

Graphical abstract

Keywords: SARS-CoV-2, Coronavirus (CoV), Orf3a, M protein, Protein superfamily, Structure prediction, Virus evolution
Abstract
The genome of SARS-CoV-2, the coronavirus responsible for the Covid-19 pandemic, encodes a number of accessory genes. The longest accessory gene, Orf3a, plays important roles in the virus lifecycle indicated by experimental findings, known polymorphisms, its evolutionary trajectory and a distinct three-dimensional fold. Here we show that supervised, sensitive database searches with Orf3a detect weak, yet significant and highly specific similarities to the M proteins of coronaviruses. The similarity profiles can be used to derive low-resolution three-dimensional models for M proteins based on Orf3a as a structural template. The models also explain the emergence of Orf3a from M proteins and suggest a recent origin across the coronavirus lineage, enunciated by its restricted phylogenetic distribution. This study provides evidence for the common origin of M and Orf3a families and proposes for the first time a working model for the structure of the universally distributed M proteins in coronaviruses, consistent with the properties of both protein families.
1. Introduction
A new coronavirus of zoonotic origin found in China and able to transmit from human to human [1], was shown to be most similar to virus strains from the bat species Rhinolophus affinis, following the sequencing of its genome [2], [3]. The strain, named SARS-CoV-2 [4], is the causal agent for the Covid-19 pandemic and related to coronavirus strains responsible for the SARS and MERS epidemics [5]. The SARS-CoV-2 genome encodes ten well-defined genes [6], five of which have known roles (Orf1a/b, S-spike, E-envelope, M-membrane, N-nucleoprotein) and five ‘accessory’ genes (Orf3a, Orf6, Orf7a/-b, Orf8, Orf10), with less well-understood functions [7]. Other animal groups such as pangolins [8] have also been considered as reservoir species, based on irregularities or recombination events [6], [9] for parts of certain genes and the genome.
For a number of accessory genes, the three-dimensional structure but not an exact molecular function is known: these include Orf3a [10] and Orf7a [11]. For some genes of known roles, the reverse holds true, e.g. for the membrane protein M (herein called M protein), there is no known structure [12]. It is remarkable that Orf3a is a variable (and the longest) accessory gene in the SARS lineage, found to exhibit polymorphisms, along with Orf8 (Orf8a/Orf8b in SARS-CoV-1) [5], even within a single bat colony [13]. Genes Orf3a and Orf8 are considered to drive the evolution of SARS-CoV-2 during the 2020 pandemic under positive selection [14], with the well-known mutations Q57H and G251V for Orf3a and L84S for Orf8 [15].
Within a wider effort to characterize the unknown relationships of accessory proteins in SARS-CoV-2, we have examined the evolutionary path of Orf3a in systematic sequence database searches and its blueprint in functional genomics resources. Orf3a is present in SARS-CoV-1 and two additional CoV ‘subgenera’, where it is also known as Orf3, X1 or U274 [7]. Orf3a has been presumed to contain an N-terminal ectodomain (positions 1–34), three transmembrane regions (35–125), three motifs – a Cys-rich region (127–133), YxxΦ (Φ: a bulky hydrophobic residue YNSV, 160–163), a diacidic peptide EGD (171–173, in SARS-CoV-1, not conserved in SARS-CoV-2) – and a C-terminal ectodomain (209–264) [7]. The N- and C-terminal domains are now confirmed by the recently announced cryo-EM structure determination of Orf3a [10] containing three α helices and eight β strands (PDB identifier: 6xdc), further to the short motifs: the Cys-rich region is seen at the end of α3 (TM3), the YxxΦ pattern at the end of β2 and the ‘diacidic’ peptide (SGD in SARS-CoV-2) at the end of strand β3 [10]. The latter is required for the intracellular transport of Orf3a [16], as the protein induces downregulation of IFNAR1, implying a role in attenuating interferon response [17]. Orf3a interacts with structural proteins S, E and M with ion channel activity [18], performing a ‘viroporin’ cellular role [19], and classified by topological – not sequence-based – criteria as a Type IIA viroporin [10].
Comparison of SARS-CoV-1 Orf3a to its homolog in SARS-CoV-2 returns a 72% sequence identity, in contrast to M (membrane) protein or N (nucleoprotein) homolog pairs, which exhibit > 90% identity, indicating a faster change for Orf3a [20], despite likely different origins [21]. Here, we explore the trajectory of Orf3a to show that it has originated recently from M proteins with implications for structure prediction for the latter, leading towards a deeper understanding of their structural and functional relationships.
2. Results
2.1. Sequence comparisons reveal homology between Orf3a and M protein families
Supervised sensitive sequence database searches using Orf3a from SARS-CoV-2 as a query detect a set of > 3000 homologs, that include Orf3a (aliases: Orf3, X1, U274) and weak similarities to the M protein of Coronaviridae (Table S1). The first detected M protein homologs from feline alpha CoVs (ACI13271.1, AIN55875.1, ACI13308.1, AAW66660.1) are seen at step 4 (which are further confirmed independently, with reverse searches) and enrich the profile at step 5 (Fig. S1). The final step incorporates sequence homologs at identity levels as low as 10% (some afflicted by shorter matches and lower scores, subsequently excluded, see Methods). The findings reported herein represent one outcome of multiple iterations with different starting points and parameters; all results chosen for this version are provided in SI to ensure reproducibility.
Following filtering, we maintain a reference alignment with 715 sequences and 275 positions, 29 of which are defined as ‘(quasi)-conserved’ with reference to Orf3a (equivalent positions might have different residue type in other homologs, including M proteins) – (see Methods). The conservation percentage 10.5% (29/275) reflects the low levels of similarity, demonstrating a distant relationship between Orf3a and M proteins of the Coronaviridae (Fig. 1). There are three areas of non-terminal gaps attributed to the longer length of Orf3a (Fig. 1a), at positions 82–96, 123–131, 163–167 with < 50% occupancy and a total length 29 residues (see Methods). Inspection of the alignment also uncovers well-known non-silent mutations in Orf3a reported in various studies during the real-time SARS-CoV-2 evolution, none of which impacts the cross-family positions defined as conserved (Fig. S2). Similarity matrices in an all-against-all comparison of the filtered sequences attest the clear presence of four major groups, largely corresponding to the structural and phylogenetic properties of the alignment (Fig. 1b). The first block in the similarity matrix corresponds to 119 M proteins from the SARS lineage (1–119), while the second block to 70 M proteins from other beta CoVs (120–189). The third block in the matrix contains 305 M proteins from the alpha/gamma groups (190–494, the delta group is absent in this dataset as none of the sequences fulfill the filter criteria – these are available in the raw data only). Finally the last block represents 221 Orf3a CoV homologs from (495–715), 216 of which are from SARS-CoV-2 (shown in Fig. S2) and five sequences from other groups.
Fig. 1.

Sequence relationships of the Orf3a-M protein superfamily. (a): Pictorial representation of the reference alignment: sequence logo displayed at the bottom, bars signify gaps and conservation; secondary structure elements of Orf3a are shown at the top (helices in yellow, strands in green) and labeled; conserved positions are marked by circles, red for those present and pink for those unavailable in the structure. (b): Similarity matrix for the superfamily, annotated on the right, and color-coded with a vertical bar as follows: blue for M protein in the beta group, green for M protein in the alpha and gamma groups and orange for Orf3a member; bar sections with a brush stroke correspond to homologs within the SARS lineage for M protein (blue) and Orf3a (orange). (c): Structure of the sequence alignment depicted in MView color scheme, white sections correspond to gap regions. (d): Sequence space embedding of the reference alignment and annotations: color-coding as in (b) with the exception of the gamma group members; ‘sars-1′/‘sars-2′ denote SARS-CoV-1/-2, for brevity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
An alignment glyph conveys the fine structure of the reference multiple sequence alignment with the three gap regions readily detectable as well as the four aforementioned groups (Fig. 1c). Full alignments (initial and reference) are provided in the SI. Finally, a multi-dimensional projection of sequence similarities in a 3D sequence space unveils the intricate relationships of the groups, with Orf3a homologs positioned between the M protein clusters from alpha and beta CoV groups (the most extensively sampled groups in the lineage). It should be noted that the sort order of the alignment is not kept as in the original progression of detected homologs and arranged by similarity; thus, the gamma group is intermingled with the alpha group, unlike the sequence space representation where it is clearly seen as distinct and separate (Fig. 1d).
2.2. Structural modeling suggests a topology for M proteins of coronaviruses
The structure of SARS-CoV-2 Orf3a has been determined by cryo-EM recently [10], providing insights for its coronavirus homologs. The Orf3a family per se appears as being relatively conserved and does not exhibit a wider variation seen for other accessory genes, e.g. Orf8 [14]. By extending the similarity of Orf3a to its M protein homologs, the variation is indeed unraveled, as fewer positions remain conserved across the now-expanded superfamily. The implications of the detected similarity are profound, as the Orf3a structure allows the low-resolution modeling of M protein for the first time. We have selected a number of targets, including the native structure as a control and without the gap-positioned regions that are excluded (Fig. S3), with Orf3a as the model template (PDB identifier: 6xdc) (Fig. 2). We have not attempted to model likely multimeric forms, as the poor quality of the monomer interface would lead to possible misinterpretations. We opted for representative members with significant, but not too high, divergence, starting from Orf3a. These were selected on the basis of a phylogenetic tree, three of which being Orf3a sequences and the other six M proteins from the beta CoV group only (Fig. 2a). Apart from the native sequence as a target, two Orf3a entries correspond to the Hibecovirus Zaria bat and Bat Hp CoV ‘Orf3′ homologs; selected M proteins are two from SARS-CoV-2, one from pangolin and three from bat viruses, two of which being of the same origin as the Orf3a entries, above (Table S2). The template-to-target (source) alignment (Fig. 2b) demonstrates that positions defined as conserved (Fig. 1) are assigned to all types of structure elements of Orf3a (i.e. helices, strands or turns), with conservation evenly distributed through the aligned positions (Fig. 2c).
Fig. 2.

Target selection and sequence-structure alignment. (a): A phylogenetic tree depicting relationships of M protein groups and Orf3a within the non-redundant alignment; characters (a-i) signify selected superfamily members (3 from Orf3a, 6 from M protein) for model building by homology, marked by different branch colors. (b): Multiple sequence alignment of targets (‘a-i’, Table S2) as in (a); conserved residues/secondary structure elements are colored and labeled as in Fig. 1; minor alpha-helical segments in some models following α4 are not labeled; grey bar plot signifies RMSD values between models; note that conservation cannot be appreciated in this alignment subset, which is solely used for model building. (c): Orf3a structure alignment between the native structure (green) and model {a} (native structure, excluding the three low-occupancy regions, see also text and Methods); secondary structure elements are labeled as in Fig. 1; this orientation is used elsewhere (Figure S4/S6). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The nine models are of moderate quality at such low sequence similarity levels, yet they have been generated to support the interpretation of the multiple sequence alignment and the evolutionary history of the superfamily, guiding further research efforts into the structural analysis of M protein for SARS-CoV-2. Remarkably, Orf3a and M protein of both Zaria {b,i} and Hp bat {c,h} –respectively– (Hibecovirus) CoVs exhibit low similarity to their counterparts, as these strains are distantly related to other beta (Sarbecovirus) CoVs (also seen in the phylogeny, Fig. 2a). This distance is also reflected by the low QMEAN scores for these models (Table S2). The sequence similarity is ~ 20% for the Orf3a homologs and ranges between 6 and 16% for M protein homologs, with regard to the template (Table S2). The solvation measure is lowest for the Orf3a homologs, suggesting that M protein models are more compatible with regard to solvent exposure of their hydrophobic side chain elements, thus supporting the cross-family match as realistic (Table S2). It should be noted that some quality control metrics of the native structure as template have values comparable to those of the targets, corroborating the validity of the models even at this level of similarity (Table S2).
All constructed models are derived from the alignment version that excludes low-occupancy gaps, displayed at the primary-secondary structure levels (Fig. S3) and clearly seen in model {a}, the native (trimmed) target versus the template structure. The Orf3a as well as the M protein targets are all shorter than the template sequence, which presents no gaps (Table S2). The alpha helical region is predicted in all models, with the exception of model {b} where α3 cannot be formed, due to the shorter length of the region (Fig. 2b, Fig. S3). There is greater variation in the cytosolic domain (CD) formed by the two beta sheets, which is discussed below in conjunction with the topology of the models, with Orf3a as template. Tertiary structure diagrams with the conserved positions highlighted enhance the credibility of the sequence-based structure prediction by homology (Fig. S4). As noted in the original report [10], the topology of Orf3a is unique, as DALI [22] does not detect similar arrangements other than alpha-helical segments (not shown).
As reported for Orf3a, the CD in the monomeric state forms a eight stranded beta sandwich, with the outer beta sheet built by β2/β1/β6/β7(N-terminal) and the inner beta sheet by β5/β4/β3/β8/β7(C-terminal). The topology diagrams for the nine predicted models reveal a likely variation of beta strand potential, based on the source alignment (Fig. S5). With the exception of the native structure (model {a}), the representative targets for Orf3a and M protein of coronaviruses exhibit consistent strand variation. Both non-native Orf3a targets miss β3 (with gaps, Fig. 2b), and in one case β2 (model {c}), yet maintaining the β4/β5 pair. In contrast, M protein targets all lack the β4/β5 pair (again due to gaps) with one exception (model {i}), and also β2 with two exceptions (models {h,i}) (Fig. S5). This suggests that the outer sheet might primarily be maintained by β1/β6/β7 and the inner sheet by β3/β7/β8, as β1/β6/β7/β8, and in the case of M proteins also β3, are predicted as shared and conserved across all target models. The loop between β1/β2 is involved in dimer-dimer contacts to form multimers [10] and the β8 in dimer formation [10]. The absence of β3 in Orf3a homologous templates might be offset by the β4/β5 pair, in terms of maintaining the beta sheet, despite its central position in comparison with the strand pair. An alternative explanation may also be a local error in the source alignment (Fig. 2b). Models are also provided in superposition mode (Fig. S6).
2.3. The role of conserved residues in a structural context
Based on the overall conservation in the reference alignment, 29 positions are defined as conserved (Fig. 1a), five of which are not available in the reported structure [10]. The remaining 24 positions are mapped onto the template-to-target alignment (Fig. 2b) and further classified into highly and moderately conserved residues, only for the interpretation of their likely roles in a structural context (Table 1, Fig. 3).
Table 1.
List of 24 (quasi)-conserved residues across Orf3a and M protein homologs.
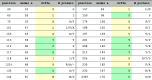 |
Fig. 3.

The role of conserved residues for the superfamily. Residues are color-coded in green (conserved) and yellow (quasi-conserved) (Table 1, [57]) with side chains shown only for those. Numbering follows the reference alignment. (a): the alpha-helical ectodomain of Orf3a, with the second subunit in blue to depict dimer configuration (orientation differs from Fig. 2b, to optimize labeling of residues); two non-conserved residues discussed in connection with the presence of G49 are colored in red. (b): the beta-sheet endodomain of Orf3a, starting at bottom (with N144) and ending at top (with D238); the outer (left) and inner (right) sheets are seen: strand β7 (starting at T208) participates in both; the α-helical turn following β7 is not marked as a helix (Fig. 2b). In both panels, two residues (R122 and D238) are marked by an *asterisk to signify detected steric clash issues. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Of the 24 positions, 11 are observed in the helical part of the Orf3a monomer, with G49 (position 10 in the template-target alignment – Fig. 2b) being quasi-conserved due to the presence of two opposing (Orf3a L84/V88) hydrophobic residues (Fig. 3a). The rest of the quasi- or conserved residues do not appear to make subunit contacts and are seen as conserved to maintain helical structure, turns and hydrophobic exposure in α3 (e.g. YFlqS in Orf3a) (Fig. 3a), consistent with mutagenesis M protein experiments [23]. Consequently, position S117 is conserved due to steric hindrance with the conserved hydrophobic side chain of F114 (e.g. YFiaS in M protein models {d-e-f}), where low case signifies a non-conserved family-specific residue. This region also contains the previously identified conserved motif of M proteins [24] (positions 135–146 in Fig. 1a, or 72–83 in Fig. 2b), corresponding to the predicted loop between α3 and β1 (Fig. 2b). This peptide starts with the cross-family conserved residue S135 (also S in Orf3a) which, when mutated to alanine in tandem with three consecutive residues downstream, affects virus growth [24] (Fig. 3a). The predominantly negatively charged ‘E121′ (E142) [24] is substituted by D142 in the Orf3a family (Fig. 1a, Fig. S2). Interestingly, in 7 cases of a Wencheng Sm shrew M protein, glutamate is replaced by serine having an aspartate residue two positions upstream as a possible compensating mutation (Fig. 1).
The cytosolic part of the monomer contains the remaining 13 (quasi)-conserved residues of the superfamily that play a role in maintaining the beta sheet structure and turns (see also Fig. 2b). As above, the quasi-conserved N144 (also predominantly N in Orf3a), when mutated to alanine in tandem with three consecutive residues downstream, affects virus growth [24]. At the outer beta sheet, positions L147, V201 and Y212 seem to maintain the integrity of the strands through the typical strand periodicity. Proline P159 (position 96 in Fig. 2b) appears to mark the end of strand β2 (Fig. 3b). This is consistent with the occasional absence of β2 in the predicted M protein models which might not be able to form β2 but would require a sharp turn to form the critical β3 as part of the inner sheet (see above). The glycines G174 and G188 (positions 106 and 120 in Fig. 2b) are not always conserved but they may be required to mark the end of β3/β4 strands, when those are formed (see above). A similar situation occurs with G224, just before strand β8. Finally, strand β8 contains three conserved residues (F230, K235 and D238) with F230 participating in the formation of the hydrophobic core between subunits [10] and K235/D238 exposed to the solvent and potentially important for virus-host protein interactions. In all, the detection of homology between CoV Orf3a and M protein is consistent with previous work on structural properties of M proteins and our own analysis, outlined here.
2.4. The distribution of Orf3a points to recent divergence from M protein
Orf3a is known to be restricted within the beta group of coronaviruses, raising crucial questions regarding its origins and molecular function. Bat CoVs have been found in four of the five subgroups of the beta group, namely Sarbecovirus, Merbecovirus, Nobecovirus and Hibecovirus but not in the Embecovirus group [25]. The two ‘outlier’ Zaria/Bat Hp Orf3a homologs belong to the Hibecovirus group; Orf3a homologs are not detectable in Merbecovirus, thus found only in the other three CoV groups known to exist in bats, as above. In a representative, genome-based phylogeny of Coronoviridae [26], M protein is seen as universally present in all strains, with Orf3a restricted to the Bat/Civet SARS-CoV-1 and Bat/Pangolin SARS-CoV-2 section of the beta group along with the outlier members (Fig. 4). When considered as a single family, Orf3a appears to be conserved and could be seen as the product of a long evolutionary process with strong structural constraints and limited diversity (Fig. S7).
Fig. 4.

Phylogenetic profiling of superfamily members. Distribution of M protein and Orf3a homologs across a representative tree of Coronaviridae. (a): Genome-based tree of 89 representative strains of coronaviruses, 89 nodes are shown and only 45 labels, for clarity; groups and corresponding nodes are color-coded as grey (alpha), orange (beta), yellow (gamma) and lavender (delta), coded in a vertical bar labeled as ‘group’; the eleven strains of the beta group containing both M protein and Orf3a homologs are coded in the vertical bar in cyan and labeled as ‘bothMOrf3a’. (b): Depiction of the representative tree with a zoom focus on the 11 strains that contain both families. (c): Section of the tree with the 11 strains that contain both families listed. For all labels, sequence identifier precedes description of the genome sequence for the corresponding strain. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
However, given the restricted phylogenetic distribution and the detected homology to the M protein family of coronaviruses, a different interpretation arises. A copy of M protein of a viral strain in bats or other species such as civets or pangolins might have been evolving rapidly as a viral paralog with less selective pressure and to a point beyond recognition, offering additional opportunities to this beta CoV lineage to efficiently infect and propagate within host cells. Thus, an alternative outlook of the superfamily that encompasses M protein and Orf3a from coronaviruses suggests that this phylogenetic trajectory may indeed be a likely scenario for the recent emergence of Orf3a in the lineage (Fig. S7). This claim is further supported by the low similarity of outgroup Orf3a sequences, e.g. Zaria ZBCoV Orf3 with < 25% identity to its SARS-CoV-2 homolog, compared to > 55% identity between the nucleoprotein (protein N) of the respective strains (protein identifiers: IDADY17917.1 for ZBCoV vs. QHD43423.2 for SARS-CoV-2). Thus, the apparent conservation of Orf3a can be attributed to its recent origin within the beta CoV group from a homologous M protein elsewhere, and not to structural or functional constraints that characterize typical non-viral protein evolution.
3. Discussion
Orf3a is considered to belong to the general class of viroporins and have an ion channel function, effected via its transmembrane domain [7]. It is primarily found in the Golgi complex before its insertion into the plasma membrane where it performs its function in multimeric forms [27]. To date, Orf3a has been seen as a single family [28], with the structure of Orf3a from SARS-CoV-2 recently solved by cryo-EM [10]. Here, we show that Orf3a of SARS-CoV-2 and its homologs share significant similarities with membrane proteins (M protein) from coronaviruses, suggesting a recent origin from a member of the M protein family.
The implication of this observation is that M protein might possess a structure similar to the unique Orf3a alpha–beta fold. Despite its abundance in the CoV particle [29], the function of M protein is not fully understood, only glimpsed from its interacting partners in SARS-CoV-1 [30]. M protein is known to share features with Orf3a, such as the N-terminal transmembrane domain and similar co-localization patterns [31], also forming dimers [29]. Although we have not attempted to model multimers, it is possible that Orf3a has inherited quaternary structure properties from M protein. Our findings are supported by pre-computed listings of profile-profile FFAS comparisons (Fig. S8), where seven positions are found as properly aligned with respect to the reference sequence alignment herein; others, especially upstream, are shifted due to gaps, and low-quality matches [32]. As the reported FFAS-based similarity has been available from previous comparisons with SARS-CoV-1 sequences yet never properly interpreted, it does lend further independent support to our observations, namely that Orf3a represents an aberrant M protein homolog in the beta CoV group lineage that has emerged recently in this group, demonstrated by the evidence provided in this study.
It is not surprising that while the Cys-rich motif is somewhat conserved in Orf3a members, the YxxΦ/diacidic motifs are not [28]. These motifs are specific adaptations of the Orf3a family and do not play a role in the structural integrity of the fold, as described in the original cryo-EM Orf3a structure report [10]. The deceivingly ‘distant’ relationship of Orf3a with a restricted distribution to the universally present M proteins in CoVs can be explained by two mechanisms only: either there is a long evolutionary history of the two genes with persistent gene loss in the other groups where Orf3a is not found, or Orf3a has emerged recently from CoV M proteins and evolved rapidly, exhibiting low sequence similarities that are now detected. Our study clearly favors the latter explanation, as an expected outcome especially in a fast-evolving virus group.
In analogy with the recently discovered similarity of Orf7a – a ‘conserved’ protein family of known structure [11], with Orf8 [33]/(Neches et al., submitted), a hypervariable protein with a recently announced structure confirming the sequence similarity [34], SARS-CoV-2 and its relatives seem to maintain a reservoir of genetic material as a constant and vary a ‘paralog’ gene. As in the Orf7a/Orf8 case, M protein is conserved, while Orf3a attained divergence to a point of being unrecognizable as a M protein homolog. The reported findings point to a credible model for the M protein, based on the structure of Orf3a with a unique fold [10].
A limitation of this study is that the detected similarity is predicated on the few weak, yet highly significant similarities of the Orf3a family to feline CoV M proteins that later enrich the iterative search that converges to a specific superfamily profile. The search, however, is reproducible given the specific parameters, well-documented and able to return similar results with other, carefully selected starting points. The reverse search with M proteins as queries does not identify Orf3a family members: this is most likely due to the greater sequence variation of M proteins across CoV strains, their much wider phylogenetic distribution and, critically, their shorter lengths. Conversely, the Orf3a family is narrowly defined both in terms of sequence and phylogeny, thus amplifying the conserved positions that subsequently detect M protein. The facts that the entire process generates profiles specific to CoVs and the conservation pattern is consistent with structure predictions by homology, provide strong evidence for a common origin and a highly divergent Orf3a form, from M proteins. The intriguing discovery of Orf3a/M protein homology can yield further insights into the evolution and structure of M protein and its potential role in SARS-CoV-2 biology, in connection to Orf3a.
It is expected that certain functional properties peculiar to Orf3a are not conserved, despite the common fold. As a matter of fact, the viral strategy is indeed to differentiate the molecular function of its limited gene repertoire. From functional genomics data, there is little evidence for the reported interaction of Orf3a with caveolin-1 in Golgi [35]. The number of detected interactions of Orf3a are limited (just 8) while those of M protein are far more extensive (namely 27) [36]. Finally, drug or small-molecule interactions differ across the two families, in various recent studies [37], [38]. From a structural perspective, the predicted conservation of the fold between M proteins and Orf3a in CoVs dictates shared structural features with diversified functions across the coronavirus lineage. The spillover effects of bat coronaviruses into human pathogens [39] are supported by this mode of viral evolution for SARS-CoV2 Orf3a and a similar phenomenon for Orf8.
4. Methods
4.1. Database searches and documentation
Orf3a (QHD43417.1) was filtered for compositional bias by CAST (threshold 30) [40] and used as a query to search the Virus section (taxid: 10239) of NCBI’s nr collection [41] (posted date 5-Aug-2020, number of entries 3,063,868). Searches were performed by PSI-BLAST [42], with default parameters – except: word size 2, no compositional adjustment, filter none, max hits 20000, and threshold 0.1, a permissive E-value in supervised sequence space walk mode, as described [43]. Convergence was achieved after nine iterations, with a total number of significant hits at 3025 (Fig. S1). No spurious hits were detected, i.e. only members of the two families were admitted, at 100% accuracy (no false positives). Matches to Pfam entries [44] were recorded (PF11289 for Orf3a, PF01635 for M protein, Table S1). The final PSSM profile was further used to query the full nr excluding Coronaviridae, without returning any additional hits, thus considered as highly specific, at this critical quality control step. All runs have been recorded as triplets of hit lists/alignment/PSSM files, available in SI as data supplement (DS01). The raw alignment with 3025 sequences is also available as data supplement (DS02).
4.2. Filtering strategy
Different strategies were used for the filtering step, given the original sequence query and the one processed by CAST [40]. The filtered query was:
>QHD43417.1 ORF3a protein [Severe acute respiratory syndrome coronavirus 2]
L-rich region from 83 to 129 corrected with score 32
MDLFMRIFTIGTVTLKQGEIKDATPSDFVRATATIPIQASLPFGWLIVGVALLAVFQSAS
KIITLKKRWQLALSKGVHFVCNXXXXFVTVYSHXXXVAAGXEAPFXYXYAXVYFXQSINF
VRIIMRXWXCWKCRSKNPLLYDANYFLCWHTNCYDYCIPYNSVTSSIVITSGDGTTSPIS
EHDYQIGGYTEKWESGVKDCVVLHSYFTSDYYQLYSTQLSTDTGVEHVTFFIYNKIVDEP
EEHVQIHTIDGSSGVVNPVMEPIYDEPTTTTSVPL
where X denotes a masked residue.
Against the nr, the iterative search converges at earlier steps, with or without compositional adjustment and/or compositional bias masking; against the Virus section, false positives with the unmasked query arise, at subsequent steps of the iterative search process (not shown). The only case which yields convergence by detecting M protein homologs is the masked query by CAST against the Virus section, without adjustment.
4.3. Confirmation with an outlier sequence
The M protein homologs can also be detected by Zaria bat virus Orf3a (ADY17912.1) in an equivalent search, with the same outcome (aligned positions), and multiple (over 40) iterations (not shown).
This sequence is also masked by CAST, with threshold 30 – below:
>ADY17912.1 putative ORF3 protein [Zaria bat coronavirus]
I-rich region from 50 to 123 corrected with score 35 (replaced by X):
MDYFKFWSFGLVNIHKPDPVYEPVVARQSFIPHGTTISPTEHPTMLAGDXCLTXFLLVYX
XSWFLRADSXPAVXLRFALTLXTGTFLVXGLFLEQPSLVLKXAVAVTXVAYVGCXSLRLA
LAXRCVSWLPLMADDDCFVCYQSGGYNYCFPYDPNGPYVTLTVHNNGVTCGSHTIYGSVS
VADRIQVVTIRNKNSYILQNTFDTGICIIAFYIADIAVVENHTVVGDLPKSCPEYHIYDE
PRATINVPL
as above.
4.4. Alignment editing and quality control
The original matches from the profile-driven searches was retained, as various multiple sequence alignment algorithms do not reproduce the detected sequence similarities. From the raw alignment (DS02), entries with sequence length ≤ 215 residues were removed, as a proxy action to exclude partial sequences, along with all sequences with undefined positions (typically marked by the X symbol in FASTA files). Removal of single undefined positions is critical, as we aim at maximum accuracy for model building, and does not affect results, due to sufficient redundancy in the generated dataset. This edit operation results in a version of 715 sequences, with description lines modified to eliminate alternative protein names (DS03, Fig. 1). There are 494 M protein and 221 Orf3a entries in the reference alignment of 715 sequences (see also “Coverage”). For visualization, editing and trimming of multiple sequence alignments, JalView 2 was used [45].
4.5. Multiple alignment processing
In the reference alignment (DS03), 29 positions are defined as ‘conserved’ at 60% identity level: M5*, T9*, I10*, Q38*, G49, L65, A72, L101, A103, Y113, F114, S117, I118, R122, S135, N144, L147, P159, G174, T176*, G188, V197, V201, T208, Y212, G224, F230, K235, D238 (5 of which are not available in the structure and are marked by an *asterisk above – residues named according to Orf3a). Thus conservation across the total alignment length (275 residues) is ~ 10% (29/275). Notably, as Orf3a sequences are typically 275-residues long, they present no gaps, facilitating the numbering scheme. Trimming of N-terminus (39 residues) and C-terminus (53 residues), to maintain only residues available both in the 3D structure entry (PDB identifier: 6xdc) and some partial homologs, results in an alignment of 183 residues (275–39-53 = 183); the gapped regions with a total length of 29 residues with occupancy < 50% (see Results) yield an alignment 183–29 = 154 residues long – saved as a JalView session file (DS04), with gapped regions hidden or shown, according to user preference. The trimmed alignment is offered only for experts as a basis for future research.
4.6. Coverage analysis
With specificity at 100% (no false positives), assessed by visual inspection and checks of full sequence records, sensitivity was also addressed. There are 3980 entries in the NCBI Protein database annotated in Pfam as Orf3a, of which only 11 are in nr (due to high redundancy). Of the 221 Orf3a entries in the reference alignment, the 11 entries in nr are detected, along with an additional 210 homologs which are not annotated by Pfam (DS05). There are also 7724 entries in the NCBI Protein database annotated in Pfam as M protein, of which 330 are in nr. Of the 494 M protein entries in the reference alignment, the 330 entries in nr are detected, along with an additional 164 homologs which are not annotated by Pfam (DS06). Thus, coverage is also 100%, extended by new hits. In all, we detect 210 and 164 ‘novel’ homologs for Orf3a and M protein, respectively. The reference alignment contains all representative members of the two connected families.
4.7. Phylogenetic tree
When 100% redundancy (i.e. identical sequences) is eliminated, the number of sequences drops to 492 (DS07, Fig. 2). FastTree was used to calculate phylogenetic relationships with LG/gamma options [46] on the NGPhylogeny servers [47], from the trimmed, non-redundant alignment (DS08). Due to the criteria imposed for quality control, no entries from the Delta group of coronaviruses are kept: those are recoverable from the raw alignment (DS02). Trees were visualized and processed by IcyTree [48] and iTOL [49].
4.8. Cross-family similarities
To check whether the original alignment can be consistently generated, it was split into two groups (494 M protein and 221 Orf3a sequence entries) and submitted to profile-profile matching with MAFFT [50], that also identifies the same conserved regions. Cross-family similarities were computed with AlignmentViewer [51] and dimensionality reduction was aided by UMAP [52] as implemented in AlignmentViewer. Color-coding for sequence glyphs was selected according to MView [53], using a residue width and height of 1 pixel. Annotations and the reference alignment processed for input to AlignmentViewer are available (DS09).
4.9. Model generation
Representative members of Orf3a and M protein families were selected as targets for model building on the basis of their similarity to the native structure that was used as a template (Fig. 2a). The nine models (three for Orf3a, including the template with missing gaps as a control, and six M proteins from the beta coronavirus group) are named as {a-i} throughout this study, with their sequence identifiers listed (Table S2). Model building was based on the trimmed, non-redundant alignment and executed with Swiss-model [54]. Structures were visualized with UCSF Chimera [55]. All models are made available in PDB format (DS10).
4.10. Quality control
Protein structure model quality parameters were recorded, including QMEAN [56], a size-independent metric that integrates key model descriptors in a linear combination. Beta-carbon and all-atom interaction potentials, solvation parameters and torsion angle potential metrics are listed for comparison (Table S2). Numbers are all negative, indicating inferior quality of models for detailed structural work; however, as stated elsewhere, models at this level of resolution and quality can be useful to interpret sequence variation. The low average QMEAN reflects all regions of the model, yet local quality of specific residues may be high.
4.11. Conserved residues for models
Of the 29 residues defined as (quasi)-conserved (DS11), 5 are not available in the template structure (N-terminal, as stated; one is missing from the loop between β3/β4). The remaining 24 equivalent positions for Orf3a and M protein homolog models are based on the trimmed alignment (DS07) and sub-divided into quasi- and conserved (Table 1). As model {a} does not contain any gaps, the numbering scheme holds for all models (Fig. 2b), but actual residue coordinates at corresponding positions might differ for models {b-i}. Also, for M protein homologs {d-i}, residue types can be different (Table 1), as models represent select cases not necessarily having the majority features of M proteins elsewhere (e.g. alpha CoV group).
4.12. Model interpretation
Topology diagrams were generated using Pro-Origami [58] and processed / annotated for detailed structure interpretation (Figure S5). In all cases, secondary structure elements were defined by DSSP [59].
4.13. Phylogenetic view
To visualize phylogenetic profiles for the two families, MicroReact was used [60], with a tree generated for 89 representative genomes of coronaviruses [26]. Corresponding input files for MicroReact (csv, nwk) are available (DS12, Fig. 4).
4.14. Functional genomics
For Orf3a and M protein of SARS-CoV-2, protein interaction data was obtained from recent systematic experimental investigations [36]. Additional protein-small molecule associations were extracted from the gene and drug set library [37] and the virus-host-drug interactome [38].
4.15. Data availability
All data are available as Supplementary Information on FigShare.
5. Ethics declarations
None.
CRediT authorship contribution statement
Christos A. Ouzounis: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing - original draft, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
CAO acknowledges support by Elixir-GR, a project implemented under the Action ‘Reinforcement of the Research & Innovation Infrastructure’, funded by the Operational Programme ‘Competitiveness, Entrepreneurship and Innovation’ (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund). Thanks to many colleagues and members of BCPL for comments.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.11.047. Data Supplements (DS in text) available on FigShare – http://dx.doi.org/10.6084/m9.figshare.13046111.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B. A novel coronavirus from patients with pneumonia in china, 2019. N Engl J Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coronaviridae Study Group The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boni M.F., Lemey P., Jiang X., Lam T.T., Perry B.W. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 2020 doi: 10.1038/s41564-020-0771-4. [DOI] [PubMed] [Google Scholar]
- 7.McBride R, Fielding BC (2012) The role of severe acute respiratory syndrome (SARS)-coronavirus accessory proteins in virus pathogenesis. Viruses 4: 2902-2923. [DOI] [PMC free article] [PubMed]
- 8.Liu P, Jiang JZ, Wan XF, Hua Y, Li L, et al. (2020) Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog 16: e1008421. [DOI] [PMC free article] [PubMed]
- 9.Li X., Giorgi E.E., Marichannegowda M.H., Foley B., Xiao C. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci Adv. 2020;6(27):eabb9153. doi: 10.1126/sciadv.abb9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kern DM, Sorum B, Hoel CM, Sridharan S, Remis JP, et al. (2020) Cryo-EM structure of the SARS-CoV-2 3a ion channel in lipid nanodiscs. bioRxiv: 2020.2006.2017.156554.
- 11.Nelson C.A., Pekosz A., Lee C.A., Diamond M.S., Fremont D.H. Structure and Intracellular Targeting of the SARS-Coronavirus Orf7a Accessory Protein. Structure. 2005;13(1):75–85. doi: 10.1016/j.str.2004.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Donoghue S, Schafferhans A, Sikta N, Kaur S, Stolte C, et al. (2020) Systematic modeling of SARS-CoV-2 protein structures. bioRxiv: 2020.2007.2016.207308.
- 13.Hu B, Zeng LP, Yang XL, Ge XY, Zhang W, et al. (2017) Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog 13: e1006698. [DOI] [PMC free article] [PubMed]
- 14.Velazquez-Salinas L, Zarate S, Eberl S, Gladue DP, Novella I, et al. (2020) Positive selection of ORF3a and ORF8 genes drives the evolution of SARS-CoV-2 during the 2020 COVID-19 pandemic. bioRxiv: 2020.2004.2010.035964. [DOI] [PMC free article] [PubMed]
- 15.Zhao J, Sun J, He W-T, Ji X, Gao Q, et al. (2020) Snapshot of the evolution and mutation patterns of SARS-CoV-2. bioRxiv: 2020.2007.2004.187435.
- 16.Minakshi R., Padhan K. The YXXPhi motif within the severe acute respiratory syndrome coronavirus (SARS-CoV) 3a protein is crucial for its intracellular transport. Virol J. 2014;11:75. doi: 10.1186/1743-422X-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minakshi R, Padhan K, Rani M, Khan N, Ahmad F, et al. (2009) The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One 4: e8342. [DOI] [PMC free article] [PubMed]
- 18.Tang X., Li G., Vasilakis N., Zhang Y., Shi Z., Zhong Y., Wang L.-F., Zhang S. Differential stepwise evolution of sars coronavirus functional proteins in different host species. BMC Evol Biol. 2009;9(1):52. doi: 10.1186/1471-2148-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nieva J.L., Madan V., Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10(8):563–574. doi: 10.1038/nrmicro2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J, Zhao S, Teng T, Abdalla AE, Zhu W, et al. (2020) Systematic Comparison of Two Animal-to-Human Transmitted Human Coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 12. [DOI] [PMC free article] [PubMed]
- 21.Song H.-D., Tu C.-C., Zhang G.-W., Wang S.-Y., Zheng K. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci. 2005;102(7):2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm L. DALI and the persistence of protein shape. Protein Sci. 2020;29(1):128–140. doi: 10.1002/pro.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng Y-T, Chang C-H, Wang S-M, Huang K-J, Wang C-T (2013) Identifying SARS-CoV membrane protein amino acid residues linked to virus-like particle assembly. PLoS One 8: e64013-e64013. [DOI] [PMC free article] [PubMed]
- 24.Arndt A.L., Larson B.J., Hogue B.G. A Conserved Domain in the Coronavirus Membrane Protein Tail Is Important for Virus Assembly. JVI. 2010;84(21):11418–11428. doi: 10.1128/JVI.01131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong ACP, Li X, Lau SKP, Woo PCY (2019) Global Epidemiology of Bat Coronaviruses. Viruses 11: 174. [DOI] [PMC free article] [PubMed]
- 26.Ou Z, Ouzounis C, Wang D, Sun W, Li J, et al. (2020) A path towards SARS-CoV-2 attenuation: metabolic pressure on CTP synthesis rules the virus evolution. bioRxiv: 2020.2006.2020.162933. [DOI] [PMC free article] [PubMed]
- 27.Lu W., Zheng B.-J., Xu K., Schwarz W., Du L., Wong C.K.L., Chen J., Duan S., Deubel V., Sun B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci. 2006;103(33):12540–12545. doi: 10.1073/pnas.0605402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Issa E, Merhi G, Panossian B, Salloum T, Tokajian S (2020) SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 5. [DOI] [PMC free article] [PubMed]
- 29.J Alsaadi E.A., Jones I.M. Membrane binding proteins of coronaviruses. Future Virology. 2019;14(4):275–286. doi: 10.2217/fvl-2018-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsieh Y.-C., Li H.-C., Chen S.-C., Lo S.-Y. Interactions between M protein and other structural proteins of severe, acute respiratory syndrome-associated coronavirus. J Biomed Sci. 2008;15(6):707–717. doi: 10.1007/s11373-008-9278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan X., Li J., Shan Y., Yang Z., Zhao Z. Subcellular localization and membrane association of SARS-CoV 3a protein. Virus Res. 2005;109(2):191–202. doi: 10.1016/j.virusres.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaroszewski L., Li Z., Cai X.-H., Weber C., Godzik A. FFAS server: novel features and applications. Nucleic Acids Res. 2011;39(suppl):W38–W44. doi: 10.1093/nar/gkr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan Y, Schneider T, Leong M, Aravind L, Zhang D (2020) Novel Immunoglobulin Domain Proteins Provide Insights into Evolution and Pathogenesis of SARS-CoV-2-Related Viruses. mBio 11. [DOI] [PMC free article] [PubMed]
- 34.Flower TG, Buffalo CZ, Hooy RM, Allaire M, Ren X, et al. (2020) Structure of SARS-CoV-2 ORF8, a rapidly evolving coronavirus protein implicated in immune evasion. bioRxiv: 2020.2008.2027.270637.
- 35.Padhan K, Tanwar C, Hussain A, Hui PY, Lee MY, et al. (2007) Severe acute respiratory syndrome coronavirus Orf3a protein interacts with caveolin. J Gen Virol 88: 3067-3077. [DOI] [PubMed]
- 36.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020 doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuleshov M., Clarke D.J.B., Kropiwnicki E., Jagodnik K., Bartal A. The COVID-19 Gene and Drug Set Library. SSRN. 2020 doi: 10.1016/j.patter.2020.100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadegh S., Matschinske J., Blumenthal D.B., Galindez G., Kacprowski T., List M., Nasirigerdeh R., Oubounyt M., Pichlmair A., Rose T.D., Salgado-Albarrán M., Späth J., Stukalov A., Wenke N.K., Yuan K., Pauling J.K., Baumbach J. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat Commun. 2020;11(1) doi: 10.1038/s41467-020-17189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Letko M., Seifert S.N., Olival K.J., Plowright R.K., Munster V.J. Bat-borne virus diversity, spillover and emergence. Nat Rev Microbiol. 2020;18(8):461–471. doi: 10.1038/s41579-020-0394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Promponas V.J., Enright A.J., Tsoka S., Kreil D.P., Leroy C., Hamodrakas S., Sander C., Ouzounis C.A. CAST: an iterative algorithm for the complexity analysis of sequence tracts. Bioinformatics. 2000;16(10):915–922. doi: 10.1093/bioinformatics/16.10.915. [DOI] [PubMed] [Google Scholar]
- 41.Sayers E.W., Beck J., Brister J.R., Bolton E.E., Canese K. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2020;48:D9–D16. doi: 10.1093/nar/gkz899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Promponas V.J., Katsani K.R., Blencowe B.J., Ouzounis C.A. Sequence evidence for common ancestry of eukaryotic endomembrane coatomers. Sci Rep. 2016;6(1) doi: 10.1038/srep22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res 47: D427-D432. [DOI] [PMC free article] [PubMed]
- 45.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price MN, Dehal PS, Arkin AP (2010) FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5: e9490. [DOI] [PMC free article] [PubMed]
- 47.Lemoine F., Correia D., Lefort V., Doppelt-Azeroual O., Mareuil F. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019;47:W260–W265. doi: 10.1093/nar/gkz303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaughan TG (2017) IcyTree: rapid browser-based visualization for phylogenetic trees and networks. Bioinformatics 33: 2392-2394. [DOI] [PMC free article] [PubMed]
- 49.Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47: W256-W259. [DOI] [PMC free article] [PubMed]
- 50.Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20: 1160-1166. [DOI] [PMC free article] [PubMed]
- 51.Reguant R., Antipin Y., Sheridan R., Dallago C., Diamantoukos D. AlignmentViewer: Sequence Analysis of Large Protein Families. F1000Res. 2020;9:213. doi: 10.12688/f1000research.22242.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I.W.H. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2019;37(1):38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 53.Brown N.P., Leroy C., Sander C. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics. 1998;14(4):380–381. doi: 10.1093/bioinformatics/14.4.380. [DOI] [PubMed] [Google Scholar]
- 54.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, et al. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296-W303. [DOI] [PMC free article] [PubMed]
- 55.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 56.Benkert P, Biasini M, Schwede T (2010) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27: 343-350. [DOI] [PMC free article] [PubMed]
- 57.Sehnal D, Rose AS, Kovca J, Burley SK, Velankar S. Mol*: towards a common library and tools for web molecular graphics. In: Byska J, Krone M, Sommer B, editors; 2018. The Eurographics Association.
- 58.Stivala A., Wybrow M., Wirth A., Whisstock J.C., Stuckey P.J. Automatic generation of protein structure cartoons with Pro-origami. Bioinformatics. 2011;27(23):3315–3316. doi: 10.1093/bioinformatics/btr575. [DOI] [PubMed] [Google Scholar]
- 59.Kabsch W., Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 60.Argimon S, Abudahab K, Goater RJE, Fedosejev A, Bhai J, et al. (2016) Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb Genom 2: e000093. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available as Supplementary Information on FigShare.