. | Focal right hand seizures, ataxia, rigidity, decreased movement and strength. | NR | NR | NR | NR |
| Brain MRI | Bilateral T2 hyperintensities in the putamina without mass effect or contrast enhancement. No white matter abnormalities. | T2 hyperintensities in putamen, dentate nuclei, superior cerebellar peducle and caudate nucleus. Hypotrophic Putamen | Bilateral T2 hyperintensities in dentate nucleus and superior cerebellar peduncle. | Bilateral T2 intensities (and T1 hypointensities) in utamen, caudate nuclei, dentate nuclei. Gradual putaminal cavitation. | Bilateral T2 hyperintensities in putamen. Putaminal hypotrophy (cavitation). | Bilateral T2 hyperintensities in putamen. Caudate spared. | Bilateral T2 hyperintensities in putamen, and partly in caudate. Mild putaminal volume loss (cavitation). | Bilateral T2 hyperintensities and volume loss in putamen and caudate. | “Neuroimaging consistent with Leigh syndrome.” | “Neuroimaging consistent with Leigh syndrome.” | NR | Abnormalities of the basal ganglia on brain MRI. | NR | Abnormalities of the basal ganglia on brain MRI. |
| State of life at last consultation, age | Alive (adult) | Alive, 5 years old | Alive, 3 years old | Alive, 8.5 years old | Alive, 11 years old | Alive, 11 years old | Alive, 8 years old | Alive, 6 years old | Died at 34 months (pneumonia) | Alive, 34 months | NR | NR | NR | NR |
| Complex I deficiency - muscle | NR | Reduced, 42% | NR | Normal | Normal | Reduced, 60% | Reduced, not quantified | Reduced, not quantified | Reduced, 36% | NR | NR | NR | NR | NR |
| Complex I deficiency - fibroblasts | 126% | Reduced, 38% | NR | Reduced, 25% | Normal | NR | NR | Reduced, not quantified | Reduced, 14% | Reduced, 14% | Reduced, < 30% | Reduced, < 30% | Reduced, < 40% | Reduced, < 30% |
| Serum lactate; normal range is 0.580–2.10 mmol/L | Normal | Initially normal then elevated; 2.818 mmol/L | Elevated; 2.293 mmol/L | Normal, value not reported | Normal, value not reported | Normal; 1.63 mmol/L | 1.59 mmol/L | NR | Elevated, value not reported | Elevated, value not reported | Elevated, value not reported | NR | NR | NR |
| Serum pyruvate; normal range is 55–145 umol/L. | Normal | Initially normal then elevated; 181 umol/L | Elevated; 146 umol/L | Normal, value not reported | Normal, value not reported | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| Consanguinity | No | No | No | No | No | No | No | No | Yes | Yes | No | No | No | No |
| Paternal NDUFAF6c variant | c.371 T > C (p.Ile124Thr) | c.532G > C (p.Ala178Pro) | c.532G > C (p.Ala178Pro) | c.532G > C (p.Ala178Pro) | NM_152416.3 c.532G > C (p.Ala178Pro) | c.371 T > C (p.Ile124Thr) | c.371 T > C (p.Ile124Thr) | c.371 T > C (p.Ile124Thr) | c.296A > G (p.Gln99Arg) | c.296A > G (p.Gln99Arg) | c.226 T > C (p.Ser76Pro) | c.371 T > C (p.Ile124Thr) | c.805C > G (p.His269Asp) | c.820A > G (p.Arg274Gly) |
| Maternal NDUFAF6c variant | c.420 + 2dup (produces abberant isoforms) | c.420 + 784C- > T (p.?; intron retention of ~124 nucleotides) | c.420 + 784C- > T (p.?; intron retention of ~124 nucleotides) | c.420 + 784C > - T (p.?; intron retention of ~124 nucleotides) | c.420 + 784C > - T (p.?; intron retention of ~124 nucleotides) | c.554_558delTTCTT (p.Tyr187AsnfsTer65) | c.554_558delTTCTT (p.Tyr187AsnfsTer65) | c.554_558delTTCTT (p.Tyr187AsnfsTer65) | c.296A > G (p.Gln99Arg) | c.296A > G (p.Gln99Arg) | c.805C > G (p.His269Asp) | c.206A > T (p.Asp69Val) | c.371 T > C (p.Ile124Thr) | c.820A > G (p.Arg274Gly) |
“+” denotes the presence of a feature; “− “denotes the absence of a feature; “NR” represents not reported.
, Patient B initially reported in Biancardi et al. 2016 [17]; the second variant and biochemical abnormalities are reported by Catania et al.
, also reported in McKenzie et. al. 2011 [22].
, Variant nomenclature in NDUFAF6 based on NM_152416.3.
LS diagnosis may be missed in other individuals with mild phenotypes. Identifying patients with NDUFS3 and NDUFAF6 variants and additional cases with milder or oligosymptomatic presentations of LS would help clinicians understand the phenotypic spectrum of this syndrome and the factors that influence severity. This information could provide knowledge generalizable to the entire spectrum of LS presentations.
Supplementary Material
Acknowledgment
The authors do not have any conflict of interest to declare. We thank the probands and their families for their participation in this study as well as their referring physicians. This work was supported by the Intramural Research Program of the National Human Genome Research Institute and the Common Fund of the NIH Office of the Director.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymgme.2020.09.008.
Web resources
ExAC Browser, http://exac.broadinstitute.org/, accessed September 28, 2020.
gnomAD Browser, https://gnomad.broadinstitute.org/, accessed September 28, 2020.
Mutation Taster: http://www.mutationtaster.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.bii.a-star.edu.sg/
ESEfinder, http://krainer01.cshl.edu/cgi-bin/tools/ESE3/, accessed July 1, 2019.
CADD, https://cadd.gs.washington.edu/snv, accessed July 3, 2019.
References
- [1].Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR, Leigh syndrome: clinical features and biochemical and DNA abnormalities, Ann Neurol 39 (1996) 343–351. [DOI] [PubMed] [Google Scholar]
- [2].Lake NJ, Compton AG, Rahman S, Thorburn DR, Leigh syndrome: One disorder, more than 75 monogenic causes, Ann. Neurol 79 (2016) 190–203. [DOI] [PubMed] [Google Scholar]
- [3].Frazier AE, Thorburn DR, Compton AG, Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology, J. Biol. Chem 294 (2019) 5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jaokar TM, Patil DP, Shouche YS, Gaikwad SM, Suresh CG, Human mitochondrial NDUFS3 protein bearing Leigh syndrome mutation is more prone to aggregation than its wild-type, Biochimie 45 (2013) 2392–2403. [DOI] [PubMed] [Google Scholar]
- [5].Andrews B, Carroll J, Ding S, Fearnley IM, Walker JE, Assembly factors for the membrane arm of human complex I, Proc Natl Acad Sci U S A 110 (2013) 18934–18939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Aulbert W, Weigt-Usinger K, Thiels C, Kohler C, Vorgerd M, Schreiner A, Hoffjan S, Rothoeft T, Wortmann SB, Heyer CM, Podskarbi T, Lucke T, Long survival in Leigh syndrome: new cases and review of literature, Neuropediatrics 45 (2014) 346–353. [DOI] [PubMed] [Google Scholar]
- [7].Chen Z, Zhao Z, Ye Q, Chen Y, Pan X, Sun B, Huang H, Zheng A, Mild clinical manifestation and unusual recovery upon coenzyme Q(1)(0) treatment in the first Chinese Leigh syndrome pedigree with mutation m.10197 G > A, Mol Med Rep 11 (2015) 1956–1962. [DOI] [PubMed] [Google Scholar]
- [8].Sofou K, De Coo IF, Isohanni P, Ostergaard E, Naess K, De Meirleir L, Tzoulis C, Uusimaa J, De Angst IB, Lonnqvist T, Pihko H, Mankinen K, Bindoff LA, Tulinius M, Darin N, A multicenter study on Leigh syndrome: disease course and predictors of survival, Orphanet J Rare Dis 9 (2014) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Levy RJ, Rios PG, Akman HO, Sciacco M, Vivo DC, DiMauro S, Long survival in patients with leigh syndrome and the m.10191T > C mutation in MT-ND3 : a case report and review of the literature, J Child Neurol 29 (2014) NP105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].van der Werf IM, Kooy RF, Vandeweyer G, A robust protocol to increase NimbleGen SeqCap EZ multiplexing capacity to 96 samples, PLoS One 10 (2015) e0123872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gahl WA, Tifft CJ, The NIH Undiagnosed Diseases Program: lessons learned, JAMA 305 (2011) 1904–1905. [DOI] [PubMed] [Google Scholar]
- [12].Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, Landis DM, Yang S, Madeo A, Mullikin JC, Boerkoel CF, Tifft CJ, Adams D, The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases, Genet Med 14 (2012) 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, Adams DR, Tifft Udn CJ, The NIH Undiagnosed Diseases Program and Network: Applications to modern medicine, Mol Genet Metab 117 (2016) 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA, Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, Genet Med 17 (2015) 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Benit P, Slama A, Cartault F, Giurgea I, Chretien D, Lebon S, Marsac C, Munnich A, Rotig A, Rustin P, Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome, J Med Genet 41 (2004) 14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lou X, Shi H, Wen S, Li Y, Wei X, Xie J, Ma L, Yang Y, Fang H, Lyu J, A Novel NDUFS3 mutation in a Chinese patient with severe Leigh syndrome, J. Hum. Genet 63 (2018) 1269–1272. [DOI] [PubMed] [Google Scholar]
- [17].Bianciardi L, Imperatore V, Fernandez-Vizarra E, Lopomo A, Falabella M, Furini S, Galluzzi P, Grosso S, Zeviani M, Renieri A, Mari F, Frullanti E, Exome sequencing coupled with mRNA analysis identifies NDUFAF6 as a Leigh gene, Mol. Genet. Metab 119 (2016) 214–222. [DOI] [PubMed] [Google Scholar]
- [18].Catania A, Ardissone A, Verrigni D, Legati A, Reyes A, Lamantea E, Diodato D, Tonduti D, Imperatore V, Pinto AM, Moroni I, Bertini E, Robinson A, Carrozzo R, Zeviani M, Ghezzi D, Compound heterozygous missense and deep intronic variants in NDUFAF6 unraveled by exome sequencing and mRNA analysis, J. Hum. Genet 63 (2018) 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Haack TB, Haberberger B, Frisch EM, Wieland T, Iuso A, Gorza M, Strecker V, Graf E, Mayr JA, Herberg U, Hennermann JB, Klopstock T, Kuhn KA, Ahting U, Sperl W, Wilichowski E, Hoffmann GF, Tesarova M, Hansikova H, Zeman J, Plecko B, Zeviani M, Wittig I, Strom TM, Schuelke M, Freisinger P, Meitinger T, Prokisch H, Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing, J. Med. Genet 49 (2012) 277–283. [DOI] [PubMed] [Google Scholar]
- [20].Baide-Mairena H, Gaudo P, Marti-Sanchez L, Emperador S, Sanchez-Montanez A, Alonso-Luengo O, Correa M, Grau AM, Ortigoza-Escobar JD, Artuch R, Vazquez E, Del Toro M, Garrido-Perez N, Ruiz-Pesini E, Montoya J, Bayona-Bafaluy MP, Perez-Duenas B, Mutations in the mitochondrial complex I assembly factor NDUFAF6 cause isolated bilateral striatal necrosis and progressive dystonia in childhood, Mol Genet Metab 126 (2019) 250–258. [DOI] [PubMed] [Google Scholar]
- [21].Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK, A mitochondrial protein compendium elucidates complex I disease biology, Cell 134 (2008) 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McKenzie M, Tucker EJ, Compton AG, Lazarou M, George C, Thorburn DR, Ryan MT, Mutations in the gene encoding C8orf38 block complex I assembly by inhibiting production of the mitochondria-encoded subunit ND1, J Mol Biol 414 (2011) 413–426. [DOI] [PubMed] [Google Scholar]
- [23].Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, Mizuno Y, Hirata T, Yatsuka Y, Yamashita-Sugahara Y, Nakachi Y, Kato H, Okuda A, Tamaru S, Borna NN, Banshoya K, Aigaki T, Sato-Miyata Y, Ohnuma K, Suzuki T, Nagao A, Maehata H, Matsuda F, Higasa K, Nagasaki M, Yasuda J, Yamamoto M, Fushimi T, Shimura M, Kaiho-Ichimoto K, Harashima H, Yamazaki T, Mori M, Murayama K, Ohtake A, Okazaki Y, A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies, PLoS Genet 12 (2016) e1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.