

Abstract
Human immunodeficiency virus (HIV) remains incurable due to latent viral reservoirs established in non-activated CD4 T cells that cannot be eliminated via antiretroviral therapy. Current efforts to cure HIV are focused on identifying drugs that will induce viral gene expression in latently infected cells, commonly known as latency reversing agents (LRAs). Some drugs have been shown to reactivate latent HIV but do not cause a reduction in reservoir size. Therefore, finding new LRAs or new combinations or increasing the round of stimulations is needed to cure HIV. However, the effects of these drugs on viral rebound after prolonged treatment have not been evaluated. In a previous clinical trial, antiretroviral therapy intensification with maraviroc for 48 weeks caused an increase in residual viremia and episomal two LTR-DNA circles suggesting that maraviroc could reactivate latent HIV. We amended the initial clinical trial to explore additional virologic parameters in stored samples and to evaluate the time to viral rebound during analytical treatment interruption in three patients. Maraviroc induced an increase in cell-associated HIV RNA during the administration of the drug. However, there was a rapid rebound of viremia after antiretroviral therapy discontinuation. HIV-specific T cell response was slightly enhanced. These results show that maraviroc can reactivate latent HIV in vivo but further studies are required to efficiently reduce the reservoir size.
Subject terms: Drug discovery, HIV infections
Introduction
The human immunodeficiency virus type 1 (HIV) currently affects 37.9 million people worldwide according to the United Nation’s HIV program, UNAIDS1. Antiretroviral therapy (ART) suppresses viral replication, reduces plasma viral load, and delays clinical disease progression2 but it does not cure the infection. Lifetime treatment is required due to viral persistence in latent reservoirs that are inaccessible to current treatments and undetectable by the immune system3,4. This life-long latent reservoir is quickly established in vivo after infection and consists mainly of memory resting (r)CD4 T cells harboring the viral genome integrated into their DNA5,6. The reservoir size in patients on ART is estimated at 10 to 100 replication-competent latent proviruses per million rCD4 T cells7, and these latent proviruses are sufficient to rapidly restore the infection after an ART interruption8.
Efforts to eradicate the HIV reservoir include use of pharmacological agents, known as latency reversing agents (LRAs)9,10, in an attempt to reverse proviral latency through activation of the viral gene expression without inducing T cell activation. Although HIV induces cytopathic effects, these are not potent enough to kill infected cells and therefore, viral reactivation should occur along with the action of cytotoxic effector cells, such as CD8 T lymphocytes, in order to remove HIV-expressing cells11. Overall the strategy is commonly known as “shock and kill”10. Using a stochastic model of infection dynamics, it has been estimated that a reservoir reduction greater than 10,000-fold is required to prevent rebound12, but how long an LRA has to be administered to ensure safe ART discontinuation remains unclear and has to be experimentally tested for each drug. To date, major problems following LRA exposure exist because ex vivo assays have shown a great diversity in HIV reactivation responses13,14 and have not always correlated with the in vivo ones15. Carefully designed clinical trials have shown the capacity of some LRAs to reactivate latent HIV in vivo5,11–13. Several LRAs, such as disulfiram, panobinostat, or romidepsin induces the activation of HIV transcription in ART-patients as observed by the increase in cell-associated unspliced HIV RNA (CA-US-RNA), but the decrease of the reservoir size could not be demonstrated14–20. These trials have included single-dose administration of a drug or, at most, multiple doses over short periods. Accordingly, a treatment modeling shows that short-term stimulation with single or combined LRAs may remove a few percentage of the latent reservoir21. As it is unlikely that a single treatment would significantly reduce the reservoir size, it has been suggested that multiple rounds of treatment could have a more profound effect over time, assuming that re-stimulations cumulatively reduce the frequency of latently infected cells22. Furthermore, the need for including analytical treatment interruption (ATI) in order to evaluate the efficacy of any therapeutic strategy design to achieve HIV eradication has been designated due to the lack of cellular biomarkers of the reservoir23,24.
Maraviroc is an inhibitor of HIV entry that has been approved worldwide to treat patients infected with R5-tropic HIV25. In the large majority of patients, R5-tropic viruses are selected during transmission and persist during primary HIV infection when the latent reservoir is established26. Therefore, ART intensification with maraviroc was suggested as a potential strategy for limiting reservoir size although it did not work as predicted in either chronic or acute HIV patients27–29. Our group conducted an open-label phase II clinical trial in order to evaluate the effects of intensification of suppressive ART with maraviroc for 48 weeks on the cellular HIV reservoir and patient follow-up was expanded for an additional 24 weeks after drug discontinuation27,30 (Registration: ClinicalTrials.gov NCT00795444; EudraCT 2007-003995-21). In that study, we showed that maraviroc intensification was associated with a trend to decrease the HIV reservoir size in memory CD4 T cells, to transiently increase residual viremia, and to enhance the levels of episomal two long terminal repeat (LTR) DNA circles (2-LTR-DNA), which are regarded as markers of recent infection events27. These effects persisted up to 24 weeks after discontinuation of maraviroc30 and raised the hypothesis that maraviroc could increase transcriptional activation of the latent virus and could be regarded as new LRA. Later, it was confirmed in an additional clinical trial (Trial registration: EudraCT 2012-003215-66) that maraviroc activates HIV replication in vitro31 and more notably, caused an increase in the expression of CA-US-RNA in rCD4T cells from HIV-infected patients on ART32,33. This action was mediated through the activation of the transcription factor nuclear factor (NF)-κB. Maraviroc binds to chemokine receptor (CCR)5, changes receptor conformation, and inhibits interactions with HIV, and therefore its entry into the host cell34,35. The effect of maraviroc on calcium flow was the only signaling pathway downstream of CCR5 receptor that was studied during commercial development of the drug34; therefore, its effects on NF-κB and subsequently on HIV replication were unexpected but are compatible with maraviroc interactions with signaling pathways.
In this work, we further evaluate the effects of maraviroc on HIV latent infection in vivo. In this regard, the above-mentioned clinical trial (ClinicalTrials.gov NCT00795444) was amended in order to study additional virologic parameters in stored samples and, more notably, time to viral rebound during an analytical treatment interruption (ATI) in three patients.
Results
Study design and subjects
This study included patients who had participated in a pilot open-label phase II clinical trial of intensification with maraviroc conducted from March 2008 to January 2015 (Trial registration: EudraCT2007-003995-21; Registration date: 24/09/2007). As described earlier, the open-label phase II clinical trial of intensification with maraviroc initially included intensification of suppressive ART with maraviroc (donated by Pfizer Inc.) for 48 weeks and patient follow-up for an additional 24 weeks after maraviroc discontinuation27,30. After that, the clinical trial was amended to include additional virologic measurements using the stored samples and to include an ATI of the ART that patients were receiving in order to determine the impact of maraviroc on the latent HIV infection in vivo and on viral rebound, respectively. ATI was based on the demonstration that maraviroc is capable of effectively disrupting HIV latency in vivo through NF-κB activation32. Study participation required that no replicative HIV could be detected in a quantitative viral outgrowth assay (QVOA) just before starting the ATI. Only three patients included in the initial clinical trial agreed to participate in the ATI substudy. The median time between maraviroc discontinuation and ART interruption was 2.5 years.
Before being included in the study, the three patients were undergoing ART with at least three drugs for no less than two years, had undetectable plasma HIV RNA (< 50 copies HIV RNA/mL) for at least two years, and the CD4 T lymphocyte count was above 350 cells/µL. This situation of adequate virologic and immunological control was maintained in every patient at the time of the ATI. The socio-demographic and clinical characteristics of the study patients at baseline are summarized in Table 1. Briefly, two patients were men, and one was a woman; the median age was 50 years [absolute range (AR): range: 51–35]. The median HIV infection time was 18.0 years (AR: 18–14), and the median time on suppressive ART was 9.75 years (AR: 8.50–16.83). Regarding the ART regimens before ATI, two patients were treated with a combination of efavirenz, emtricitabine, and tenofovir, and the other one was treated with darunavir/ritonavir, emtricitabine, and tenofovir (Table 1). All patients showed CD4 T cell counts > 500 cells/µL. The median CD4 nadir was 230 cells/µL (AR: 246–171). The median CD4 T and CD8 T cell counts were 746 cells/µL (AR: 552–989) and 857 cells/µL (AR: 590–883), respectively. The CD4/CD8 ratio was 0.8 (AR: 0.6–1.6). The median percentage of CD38+/HLA-DR+/CD4+ T cells and CD38+/HLA-DR+/CD8+ T cells was 2.3 (AR: 1.7–2.9) and 2.9 µL (AR: 2.1–5.6), respectively.
Table 1.
Socio-demographic and clinical characteristics of the study patients at the time of starting the analytical treatment interruption.
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Gender | Female | Male | Male |
| Age (years) | 51 | 50 | 35 |
| Time of HIV infection (years) | 18 | 18 | 14 |
| Time on ART (years) | 9.75 | 16.83 | 8.50 |
| Current ART regimen | EFV + FTC + TDF | DRVr + FTC + TDF | EFV + FTC + TDF |
| Nadir CD4 (cells/µL) | 171 | 246 | 230 |
| CD4+ T cell counts (cells/µL) | 746 | 989 | 552 |
| CD8+ T cell counts (cells/µL) | 857 | 590 | 883 |
| CD4:CD8 Ratio | 0.8 | 1.6 | 0.6 |
| CD38+/HLA-DR+/CD4+ T cells (%) | 1.7 | 2.3 | 2.9 |
| CD38+/HLA-DR+/CD8+ T cells (%) | 2.1 | 2.9 | 5.6 |
| IUPM | < 0.012 | < 0.012 | < 0.012 |
| Episomal2 LTR DNA circles | Undetectable | Undetectable | Undetectable |
| HIV tropism | R5 | R5 | R5 |
ART antiretroviral treatment, DRVr ritonavir-boosted darunavir, EFV efavirenz, FTC emtricitabine, TDF tenofovir, IUPM infections units per million cells.
The number of memory CD4 T lymphocytes that were latently infected with replication-competent HIV was measured at baseline using a QVOA as previously described3,27,36,37. Results confirmed that the reservoir size persisted below the limits of detection, which was 0.012 infections units per million cells (IUPM) in the three patients (Table 1). At baseline, no patient showed the 2-LTR-DNA. All of the patients had wild-type CCR5 genotypes (Table 1), which was a requisite to be included in the initial study.
Cell-associated unspliced HIV RNA is enhanced during maraviroc intensification but reduced after treatment discontinuation
CA-US-RNA has been appointed as an indicator of viral transcription and ongoing viral production from latently infectedcells38. CA-US-RNA was measured in human peripheral blood mononuclear cells (PBMCs) isolated before intensification with mararivoc, after intensification for 12 and 48 weeks, and again at 12 weeks after maraviroc discontinuation. Based on this schedule, CA-US HIV RNA was measured before starting ATI. Unfortunately, the baseline of patient-3 could not be measured due to sample limitations. The expression of CA-US-RNA, expressed as copies per million PBMCs, showed a slight increase during maraviroc intensification for 12 weeks in patient-1 and patient-2 and was further increased throughout the follow-up in patient-2 and patient-3 (Fig. 1A). Relative data, shown as fold increase relative to baseline, are summarized for patient-1 and patient-2 (Fig. 1B); the fold increase relative to 12 weeks of intensification is also shown for the three patients (Fig. 1C). Briefly, the CA-US-RNA expression was enhanced 6.11-fold after 12 weeks in patient-1, but it reached levels lower than those shown at baseline at 48 weeks of maraviroc intensification. Although CA-US-RNA levels increased only 2.04-fold compared to the baseline in patient-2, it was further enhanced up to 4.30-fold after 48 weeks of drug treatment. In patient-3, CA-US-RNA expression levels at 48 weeks were 3.65-fold higher than at 12 weeks of maraviroc exposure. Considerably, there was a decrease in CA-US-RNA after maraviroc discontinuation in patient-1 and patient-2, as levels were 0.19- and 0.42-fold reduced, respectively, in comparison to baseline levels. In patient-3, levels were 0.35-fold reduced in comparison to levels at 12 weeks of intensification. Similarly, CA-US-RNA expression was reduced 0.03- and 0.21-fold in patient-1 and patient-2, respectively, when compared versus 12 weeks of drug exposure. These results confirmed previous data indicating that administration of maraviroc can increase HIV transcription from latency in HIV patients and therefore may potentially decrease the latent reservoir in vivo.
Figure 1.

Levels of cell associated unspliced HIV RNA in PBMCs isolated from HIV-infected patients on ART who were under intensification with maraviroc. (A) Copies per million peripheral blood mononuclear cells (PBMCs), (B) relative fold change with respect to baseline and (C) relative fold change with respect to 12 weeks of treatment intensification. The blue shaded box shows the time on maraviroc intensification. BL, baseline; 12w, 12 weeks on maraviroc; 48w, 48 weeks on maraviroc; 12d, 12 weeks after discontinuation of maraviroc. *Baseline sample in patient 3 was not available.
HIV rapidly rebounded in the maraviroc-treated patients included in the analytical treatment interruption
The three patients underwent ATI once that it was confirmed that maraviroc could disrupt HIV latency and that replicative-competent HIV was undetectable. Patients were closely monitored during the ATI, including a plasma HIV RNA test once a week to detect viral rebound as soon as possible. In the case of HIV rebound, it was scheduled to restart the previous ART immediately after confirming a viral load determination higher than 200 copies/mL (2.30 log10). The detection limit of the assay was 1.57log10 HIVRNA copies/mL. Viremia rebound was rapidly detected in all of the patients, at weeks 3, 2, and 5 after ART interruption for patient-1, patient-2 and patient-3, respectively (Fig. 2). Therefore, the median time of HIV rebound was 3 weeks (21 days). The highest values of viremia before re-initiation of the previous ART were 5.8, 6.3, and 4.7 log10 HIV RNA copies/mL at 4, 4, and 6 weeks in patient-1, patient-2 and patient-3, respectively (Fig. 2). CD4 T cell counts just before starting ATI before were 746, 981 and 552, respectively for patient-1, patient-2 and patient-3 (Fig. 2). CD4 T cell counts at the end of the study were 923, 710 and 459, respectively for patient-1, patient-2 and patient-3. Plasma levels of antiviral drugs were undetectable one week after drug discontinuation in all cases. After the confirmation of viral rebound, the three patients resumed previous ART with good response. No patient developed an acute retroviral syndrome. Genotyping of the rebounded HIV showed no resistance mutations and therefore starting the previous ART led to the subsequent control of plasma viremia within the following 4 weeks in all of the patients.
Figure 2.
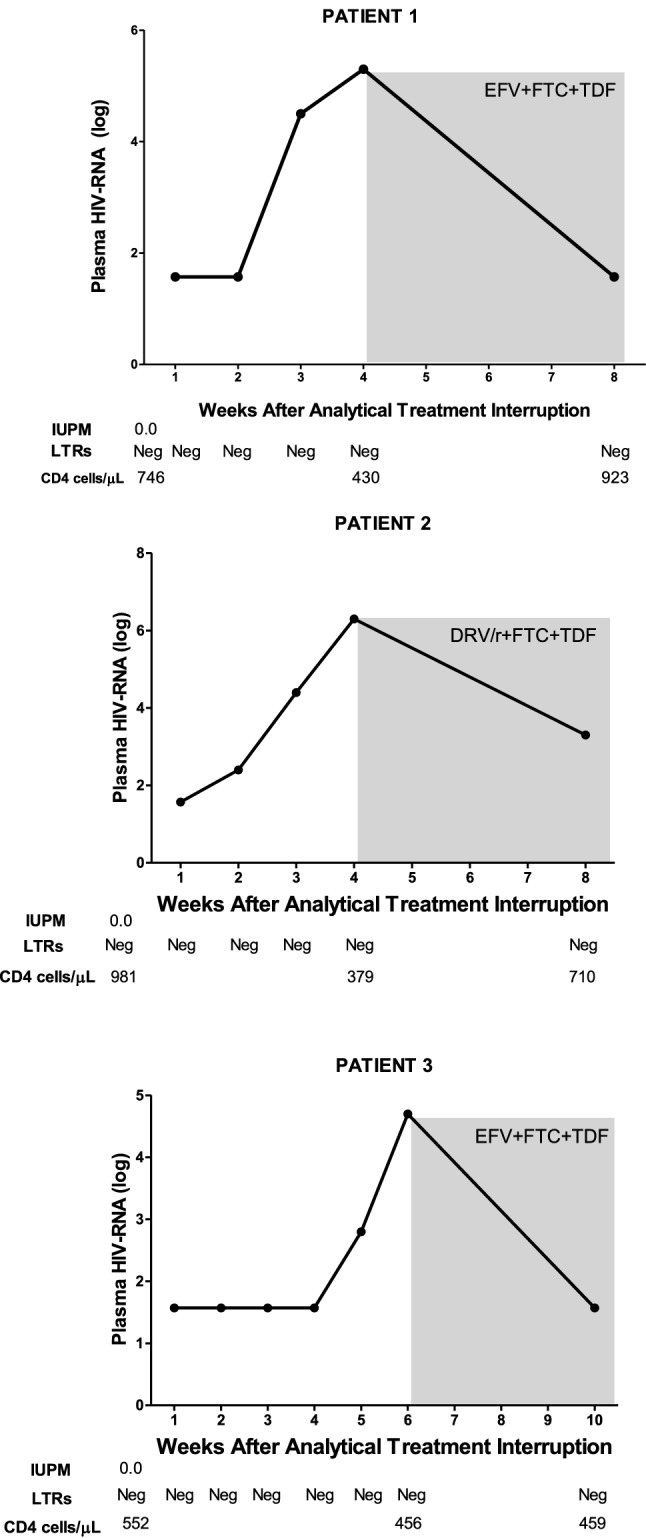
Virologic evolution of HIV rebounded in the maraviroc-treated patients who underwent into the analytical treatment interruption shown in terms of conventional viral load (log plasma HIV RNA), infections units per million cells (IUPM), episomal two LTR-DNA circles (2LTRs), and total CD4 T cell count (CD4 cells/µL). Measurements for each parameter were done at the indicated time. The grey shaded box shows the time on ART restart. EFV, efavirenz; FTC, emtricitabine; TDF, tenofovir; DRV/r, ritonavir-boosted darunavir.
T cell specific responses against some peptide pools of HIV was weak but increased at the end of the study in two out of three patients
After reversal of HIV latency, the cytopathic effects of the virus were not potent enough to kill infected cells11 and therefore the removal of HIV-expressing cells must have occurred via the action of an immune cytotoxic effector, such as CD8 T lymphocytes11,39. Therefore, the specific responses of CD8T lymphocytes against HIV was studied in these patients using an ELISpot assay that quantifies interferon-γ secretion against different peptide pools covering p17, p24, sp1, sp2, gag, pol, nef, and env sequences. The methodology used involved at least 85% of CD8 T cell responses40,41. However, all studies were conducted with unfractionated PBMCs and, therefore, responses are described as HIV-specific T cells. Measurements were performed at two time points: (1) before the intensification with maraviroc during the initial study and (2) the point at which ART was resumed after the ATI during the amended study. The three patients had very weak HIV-specific T cell responses before intensification with maraviroc (Fig. 3). Although low, these responses had increased at the end of the study in some peptide pools, such as sp2, nef and gag, mainly in patient-2 and patient-3 but not in patient-1 (Fig. 3). Briefly, specific responses against p17.1, sp2, nef, env, and gag were enhanced 23.13-, 58.50-, 73.75-, 38.50-, and 81.30-fold, respectively, after maraviroc exposure and ATI intervention in patient-2. Similarly, specific responses against p24.1, sp2, nef, pol, and gag were enhanced 56.67-, 29.33-, 214.33-, 31.00- and 86.00-fold, respectively, in patient-3.
Figure 3.

HIV specific T lymphocyte response. The ELISpot assay, that quantifies interferon-γ secretion against different peptide pools covering p17, p24, sp1, sp2, gag, pol, nef and env sequences, was used. Samples were taken before the intensification with maraviroc during the initial study (grey bars) and when antiretroviral treatment was resumed after the ATI during the amended study (black bars). Data are are shown as the number of cells secreting interferon-γ against the indicated peptide per 106 peripheral blood mononuclear cells (PBMCs). The dotted grey line indicates the threshold for positivity (50 SFC per million PBMCs). SFC, spot forming cells.
HIV clones found in PBMCs were not present in plasma at the time of viral rebound
The diversity of the latent pro-viruses contributing to HIV rebound after ATI was evaluated through a phylogenetic analysis based on deep sequencing of the protease region. The analysis was done on cell-associated HIV DNA from frozen PBMCs obtained from patients while they were on ART during maraviroc intensification as well as on plasma HIVRNA obtained at two time points: (1) just after HIV rebound and (2) a week later when viral rebound was confirmed. Lower diversity was observed (average pairwise distance [APD]) in HIV genomes sequenced from plasma than from cells in patient-1 and patient-2 but not in patient-3 (Fig. 4). Many HIV clones found in PBMCs were not present in plasma at the time of viral rebound; this observation was especially significant for patient-1 and patient-2.
Figure 4.

Phylogenetic analysis of the latent pro-viruses contributing to HIV rebound after analytical treatment interruption. (A) Patient 1; (B) Patient 2; (C) Patient 3. HIV protease sequences were obtained from DNA isolated from peripheral blood mononuclear cells obtained while patients were on antiretroviral treatment and maraviroc intensification during the initial study (green) and from plasma RNA after viral rebound at analytical treatment interruption during the amended study (red). Open squares represent the HIV reference sequence HXB2. Histograms represent the average pairwise distance (APD) between HIV sequences on cellular and plasma samples.
Discussion
HIV infection remains currently incurable due to viral persistence in latent reservoirs and therefore, patients require lifelong antiretroviral treatment. Clinical trials are the only experimental approach for investigating whether drugs aimed at disrupting HIV latency in vivo are effective or not18. The results shown here confirm that therapeutic doses of maraviroc can disrupt the quiescence of latent HIV proviruses within rCD4 T cells, although the sample size of our study was quite limited. It quite unlikely that results are stochastic because the increase of HIV transcription after maraviroc administration in patients has already been described in previous clinical trials conducted by our group and others32,33. Traditionally, the HIV reservoir mainly consists of several populations of memory rCD4 T cells, such as stem central, transitional memory and effector memory cells5,6. However, recent studies have suggested that viral latency is not restricted to non-activated and quiescent CD4 T cells and may also be established in activated and proliferating cells42, suggesting that the number and type of CD4 T cells capable of supporting viral latency is larger than initially assumed. Unfortunately, this hypothesis arose after our study and could not be tested in our patients.
Multiple rounds of treatment with LRAs have been suggesting as a strategy to cumulatively reduce the reservoir size22. However, most studies have included only single-dose administration of a drug or, at most, multiple doses over short periods16–19. Prolonged administration of maraviroc for 48 weeks increased HIV transcription as measured via CA-US HIV-1 RNA levels, confirming data previously observed by our group and others32,33,43. This increase was within the range of that observed in studies with some histone deacetylase inhibitors (HDACi), such as vorinostat or panobinostat16,18,19. Moreover, maraviroc-mediated HIV reactivation persisted during the administration of the drug, increasing in two out of three patients until week 48 of administration and then decreased when the drug was discontinued. These findings may be important for the design and evaluation of maraviroc as an LRA in clinical trials as each drug may display a different pattern. For example, reactivated HIV persisted after 8 weeks of panobinostat treatment although intermittent administration of the drug was scheduled in this clinical trial18. However, the decrease in the reservoir size could not be demonstrated, similar to what has been shown with most tested LRAs17–20,44,45. Consequently, at present, it is assumed that a combination of LRAs that exploit different cellular pathways is required to induce the complete expression of replication-competent proviral genomes and therefore, to effectively reduce the size of the HIV reservoir in vivo13,46,47. The well-known tolerability and safety of maraviroc make it a good candidate for safe and active combinations of LRAs aimed at the cure of HIV infection.
Furthermore, the so-called “shock and kill” strategy10 aimed at curing HIV includes the administration of any LRA in combination with an additional action that enhances CD8 T lymphocytes so that HIV-expressing cells are removed. It is believed that CD8 T cell exhaustion prevents the control of the infection in HIV-infected patients48–51. Therefore, it is essential to describe the effects of LRAs on CD8 T lymphocyte functions so that their therapeutic potential can be determined52,53. In our study, the HIV-specific T cell responses, assumed to be mainly from CD8 T lymphocytes40,41, slightly increased in two out three patients after maraviroc intensification and ATI intervention. This may not directly be due to maraviroc but to active viral replication. Because the median time between maraviroc discontinuation and ART interruption was 2.5 years we cannot reject HIV-specific immunity had already been vanished. However, it is also plausible that the effects of maraviroc on CD8 T cells may persist during long periods through the persistence of long-lasting memory cells that acquire multiple unique functional features which make them able not only to respond to, but also to actively provide different signals ultimately culminating in host protection54. A remarkable issue is that HIV-specific T cell responses are not reduced after maraviroc intensification, in agreement with data from in vitro assays that we have recently published55. This confers maraviroc an important advantage over other well-known LRAs as panobinostat or disulfiram as both of them dramatically reduces in vitro the cellular viability of CD8 T cells isolated from both healthy donors52,56 and HIV-infected patients on ART55, even after a short-term stimulation. As a result, the specific antiviral function of CD8 T cells is inhibited by some histone deacetylase inhibitors, including panobinostat57, which is a profound limitation for the implementation of in vivo trials.
Rapid plasma viral rebound occurs upon ART interruption in the vast majority of infected individuals58. Plasma viremia levels remain the only clinically relevant virologic marker that is currently available and therefore, the efficacy of any therapeutic strategies designed to achieve ART-free virologic remission can only be adequately assessed with ATI with extensive monitoring of virologic and immunological parameters23,59. Recent studies have demonstrated that short-term ATI followed by immediate re-initiation of ART after HIV rebounded did not lead to permanent expansion of the HIV reservoir or irreversible damage to the immune system of infected individuals60–64 and therefore, it is justified and safe for clinical trials of potentially curative interventions to include ATI59. In this study, QVOA could not be performed to quantify HIV replication due to sample limitations. Therefore, including ATI in this clinical trial was required to evaluate the efficacy of maraviroc as an LRA in vivo. In order to do that, a previous open-label phase II clinical trial of ART intensification with maraviroc for 48 weeks was amended to include ATI and to use the remaining samples before ATI, as it has been done in other studies62.
During ATI, reactivation of a single latently infected cell can lead to HIV rebound. Control of viremia of the rebounded viruses depends on both the strength of cytotoxic T lymphocytes and the size of the HIV reservoir65. Furthermore, the total inducible reservoir is significantly smaller in women than in men66. Virologic rebound was detected in the three patients evaluated, all of the men, soon after ART cessation, and the time to rebound was similar to previous studies in cohorts of HIV infected individuals59,61,64 or, for instance, in one study using panobinostat as an LRA18. In our study, this observation may be explained from at least three different perspectives: Firstly, it is very likely that maraviroc as an LRA is not potent enough and higher doses or combinations of drugs should be used as discussed above. Secondly, there could be lack of capability of targeting the pool of latently infected cells67, and accordingly, maraviroc only could reactivate a minority of latent pro-viruses. Supporting this argument, we have found a low diversity of viral clones in plasma after the rebound in two out of three patients analysed. Patients included in the primary study were infected with wild-type CCR5 genotypes according to the fact that most of the founder viruses establishing the latent reservoir during early infection are CCR5-tropic26,68 and persisted when the amended study started. However, HIV clones found in PBMCs were not present in plasma at the time of rebound, especially in two patients, suggesting that rebounded viruses do not come from circulating blood but from lymphoid nodes or gut-associated lymphoid tissue (GALT)69, that are regarded as the main anatomical HIV reservoir in ART-treated patients due to low ART concentrations in these areas70,71. Thirdly, HIV-specific immune responses, required for the clearance of the latent reservoir11,39, seems not to be potent enough as ELISpot results showed only an slight increase in these responses. However, the assay used involved PBMCs instead of purified CD8 T cells due to sample limitations and therefore cytotoxic responses may come also from other cell types as, for instance, Natural Killer cells. Anyhow, combining maraviroc with a treatment-enhancing cytotoxic functions seems necessary in order to efficiently reduce the reservoir size after reactivation.
The results presented in this study show that maraviroc reverses HIV latency in vivo and confirm that it may be considered a new LRA. Although the effect persists during the administration of maraviroc, prolonged administration of the drug fails to impact the time to viremia rebound after ART interruption, which can be due to either insufficient potency of the drug, the lack of a strong HIV specific response, or both. Furthermore, time to rebound after ATI was similar to that observed in ART-treated patients who have not received any therapeutic intervention, as discussed above59,61,64, showing that long-term intensification with maraviroc did not significantly reduce the HIV reservoir size. Because changes in HIV transcription were also modest, it seems that the reservoir size was balanced from maraviroc intensification to ATI, probably through homeostatic proliferation. Considering the results shown is this work, ATI should take place earlier in future studies. Higher cell-associated unspliced HIV RNA (CA-US-RNA) levels correlated with faster time to rebound72. Therefore, ATI cannot be implemented just after maraviroc intensification when CA-US-RNA are the highest due to viral reactivation but at least 12 weeks after the intervention when CA-US-RNA were similar to levels at baseline.
The effect of maraviroc on initiating HIV transcription from latent reservoirs was similar after administrating the drug for 10 days32,33 or for 48 weeks and longer administration did not result in HIV reservoir disruption. Therefore, it seems that combining maraviroc with other LRA or with other experimental strategy will be more suitable than extending maraviroc administration longer in order to disrupt latency. So far, in vitro studies looking for combination of maraviroc with other LRAs has not been successful31,55.
A major limitation of our study is that it only included three participants. Therefore, clinical trials including a higher number of patients treated with maraviroc fare needed to confirm the results shown here. The strength of the results would have been also increased using a control group or samples from the patients included in the initial study who did not agree to participate in the amended one, but it could not be done due to ethics concerns. Further characterizing the reservoir size and the proportion that is able to induce new infections in maraviroc treated patients as well as determining the changes induced in the genomic profile and the functional effects of these changes in immune system may be also relevant but represent themselves new clinical trials that should be performed in the future.
Maraviroc is a CCR5 inhibitor approved for clinical use as an antiretroviral drug and therefore, a new use for this drug can be rapidly authorized. An additional advantage of maraviroc is that specific T cell responses are not reduced after long-term exposure contrary to other common LRAs. Additionally, further clinical trials combining maraviroc with other LRAs and/or with strategies to enhanced cytotoxic responses are warranted.
Materials and methods
Plasma HIV RNA measurement
The plasma viral load was quantified using the VERSANT HIV RNA kPCR system (Siemens Healthcare Diagnostics Inc., Tarrytown, NY) with an assay limit of quantization of 37 copies HIV RNA/mL, equivalent to 1.57 log10 HIV RNA copies/mL.
Quantification of CD4 T cells harbouring replication-competent HIV
The detection of CD4 T cells harbouring latent and replication-competent HIV was determined via a QVOA as previously described3,37, with some minor modifications27,36. This assay involves an enhanced culture of highly enriched rCD4 T cells and provides a precise minimal estimation of the HIV reservoir in rCD4 T cells, which constitutes the principal viral reservoir5,6. The QVOAis widely accepted and considered as the gold-standard formeasuring the frequency of productively- and latently-infected cells in clinical settings73. rCD4 T cells were isolated from total PBMCs via negative selection of CD3+/CD4+/HLA-DR−/CD25− T cells using magnetic beads according to manufacturer’s recommendation (MiltenyeBiotec, Bergisch Gladbach, Germany). Cells were then placed in a duplicate five-fold serial dilution cultures, ranging from 25 × 106 to 320 cells. Irradiated PBMCs from healthy donors were added at ten-fold to each culture with phytohaematoglutinin (PHA, 1 mg/mL) (Sigma-Aldrich, St. Louis, MO, USA) and recombinant interleukin 2 (IL-2, 100 U/mL) (Sigma-Aldrich) in order to achieve efficient cell activation. On days 15 and 21, culture supernatants were tested for HIV replication trough the presence of viral antigen using the HIV p24 antigen assay kit (Innogenetics, Barcelona, Spain). The frequency of infected cells was determined using the maximum likelihood method and was expressed as infectious units per million (IUPM) of rCD4 T cells with a limit of detection of 0.012 IUPM.
Quantification of cell-associated unspliced HIV RNA
A semi-nested quantitative PCR was performed to quantify CA-US-RNA as previously described74. Briefly, total RNA was extracted from stored PBMCs using RNeasy Mini Kit Qiagen (Hilden, Germany), and the eluted cellular RNA was directly subject to two rounds of PCR amplification. The primer pair used in the first PCR consisted of MH535 (5′-AACTAGGGAACCCACTGCTTAAG-3′) and SL20 (5′-TCTCCTTCTAGCCTCCGCTAGTC-3′) and amplified a region within the HIV gag gene. The first PCR round was performed using these conditions: (1) 95 °C for 10 min; (2) 15 cycles of 94 °C for 20 s; (3) 55 °C for 40 s; and (4) 72 °C for 40 s. The product of the first PCR was subsequently used as a template for the second semi-nested PCR amplification, which was performed on a real-time LightCycler 480 machine (Roche Life Sciences, Penzberg, Germany) using SYBR Green detection. The primer pair was SL20 and SL19 (5′-TCTCTAGCAGTGGCGCCCGAACA-3′). Triplicates were conducted for each tested experimental condition. The second PCR settings were as follows: (1)95 °C for 10 min; (2)45 cycles of 94 °C for 20 s; and (3) 55 °C for 40 s. Synthetic runoff RNA transcripts, corresponding to the HIV gag region, were used as external standards. These RNA standards were kindly gifted by Professor Sharon R. Lewin (Doherty Institute, Melbourne, Australia). Serial dilutions of standards between 1 and 4, 4 × 1011 input copies were made. The amplicon sizes were 286 bp for the first PCR and 160 bp for the second one. HIV RNA copy numbers were standardised to cellular equivalents using RNA concentration (assuming that 1 ng RNA corresponds to 1000 cells75, which has been shown to correlate with levels of glyceraldehyde phosphate dehydrogenase [GAPDH] RNA76). PCR results are expressed in CA-US-RNA copies per million PBMCs.
Detection of episomal two LTR DNA circles
The existence of 2-LTR-DNA was checked via nested PCR as described previously27. DNA was extracted from 5 million PBMCs using QIAprep Spin Miniprep following manufacturer’s protocol for low copy number plasmids in order to maximize the recovery of 2-LTR-DNA77. A nested PCR flanking the junction of the 2-LTR-DNAwas designed. In the first round, episomal DNA was amplified using the following pair of primers: forward, 5′-TAAGATGGGTGGCAAGTGGTCA-3′ and reverse, 5′-TCTACTTGTCCATGCATGGCTT-3′. The second PCR round used a set primers spanning the unique junction formed by ligation of 5′ and 3′ LTR sequences: forward, 5´-AATCTCTAGCAGTACTGGAAG-3′ and reverse, 5′-GCGCTTCAGCAAGCCGAGTCCT-3′. PCR products were analyzed on a 1% agarose gel stained with GelRed (Biotium, Hayward, California, USA).
Measurement of antiretroviral drug levels in plasma
Five milliliters of total blood were collected without anticoagulant for pharmacokinetics assays before starting the ATI and weekly thereafter. Blood samples were centrifuged and plasma was stored at − 80 °C until posterior analysis. Concentrations of the antiretroviral drugs included in the treatment regimens were measured by high-performance liquid chromatography (HPLC).
ELISpot assay
ELISpot assay was performed to measure the numbers of PBMCs producing interferon (IFN)-γ as a response against HIV sequences78. Briefly, experiments were performed with cryopreserved PBMCs using 10 pools of peptides, each consisting of 15-mers overlapping 11 peptides, grouped in pools of 10 to 12 peptides each, and covering the whole HIV gag, nef, pol, and env-gp41 sequences. Negative control responses were obtained with non-stimulated PBMCs. Positive controls were PBMCs stimulated with PHA and a pool containing 32 human leukocyte antigen (HLA) class I peptides from cytomegalovirus (CMV), Epstein-Barr virus (EBV), and influenza virus (CEF pool obtained from the National Institutes of Health (NIH) AIDS Research and Reference Research Program). The assay was done in 96-well polyvinyl difluoride (PVDF) microtiter plates coated overnight with a mAb specific for human IFN-γ (mAb B-B1, Diaclone, BioNova Cientifica, Spain). PBMCs resuspended in RPMI medium plus 10% fetal bovine serum were plated in the presence of different peptide pools (2 μg/mL, final concentration) and incubated overnight at 37 °C, 5% CO2. Plates were developed using biotinylated anti-human IFN-γ streptavidin conjugated to alkaline phosphatase (Amersham Biosciences, UK) and chromogenic substrate BCIP/NBT (Sigma-Aldrich). Spot-forming cells (SFC) were counted using an AID ELISPOT reader (Autoimmun Diagnostic GmHb, Germany). Results were expressed as the number of SFC per million of PBMCs after subtracting the background. The positivity threshold for each peptide pool or antigen was defined at 50 SFC per million PBMCs as previously reported40,41. By using this methodology, at least 85% of the responding cells are CD8 T cells, but as all studies were conducted with unfractionated PBMCs, responses are described as HIV-specific T cells.
Genome analysis of protease gene
A fragment corresponding to the HIV protease gene was obtained by PCR or RT-PCR from DNA isolated from PBMCs or from RNA isolated from plasma using commercial assays (Qiagen) and then deep-sequenced in a GS-Junior instrument using 454 sequencing technology (Roche Life Sciences). On average, 200 sequences by sample were analyzed25. Phylogenetic trees (Neiborgh-joining) and average pairwise distances (APD) were estimated with Mega v6.2 software (https://www.megasoftware.net/).
Ethical statement
Appropriate informed consent was obtained from each subject in accordance with the Spanish legislation in compliance with the confidentiality and privacy rules. The procedures of this study were approved by the Institute Ramón y Cajal for Health Research (IRYCIS) Ethics Committee and by the Spanish Agency for Medications and Health Products (AEMPS) and were carried out in compliance with the Helsinki Declaration.
Acknowledgements
The authors thank Ms. Ester Dominguez and Ms. María Coronel for technical assistance. We thank Dr. Sharon Lewin and Dr Ajantha Solomon for their assistance with the establishment of the cell-associated unspliced RNA measurement. This work was supported by: RD12/0017/0017 project as part of the Plan Estatal I+D+I that is co-financed by Instituto de Salud Carlos III – Subdirección General de Evaluación y Fomento de la Investigación – Fondo Europeo de Desarrollo Regional (FEDER) (European Regional Development Fund); Instituto de Salud Carlos III Grants (FIS-PI080958, CP08/00046, PI12/00969 and FIS PI1400708); HIV Vaccine Research and Development Programme in Catalonia (M.P); Foundation of Investigation and prevention of AIDS (FIPSE-36-0844/09). C.G. and N.M.E. are supported by SPANISH AIDS Research Network (RIS) (RD12/0017/0017; RD16/0025/0001). M.P. is a researcher from the Institutd’ Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) and is supported by the Instituto de Salud Carlos III and the Health Department of the Catalan Government (Generalitat de Catalunya). R.D. is supported by Instituto de Salud Carlos III (FIS PI 1801007), European Commission Horizon 2020 Framework Programme (Project VIRUSCAN FETPROACT-2016: 731868) and Fundación Caixa-Health Research (Project StopEbola).
Author contributions
C.G., N.M., B.H.N. and S.M. designed experiments; O.S., R.D., M.P. and M.A.M.F. performed experiments; R.R. and S.M. managed study participant recruitment and follow-up; M.R.L.H., C.G., N.M., R.D., M.P., M.A.M.F. and S.M. analysed the data; C.G., N.M. and S.M. wrote the first version of the manuscript. M.R.L.H. drafted the manuscript and wrote its final version. All the authors have approved the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: María Rosa López-Huertas and Carolina Gutiérrez.
References
- 1.UNAIDS . Fact sheet—Global AIDS update 2019. Geneva: UNAIDS; 2019. [Google Scholar]
- 2.Trickey A, et al. Survival of HIV-positive patients starting antiretroviral therapy between 1996 and 2013: A collaborative analysis of cohort studies. Lancet HIV. 2017;4:e349–e356. doi: 10.1016/S2352-3018(17)30066-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 4.Coiras M, López-Huertas MR, Pérez-Olmeda M, Alcamí J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 2009;7:798–812. doi: 10.1038/nrmicro2223. [DOI] [PubMed] [Google Scholar]
- 5.Buzon MJ, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat. Med. 2014;20:139–142. doi: 10.1038/nm.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crooks AM, et al. Precise quantitation of the latent HIV-1 reservoir: Implications for eradication strategies. J. Infect. Dis. 2015;212:1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henrich TJ, et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 2014;161:319–327. doi: 10.7326/M14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richman DD, et al. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 10.Deeks SG. HIV: Shock and kill. Nature. 2012;487:439–440. doi: 10.1038/487439a. [DOI] [PubMed] [Google Scholar]
- 11.Shan L, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henrich TJ, et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 2017;14:e1002417. doi: 10.1371/journal.pmed.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: An update. Retrovirology. 2013;10:67. doi: 10.1186/1742-4690-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darcis G, et al. Reactivation capacity by latency-reversing agents ex vivo correlates with the size of the HIV-1 reservoir. AIDS. 2017;31:181–189. doi: 10.1097/QAD.0000000000001290. [DOI] [PubMed] [Google Scholar]
- 15.Laird GM, et al. Ex vivo analysis identifies effective HIV-1 latency—Reversing drug combinations. J. Clin. Invest. 2015;125:1901–1912. doi: 10.1172/JCI80142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Archin NM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012 doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Archin NM, et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 2014;210:728–735. doi: 10.1093/infdis/jiu155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen TA, et al. Panobinostat, a histone deacetylase inhibitor, for latent virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV. 2014;1:e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 19.Elliott JH, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014;10:e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott JH, et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV. 2015 doi: 10.1016/S2352-3018(15)00226-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petravic J, Rasmussen TA, Lewin SR, Kent SJ, Davenport MP. Relationship between measures of HIV reactivation and decline of the latent reservoir under latency-reversing agents. J. Virol. 2017 doi: 10.1128/jvi.02092-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davenport MP, et al. Functional cure of HIV: The scale of the challenge. Nat. Rev. Immunol. 2019 doi: 10.1038/s41577-018-0085-4. [DOI] [PubMed] [Google Scholar]
- 23.Wen Y, Bar KJ, Li JZ. Lessons learned from HIV antiretroviral treatment interruption trials. Curr. Opin. HIV AIDS. 2018;13:416–421. doi: 10.1097/COH.0000000000000484. [DOI] [PubMed] [Google Scholar]
- 24.Kutzler MA, Jacobson JM. Treatment interruption as a tool to measure changes in immunologic response to HIV-1. Curr. Opin. HIV AIDS. 2008;3:131–135. doi: 10.1097/COH.0b013e3282f54cde. [DOI] [PubMed] [Google Scholar]
- 25.Woollard SM, Kanmogne GD. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 2015;9:5447–5468. doi: 10.2147/DDDT.S90580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierson T, et al. Characterization of chemokine receptor utilization of viruses in the latent reservoir for human immunodeficiency virus type 1. J. Virol. 2000;74:7824–7833. doi: 10.1128/JVI.74.17.7824-7833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutiérrez C, et al. Intensification of antiretroviral therapy with a CCR5 antagonist in patients with chronic HIV-1 infection: Effect on T cells latently infected. PLoS ONE. 2011;6:e27864. doi: 10.1371/journal.pone.0027864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lafeuillade A, et al. Failure of combined antiretroviral therapy intensification with maraviroc and raltegravir in chronically HIV-1 infected patients to reduce the viral reservoir: The IntensHIV randomized trial. AIDS Res. Ther. 2014;11:33. doi: 10.1186/1742-6405-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostrowski M, et al. Intensifying antiretroviral therapy with raltegravir and maraviroc during early human immunodeficiency virus (HIV) infection does not accelerate HIV reservoir reduction. Open Forum Infect. Dis. 2015;2:ofv138. doi: 10.1093/ofid/ofv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutiérrez C, et al. Dynamics of the HIV-1 latent reservoir after discontinuation of the intensification of antiretroviral treatment: Results of two clinical trials. AIDS. 2013;27:2081–2088. doi: 10.1097/QAD.0b013e328361d0e1. [DOI] [PubMed] [Google Scholar]
- 31.López-Huertas MR, et al. The CCR5-antagonist maraviroc reverses HIV-1 latency in vitro alone or in combination with the PKC-agonist bryostatin-1. Sci. Rep. 2017;7:2385. doi: 10.1038/s41598-017-02634-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madrid-Elena N, et al. Maraviroc is associated with latent HIV-1 reactivation through NF-κB activation in resting CD4+ T cells from HIV-infected individuals on suppressive antiretroviral therapy. J. Virol. 2018;92:e01931–e2017. doi: 10.1128/JVI.01931-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Symons, J. et al. Maraviroc induces HIV production in RCT and in vitro, potentially via the NFkB pathway. In Conference on Retroviruses and Opportunistic Infections (CROI) Abstract Number: 549 (2015).
- 34.Dorr P, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Perez J, et al. New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J. Biol. Chem. 2011;286:4978–4990. doi: 10.1074/jbc.M110.168955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vallejo A, et al. The effect of intensification with raltegravir on the HIV-1 reservoir of latently infected memory CD4 T cells in suppressed patients. AIDS. 2012;26:1885–1894. doi: 10.1097/QAD.0b013e3283584521. [DOI] [PubMed] [Google Scholar]
- 37.Siliciano JD, Siliciano RF. Enhanced culture assay for detection and quantitation of latently infected, resting virus in HIV-1-infected individuals. Methods Mol. Biol. 2005;304:3–15. doi: 10.1385/1-59259-907-9:003. [DOI] [PubMed] [Google Scholar]
- 38.Lewin SR, Rouzioux C. HIV cure and eradication: How will we get from the laboratory to effective clinical trials? AIDS. 2011;25:885–897. doi: 10.1097/QAD.0b013e3283467041. [DOI] [PubMed] [Google Scholar]
- 39.Deng K, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517:381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinloch-de Loes S, et al. Impact of therapeutic immunization on HIV-1 viremia after discontinuation of antiretroviral therapy initiated during acute infection. J. Infect. Dis. 2005 doi: 10.1086/432002. [DOI] [PubMed] [Google Scholar]
- 41.Martinez V, et al. Combination of HIV-1-specific CD4 Th1 cell responses and IgG2 antibodies is the best predictor for persistence of long-term nonprogression. J. Infect. Dis. 2005 doi: 10.1086/430320. [DOI] [PubMed] [Google Scholar]
- 42.Hosmane NN, et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017;214:959–972. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Symons, J. et al. The antiretroviral CCR5-inhibitor maraviroc effectively reverses HIV latency by phosphorylation of Nf-κB. In 22nd International Aids Conference 346 (2018).
- 44.Barton K, et al. Broad activation of latent HIV-1 in vivo. Nat. Commun. 2016;7:12731. doi: 10.1038/ncomms12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Søgaard OS, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 2015;11:e1005142. doi: 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Margolis DM, Hazuda DJ. Combined approaches for HIV cure. Curr. Opin. HIV AIDS. 2013;8:230–235. doi: 10.1097/COH.0b013e32835ef089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu C, Ma X, Liu B, Chen C, Zhang H. HIV-1 functional cure: Will the dream come true? BMC Med. 2015;13:284. doi: 10.1186/s12916-015-0517-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuchroo VK, Anderson AC, Petrovas C. Coinhibitory receptors and CD8 T cell exhaustion in chronic infections. Curr. Opin. HIV AIDS. 2014;9:439–445. doi: 10.1097/COH.0000000000000088. [DOI] [PubMed] [Google Scholar]
- 49.Shankar P, et al. Impaired function of circulating HIV-specific CD8(+) T cells in chronic human immunodeficiency virus infection. Blood. 2000;96:3094–3101. doi: 10.1182/blood.V96.9.3094. [DOI] [PubMed] [Google Scholar]
- 50.Trautmann L, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto T, et al. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood. 2011;117:4805–4815. doi: 10.1182/blood-2010-11-317297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao M, et al. T cell toxicity of HIV latency reversing agents. Pharmacol. Res. 2018 doi: 10.1016/j.phrs.2018.10.023. [DOI] [PubMed] [Google Scholar]
- 53.Walker-Sperling VE, Pohlmeyer CW, Tarwater PM, Blankson JN. The effect of latency reversal agents on primary CD8+ T cells: Implications for shock and kill strategies for human immunodeficiency virus eradication. EBioMedicine. 2016;8:217–229. doi: 10.1016/j.ebiom.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lauvau G, Soudja SM. Mechanisms of memory T cell activation and effective immunity. Adv. Exp. Med. Biol. 2015 doi: 10.1007/978-3-319-15774-0_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.López-Huertas MR, et al. Maraviroc reactivates HIV with potency similar to that of other latency reversing drugs without inducing toxicity in CD8 T cells. Biochem. Pharmacol. 2020 doi: 10.1016/j.bcp.2020.114231. [DOI] [PubMed] [Google Scholar]
- 56.Garrido C, et al. HIV latency-reversing agents have diverse effects on natural killer cell function. Front. Immunol. 2016;7:356. doi: 10.3389/fimmu.2016.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones RB, et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic t-lymphocytes. PLoS Pathog. 2014;10:e1004287. doi: 10.1371/journal.ppat.1004287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chun TW, Moir S, Fauci AS. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat. Immunol. 2015 doi: 10.1038/ni.3152. [DOI] [PubMed] [Google Scholar]
- 59.Huiting ED, et al. Impact of treatment interruption on HIV reservoirs and lymphocyte subsets in individuals who initiated antiretroviral therapy during the early phase of infection. J. Infect. Dis. 2019 doi: 10.1093/infdis/jiz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clarridge KE, et al. Effect of analytical treatment interruption and reinitiation of antiretroviral therapy on HIV reservoirs and immunologic parameters in infected individuals. PLoS Pathog. 2018 doi: 10.1371/journal.ppat.1006792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Papasavvas E, et al. Analytical antiretroviral therapy interruption does not irreversibly change preinterruption levels of cellular HIV. AIDS. 2018 doi: 10.1097/QAD.0000000000001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salantes DB, et al. HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J. Clin. Invest. 2018;128:3102–3115. doi: 10.1172/JCI120194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strongin Z, et al. Effect of short-term antiretroviral therapy interruption on levels of integrated HIV DNA. J. Virol. 2018 doi: 10.1128/jvi.00285-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Colby DJ, et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection brief-communication. Nat. Med. 2018 doi: 10.1038/s41591-018-0026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conway JM, Perelson AS. Post-treatment control of HIV infection. Proc. Natl. Acad. Sci. 2015 doi: 10.1073/pnas.1419162112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das B, et al. Estrogen receptor-1 is a key regulator of HIV-1 latency that imparts gender-specific restrictions on the latent reservoir. Proc. Natl. Acad. Sci. USA. 2018 doi: 10.1073/pnas.1803468115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill AL, Rosenbloom DIS, Fu F, Nowak MA, Siliciano RF. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc. Natl. Acad. Sci. 2014;111:13475–13480. doi: 10.1073/pnas.1406663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keele BF, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rothenberger MK, et al. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc. Natl. Acad. Sci. 2015 doi: 10.1073/pnas.1414926112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fletcher CV, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. 2014;111:2307–2312. doi: 10.1073/pnas.1318249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lorenzo-Redondo R, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature. 2016;530:51–56. doi: 10.1038/nature16933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li JZ, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS. 2016 doi: 10.1097/QAD.0000000000000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hodel F, Patxot M, Snäkä T, Ciuffi A. HIV-1 latent reservoir: Size matters. Future Virol. 2016;11:785–794. doi: 10.2217/fvl-2016-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lewin SR, et al. Use of real-time PCR and molecular beacons to detect virus replication in human immunodeficiency virus type 1-infected individuals on prolonged effective antiretroviral therapy. J. Virol. 1999;73:6099–6103. doi: 10.1128/JVI.73.7.6099-6103.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fischer M, et al. Highly sensitive methods for quantitation of human immunodeficiency virus type 1 RNA from plasma, cells, and tissues. J. Clin. Microbiol. 1999;37:1260–1264. doi: 10.1128/JCM.37.5.1260-1264.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yukl SA, et al. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J. Infect. Dis. 2010 doi: 10.1086/656722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharkey ME, et al. Persistence of episomal HIV-1 infection intermediates in patients on highly active anti-retroviral therapy. Nat. Med. 2000 doi: 10.1038/71569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guardo AC, et al. Use of RT-defective HIV virions: New tool to evaluate specific response in chronic asymptomatic HIV-infected individuals. PLoS ONE. 2013 doi: 10.1371/journal.pone.0058927. [DOI] [PMC free article] [PubMed] [Google Scholar]