

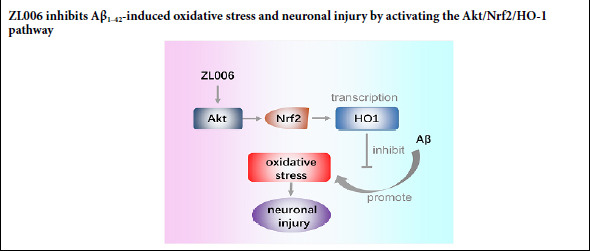
Keywords: Akt, Alzheimer's disease, amyloid-beta, apoptosis, heme oxygenase-1, neurotoxicity, Nrf2, oxidative stress, treatment, ZL006
Abstract
Amyloid beta (Aβ)-induced neurotoxicity and oxidative stress plays an important role in the pathogenesis of Alzheimer’s disease (AD). ZL006 is shown to reduce over-produced nitric oxide and oxidative stress in ischemic stroke by interrupting the interaction of neuronal nitric oxide synthase and postsynaptic density protein 95. However, few studies are reported on the role of ZL006 in AD. To investigate whether ZL006 exerted neuroprotective effects in AD, we used Aβ1–42 to treat primary cortical neurons and N2a neuroblastoma cells as an in vitro model of AD. Cortical neurons were incubated with ZL006 or dimethyl sulfoxide for 2 hours and treated with Aβ1–42 or NH3•H2O for another 24 hours. The results of cell counting Kit-8 (CCK-8) assay and calcein-acetoxymethylester/propidium iodide staining showed that ZL006 pretreatment rescued the neuronal death induced by Aβ1–42. Fluorescence and western blot assay were used to detect oxidative stress and apoptosis-related proteins in each group of cells. Results showed that ZL006 pretreatment decreased neuronal apoptosis and oxidative stress induced by Aβ1–42. The results of CCK8 assay showed that inhibition of Akt or NF-E2-related factor 2 (Nrf2) in cortical neurons abolished the protective effects of ZL006. Moreover, similar results were also observed in N2a neuroblastoma cells. ZL006 inhibited N2a cell death and oxidative stress induced by Aβ1–42, while inhibition of Akt or Nrf2 abolished the protective effect of ZL006. These results demonstrated that ZL006 reduced Aβ1–42-induced neuronal damage and oxidative stress, and the mechanisms might be associated with the activation of Akt/Nrf2/heme oxygenase-1 signaling pathways.
Chinese Library Classification No. R459.9; R453; R364
Introduction
More than 47 million people worldwide have Alzheimer’s disease (AD), which is a major cause of dementia in the elderly (Alzheimer’s Association, 2016). The neuropathological hallmarks of AD include extracellular plaques consisting of amyloid beta (Aβ) and intraneuronal neurofibrillary tangles (Selkoe and Hardy, 2016). The generation and accumulation of Aβ is shown to initiate the pathological cascade in AD, leading to the dysfunction and death of neuronal cells (Hardy and Higgins, 1992; Selkoe and Hardy, 2016). Furthermore, reducing Aβ levels alleviates Aβ-induced apoptosis and cognitive impairment (Aminyavari et al., 2018; Gan et al., 2018).
The production of free radicals and oxidative stress is a key factor in the progression of AD (Querfurth and LaFerla, 2010). Aβ has been shown to induce the generation of H2O2 in vitro (Huang et al., 1999). Accumulated Aβ leads to dysfunction of mitochondria and induces extensive production of reactive oxygen species (ROS), followed by lipid peroxidation, protein oxidation, and DNA/RNA oxidation (Yu et al., 2018). Furthermore, increased oxidative stress results in impaired degradation of proteins in AD (Haynes et al., 2004). We have shown that several herb extracts, including Hopeahainol A, Diammonium glycyrrhizinate and Orientin, exert anti-oxidative effects and protect against Aβ-induced neuronal death and cognitive dysfunction (Zhu et al., 2012, 2013; Yu et al., 2015). Although the use of antioxidant supplements as primary prevention of AD is controversial, several clinical trials have shown that antioxidant might be associated with the reduced risk and progression of AD (Morris et al., 2002a, b; Barnes and Yaffe, 2005; Turner et al., 2015; Kryscio et al., 2017).
Akt pathway plays an essential role in the regulation of cell apoptosis, and is downregulated in the brains of AD rats (Wang et al., 2016). Accumulating evidence suggests that activation of Akt pathway protects neuronal cells from Aβ-induced detrimental effects (Chong et al., 2007; Yi et al., 2018). The nuclear transcription factor NF-E2-related factor 2 (Nrf2) is a downstream target of Akt, and it regulates the redox status in the central nervous system (de Vries et al., 2008). Under oxidative stress, Nrf2 translocates into the nucleus and binds to heme oxygenase-1 HO-1 (Tan et al., 2013). The levels of nuclear Nrf2 are decreased in the hippocampal CA1 region of AD patients (Rojo et al., 2017). In addition, the levels of oxidative stress are increased in the hippocampus of Nrf2–/– APP mice (Rojo et al., 2017). Therefore, the Akt/Nrf2/HO-1 pathway is a potential target for the treatment of AD.
ZL006 is designed and synthesized to disrupt the interaction of neuronal nitric oxide synthase (nNOS) and postsynaptic density 95 (PSD95) and shows promising therapeutic effects in several neurological disorders (Zhou et al., 2010; Cai et al., 2018; Lin et al., 2018; Qu et al., 2020). However, whether ZL006 protects neuronal cells against Aβ-induced neurotoxicity remains unknown. In this study, we investigated the neuroprotective effects of 5-(4, 5-dichloro-2-hydroxybenzylamino)-2-hydroxybenzoic acid (ZL006) on Aβ1–42-treated primary cortical neurons and N2a neuroblastoma cells.
Materials and Methods
Cell culture
Mouse neuroblastoma N2a cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and treated with Dulbecco’s modified Eagle medium (DMEM; Gibco, Shanghai, China) containing 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco) at 37°C in a humidified 5% CO2 incubator. Primary cortical neurons were prepared from mouse embryos at the embryonic days 16–17 (E16–E17) as described previously (Xu and Tao, 2004). In brief, the cortexes of mouse embryos were dissected on ice, and digested with trypsin at 37°C in a humidified 5% CO2 incubator for 10 minutes to prepare cell suspension. Cells were seeded at 5 × 105 cells/mL and maintained in neurobasal media (Gibco) supplemented with B27 (Gibco) and 25 nM glutamine (Gibco) for 10–12 days in poly-D-lysine pre-coated plates.
ZL006 treatment
ZL006, a kind gift from Dr. Fei Li (Nanjing Medical University, China), was described previously (Zhou et al., 2010) and the chemical structure is shown in Figure 1A. ZL006 was dissolved in DMSO, and Aβ1–42 (Millipore, Boston, MA, USA, AG968-1MG) was dissolved in NH3•H2O as described previously (Zhu et al., 2014). Akt inhibitor VIII, an Akt inhibitor, was obtained from Beyotime Biotechnology (Nanjing, China; SF2784). ML385, which interacts with Nrf2 protein and inhibits the transcriptional activity of Nrf2 (Singh et al., 2016), was purchased from Selleck (CA, USA, S8790).
Figure 1.
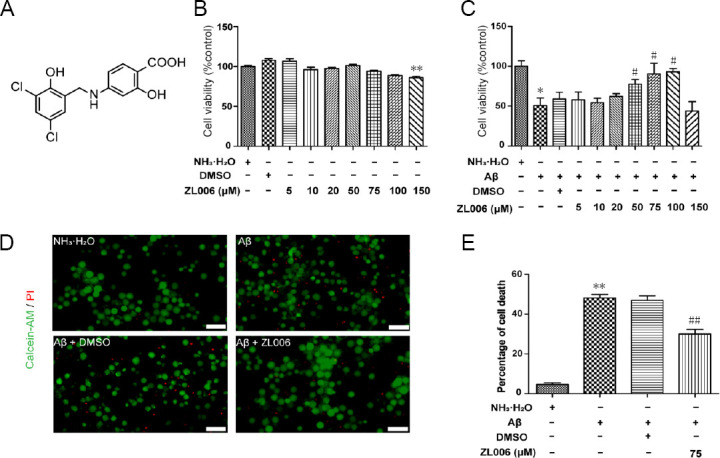
ZL006 protects against Aβ1–42-induced neuronal cell death.
(A) The chemical structure of ZL006. (B) Primary cortical neurons at DIV 10–12 were treated with ZL006 (0–150 μM) or DMSO vehicle (< 1‰) for 24 hours and CCK8 assay was performed. n = 8–11 cells per group. (C) Primary cortical neurons at DIV 10–12 were pretreated with ZL006 (0–150 μM) or DMSO vehicle (< 1‰) for 2 hours and then treated with Aβ1–42 (2 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, and cell viability was accessed. n = 6 cells per group. (D) Primary cortical neurons at DIV 10–12 were pretreated with ZL006 (75 μM) or DMSO vehicle (< 1‰) for 2 hours and then treated with Aβ1–42 (2 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, and calcein-acetoxymethylester/ PI staining was performed. Representative images of neurons stained by calcein-AM/PI were shown. Original magnification, 20×, scale bar: 50 μm. (E) Quantitative analysis of the PI+ cells, n = 6 cells per group. Data were expressed as mean ± SEM (oneway analysis of variance followed by Bonferroni's post hoc test). *P < 0.05, **P < 0.01, vs. NH3•H2O vehicle group; #P < 0.05, ##P < 0.01, vs. Aβ1–42 + DMSO vehicle group. Aβ: Amyloid-beta; AM: acetoxymethylester; DIV: day- in-vitro; DMSO: dimethyl sulfoxide; PI: propidium iodide.
To investigate the toxic dose of ZL006 to cells, we treated cells with ZL006 in a concentration gradient (NH3•H2O group; DMSO group; ZL006-treated groups: 5, 10, 20, 50, 75, 100, 150 μM). To select the effective dose of ZL006, we also set a concentration gradient (NH3•H2O group; Aβ1–42 group; DMSO group; ZL006-treated groups: 5, 10, 20, 50, 75, 100, 150 μM). Then the cells were divided into four groups: NH3•H2O group (NH3•H2O, < 1 ‰), Aβ1–42 group (neurons: 2 μM Aβ1–42; N2a neuroblastoma cells: 20 μM Aβ1–42), DMSO group (neurons: 2 μM Aβ1–42+ < 1 ‰ DMSO; N2a neuroblastoma cells: 20 μM Aβ1–42+ < 1 ‰ DMSO), and ZL006-treated groups (neurons: 75 μM ZL006 + 2 μM Aβ1–42; N2a neuroblastoma cells: 10 μM ZL006 + 20 μM Aβ1–42). The primary cortical neurons and N2a neuroblastoma cells were incubated with ZL006 or DMSO for 2 hours and treated with Aβ1–42 or NH3•H2O for another 24 hours, and then the following experiments were performed. To investigate the role of Akt/Nrf2 pathway, Akt inhibitor VIII group (neurons: 5 μM Akt inhibitor VIII + 75 μM ZL006 + 2 μM Aβ1–42; N2a neuroblastoma cells: 5 μM Akt inhibitor VIII + 10 μM ZL006 + 20 μM Aβ1–42) or ML385 group (neurons: 5 μM ML385 + 75 μM ZL006 + 2 μM Aβ1–42; N2a neuroblastoma cells: 5 μM ML385 + 10 μM ZL006 + 20 μM Aβ1–42) was included. The cells of these two groups were incubated with ZL006 and Akt inhibitor VIII or ML385 for 2 hours and treated with Aβ1–42 for another 24 hours. Then, CCK8 assay was performed.
CCK8 assay
Cell viability was determined using the CCK8 assay (Beyotime Biotechnology) according to the manufacturer’s instructions. Briefly, primary cortical neurons at day-in-vitro (DIV) 11–13 or N2a neuroblastoma cells were incubated with 10 μL CCK8 for 2 hours at 37°C in a 96-well plate, and the absorbance was measured at 450 nm in a plate reader (Bio-Rad, Hercules, CA, USA). Cell viability was expressed as the percentage live cells over the control ones (NH3•H2O group).
Cell apoptosis assay
The apoptotic rate of neurons or N2a neuroblastoma cells was determined by an Annexin V-FITC/propidium iodide (PI) kit (Vazyme, Nanjing, China). Briefly, the neurons at DIV 11–13 were incubated with the binding buffer containing Annexin V-FITC for 5 minutes in the dark, and the fluorescence was detected using a fluorescence microscope (Olympus, Tokyo, Japan). The N2a neuroblastoma cells were collected and suspended in 0.5 mL binding buffer containing 5 μL Annexin V and 10 μL PI, and incubated for 5 minutes at 37°C in the dark. The apoptotic rates including early apoptotic (AV+/PI–) and late apoptotic (AV+/PI+) N2a neuroblastoma cells were analyzed using a flow cytometer (BD Biosciences, Carlsbad, CA, USA).
Western blot assay
Western blot assay was performed as described previously (Yu et al., 2017). Primary cortical neurons or N2a neuroblastoma cells were pretreated with ZL006 or DMSO vehicle for 2 hours and then treated with Aβ1–42 or NH3•H2O vehicle for 24 hours. Then total proteins were extracted using RIPA lysis buffer (Beyotime, Nanjing, China; P0013C) and 30 μg protein of each group was separated by 10% sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred onto polyvinylidene fluoride membranes. These membranes were blocked with 5% non-fat milk for 1 hour and incubated overnight at 4°C with the following primary antibodies: rabbit anti-Bax monoclonal antibody (1:1000; Cell Signaling Technology, Danvers, MA, USA), rabbit anti-Bcl-2 monoclonal antibody (1:1000; Cell Signaling Technolog), rabbit anti-Phospho-Akt monoclonal antibody (Ser473) (1:1000; Cell Signaling Technology), rabbit anti-Akt monoclonal antibody (1:1000; Cell Signaling Technology), rabbit anti-Nrf2 monoclonal antibody (1:1000, Abcam, Cambridge, MA, USA), mouse anti-HO-1 polyclonal antibody (1:2000, Abcam), rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) polyclonal antibody (1:5000; Bioworld, Louis Park, MN, USA) and rabbit anti-β-tubulin polyclonal antibody (1:2000; Bioworld). After washed three times with TBS/Tween 20, the membranes were incubated with corresponding secondary antibodies (horseradish peroxidase-conjugated goat anti-rabbit IgG and horseradish peroxidase-conjugated goat anti-mouse IgG) at room temperature for 2 hours. The proteins were visualized with the ECL kit (Millipore, Boston, MA, USA). The intensity of bands was quantified using ImageJ (https://imagej.nih.gov/ij/, NIH, Bethesda, USA). The relative protein expression was expressed as the band intensity of each group/ the band intensity of the loading control β-tubulin or GAPDH.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was measured using JC-1 fluorescence assay (Beyotime, Nanjing, China) as described previously (Zhu et al., 2012). Briefly, the neurons at DIV 11–13 were incubated with the JC-1 working solution for 20 minutes in the dark, and the fluorescence was detected using a fluorescence microscope (Olympus). The monomer form of JC-1 (green) represents decreased MMP and aggregate form (red) represents relatively intact MMP. The N2a neuroblastoma cells were collected and suspended in JC-1 staining buffer, and incubated for 20 minutes at 37°C in the dark. The MMP of N2a neuroblastoma cells was examined using a flow cytometer (BD Biosciences, San Diego, CA, USA).
ROS detection
The intracellular ROS of primary cortical neurons was detected using a ROS assay kit (Genmed, Shanghai, China). Briefly, the neurons at DIV 11–13 were incubated with the staining solution (mixture of reagent B and reagent C) for 30 minutes in the dark and washed with Reagent D. Then the fluorescence was detected using a fluorescence microscope (Olympus). The intracellular ROS of N2a neuroblastoma cells was detected using a ROS assay kit (Jiancheng Bioengineering, Nanjing, China). N2a neuroblastoma cells were incubated with 10 μM 2,7-dichlorodi-hydrofluorescein diacetate at 37°C for 30 minutes in the dark, and subjected to the measurement of 2,7-dichlorodi-hydrofluorescein diacetate fluorescence by a fluorescence microplate reader (Hitachi, Tokyo, Japan).
Calcein acetoxymethylester/propidium iodide AM/PI staining assay
The viability of neurons was also determined by a calcein-acetoxymethylester/propidium iodide (AM/PI) Double Stain Kit (Invitrogen, Carlsbad, CA, USA). Cortical neurons at DIV 11–13 were incubated with the calcein-AM and PI buffer at 37°C for 15 minutes in the dark, and the viable cells (green fluorescence) and dead cells (red fluorescence) were observed by an inverted fluorescence microscope (Olympus) and the ratio of viable cells (green fluorescence) to total cells (green fluorescence and red fluorescence) was counted.
Immunostaining
The cells were fixed in 4% paraformaldehyde for 20 minutes, washed with PBS-T for 30 minutes, and blocked with 2% bovine serum albumin for 2 hours. Then the cells were incubated with anti-cleaved caspase-3 polyclonal antibody (1:200; Cell Signaling Technology) and mouse anti-microtubule-associated protein-2 (MAP2) monoclonal antibody (1:500; Abcam) at 4°C overnight, and washed with PBS-T for 30 minutes, then incubated with Alexa Fluor Plus 488, goat anti-mouse IgG (H+L) secondary antibody and Alexa Fluor Plus 594, goat anti-mouse IgG (H+L) secondary antibody (1:500; Invitrogen, Carlsbad, CA, USA) for 60 minutes at room temperature. DAPI reagent (1:1000; Bioworld, Louis Park, MN, USA) was used for the nucleus staining. Images were taken using a fluorescence microscope (Olympus, Japan) and the ratio of cleaved-caspase3 positive cells (red fluorescence) to MAP2 positive cells (green fluorescence) was counted.
Nitrite analysis
The concentration of nitric oxide was measured by a notric oxide detection kit (Beyotime, Nanjing, China). The supernatant of primary cortical neurons was collected and added with Griess reagent, and the absorbance was obtained at 540 nm with sodium nitrite as a standard curve.
Statistical analysis
Results were expressed as the mean ± SEM and analyzed by SPSS 16.0 statistical analytical software (SPSS, Chicago, IL, USA). Statistical analysis among groups was performed using one-way analysis of variance followed by Bonferroni’s post hoc test and P < 0.05 was considered statistically significant.
Results
ZL006 protects against Aβ1–42-induced primary cortical neuronal cell death
To determine the potential neurotoxicity of ZL006, different concentrations of ZL006 were added to the media of neurons. As shown in Figure 1B, ZL006 moderately decreased the viability of primary cortical neuron at the concentration of 150 μM, and it did not show any cytotoxicity at lower concentrations. ZL006 (50–100 μM) significantly increased the cell viability in Aβ1–42-treated neurons (P < 0.05; Figure 1C). In addition, Aβ1–42 treatment induced extensive neuronal cell death as shown by PI staining, and ZL006 pretreatment partially rescued the neurotoxicity (P < 0.01; Figure 1D and E).
ZL006 ameliorates Aβ1–42-induced apoptosis in primary neurons
To access whether ZL006 protected against Aβ1–42-induced apoptosis, Annexin V staining was performed. As shown in Figure 2A and B, AnnexinV-FITC+ neurons were significantly increased after Aβ1–42 treatment, while pretreatment of ZL006 significantly decreased the AnnexinV-FITC+ neurons (P < 0.01). In addition, the level of cleaved caspase-3 was significantly reduced in ZL006-pretreated neurons (P < 0.05; Figure 2C and D). ZL006 decreased the level of Bax (an apoptotic activator) and increased the level of Bcl-2 (an anti-apoptotic protein) in Aβ1–42-treated neurons (Figure 2E and F). These data demonstrated that ZL006 attenuated Aβ1–42-induced apoptosis in primary neurons.
Figure 2.

ZL006 ameliorates Aβ1–42-induced apoptosis in primary neurons.
Primary neurons at DIV 10–12 were pretreated with ZL006 (75 μM) or DMSO vehicle (< 1‰) for 2 hours and then treated with Aβ1–42 (2 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, and Annexin V-FITC staining analysis was performed. Original magnification, 40×, scale bar: 20 μm. (B) Quantitative analysis of green fluorescence intensity. n = 3 cells per group. (C) The level of cleaved caspase-3 and MAP2 of primary cortical neurons at DIV 11–13 was determined by immunostaining. Original magnification, 20×, scale bar: 50 μm. (D) Quantitative analysis of the level of cleaved caspase-3. n = 4 cells per group. (E) The protein expressions of Bax and Bcl-2 in primary neurons at DIV 11–13 were examined by western blot assay. n = 4 cells per group. Data were expressed as mean ± SEM (one-way analysis of variance followed by Bonferroni's post hoc test). *P < 0.05, **P < 0.01, vs. NH3•H2O group; #P < 0.05, ##P < 0.01, vs. Aβ1–42 + DMSO group. Aβ: Amyloid-beta; DAPI: 4',6-diamidino-2-phenylindole; DIV: day- in-vitro; DMSO: dimethyl sulfoxide; FITC: fluoresceine isothiocyanate; MAP2: microtubule-associated protein 2.
ZL006 attenuates Aβ1–42-induced oxidative stress in primary cortical neurons
To evaluate the potential anti-oxidative effects of ZL006, the levels of MMP and intracellular ROS were examined in Aβ1–42-treated primary cortical neurons. As shown in (Figure 3A and B), Aβ1–42 significantly decreased the MMP in primary cortical neurons, while ZL006 pretreatment partially reversed the MMP loss (P < 0.01). Intracellular ROS was increased after Aβ1–42 treatment, and ZL006 pretreatment significantly inhibited the ROS generation in neurons (P < 0.01; Figure 3C and D). In addition, Aβ1–42 treatment induced a significant increase of NO release in the supernatant (P < 0.05). However, the level of NO was decreased by ZL006 pretreatment (P < 0.05; Figure 3E). These results suggested that ZL006 decreased the oxidative stress in Aβ1–42-treated neurons.
Figure 3.
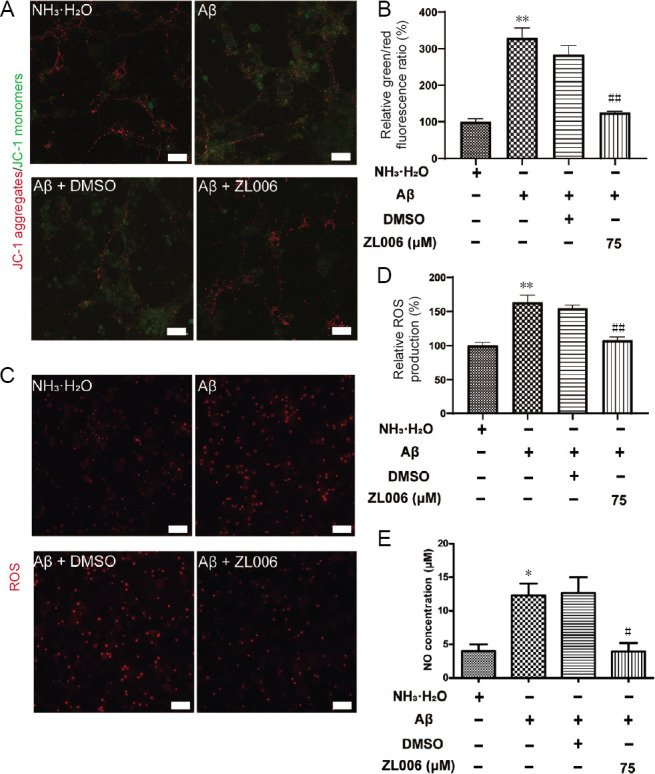
ZL006 attenuates Aβ1–42-induced oxidative stress in primary cortical neurons.
(A) Cortical neuronal cells at DIV 10–12 were pretreated with ZL006 (75 μM) or DMSO vehicle (< 1‰) for 2 hours and then treated with Aβ1–42 (2 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, and MMP was measured using JC-1 fluorescence assay (JC-1 fluorescent probe; green: JC-1 monomers; red: JC-1 aggregates). Original magnification, 40×, scale bar: 20 μm. (B) Quantitative analysis of green/red fluorescence ratio. n = 3 cells per group. (C) The level of intracellular ROS in neurons at DIV 11–13 was measured by fluorescence assay. Original magnification, 20×, scale bars: 50 μm. (D) Quantitative analysis of ROS. n = 4 cells per group. (E) The NO release in the supernatants of neurons at DIV 11–13 was examined by Griess reaction. n = 3–4 cells per group. Data were expressed as mean ± SEM (one-way analysis of variance followed by Bonferroni's post hoc test). *P < 0.05, **P < 0.01, vs. NH3•H2O group; #P < 0.05, vs. ##P < 0.01, vs. Aβ1–42 + DMSO group. Aβ: Amyloid-beta; DIV: day- in-vitro; DMSO: dimethyl sulfoxide; JC-1: 5,5', 6,6'-tetrachloro-1,1', 3,3'-tetraethyl-imidacarbocyanine; NO: nitric oxide; ROS: reactive oxygen species.
ZL006 protects against Aβ1–42-induced neurotoxicity partially by Akt/Nrf2/HO-1 pathway in primary cortical neurons
Given that Akt/Nrf2/HO-1 pathways played an important role in the pathogenesis of AD, we examined whether ZL006 could modulate the Akt/Nrf2/HO-1 pathway. As shown in (Figure 4A and B), the level of p-Akt was significantly decreased after Aβ1–42 treatment (P < 0.01), while ZL006 pretreatment induced the phosphorylation of Akt (P < 0.01). In addition, ZL006 pretreatment increased the levels of Nrf2 and HO-1 in Aβ1–42-treated primary neurons (P < 0.05; Figure 4A and C). Furthermore, Akt inhibitor VIII (Figure 4D) or ML385 (Figure 4E) pretreatment reversed the beneficial effects of ZL006 in Aβ1–42-treated neurons, suggesting that ZL006 protects primary cortical neurons partially by activating the Akt/Nrf2/HO-1 pathway.
Figure 4.

ZL006 protects against Aβ1–42-induced neurotoxicity partially by the Akt/Nrf2/HO-1 pathway.
(A) Primary neurons at DIV 10–12 were pretreated with ZL006 (75 μM) or DMSO vehicle (< 1‰) for 2 hours and then treated with Aβ1–42 (2 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, and the levels of p-Akt/Akt, Nrf2 and HO-1 were determined by western blot assay. (B, C) Quantitative analysis of the relative levels of p-Akt/Akt. n = 4 cells per group. (D) Primary neurons at DIV 10–12 were pretreated with ZL006 (75 μM) or DMSO vehicle (< 1‰) with or without Akt inhibitor VIII (5 μM) for 2 hours and then treated with Aβ1–42 (2 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, and cell viability was determined by CCK8 assay. n = 12 cells per group (analysis of variance with Bonferroni's post hoc test). (E) Primary neurons at DIV 10–12 were pretreated with ZL006 (75 μM) or DMSO vehicle (< 1‰) with or without ML385 (5 μM) for 2 hours and then treated with Aβ1–42 (2 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, and cell viability was determined by CCK8 assay. n = 12 cells per group. Data were expressed as mean ± SEM (one-way analysis of variance followed by Bonferroni's post hoc test). *P < 0.05, **P < 0.01, vs. NH3•H2O vehicle group; # P< 0.05, ##P < 0.01, vs. Aβ1–42+ DMSO group; &&P < 0.01, vs. Aβ1–42+ ZL006 group. Aβ: Amyloid-beta; DMSO: dimethyl sulfoxide; Nrf2: NF-E2-related factor 2; HO-1: heme oxygenase-1.
ZL006 inhibits Aβ1–42-induced neurotoxicity and oxidative stress by Akt/Nrf2/HO-1 pathway in N2a cells
To further confirm the neuroprotective effects of ZL006 against Aβ, N2a neuroblastoma cells were used. Similarly, Aβ1–42 treatment resulted in cell viability reduction in N2a neuroblastoma cells, and ZL006 partially rescued the detrimental effects (Figure 5A). In addition, ZL006 significantly reduced the apoptotic rate in Aβ1–42-treated N2a neuroblastoma cells (Figure 5B and C). The MMP was accessed by JC-1 staining, and the results demonstrated that ZL006 protected against Aβ1–42 induced MMP loss (Figure 5D and E). The ROS level was significantly increased in Aβ1–42-treated N2a neuroblastoma cells, while ZL006 pretreatment reduced the ROS production (Figure 5F). Next we examined the effects of ZL006 on Akt/Nrf2/HO-1 pathway in Aβ1–42-treated N2a neuroblastoma cells. As expected, ZL006 induced the phosphorylation of Akt, and upregulated the expressions of Nrf2 and HO-1 in Aβ1–42-treated N2a neuroblastoma cells (Figure 5G–I). Aβ1–42 exposure decreased the viability of N2a neuroblastoma cells, while Akt inhibitor VIII or ML385 pretreatment counteracted the protective effects of ZL006 (Figure 5J). These results showed that ZL006 protected N2a neuroblastoma cells against Aβ1–42-induced neurotoxicity and oxidative stress, and activated the Akt/Nrf2/HO-1 pathway, which suggested that ZL006 might be an alternative compound for AD treatment.
Figure 5.
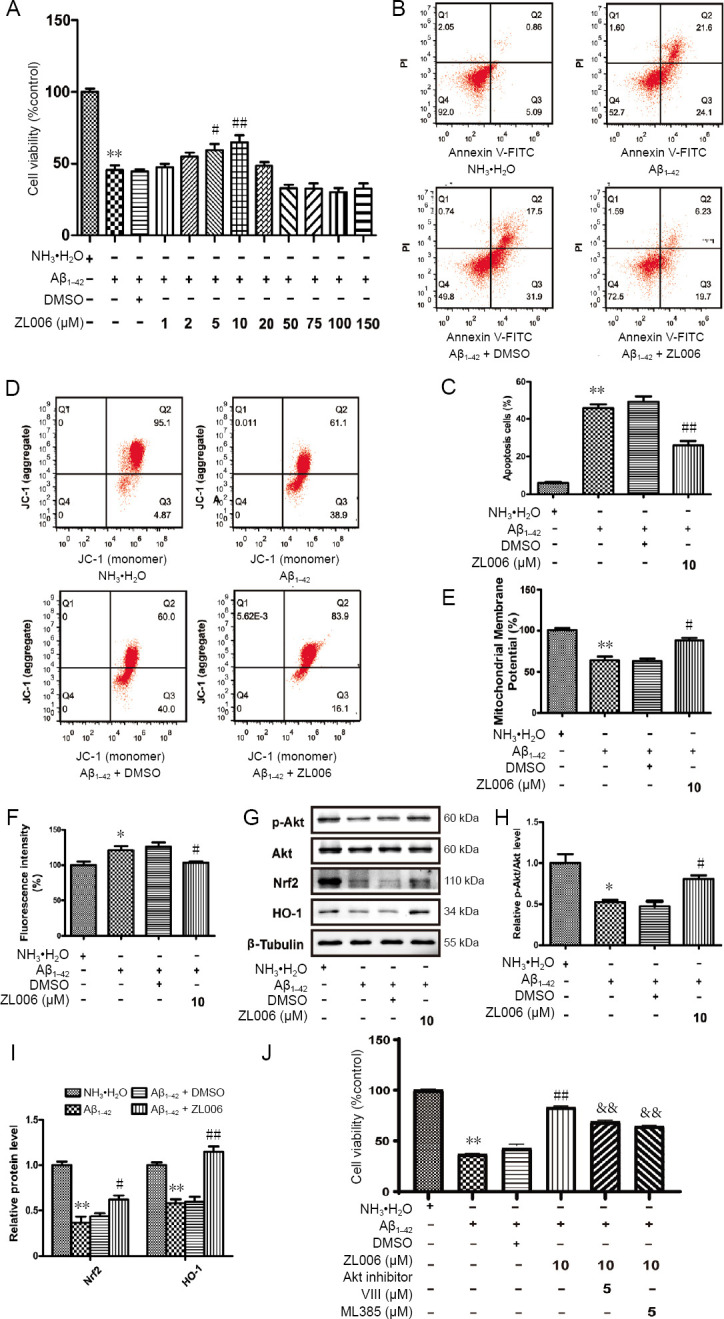
ZL006 inhibits Aβ1–42-induced neurotoxicity and oxidative stress by Akt/Nrf2/HO-1 pathway in N2a neuroblastoma cells.
(A) N2a neuroblastoma cells were pretreated with ZL006 (10 μM) or DMSO vehicle (< 1‰) for 2 hours and then treated with Aβ1–42 (20 μM) or NH3•H2O vehicle (< 1‰) for 24 hours, the cell viability was determined using the CCK8 assay. n = 8–12 cells per group. (B) The apoptotic rate of N2a neuroblastoma cells (early apoptotic (AV+/PI–) and late apoptotic (AV+/PI+) was accessed using AnnexinV/ PI flow cytometry. (C) Quantitative analysis of the apoptotic rate. The apoptotic cells included the AnnexinV-FITC+/PI– and AnnexinV-FITC+/PI+ cells. n = 4 cells per group. (D) The MMP of N2a neuroblastoma cells was examined by JC-1 flow cytometry. (E) Quantitative analysis of JC-1 assay. n = 3 cells per group. (F) The level of ROS in N2a neuroblastoma cells was measured by flow cytometry. n = 4 cells per group. (G) The protein levels of p-Akt/Akt, Nrf2 and HO-1 in N2a neuroblastoma cells were determined by western blot assay. (H, I) Quantitative analysis of the relative protein levels of p-Akt/Akt. n = 3 cells per group. (J) N2a cells were pretreated with ZL006 (10 μM) with or without Akt inhibitor VIII (5 μM) or ML385 (5 μM) for 2 hours and then treated with Aβ1–42 (20 μM) for 24 hours, and cell viability was determined by the CCK8 assay. n = 10–12 cells per group. Data were expressed as the mean ± SEM (one-way analysis of variance followed by Bonferroni's post hoc test). *P < 0.05, **P < 0.01, vs. NH3•H2O group; #P < 0.05, ##P < 0.01, vs. Aβ1–42 + DMSO group; &&P < 0.01, vs. Aβ1–42+ ZL006 group. Aβ: Amyloid-beta; DMSO: dimethyl sulfoxide; JC-1: 5,5', 6,6'-tetrachloro- 1,1', 3,3'-tetraethyl-imidacarbocyanine; Nrf2: NF-E2-related factor 2; HO-1: heme oxygenase-1.
Discussion
Oxidative stress plays an important role in the progression of AD, making it a potential target for the treatment (Li et al., 2018a; Onyango, 2018; Weinstein, 2018). In this study, we found that Aβ1–42 reduced neuronal viability and promoted neuronal apoptosis and oxidative stress in primary cortical neurons and N2a neuroblastoma cells. However, ZL006 treatment decreased neuronal apoptosis and oxidative stress induced by Aβ1–42. In addition, ZL006 protected against Aβ1–42-induced neurotoxicity partially by the activation of the Akt/Nrf2/HO-1 pathway.
The neuroprotective effects of ZL006 have been extensively studied in ischemic stroke, traumatic brain injury, Parkinson’s disease and hemorrhage-induced thalamic pain (Zhou et al., 2010; Hu et al., 2014; Cai et al., 2018; Qu et al., 2020). ZL006 is designed to selectively interrupt the interaction between PSD95 and nNOS, which plays a critical role in synaptic transmission and neuronal functions. ZL006 treatment ameliorates ischemic injury in oxygen-glucose deprivation (OGD) treated primary cortical neurons and in middle cerebral artery-occluded mice and rats (Zhou et al., 2010). In addition, ZL006 treatment promotes the migration and differentiation of transplanted neural stem cells in ischemic stroke models by upregulating the activity of cAMP responsive element binding protein (Wang et al., 2017). ZL006 treatment attenuates apoptosis and neurological deficits in the controlled cortical impact mouse model, indicating that ZL006 might be used to treat traumatic brain injury (Qu et al., 2020). Hu et al. (2014) reported that ZL006 reduced neuronal apoptosis and oxidative stress by upregulating sirtuin 3 in 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPP+)-treated neurons. Moreover, systemic or intra-amygdala treatment of ZL006 alleviated conditioned fear, while it did not affect object recognition memory and spatial memory, suggesting that ZL006 is a potential drug for the treatment of fear-related disorders (Li et al., 2018b; Song et al., 2019). In this study, we found that ZL006 ameliorated Aβ1–42-induced oxidative stress and neuronal apoptosis. Interestingly, ZL006 did not affect spatial memory, which was compromised in the early stage of AD, in normal conditions (Zhou et al., 2010; Li et al., 2018b). Whether ZL006 treatment improves spatial memory in AD models needs further investigations.
Neuronal apoptosis plays a critical role in the development and homeostasis of the central nervous system, and Aβ induced neuronal apoptosis contributes to the pathogenesis of AD (Obulesu and Lakshmi, 2014). Although the underling mechanisms are not fully defined, it is generally considered that Aβ directly or indirectly targets mitochondria, and induces the depolarization of MMP, which results in the release of cytochrome c and activation of caspase-9 and caspase-3. Aβ treatment also triggers the upregulation of pro-apoptotic protein including Bax and downregulation of anti-apoptotic protein including Bcl-2. Humanin is able to bind to Bax and prevents the translocation of Bax to the mitochondria to inhibit cellular apoptosis. In addition, humanin and its derivatives protect against learning and memory deficits in Aβ-induced AD models (Li et al., 2013; Chai et al., 2014; Romeo et al., 2017). A point mutation of Beclin 1 (F121A) disrupts the interaction of Beclin 1 and Bcl-2, and attenuates cognitive impairment in AD mice (Rocchi et al., 2017). Several chemicals, including Sulforaphane, Tanshinone IIA, and Dalesconol B, have been reported to decrease Aβ-induced neuronal apoptosis and memory decline in vitro and in vivo (Zhu et al., 2014; Li et al., 2016; Hou et al., 2018). Here, we showed that ZL006 mitigated Aβ1–42-induced neuronal apoptosis and promoted the survival of neurons, which might contribute to the neuroprotective effects of ZL006.
It is well known that increased Aβ induces extensive oxidative stress, which in turn contributes to the accumulation of amyloid plaques through the amyloidogenic pathway (Gu et al., 2013; Zuo et al., 2015). The generation of ROS and antioxidant molecules and enzymes is imbalanced in AD, which leads to the aggravation of oxidative stress (Zuo et al., 2015). In addition, Aβ-induced oxidative stress contributes to the pathology of tau phosphorylation by activating the JNK/ p38 mitogen-activated protein kinase pathway, suggesting the association between oxidative stress and tau hyperphosphorylation (Giraldo et al., 2014). Moreover, inhibition of oxidative stress seems to be an attractive strategy for AD treatment (Cheignon et al., 2018; Wojsiat et al., 2018). Resveratrol upregulates the activity of sirtuin 1 and attenuates spatial memory impairment and synaptic dysfunctions in Aβ1–42-treated rats (Gomes et al., 2018). Our previous results also demonstrate that Hopeahainol A and Diammonium glycyrrhizinate decrease the oxidative response and improve spatial memory in AD mice (Zhu et al., 2012, 2013). Here, we identified that ZL006 pretreatment could significantly inhibit the oxidative stress in Aβ1–42-treated neurons, which was consistent with previous studies showing the antioxidant effects of ZL006 (Hu et al., 2014; Liu et al., 2017).
Increasing evidence shows that activation of the Akt pathway is a potential therapeutic strategy for AD treatment (Heras-Sandoval et al., 2014; Kitagishi et al., 2014; Rai et al., 2019). The level of Akt phosphorylation and the activity of Akt1 are decreased in the brain samples of AD patients (Lee et al., 2009). Constitutive activation of Akt protects hippocampal neurons against mutant presenilin-1-induced cell death (Weihl et al., 1999). Our previous data show that Dalesconol B attenuates memory deficits and Aβ1–42-induced neuronal apoptosis by activating the Akt pathway (Zhu et al., 2014). However, the effects of Aβ on the Akt pathway are still controversial. Akt activity and phosphorylation are decreased in human neuroblastoma SH-SY5Y cells expressing Aβ, while the PI3K activity is not affected (Lee et al., 2009). Acute Aβ1–42 exposure induces phosphorylation of Akt in primary neurons, which is dependent of the activation of both NMDA receptor and 7 nicotinic receptor. The level of Akt phosphorylation is compromised in the hippocampus of 13-month-old AD transgenic (TAS10) mice (Abbott et al., 2008). We have demonstrated that Aβ1–42 treatment inhibits the Akt activity and phosphorylation in SH-SY5Y cells and in Aβ1–42 treated mice (Zhu et al., 2014). In this study, the phosphorylation of Akt was inhibited in Aβ1–42-treated primary cortical neurons and N2a neuroblastoma cells, which might be due to the diversity of Aβ species.
Akt modulates the pathogenesis of AD by regulating downstream kinases including GSK3β, which has been extensively studied as a tau kinase (Hernandez et al., 2013; Bhat et al., 2018). Nrf2/HO-1 axis is a downstream target of the Akt pathway (Martin et al., 2004; Dai et al., 2007; Rong et al., 2018; Wahyudi et al., 2018). Nrf2 is decreased in the brains of AD mice, and activation of Nrf2 pathway or Nrf2 overexpression protects neurons against Aβ-induced neurotoxicity (Kanninen et al., 2008). Nrf2 deficiency exacerbates Aβ or Tau-associated neuropathology and spatial memory deficits in APP or Tau transgenic mice (Rojo et al., 2017). Furthermore, HO-1 is localized with neurofibrillary tangles and senile plaque in the brains of AD patients, and is associated with the neurofibrillary pathology of AD (Smith et al., 1994). The plasma level of HO-1 is significantly decreased in AD patients (Schipper et al., 2000; Ishizuka et al., 2002), indicating that HO-1 might be a biomarker and therapeutic target for AD. Sulforaphane activates the Nrf2/HO-1 pathway to exert its anti-inflammatory effect against Aβ (An et al., 2016). Orientin promotes Nrf2 translocation from cytoplasm to nucleus and attenuates spatial memory deficits in Aβ1–42-induced AD mice (Yu et al., 2015). In this study, it was shown that ZL006 pretreatment activated the Akt/Nrf2/HO-1 pathway in Aβ1–42-treated neuronal cells, which contributed to the neuroprotective effects of ZL006.
In conclusion, the current study demonstrated that ZL006 could protect neurons against Aβ1–42-induced neurotoxicity and inhibit oxidative stress. In addition, ZL006 activated the Akt/Nrf2/HO-1 pathway in Aβ1–42-treated neurons, which might be associated with its neuroprotective effects. Thus, these results suggest that ZL006 might be a potential compound for AD treatment. Since ZL006 is able to cross the blood-brain barrier quickly without major side effects (Zhou et al., 2010), further studies are needed to investigate the neuroprotective effects of ZL006 and the potential role of the Akt/Nrf2/HO-1 pathway in AD models.
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support: This study was supported by the National Nature Science Foundation of China, Nos. 81671055, 81971009 (to XLZ), the Key Research and Development Program of Jiangsu Province of China, No. BE2016610 (to YX), the National Key Research and Development Program of China, No. 2016YFC1300504 (to YX) and No. 2018YFC1704405 (to XLZ), Jiangsu Province Key Medical Discipline, No. ZDXKA2016020 (to YX), Jiangsu Province Medical Youth Talent, No. QNRC2016024 (to XLZ), and Young Talent Support Program from Jiangsu Association for Science and Technology (to XLZ). The funding bodies played no role in the study design, collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
Institutional review board statement: No ethical issue is considered because PC12 cells are commercially available.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This study was supported by the National Natural Science Foundation of China, Nos. 81671055, 81971009 (to XLZ), the Key Research and Development Program of Jiangsu Province of China, No. BE2016610 (to YX), the National Key Research and Development Program of China, No. 2016YFC1300504 (to YX) and No. 2018YFC1704405 (to XLZ), Jiangsu Province Key Medical Discipline, No. ZDXKA2016020 (to YX), Jiangsu Province Medical Youth Talent, No. QNRC2016024 (to XLZ), and Young Talent Support Program from Jiangsu Association for Science and Technology (to XLZ).
C-Editor: Zhao M; S-Editor: Wang J; L-Editor: Song LP; T-Editor: Jia Y
References
- 1.Abbott JJ, Howlett DR, Francis PT, Williams RJ. Abeta (1-42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiol Aging. 2008;29:992–1001. doi: 10.1016/j.neurobiolaging.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer’s Association (2016) 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Aminyavari S, Zahmatkesh M, Farahmandfar M, Khodagholi F, Dargahi L, Zarrindast MR. Protective role of Apelin-13 on amyloid beta25-35-induced memory deficit; Involvement of autophagy and apoptosis process. Prog Neuropsychopharmacol Biol Psychiatry. 2018;89:322–334. doi: 10.1016/j.pnpbp.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 4.An YW, Jhang KA, Woo SY, Kang JL, Chong YH. Sulforaphane exerts its anti-inflammatory effect against amyloid-beta peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiol Aging. 2016;38:1–10. doi: 10.1016/j.neurobiolaging.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Barnes DE, Yaffe K. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;353:951–952. doi: 10.1056/NEJMc051856. [DOI] [PubMed] [Google Scholar]
- 6.Bhat RV, Andersson U, Andersson S, Knerr L, Bauer U, Sundgren-Andersson AK. The conundrum of GSK3 inhibitors: is it the dawn of a new beginning. J Alzheimers Dis. 2018;64:S547–554. doi: 10.3233/JAD-179934. [DOI] [PubMed] [Google Scholar]
- 7.Cai W, Wu S, Pan Z, Xiao J, Li F, Cao J, Zang W, Tao YX. Disrupting interaction of PSD-95 with nNOS attenuates hemorrhage-induced thalamic pain. Neuropharmacology. 2018;141:238–248. doi: 10.1016/j.neuropharm.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chai GS, Duan DX, Ma RH, Shen JY, Li HL, Ma ZW, Luo Y, Wang L, Qi XH, Wang Q, Wang JZ, Wei Z, Mousseau DD, Wang L, Liu G. Humanin attenuates Alzheimer-like cognitive deficits and pathological changes induced by amyloid beta-peptide in rats. Neurosci Bull. 2014;30:923–935. doi: 10.1007/s12264-014-1479-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong ZZ, Li F, Maiese K. Cellular demise and inflammatory microglial activation during beta-amyloid toxicity are governed by Wnt1 and canonical signaling pathways. Cell Signal. 2007;19:1150–1162. doi: 10.1016/j.cellsig.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA, Jr Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101:723–733. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 12.de Vries HE, Witte M, Hondius D, Rozemuller AJ, Drukarch B, Hoozemans J, van Horssen J. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease. Free Radic Biol Med. 2008;45:1375–1383. doi: 10.1016/j.freeradbiomed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Gan SY, Wong LZ, Wong JW, Tan EL. Fucosterol exerts protection against amyloid beta-induced neurotoxicity, reduces intracellular levels of amyloid beta and enhances the mRNA expression of neuroglobin in amyloid beta-induced SH-SY5Y cells. Int J Biol Macromol. 2018;121:207–213. doi: 10.1016/j.ijbiomac.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 14.Giraldo E, Lloret A, Fuchsberger T, Vina J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–877. doi: 10.1016/j.redox.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomes BAQ, Silva JPB, Romeiro CFR, Dos Santos SM, Rodrigues CA, Goncalves PR, Sakai JT, Mendes PFS, Varela ELP, Monteiro MC. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid Med Cell Long. 2018;2018:8152373. doi: 10.1155/2018/8152373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu X, Sun J, Li S, Wu X, Li L. Oxidative stress induces DNA demethylation and histone acetylation in SH-SY5Y cells: potential epigenetic mechanisms in gene transcription in Abeta production. Neurobiol Aging. 2013;34:1069–1079. doi: 10.1016/j.neurobiolaging.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 18.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15:767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 19.Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26:2694–2701. doi: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez F, Lucas JJ, Avila J. GSK3 and tau: two convergence points in Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S141–144. doi: 10.3233/JAD-2012-129025. [DOI] [PubMed] [Google Scholar]
- 21.Hou TT, Yang HY, Wang W, Wu QQ, Tian YR, Jia JP. Sulforaphane inhibits the generation of amyloid-beta oligomer and promotes spatial learning and memory in Alzheimer’s Disease (PS1V97L) transgenic mice. J Alzheimers Dis. 2018;62:1803–1813. doi: 10.3233/JAD-171110. [DOI] [PubMed] [Google Scholar]
- 22.Hu W, Guan LS, Dang XB, Ren PY, Zhang YL. Small-molecule inhibitors at the PSD-95/nNOS interface attenuate MPP+-induced neuronal injury through Sirt3 mediated inhibition of mitochondrial dysfunction. Neurochem Int. 2014;79:57–64. doi: 10.1016/j.neuint.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 24.Ishizuka K, Kimura T, Yoshitake J, Akaike T, Shono M, Takamatsu J, Katsuragi S, Kitamura T, Miyakawa T. Possible assessment for antioxidant capacity in Alzheimer’s disease by measuring lymphocyte heme oxygenase-1 expression with real-time RT-PCR. Ann N Y Acad Sci. 2002;977:173–178. doi: 10.1111/j.1749-6632.2002.tb04814.x. [DOI] [PubMed] [Google Scholar]
- 25.Kanninen K, Malm TM, Jyrkkanen HK, Goldsteins G, Keksa-Goldsteine V, Tanila H, Yamamoto M, Yla-Herttuala S, Levonen AL, Koistinaho J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci. 2008;39:302–313. doi: 10.1016/j.mcn.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Kitagishi Y, Nakanishi A, Ogura Y, Matsuda S. Dietary regulation of PI3K/AKT/GSK-3beta pathway in Alzheimer’s disease. Alzheimers Res Ther. 2014;6:35. doi: 10.1186/alzrt265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kryscio RJ, Abner EL, Caban-Holt A, Lovell M, Goodman P, Darke AK, Yee M, Crowley J, Schmitt FA. Association of antioxidant supplement use and dementia in the prevention of Alzheimer’s Disease by vitamin e and selenium trial (PREADViSE) JAMA Neurol. 2017;74:567–573. doi: 10.1001/jamaneurol.2016.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW. The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol Bio Cell. 2009;20:1533–1544. doi: 10.1091/mbc.E08-07-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F, Han G, Wu K. Tanshinone IIA alleviates the AD phenotypes in APP and PS1 transgenic mice. BioMed Res Int. 2016;2016:7631801. doi: 10.1155/2016/7631801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li JG, Chu J, Pratico D. Downregulation of autophagy by 12/15Lipoxygenase worsens the phenotype of an Alzheimer’s disease mouse model with plaques, tangles, and memory impairments. Mol Psychiat. 2018a doi: 10.1038/s41380-018-0268-1. doi: 101038/s41380-018-0268-1. [DOI] [PubMed] [Google Scholar]
- 31.Li LP, Dustrude ET, Haulcomb MM, Abreu AR, Fitz SD, Johnson PL, Thakur GA, Molosh AI, Lai Y, Shekhar A. PSD95 and nNOS interaction as a novel molecular target to modulate conditioned fear: relevance to PTSD. Transl Psychiat. 2018b;8:155. doi: 10.1038/s41398-018-0208-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Zhao W, Yang H, Zhang J, Ma J. S14G-humanin restored cellular homeostasis disturbed by amyloid-beta protein. Neural Regen Res. 2013;8:2573–2580. doi: 10.3969/j.issn.1673-5374.2013.27.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin YH, Liang HY, Xu K, Ni HY, Dong J, Xiao H, Chang L, Wu HY, Li F, Zhu DY, Luo CX. Dissociation of nNOS from PSD-95 promotes functional recovery after cerebral ischaemia in mice through reducing excessive tonic GABA release from reactive astrocytes. J Pathol. 2018;244:176–188. doi: 10.1002/path.4999. [DOI] [PubMed] [Google Scholar]
- 34.Liu SG, Wang YM, Zhang YJ, He XJ, Ma T, Song W, Zhang YM. ZL006 protects spinal cord neurons against ischemia-induced oxidative stress through AMPK-PGC-1alpha-Sirt3 pathway. Neurochem Int. 2017;108:230–237. doi: 10.1016/j.neuint.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 36.Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Wilson RS, Scherr PA. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA. 2002a;287:3230–3237. doi: 10.1001/jama.287.24.3230. [DOI] [PubMed] [Google Scholar]
- 37.Morris MC, Evans DA, Bienias JL, Tangney CC, Wilson RS. Vitamin E and cognitive decline in older persons. Arch Neurol. 2002b;59:1125–1132. doi: 10.1001/archneur.59.7.1125. [DOI] [PubMed] [Google Scholar]
- 38.Obulesu M, Lakshmi MJ. Apoptosis in Alzheimer’s disease: an understanding of the physiology, pathology and therapeutic avenues. Neurocheml Res. 2014;39:2301–2312. doi: 10.1007/s11064-014-1454-4. [DOI] [PubMed] [Google Scholar]
- 39.Onyango IG. Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease. Neural Regen Res. 2018;13:19–25. doi: 10.4103/1673-5374.224362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu W, Liu NK, Wu X, Wang Y, Xia Y, Sun Y, Lai Y, Li R, Shekhar A, Xu XM. Disrupting nNOS-PSD95 interaction improves neurological and cognitive recoveries after traumatic brain injury. Cereb Cortex. 2020 doi: 10.1093/cercor/bhaa002. doi: 101093/cercor/bhaa002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 42.Rai SN, Dilnashin H, Birla H, Singh SS, Zahra W, Rathore AS, Singh BK, Singh SP. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res. 2019;35:775–795. doi: 10.1007/s12640-019-0003-y. [DOI] [PubMed] [Google Scholar]
- 43.Rocchi A, Yamamoto S, Ting T, Fan Y, Sadleir K, Wang Y, Zhang W, Huang S, Levine B, Vassar R, He C. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet. 2017;13:e1006962. doi: 10.1371/journal.pgen.1006962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rojo AI, Pajares M, Rada P, Nunez A, Nevado-Holgado AJ, Killik R, Van Leuven F, Ribe E, Lovestone S, Yamamoto M, Cuadrado A. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–451. doi: 10.1016/j.redox.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romeo M, Stravalaci M, Beeg M, Rossi A, Fiordaliso F, Corbelli A, Salmona M, Gobbi M, Cagnotto A, Diomede L. Humanin specifically interacts with amyloid-beta oligomers and counteracts their in vivo toxicity. J Alzheimers Dis. 2017;57:857–871. doi: 10.3233/JAD-160951. [DOI] [PubMed] [Google Scholar]
- 46.Rong H, Liang Y, Niu Y. Rosmarinic acid attenuates beta-amyloid-induced oxidative stress via Akt/GSK-3beta/Fyn-mediated Nrf2 activation in PC12 cells. Free Radic Biol Med. 2018;120:114–123. doi: 10.1016/j.freeradbiomed.2018.03.028. [DOI] [PubMed] [Google Scholar]
- 47.Schipper HM, Chertkow H, Mehindate K, Frankel D, Melmed C, Bergman H. Evaluation of heme oxygenase-1 as a systemic biological marker of sporadic AD. Neurology. 2000;54:1297–1304. doi: 10.1212/wnl.54.6.1297. [DOI] [PubMed] [Google Scholar]
- 48.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, Nimmagadda S, Sudini K, Brimacombe KR, Gajghate S, Ma J, Wang A, Xu X, Shahane SA, Xia M, Woo J, Mensah GA, Wang Z, Ferrer M, Gabrielson E, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016;11:3214–3225. doi: 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, Wiggert B, Petersen RB, Perry G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am J Pathol. 1994;145:42–47. [PMC free article] [PubMed] [Google Scholar]
- 51.Song S, Lee J, Park S, Choi S. Fear renewal requires nitric oxide signaling in the lateral amygdala. Biochem Biophys Res Commun. 2019;523:86–90. doi: 10.1016/j.bbrc.2019.12.038. [DOI] [PubMed] [Google Scholar]
- 52.Tan M, Ouyang Y, Jin M, Chen M, Liu P, Chao X, Chen Z, Chen X, Ramassamy C, Gao Y, Pi R. Downregulation of Nrf2/HO-1 pathway and activation of JNK/c-Jun pathway are involved in homocysteic acid-induced cytotoxicity in HT-22 cells. Toxicol Lett. 2013;223:1–8. doi: 10.1016/j.toxlet.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 53.Turner RS, Thomas RG, Craft S, van Dyck CH, Mintzer J, Reynolds BA, Brewer JB, Rissman RA, Raman R, Aisen PS Alzheimer’s Disease Cooperative S (2015) A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology. 85:1383–1391. doi: 10.1212/WNL.0000000000002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wahyudi LD, Jeong J, Yang H, Kim JH. Amentoflavone-induced oxidative stress activates NF-E2-related factor 2 via the p38 MAP kinase-AKT pathway in human keratinocytes. Int J Biochem Cell Biol. 2018;99:100–108. doi: 10.1016/j.biocel.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 55.Wang DL, Qian XD, Lin YH, Tian BB, Liang HY, Chang L, Wu HY, Zhu DY, Luo CX. ZL006 promotes migration and differentiation of transplanted neural stem cells in male rats after stroke. J Neurosci Res. 2017;95:2409–2419. doi: 10.1002/jnr.24068. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Wu C, Han B, Xu F, Mao M, Guo X, Wang J. Dexmedetomidine attenuates repeated propofol exposure-induced hippocampal apoptosis, PI3K/Akt/Gsk-3beta signaling disruption, and juvenile cognitive deficits in neonatal rats. Mol Med Rep. 2016;14:769–775. doi: 10.3892/mmr.2016.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinstein JD. A new direction for Alzheimer’s research. Neural Regen Res. 2018;13:190–193. doi: 10.4103/1673-5374.226381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wojsiat J, Zoltowska KM, Laskowska-Kaszub K, Wojda U. Oxidant/antioxidant imbalance in Alzheimer’s Disease: therapeutic and diagnostic prospects. Oxid Med Cell Longev. 2018;2018:6435861. doi: 10.1155/2018/6435861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu Y, Tao YX. Involvement of the NMDA receptor/nitric oxide signal pathway in platelet-activating factor-induced neurotoxicity. Neuroreport. 2004;15:263–266. doi: 10.1097/00001756-200402090-00010. [DOI] [PubMed] [Google Scholar]
- 61.Yi JH, Baek SJ, Heo S, Park HJ, Kwon H, Lee S, Jung J, Park SJ, Kim BC, Lee YC, Ryu JH, Kim DH. Direct pharmacological Akt activation rescues Alzheimer’s disease like memory impairments and aberrant synaptic plasticity. Neuropharmacology. 2018;128:282–292. doi: 10.1016/j.neuropharm.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 62.Yu L, Liu Y, Jin Y, Cao X, Chen J, Jin J, Gu Y, Bao X, Ren Z, Xu Y, Zhu X. Lentivirus-Mediated HDAC3 inhibition attenuates oxidative stress in APPswe/PS1dE9 mice. J Alzheimers Dis. 2018;61:1411–1424. doi: 10.3233/JAD-170844. [DOI] [PubMed] [Google Scholar]
- 63.Yu L, Liu Y, Yang H, Zhu X, Cao X, Gao J, Zhao H, Xu Y. PSD-93 attenuates amyloid-beta-mediated cognitive dysfunction by promoting the catabolism of amyloid-beta. J Alzheimers Dis. 2017;59:913–927. doi: 10.3233/JAD-170320. [DOI] [PubMed] [Google Scholar]
- 64.Yu L, Wang S, Chen X, Yang H, Li X, Xu Y, Zhu X. Orientin alleviates cognitive deficits and oxidative stress in Abeta1-42-induced mouse model of Alzheimer’s disease. Life Sci. 2015;121:104–109. doi: 10.1016/j.lfs.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 65.Zhou L, Li F, Xu HB, Luo CX, Wu HY, Zhu MM, Lu W, Ji X, Zhou QG, Zhu DY. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat Med. 2010;16:1439–1443. doi: 10.1038/nm.2245. [DOI] [PubMed] [Google Scholar]
- 66.Zhu X, Chen C, Ye D, Guan D, Ye L, Jin J, Zhao H, Chen Y, Wang Z, Wang X, Xu Y. Diammonium glycyrrhizinate upregulates PGC-1alpha and protects against Abeta1-42-induced neurotoxicity. PLoS One. 2012;7:e35823. doi: 10.1371/journal.pone.0035823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu X, Wang S, Yu L, Yang H, Tan R, Yin K, Jin J, Zhao H, Guan D, Xu Y. TL-2 attenuates beta-amyloid induced neuronal apoptosis through the AKT/GSK-3beta/beta-catenin pathway. Int J Neuropsychopharmacol. 2014;17:1511–1519. doi: 10.1017/S1461145714000315. [DOI] [PubMed] [Google Scholar]
- 68.Zhu X, Ye L, Ge H, Chen L, Jiang N, Qian L, Li L, Liu R, Ji S, Zhang S, Jin J, Guan D, Fang W, Tan R, Xu Y. Hopeahainol A attenuates memory deficits by targeting beta-amyloid in APP/PS1 transgenic mice. Aging Cell. 2013;12:85–92. doi: 10.1111/acel.12022. [DOI] [PubMed] [Google Scholar]
- 69.Zuo L, Hemmelgarn BT, Chuang CC, Best TM. The role of oxidative stress-induced epigenetic alterations in amyloid-beta production in alzheimer’s disease. Oxid Med Cell Longev. 2015;2015:604658. doi: 10.1155/2015/604658. [DOI] [PMC free article] [PubMed] [Google Scholar]