

Abstract
Amyotrophic lateral sclerosis and frontotemporal lobar degeneration are multifaceted diseases with genotypic, pathological and clinical overlap. One such overlap is the presence of SQSTM1/p62 mutations. While traditionally mutations manifesting in the ubiquitin-associated domain of p62 were associated with Paget’s disease of bone, mutations affecting all functional domains of p62 have now been identified in amyotrophic lateral sclerosis and frontotemporal lobar degeneration patients. p62 is a multifunctional protein that facilitates protein degradation through autophagy and the ubiquitin-proteasome system, and also regulates cell survival via the Nrf2 antioxidant response pathway, the nuclear factor-kappa B signaling pathway and apoptosis. Dysfunction in these signaling and protein degradation pathways have been observed in amyotrophic lateral sclerosis and frontotemporal lobar degeneration, and mutations that affect the role of p62 in these pathways may contribute to disease pathogenesis. In this review we discuss the role of p62 in these pathways, the effects of p62 mutations and the effect of mutations in the p62 modulator TANK-binding kinase 1, in relation to amyotrophic lateral sclerosis-frontotemporal lobar degeneration pathogenesis.
Keywords: aggregate/inclusion body formation, amyotrophic lateral sclerosis-frontotemporal lobar degeneration, autophagy, cell signaling, mitophagy, p62/SQSTM1, protein degradation
Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
Amyotrophic lateral sclerosis (ALS), the most common form of motor neuron disease, is a severe neurodegenerative disorder marked by progressive muscle weakness and paralysis caused by selective loss of motor neurons in the brain and spinal cord. The average survival time post-diagnosis is 2–3 years, with death most commonly caused by respiratory failure (Zarei et al., 2015). ALS can be classified as either sporadic, with no previously reported family history of the disease, or as familial. Frontotemporal lobar degeneration (FTLD), the third most common cause of dementia (Bang et al., 2015), is an umbrella term that encompasses a range of diseases marked by progressive loss of neurons in either one or both the frontal and temporal lobes (Mann et al., 1993; Rosen et al., 2002). FTLD patients generally present with behavioral and personality changes, and similarly to ALS, can be classified as either familial or sporadic (Seelaar et al., 2011). Originally considered unrelated diseases the two diseases are now thought to exist on a disease continuum due to pathological, genetic and clinical overlap (Neumann et al., 2006; Vance et al., 2006; Hasegawa et al., 2008; Lillo et al., 2012; Majounie et al., 2012).
While the main symptom of ALS is progressive muscle paralysis, up to 50% of patients will also develop cognitive impairment, and up to 15% of ALS patients meet the FTLD diagnostic criteria (Lomen-Hoerth et al., 2003; Ringholz et al., 2005). Conversely, up to 40% of FTLD patients exhibit ALS symptoms, while 15% meet the diagnostic criteria for ALS (Lomen-Hoerth et al., 2002). Additionally there is overlap in protein pathology. Pathological protein inclusions containing TAR DNA-binding protein 43 (TDP-43) or tau have been identified in both ALS and FTLD patients (Lippa et al., 2000; Morris et al., 2001; Yang et al., 2003; Neumann et al., 2006; Behrouzi et al., 2016). On a genetic level, while mutations in the superoxide dismutase 1 (SOD1) gene appear to be unique to ALS and mutations in MAPT (coding for tau) are identified only in FTLD patients (Bennion Callister and Pickering-Brown, 2014), commonly associated genes have been identified. For example, the hexanucleotide repeat expansion in chromosome 9 open reading frame 72 (C9ORF72) is the most common genetic cause of both ALS and FTLD, having been identified in 30–40% of familial ALS and up to 25% of FTLD cases (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Repeat expansions have also been identified in sporadic ALS and FTLD (Renton et al., 2011; Majounie et al., 2012).
Other genes implicated in both diseases include those coding for the autophagy receptors (ARs) sequestosome 1/p62 protein (SQSTM1/p62), optinuerin (OPTN), valosin-containing protein (VCP) and ubiquilin 2 (UBQLN2) (Synofzik et al., 2012; Le Ber et al., 2013; Kwok et al., 2014; Deng et al., 2017; Blauwendraat et al., 2018; Pottier et al., 2018), the function of these ARs is to sequester and remove old or damaged proteins and organelles via the autophagy-lysosome system. In addition to variants in ARs, mutations in tank-bingling kinase (TBK1), a kinase that modifies OPTN and SQSTM1/p62 function, have also been identified in ALS and FTLD (Freischmidt et al., 2015; Le Ber et al., 2015; Cui et al., 2018; McCombe et al., 2018). We searched for the terms p62 and SQSTM1 in combination with neurodegeneration, amyotrophic lateral sclerosis, frontotemporal degeneration, ALS and FTD. Searches were carried out on September 1st, 2019.
Sequestosome 1/p62 Protein in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
SQSTM1/p62, henceforth referred to as p62, is a scaffold protein that has roles in various signaling pathways and protein degradation. Loss of p62 enhances the rate of neurodegeneration in a number of disease models; p62 knockout mice exhibited increased accumulation of hyperphosphorylated tau and subsequent neurodegeneration (Babu et al., 2008), while absence of p62 in SOD1H46R mice lead to increased motor neuron degeneration (Hadano et al., 2016), enhanced α-synuclein pathology in a Lewy body disease mouse model (Tanji et al., 2015), and exacerbated motor phenotypes and neuropathological outcomes in a Spinal and bulbar muscular atrophy mouse model (Doi et al., 2013).
Mutations in the SQSTM1 gene are commonly identified in Paget’s disease of bone (PDB) (Laurin et al., 2002; Falchetti et al., 2004; Hocking et al., 2004), but have more recently been identified in both ALS and FTLD patients (Fecto et al., 2011; Rubino et al., 2012; Hirano et al., 2013; Le Ber et al., 2013; Chen et al., 2014; Bartolome et al., 2017; Blauwendraat et al., 2018). Unlike mutations associated with PDB, which cluster within the C-terminal ubiquitin-associated (UBA) domain, ALS and FTLD-associated mutations affect various domains throughout the entire protein (Additional Table 1). Further evidence of p62 involvement in ALS and FTLD is the observation of p62-positive inclusions in patient samples (Arai et al., 2003; Mann et al., 2013). While the role of ALS and FTLD-associated p62 variants and their contribution to disease onset and progression remains unclear, recent research demonstrates that pathogenic p62 mutant proteins alter signaling pathways involved in cell survival and differentiation.
Additional Table 1.
SQSTM1/p62 variants identified in ALS or FTLD patients
| Mutation | Exon | Change (bp) | p62 Domain | ALS | FTLD | PDB | Controls |
|---|---|---|---|---|---|---|---|
| A16V | 1 | c.47C>T | – | Yesh | Yesh | – | – |
| A33V | 1 | c.98C>T | PB1 | Yesb | Yesbk | – | Yesh |
| A53T | 1 | c.157G>A | PB1 | Yesc | – | – | – |
| D80E | 1 | c.240C>G | PB1 | – | Yesh | – | – |
| E81K | 2 | c.241G>A | PB1 | Yesi | – | – | – |
| M87V | 2 | c.259A>G | PB1 | Yese | – | – | – |
| V90M | 2 | c.268G>A | PB1 | Yesdh | – | – | – |
| I99L | 2 | c.295A>C | PB1 | Yesf | – | – | – |
| K102E | 3 | c.304A>G | C-terminal to PB1 | Yesc | – | – | – |
| R107W | 3 | c.319C>T | – | Yesh | – | – | – |
| R110C | 3 | c.328C>T | – | – | Yesk | – | – |
| D129N | 3 | c.358G>A | ZZ | Yesh | – | – | – |
| V153I | 3 | c.457G>A | ZZ | Yesbd | – | – | Yesh |
| E155K | 3 | c.463G>A | ZZ | Yesg | – | – | |
| R212C | 4 | c.634C>T | – | Yesh | Yesh | – | – |
| G219V | 4 | c.656G>T | – | – | Yesh | – | – |
| S226P | 5 | c.676T>C | TBS | – | Yesh | – | – |
| P228L | 5 | c.683C>T | TBS | Yesb | – | – | Yesagk |
| P232T | 5 | c.694C>A | TBS | – | Yesh | – | – |
| V234V | 5 | c.702G>A | TBS | Yesb | – | – | – |
| K238E | 5 | c.712A>G | TBS | Yesa | Yesk | – | Yeshk |
| K238del | 5 | c.714–716delGAA | TBS | Yesb | – | – | Yesh |
| N239K | 5 | c.717C>A | TBS | Yesi | – | – | – |
| D258N | 6 | c.772G>A | – | Yesh | – | – | – |
| V259L | 6 | c.775G>C | – | – | Yesa | – | – |
| H261H | 6 | c.783C>T | Close to PEST1 | Yesb | – | – | Yesb |
| E274D | 6 | c.822G>C | PEST 1 | Yesagh | Yesahk | – | Yesahk |
| E280del | 6 | c.838-840delGAG | PEST1 | – | Yesh | – | – |
| P296P | 6 | c.888G>T | Close to PEST1 | Yesg | – | – | Yesg |
| G297S | 6 | c.889G>A | Close to PEST1 | Yesi | – | – | – |
| R312H | 6 | c.1029A>G | – | – | Yesk | – | – |
| S318P | 6 | c.952T>C | – | Yesb | – | – | |
| S318S | 6 | c.954C>T | – | Yesg | – | – | Yesg |
| E319K | 6 | c.955G>A | – | – | Yesa | – | Yesh |
| R321C | 6 | c.961C>T | – | Yesb | – | – | Yesh |
| R321H | 6 | c.962G>A | – | – | Yesh | – | – |
| D329G | 7 | c.986A>G | LIR | – | Yesh | – | – |
| D337E | 7 | c.1011C>G | LIR | Yesf | – | – | – |
| L341V | 7 | c.1021C>G | LIR | Yesf | – | – | – |
| K344E | 7 | c.1032A>G | Close to LIR/KIR | – | Yesa | – | – |
| V346V | 7 | c.1038G>A | KIR | Yesg | – | – | Yesg |
| P348L | 7 | c.1044C>T | KIR | Yesa | Yesh | – | – |
| S370P | 7 | c.1108T>C | PEST 2 | Yesb | – | – | Yesh |
| E372D | 7 | c.1116G>C | PEST2 | Yesi | – | – | – |
| A381V | 7 | c.1142C>T | Close to UBA | – | Yesk | Yes | – |
| P387L | 7 | c.1160C>T | Close to UBA | – | Yeshk | – | – |
| P388S | 7 | c.1162C>T | Close to UBA | Yesi | – | – | – |
| A390X | 7 | c.1165+1G>A | Intronic | Yese | – | Yes | – |
| P392A | 7 | c.1174C>G | UBA | – | Yesj | – | – |
| P392L | 8 | c.1175C>T | UBA | Yesbcghi | Yeshk | Yesk | Yeshk |
| G411S | 8 | c.1231G>A | UBA | Yesb | – | Yes | – |
| G425R | 8 | c.1273G>A | UBA | Yesb | – | Yes | Yesh |
| T430P | 8 | c.1288AA>C | UBA | – | Yesh | – | – |
| P438L | 8 | c.1313C>T | C-terminal region | Yesa | – | – | Yeseh |
| P439L | 8 | c.1316C>T | C-terminal region | Yesc | Yesh | – | Yesh |
ALS: Amyotrophic lateral sclerosis; FTLD: frontotemporal lobar degeneration; KIR: Keap1-interacting region; LIR: LC3-interacting region; PB1: Phox and Bem1p; PEST: proline, glutamate, serine and threonine rich sequence; TBS: TRAF6 binding sequence; UBA: ubiquitin-associated; ZZ: zinc finger. a: Rubino et al., 2012; b: Fecto et al., 2011; c: Hirano et al., 2013; d: Shimizu et al., 2013; e: Teyssou et al., 2013; f: Chen et al., 2014; g: Kwok et al., 2014; h: van der Zee et al., 2014; i: Yang et al., 2015; j: Blauwendraat et al., 2018; k: Le Ber et al., 2013.
p62 structure and function
The functions of p62 are executed through protein-protein interactions, facilitated by various domains (Figure 1). The role of p62 as an autophagy receptor is mediated through several domains; p62 first binds to ubiquitinated substrates via its UBA domain and subsequently binds to the autophagosome membrane-protein LC3 via its LC3-interacting region (LIR) (Pankiv et al., 2007). The PB1 domain of p62 mediates self-oligomerisation, which can play an important role in the degradation of specific substrates by either macroautophagy (Jain et al., 2010; Itakura and Mizushima, 2011) or the ubiquitin-proteasome system (UPS) (Seibenhener et al., 2004).
Figure 1.
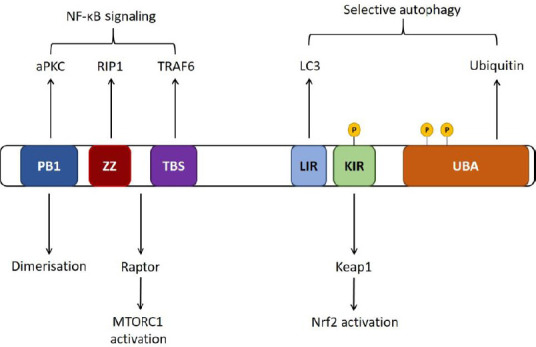
Schematic of p62 domains.
p62 self-oligomerizes via its PB1 domain. The LC3-interacting region (LIR) is required for p62 binding to the autophagosome membrane protein LC3, and p62 binds to ubiquitinated substrates via the ubiquitin associated (UBA) domain. p62 regulates Nrf2 signaling through interaction with inhibitory protein Keap1 via the Keap1 interacting region (KIR) domain, and regulates nuclear factor-kappa B (NF-κB) signaling via TRAF6 binding through the TRAF6 binding sequence (TBS) and interaction with RIP via the ZZ domain (Ichimura and Komatsu, 2018).
p62 also regulates several signaling pathways important for neuronal health, namely those that activate the transcription factors Nrf2 and nuclear factor kappa B (NF-κB). The Nrf2 pathway is the main response to oxidative stress in neurons and knockdown of Nrf2 leaves cells susceptible to neurotoxic insult (Ishii et al., 2000; Mimoto et al., 2012). Under normal conditions Kelch-like receptor protein 1 (Keap1) binds to and inhibits the action of the transcription factor, Nrf2. Upon cellular exposure to stress conditions, p62 expression is increased and p62 binds to Keap1 via the Keap1-interacting region (KIR) (Ishii et al., 2000; Copple et al., 2010; Jain et al., 2010; Lau et al., 2010). This promotes translocation of Nrf2 to the nucleus and subsequently transcription of antioxidant and protective genes such as Heme Oxygenase 1 and NAD(P)H quinone oxidase 1 (Itoh et al., 1997; Dinkova-Kostova et al., 2002; Kobayashi et al., 2004, 2006). The other main signaling pathway that p62 regulates is NF-κB, which is pathologically upregulated in ALS (Frakes et al., 2014). The role of p62 in this pathway has mainly been characterized with respect to mutations that cause PDB, a chronic and progressive skeletal disorder that involves increased osteoclastic bone resorption and deposition, leading to focal lesions of increased bone turnover. PDB-associated mutations primarily affect the UBA domain of p62, as either missense or truncating mutations that result in the removal of most or the entire domain. Very few PDB-associated mutations have been identified outside of the UBA domain, however those that have are located within approximately 50 residues of the UBA domain (Rea et al., 2014). PDB research shows that overexpressed mutant p62 proteins do not inhibit NF-κB, in contrast to over-expression of wild type p62 (Rea et al., 2006, 2009). The effect of increased NF-κB signaling due to SQSTM1/p62 mutations in an ALS and FTLD context could increase the production of pro-inflammatory cytokines with adverse effects on neuronal health (Shih et al., 2015).
It is important to note that p62 has been shown to be involved in positive regulation of NF-κB. Thus, p62 has a dual role in NF-κB regulation that may be dependent on p62 concentration or may be temporally regulated. The overall effect of these mutations on PDB pathogenesis is still under investigation, however there is strong evidence to suggest they contribute significantly to the disease, as patients with SQSTM1 mutations experience greater disease severity compared to patients without (Visconti et al., 2010; Rea et al., 2014). The main effect of SQSTM1/p62 mutations appears to be on autophagy (Hocking et al., 2004; Daroszewska et al., 2011) and NF-κB signaling (Najat et al., 2009; Rea et al., 2009), both of which play a role in the pathogenesis of ALS and FTLD.
p62 and proteostasis
The cell utilizes two systems to degrade misfolded or damaged proteins; the UPS and macroautophagy, or simply autophagy. The UPS degrades short-lived proteins, while autophagy degrades long-lived or aggregated proteins, bacteria, and organelles, both systems have the ability to degrade misfolded proteins, and both systems recognize ubiquitin as a marker for degradation. A major hallmark of ALS and FTLD is the accumulation of misfolded, ubiquitinated proteins in damaged neurons (Leigh et al., 1991; Bucciantini et al., 2002; Strong et al., 2005; Mackenzie and Neumann, 2016), indicating a role for malfunctioning protein degradation systems in pathogenesis. The accumulation of protein aggregates are linked with cell death (Giordana et al., 2010; Ticozzi et al., 2010; Brettschneider et al., 2014). p62 is a cargo protein for both systems (Seibenhener et al., 2004).
p62 and aggregate/inclusion body formation
p62 has been identified as a major component of aggregates from a number of neurodegenerative diseases; from neurofibrillary tangles in Alzheimer’s disease (Kuusisto et al., 2001a, 2002), and ubiquitin-positive inclusions in Parkinson’s disease (Kuusisto et al., 2003), FTLD (Kuusisto et al., 2002), and dementia with Lewy bodies (Kuusisto et al., 2001a), to SOD1 aggregates in ALS (Gal et al., 2007), and even Mallory bodies in hepatocytes (Zatloukal et al., 2002). While the presence of toxic protein aggregates inevitably leads to further damage to cellular organelles and subsequent cell death, it is possible that protein aggregate formation is an attempt at cellular protection. p62 was found to selectively form aggregates with mutant SOD1, but not wild-type SOD1, and these aggregates were not found to be directly toxic to the cell (Gal et al., 2007). Likewise, p62 was observed to form a shell around mutant huntingtin, and interference with p62 function lead to increased cell death (Bjorkoy et al., 2005). Depletion of p62 in spinal muscular bulbar atrophy mouse models exacerbated motor phenotypes via accumulation of toxic mutant androgen receptor protein, whereas increased p62 expression induced cytoprotective inclusion formation and subsequent improved phenotype (Doi et al., 2013). In SOD1H46R mice, p62 suppression lead to an increase in insoluble mutant SOD1 and ubiquitinated proteins (Hadano et al., 2016).
It is not clear whether p62 initiates aggregate formation, however it does appear to bind to toxic misfolded proteins in an attempt to isolate them from the cell, a process that is enhanced by substrate ubiquitination (Zatloukal et al., 2002; Gal et al., 2007). Thus, p62 may play a protective role in preventing further cell damage by toxic misfolded proteins, as cells that retain this p62 function exhibit increased survivability over cells that do not (Nakaso et al., 2004; Bjorkoy et al., 2005; Gal et al., 2007; Komatsu et al., 2007). On one hand p62 may bind to aggregates in order to prevent further accumulation of misfolded proteins to the aggregate (Zatloukal et al., 2002), or more likely, binds toxic proteins and holds them in a less active state in order to prevent them from causing further damage to the cell in their unbound form until clearance is possible.
p62, the ubiquitin proteasome system and neurodegeneration
The UPS facilitates the degradation of K48- and K63-linked polyubiquitinated substrates that are shuttled to the proteasome by cargo proteins (Seibenhener et al., 2004). UPS dysfunction is a hallmark of a number of neurodegenerative diseases (Keller et al., 2000; Seo et al., 2004; Bukhatwa et al., 2010), including ALS and FTLD (Bendotti et al., 2012; Kabashi et al., 2012; Myeku et al., 2016). Accumulation of UPS-specific substrates and proteins involved in UPS function into cellular inclusions provides evidence of UPS dysfunction (Farrawell et al., 2018). Further, aggregation of ALS-associated proteins leads to an accumulation of UPS-specific substrates, indicating that protein aggregation directly impedes UPS function (Farrawell et al., 2018) and deletion of a proteasome subunit, Rpt3, but not the critical autophagy protein Atg7, was sufficient to cause an ALS-like phenotype in mice (Tashiro et al., 2012). Thus, proteasome function may be critical to ALS and FTLD pathogenesis.
p62 is a cargo protein that shuttles the Alzheimer’s and FTLD-associated protein tau to the proteasome for degradation (Babu et al., 2005). Through the PB1 domain p62 directly interacts with the 26S proteasome (Seibenhener et al., 2004; Cohen-Kaplan et al., 2016), whereas the UBA domain of p62 recognizes ubiquitinated substrates. Thus it is possible that a mutation affecting either domain may impede p62-mediated degradation of substrates via the proteasome. Mutations to the UBA domain of p62, some of which have been identified in ALS and FTLD patients, reduce or abolish p62 ubiquitin-binding (Cavey et al., 2005; Garner et al., 2011). Thus, shuttling of ubiquitinated substrates to the UPS or autophagy is likely to be impeded. Intriguingly, either a loss of p62 (Seibenhener et al., 2004) or an increase in p62 (Korolchuk et al., 2009) can abrogate UPS function, and both p62 suppression and overexpression can cause accelerated disease onset and shortened lifespan in ALS-mouse models (Hadano et al., 2016; Mitsui et al., 2018). Together, these studies indicate a fine balance between p62 levels and both UPS function and disease pathogenesis. Upon proteasomal inhibition, p62 expression and autophagic activity is upregulated as a compensatory mechanism (Kuusisto et al., 2001b; Myeku and Figueiredo-Pereira, 2011; Choe et al., 2014; Fraiberg et al., 2017; Sha et al., 2018). ALS and FTLD-associated SQSTM1 mutations that affect the cargo function of p62, or perhaps p62 protein levels, may contribute to disease pathogenesis by upsetting this balance.
p62, autophagy and neurodegeneration
Basal autophagy is vital for maintaining neuronal health, and suppression of basal autophagy is sufficient to cause neurodegeneration in mice (Hara et al., 2006; Komatsu et al., 2006). Deficiencies in autophagy have been implicated in several neurodegenerative diseases; including FTLD (Gotzl et al., 2016), ALS (Xie et al., 2015), Alzheimer’s disease (Nixon et al., 2005), Huntingtin’s disease (Shibata et al., 2006; Fu et al., 2017), spinocerebellar ataxia type 7 (Alves et al., 2014) and across several tauopathies including corticobasal degeneration, and progressive supranuclear palsy (Lin et al., 2003; Piras et al., 2016). Inhibition of autophagy leads to accumulation of a number of proteins involved in neurodegenerative diseases including hyperphosphorylated tau (Babu et al., 2008; Tang et al., 2013), α-synuclein (Minakaki et al., 2018), and TDP-43 (Brady et al., 2011), and accelerates disease onset in SOD1G93A mice (Rudnick et al., 2017). In contrast, pharmacological enhancement of autophagy in several mouse models has proven effective at enhancing clearance of mutant SOD1 (Perera et al., 2018), as well as TDP-43 and FUS-positive inclusions (Brady et al., 2011; Wang et al., 2012; Cheng et al., 2015).
The formation of polyubiquitinated-inclusion bodies and their subsequent removal by autophagy are dependent on p62 (Pankiv et al., 2007), this includes the clearance of mutant SOD1 via autophagy, which is facilitated by p62 binding to mutant SOD1 (Gal et al., 2009). p62 itself is degraded by autophagy, but not the proteasome (Myeku and Figueiredo-Pereira 2011), and as such autophagy inhibition leads to an accumulation of p62 (Bjorkoy et al., 2009). This is problematic, as increased p62 can delay the delivery of ubiquitinated substrates to the proteasome, eventually compromising the UPS (Korolchuk et al., 2009). Unlike the ability of autophagy to compensate for a UPS failure, the UPS is unable to compensate for a failure in autophagy. The effect of p62-overexpression on protein degradation was demonstrated in p62-overexpressing SOD1H46R mice. These mice had accelerated disease onset and reduced turnover of SOD1H46R by either the UPS or autophagy (Mitsui et al., 2018). However, p62-ablation in the same mouse model lead to a worsened disease phenotype and shortened lifespan (Hadano et al., 2016). This highlights that alterations in p62 levels may affect protein degradation, overall autophagic flux, and ALS and FTLD disease progression.
Due to the significant role of p62 in autophagy, SQSTM1 mutations that may affect p62 function are of interest in ALS and FTLD. A number of mutations affecting the PB1 domain of p62 have been identified in both ALS and FTLD patients (Le Ber et al., 2013; Chen et al., 2014; Yang et al., 2015), and deletion of the PB1 domain prevents p62 binding to mutant SOD1 (Gal et al., 2009). p62 utilizes its PB1 domain to facilitate self-oligomerization during autophagy (Itakura and Mizushima, 2011). Due to the proximity of functional binding sites, p62 is not always able to bind multiple substrates at once. One such example is Keap1, a Nrf2 inhibitory protein that is degraded by autophagy in a p62-dependent manner. Due to the proximity of the Keap1-interacting region and the LC3-interacting region, one p62 monomer bound to LC3 will bind to another p62 monomer that is bound to Keap1, and in this way facilitates Keap1 degradation via autophagy (Jain et al., 2010). Mutations affecting the PB1 domain of p62 may affect functions other than dimerization; an FTLD-associated p62 variant, p.R110C, located just outside of the PB1 domain, is unable to promote Nrf2 signaling in line with p62 wild-type, and is associated with increased NF-κB signaling, while having no observed effect on dimerization (Foster et al., 2019), thus indicating further roles for the PB1 domain in p62 function. p62 can bind to LC3 on the isolation membrane through the LIR as well as ubiquitinated cargo through the UBA domain (Wurzer et al., 2015). Mutations affecting the LIR of p62 have been identified in ALS patients, p.D337E, and p.L341V (Chen et al., 2014). In vivo analyses demonstrated that a reduction in p62 LC3-binding ability leads to p62 accumulation in ubiquitinated-inclusions, yet the mutant p62 proteins were unable to be degraded by autophagy (Ichimura et al., 2008). Further, removal of the UBA domain or the LIR domain of p62 reduces the ability of p62 to clear TDP-43 aggregates (Brady et al., 2011). Thus, mutations to the LIR or UBA domains of p62, or the PB1 domain, which is also essential for autophagy, may contribute to ALS and FTLD pathology through a build-up of aggregated TDP-43.
However, upregulation of autophagy does not provide a simple solution to reducing disease progression. Inhibition of autophagy in SOD1G93A mice lead to increased survival time via reduced glial inflammation (Rudnick et al., 2017), while upregulated autophagy increased symptom progression and motor neuron degeneration via increased mitochondrial depletion and increased apoptosis in the same mouse model (Zhang et al., 2011; Perera et al., 2018). Overexpression of p62 in SOD1H46R mice lead to accelerated disease onset (Mitsui et al., 2018), indicating a fine balance between over- and underactive autophagy and p62 levels.
Additionally, p62 plays a role in the interplay between autophagy and the UPS. Of note, proteotoxic stress caused by proteasome inhibition can activate autophagy through p62 phosphorylation, however increased p62 levels can lead to delayed delivery of ubiquitinated substrates to the proteasome, causing UPS impairment despite an intact proteasome (Liu et al., 2016). This indicates that p62 variants that lead to abrogated p62 function may simultaneously affect both autophagy and the UPS by upsetting the interplay between the two. For a more in depth review of the interplay between the UPS and autophagy (Liu et al., 2016).
p62 and mitophagy
Fragmentation and changes to mitochondrial morphology have been extensively documented in ALS (Sasaki and Iwata, 2007; Jin et al., 2013), and are linked with aberrant oxidative metabolism and greater production of reactive oxygen species (ROS) (Mattiazzi et al., 2002; Jin et al., 2013). Clearance of mitochondria occurs by a form of autophagy known as mitophagy. Defects in mitophagy are observed in ALS, and lysosomal defects were observed along with accumulation of damaged mitochondria in motor neurons of SOD1G93A mice (Xie et al., 2015). The role of p62 in the removal of dysfunctional mitochondria is controversial. While some studies indicated that p62 is essential for the final clearance of mitochondria (Geisler et al., 2010; Lee et al., 2010), other studies suggest that p62 is responsible for the aggregation and autophagosomal engulfment of mitochondria, but is not essential for their ultimate removal via mitophagy (Narendra et al., 2010; Okatsu et al., 2010; Matsumoto et al., 2015). It is possible that, like its role in inclusion body formation, p62-mediated aggregation of mitochondria into tight clusters is a cytoprotective attempt at preventing further mitochondrial access to substrates and thereby limiting the spread of mitochondria-derived ROS throughout the cell (Narendra et al., 2010). Mutations that cause a loss of p62 function could prevent mitochondrial aggregation and reduce turnover of dysfunctional mitochondria, exposing the cell to further assault by ROS. Mutations in modifiers of p62, such as TBK1, may also decrease p62 function as ablation of TBK1-mediated phosphorylation of Serine residue 403 (Ser-403) of p62 results in reduced autophagosomal engulfment of mitochondria (Matsumoto et al., 2015). Thus, it is possible that TBK1-mutations lead to ALS pathogenesis partly via reduced p62 phosphorylation. Of note, the p.R110C and p.G427R mutations of p62 were reported to lead to decreased phosphorylation at Ser-403 and Ser-349, which was associated with dysregulated cell signaling (Deng et al., 2019; Foster et al., 2019).
The Effect of p62 Mutations on the Oxidative Stress Response
Nuclear erythroid 2-related factor 2 signaling
Nuclear-erythroid factor-2 (Nrf2) is a transcription factor that is normally sequestered in the cytosol, however under conditions of oxidative stress it is translocated to the nucleus where it promotes the expression of genes involved in the stress response, including glutathione-s-transferase and nuclear respiratory factor-1 (Nrf-1), Heme oxygenase 1, and NAD(P)H quinone oxidase 1 (NQO1) (Itoh et al., 1997; Ishii et al., 2000). The Nrf2 response is the primary mechanism utilised by neurons to protect against oxidative stress (Lau et al., 2010). SOD1G93A mice display impaired induction of Nrf2-induced protective genes (Mimoto et al., 2012; Guo et al., 2013), while overexpression of Nrf2 in the same mouse model delays symptom onset and extends survival time (Vargas et al., 2008). Further, Nrf2-deficient cells are more prone to cell death upon to exposure to reactive oxygen species (Ishii et al., 2000). It is therefore likely that the Keap1-Nrf2 system is a crucial pathway in ALS-FTLD pathogenesis. As an early stress response gene p62 promotes Nrf2 signaling. Under basal conditions Nrf2 is bound to the inhibitory protein Keap1, which shuttles Nrf2 to the proteasome for degradation (Ishii et al., 2000; Cullinan et al., 2004; Kobayashi et al., 2004; Furukawa and Xiong, 2005). p62 has a KIR that mediates p62 binding to Keap1 (Jain et al., 2010; Lau et al., 2010). The interaction between p62 and Keap1 is essential for p62-dependent Nrf2 signaling, and ALS and FTLD-associated mutant proteins that exhibit reduced p62-Keap1 binding also have a reduced ability to activate Nrf2 (Copple et al., 2010; Jain et al., 2010; Lau et al., 2010; Foster et al., 2019). This includes mutations affecting the KIR region of p62, such as p.P348L and p.G351A (Goode et al., 2016), and p.R110C, a mutation affecting the PB1 domain (outside of the KIR) (Foster et al., 2019). In the absence of a PB1 domain, or in the presence of point mutations within the PB1 domain (p.R21A and p.D69A), activation of the NQO1 anti-oxidant response element luciferase reporter was greatly diminished, indicating that a fully functioning PB1 domain is required for p62-dependent upregulation of Nrf2 (Jain et al., 2010). Pharmacological induction of Nrf2 was sufficient to restore NADH and FAD pools and cellular respiration in p62-deficient SH-SY5Y cells and FTLD patient fibroblasts expressing p62 variants p.A381V and p.K238del (Bartolome et al., 2017). Thus, activation of Nrf2 may be an important avenue of research for ALS and FTLD therapeutics.
Selective autophagy and p62-dependent induction of Nrf2 may also be linked to p62 phosphorylation at Serine residue 349 (Ser-349), which is phosphorylated in an mTORC1-dependent manner, and increases p62 binding affinity for Keap1 and subsequently increases Nrf2 signaling (Ichimura et al., 2013). Inhibition of mTORC kinase by rapamycin (and subsequently induction of autophagy) decreased p62 Ser-349 phosphorylation and resulted in decreased Heme oxygenase-1 expression and Nrf2 activity (Ichimura et al., 2013). Further, autophagy inhibition increases p62-dependent Nrf2 activity (Jain et al., 2010). Thus, it is clear that autophagy and the Nrf2 response are linked via p62 activity.
Mutations in genes that code for post-translational modifiers of p62, such as TBK1, may also affect p62-mediated Nrf2 signaling. The phosphorylation of Ser-349 of p62 enhances the binding affinity of p62 for Keap1 (Ichimura et al., 2013), and thus promotes Nrf2 signaling (Figure 2). A protein with the FTLD-associated variant p.R110C substitution had reduced phosphorylation of serine residues 403 and 349 (351 in mice), and while it did not demonstrate altered autophagic flux, did exhibit reduced ability to stimulate Nrf2 (Foster et al., 2019). Mutations in TBK1, which is responsible for the phosphorylation of p62 at Ser-403 (Matsumoto et al., 2011, 2015; Pilli et al., 2012), have been identified in ALS and FTLD patients (Freischmidt et al., 2015; van der Zee et al., 2017). Phosphorylation of Ser-403 (Matsumoto et al., 2011, 2015; Pilli et al., 2012) is a preceding step for Ser-349 phosphorylation (Ichimura et al., 2013), therefore mutations in kinases such as TBK1 may play a role in altered cell signaling in ALS/FTLD via changes to p62 phosphorylation.
Figure 2.
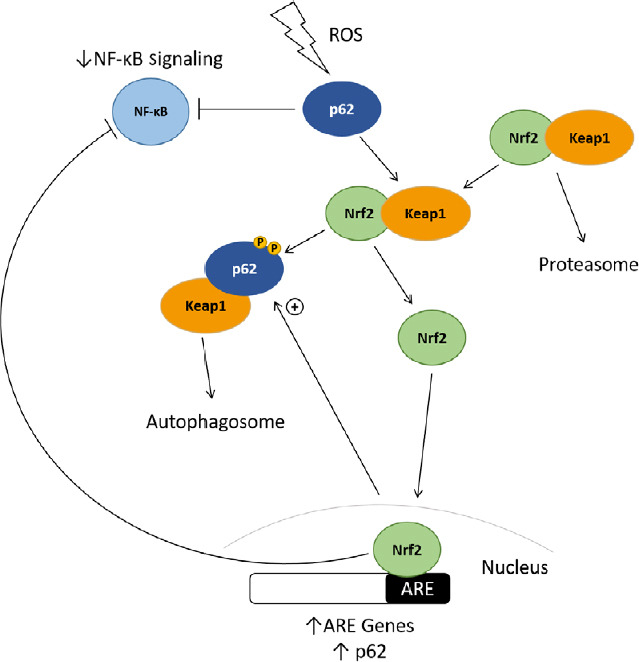
The effect of p62 on Nrf2 and NF-κB signaling.
Upon exposure to oxidative stress, p62 is phosphorylated at Serine residues 403 and 349, and binds to the Nrf2-inhibitory protein Keap1, shuttling it to the autophagosome for degradation. Nrf2 can then enter the nucleus and promote transcription of ARE-genes, including p62, which can also downregulate NF-κB signaling. Keap1: Kelch-like receptor protein 1; NF- κB: nuclear factor-kappa B; Nrf2: nuclear-erythroid factor-2; TRAF6: tumor necrosis factor receptor associated factor 6; ZZ: zinc finger.
Nuclear factor-kappa B signaling
NF-κB is a transcription factor responsible for promoting expression of both pro-survival and pro-inflammatory genes. Under basal conditions, NF-κB is bound to the inhibitory proteins IκBα, β, and ε, which prevent translocation of NF-κB into the nucleus and thereby block transcriptional effects. Under stimulation, the IκB proteins are phosphorylated and degraded via the proteasome, allowing NF-κB to move into the nucleus and promote transcription. Constitutive NF-κB signaling is required for neuronal survival and induction of NF-κB activity occurs in response to oxidative stress (Mattson et al., 2000; Bhakar et al., 2002).
The potential role of NF-κB signaling in ALS is complex. NF-κB is upregulated in the spinal cords of ALS patients and SOD1G93A mice (Swarup et al., 2011; Frakes et al., 2014). However, while inhibition of NF-κB in ALS astrocytes did not prevent motor neuron death, selective inhibition of NF-κB in microglia prevented microglial-mediated death in vitro, impaired pro-inflammatory microglial activation and increased survival time in ALS mice (Frakes et al., 2014). Constitutive expression of NF-κB in microglia is sufficient to induce gliosis and motor neuron death (Frakes et al., 2014), while increased NF-κB activity promoted survival of cells exposed to oxidative stress and NF-κB inhibition resulted in greater toxicity (Heck et al., 1999). Astrocytic NF-κB activation upregulated microglial proliferation in SOD1G93A mice, and this response delayed muscle denervation and prolonged the presymptomatic phase, while inhibition of the early microglial response resulted in acute detrimental effects (Ouali Alami et al., 2018). Interestingly, astrocytic NF-κB activation accelerated disease progression in the symptomatic phase of SOD1G93A mice (Ouali Alami, Schurr et al., 2018), however inhibition of NF-κB in astrocytes in the same mouse model did not show any change in disease onset or progression (Crosio et al., 2011). ALS-associated proteins FUS, TDP-43 (Swarup et al., 2011; Zhao et al., 2015), and UBQLN2 (Picher-Martel et al., 2015) all promote NF-κB signaling, and either astrocytic- (Kia et al., 2018) or microglial-induced motor neuron death (Zhao et al., 2015).
p62 has a dual role in the regulation of NF-κB signaling and dysregulation of NF-κB has been associated with PDB associated UBA domain p62-variants, which have a decreased ability to inhibit NF-κB compared with p62 wild type (Rea et al., 2014). While over-expressed wild type p62 inhibits NF-κB, p62 has also been reported to play an essential role in positively regulating NF-κB signaling in relation to osteoclastogenesis, bone homeostasis, and in cancer (Duran et al., 2004, 2008; Guo et al., 2011). Genetic ablation of p62 was shown to decrease IκB kinase activation and subsequently inhibited NF-κB translocation both in vitro and in vivo in mice (Duran, Serrano et al., 2004), and p62 downregulation abrogated tumour necrosis factor receptor 6 (TRAF6) and IL-1 mediated NF-κB signaling (Sanz et al., 2000), further demonstrating a role for p62 in NF-κB activation outside of neurodegeneration. Ubiquitin-binding affinity affects p62-regulated NF-κB signaling as p62 dimerization via the UBA domain and ubiquitin-binding are mutually exclusive (Isogai et al., 2011). Non-disease associated mutations within the UBA domain that prevent dimer formation increase p62 binding to polyubiquitin and subsequently decrease NF-κB signaling. Consistent with this finding, p62 UBA domain variants have a reduced ability to bind ubiquitin and impaired negative regulation of NF-κB signaling (Long et al., 2010). However, the PDB-associated non-UBA domain mutant p.P364S exhibits increased NF-κB signaling similar to p62 UBA-domain variants, despite retaining full affinity for ubiquitin-binding (Rea et al., 2009), indicating that the role of p62 in regulating NF-κB signaling is not solely due to its ability to bind ubiquitin.
p62 interacts with TRAF6, an adaptor protein and E3 ubiquitin-ligase that regulates NF-κB, via its TRAF6-binding sequence (TBS), and mutations within this domain of p62 have been identified in ALS and FTLD (Rubino et al., 2012; Le Ber et al., 2013). TRAF6 polyubiquitination is associated with increased NF-κB and is promoted by p62, as evident in p62 knockout mice, which exhibit no polyubiquitinated TRAF6 in brain tissue and also exhibit neurofibrillary tangles and neurodegeneration (Wooten et al., 2005). However, p62 also facilitates TRAF6 de-ubiquitination by forming a scaffolding complex linking the de-ubiquitinating enzyme CYLD with TRAF6 (Jin et al., 2008; Wooten et al., 2008). Expression of a p62 UBA-deletion construct abolished TRAF6 polyubiquitination, as did a p62 PB1-deletion construct. Nerve growth factor treatment of PC12 cells induces TRAF6 polyubiquitination and formation of the p62-TRAF6-IκB kinase-PKC iota complex, which leads to NF-κB activation, and inhibition of p62-TRAF6 interaction blocks both TRAF6 polyubiquitination and complex formation (Wooten et al., 2005). Deletion of the TRAF6 binding region of p62 was sufficient to block TRAF6-mediated NF-κB induction. TRAF6 exhibits a low basal level of polyubiquitination, however co-expression with p62 enhances TRAF6 polyubiquitination (Wooten et al., 2005). Therefore, TBS domain variants in ALS-FTLD may alter NF-κB signaling and thereby contribute to ALS-FTLD pathogenicity.
Concluding Remarks
In this review, we have summarized the current understanding of the role of p62 in ALS and FTLD pathogenesis, and how mutations in various domains of the protein may contribute to disease onset (Figure 3).
Figure 3.
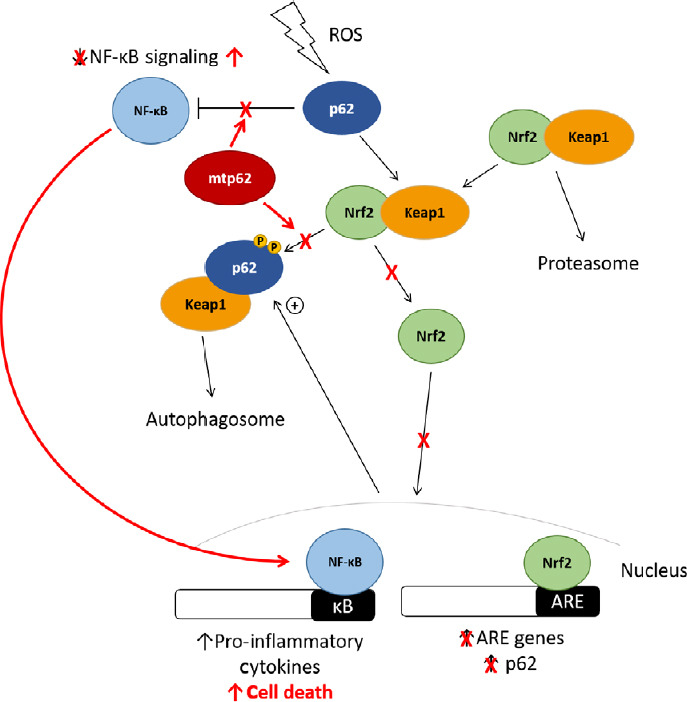
The effect of mutant p62 on Nrf2 and NF-κB signaling in ALS and FTLD pathogenesis.
Several ALS and FTLD-associated variants exhibit reduced Keap1-binding, preventing Nrf2 from entering the nucleus and promoting protective genes, predisposing the cell death upon exposure to ROS. Upregulation of Nrf2 signaling downregulates NF-κB signaling, indicating that p62 variants that fail to promote Nrf2 signaling may also lead to a concomitant increase in NF-κB signaling. Additionally, several p62 variants are unable to regulate NF-κB signaling, which could potentially lead to increased transcription of pro-inflammatory and pro-apoptotic factors, predisposing the cell to death. ALS: Amyotrophic lateral sclerosis; FTLD: frontotemporal lobar degeneration; FronNrf2: nuclear-erythroid factor-2; NF-κB: nuclear factor kappa-B; ROS: reactive oxygen species.
While SQSTM1 mutations identified in PDB cases primarily affect the UBA domain, the variants identified in ALS or FTLD patients span the entirety of the protein. As p62 is a multifunctional protein that exerts its effects through numerous functional domains, mutations affecting these domains can have an effect on both autophagy and cell signaling pathways such as the Nrf2 and NF-κB pathways, and are likely to also affect mitophagy and aggregate/inclusion body formation. ALS and FTLD-associated p62 variants may therefore contribute to ALS and FTLD pathogenesis via reduced degradation of toxic proteins as well as via a reduced ability to mount a suitable stress response, leaving cells more susceptible to neurotoxic insult. The role of p62 and modifiers such as TBK1 in a functioning autophagy lysosome system are well established, however further research into how mutations that affect mitochondrial turnover and altered NF-κB signaling in regards to neuronal health and survival is required. As failure of autophagy can disturb the UPS and lead to increased dysfunctional mitochondria and increased ROS production, further investigations into modulation of autophagy and mitophagy to identify potential therapeutic approaches for ALS-FTLD are needed.
Additional file:
Additional Table 1: SQSTM1/p62 variants identified in ALS or FTLD patients.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported by the NHMRC-ARC Dementia Research Development Fellowship Grant (AP1102977) and an Australian Government Research Training Program (RTS) Scholarship.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work was supported by the NHMRC-ARC Dementia Research Development Fellowship Grant (AP1102977) and an Australian Government Research Training Program (RTS) Scholarship.
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Alves S, Cormier-Dequaire F, Marinello M, Marais T, Muriel MP, Beaumatin F, Charbonnier-Beaupel F, Tahiri K, Seilhean D, El Hachimi K, Ruberg M, Stevanin G, Barkats M, den Dunnen W, Priault M, Brice A, Durr A, Corvol JC, Sittler A. The autophagy/lysosome pathway is impaired in SCA7 patients and SCA7 knock-in mice. Acta Neuropathol. 2014;128:705–722. doi: 10.1007/s00401-014-1289-8. [DOI] [PubMed] [Google Scholar]
- 2.Arai T, Nonaka T, Hasegawa M, Akiyama H, Yoshida M, Hashizume Y, Tsuchiya K, Oda T, Ikeda K. Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett. 2003;342:41–44. doi: 10.1016/s0304-3940(03)00216-7. [DOI] [PubMed] [Google Scholar]
- 3.Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 4.Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- 5.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386:1672–1682. doi: 10.1016/S0140-6736(15)00461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartolome F, Esteras N, Martin-Requero A, Boutoleau-Bretonniere C, Vercelletto M, Gabelle A, Le Ber I, Honda T, Dinkova-Kostova AT, Hardy J, Carro E, Abramov AY. Pathogenic p62/SQSTM1 mutations impair energy metabolism through limitation of mitochondrial substrates. Sci Rep. 2017;7:1666. doi: 10.1038/s41598-017-01678-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behrouzi R, Liu X, Wu D, Robinson AC, Tanaguchi-Watanabe S, Rollinson S, Shi J, Tian J, Hamdalla HH, Ealing J, Richardson A, Jones M, Pickering-Brown S, Davidson YS, Strong MJ, Hasegawa M, Snowden JS, Mann DM. Pathological tau deposition in Motor Neurone Disease and frontotemporal lobar degeneration associated with TDP-43 proteinopathy. Acta Neuropathol Commun. 2016;4:33. doi: 10.1186/s40478-016-0301-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bendotti C, Marino M, Cheroni C, Fontana E, Crippa V, Poletti A, De Biasi S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol. 2012;97:101–126. doi: 10.1016/j.pneurobio.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Bennion Callister J, Pickering-Brown SM. Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp Neurol. 2014;262:84–90. doi: 10.1016/j.expneurol.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J Neurosci. 2002;22:8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 13.Blauwendraat C, Wilke C, Simón-Sánchez J, Jansen IE, Reifschneider A, Capell A, Haass C, Castillo-Lizardo M, Biskup S, Maetzler W, Rizzu P, Heutink P, Synofzik M. The wide genetic landscape of clinical frontotemporal dementia: systematic combined sequencing of 121 consecutive subjects. Genet Med. 2018;20:240–249. doi: 10.1038/gim.2017.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem. 2011;116:248–259. doi: 10.1111/j.1471-4159.2010.07098.x. [DOI] [PubMed] [Google Scholar]
- 15.Brettschneider J, Arai K, Del Tredici K, Toledo JB, Robinson JL, Lee EB, Kuwabara S, Shibuya K, Irwin DJ, Fang L, Van Deerlin VM, Elman L, McCluskey L, Ludolph AC, Lee VM, Braak H, Trojanowski JQ. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014;128:423–437. doi: 10.1007/s00401-014-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 17.Bukhatwa S, Zeng BY, Rose S, Jenner P. A comparison of changes in proteasomal subunit expression in the substantia nigra in Parkinson's disease, multiple system atrophy and progressive supranuclear palsy. Brain Res. 2010;1326:174–183. doi: 10.1016/j.brainres.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 18.Cavey JR, Ralston SH, Hocking LJ, Sheppard PW, Ciani B, Searle MS, Layfield R. Loss of ubiquitin-binding associated with Paget’s disease of bone p62 (SQSTM1) mutations. J Bone Miner Res. 2005;20:619–624. doi: 10.1359/JBMR.041205. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Zheng ZZ, Chen X, Huang R, Yang Y, Yuan L, Pan L, Hadano S, Shang HF. SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2014;35:726. doi: 10.1016/j.neurobiolaging.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Cheng CW, Lin MJ, Shen CK. Rapamycin alleviates pathogenesis of a new Drosophila model of ALS-TDP. J Neurogenet. 2015;29:59–68. doi: 10.3109/01677063.2015.1077832. [DOI] [PubMed] [Google Scholar]
- 21.Choe JY, Jung HY, Park KY, Kim SK. Enhanced p62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1b expression. Rheumatology (Oxford) 2014;53:1043–1053. doi: 10.1093/rheumatology/ket474. [DOI] [PubMed] [Google Scholar]
- 22.Cohen-Kaplan V, Livneh I, 1, Avni N, 1, Fabre B, 1, Ziv T, 2, Kwon YT, 3, Ciechanover A. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci U S A. 2016;113:7490–7499. doi: 10.1073/pnas.1615455113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Copple IM, Lister A, Obeng AD, Kitteringham NR, Jenkins RE, Layfield R, Foster BJ, Goldring CE, Park BK. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J Biol Chem. 2010;285:16782–16788. doi: 10.1074/jbc.M109.096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crosio C, Valle C, Casciati A, Iaccarino C, Carrì MT. Astroglial inhibition of NF-kappaB does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS) PLoS One. 2011;6:e17187. doi: 10.1371/journal.pone.0017187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui R, Tuo M, 1, Li P, 1, Zhou C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: a meta-analysis. Neurol Sci. 2018;39:811–820. doi: 10.1007/s10072-018-3246-0. [DOI] [PubMed] [Google Scholar]
- 26.Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daroszewska A, van ‘t Hof RJ, Rojas JA, Layfield R, Landao-Basonga E, Rose L, Rose K, Ralston SH. A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget’s disease-like disorder in mice. Hum Mol Genet. 2011;20:2734–2744. doi: 10.1093/hmg/ddr172. [DOI] [PubMed] [Google Scholar]
- 28.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demishtein A, Fraiberg M, Berko D, Tirosh B, Elazar Z, Navon A. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy. 2017;13:1697–1708. doi: 10.1080/15548627.2017.1356549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng Z, Lim J, Wang Q, Purtell K, Wu S, Palomo GM, Tan H, Manfredi G, Zhao Y, Peng J, Hu B, Chen S, Yue Z. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2019 doi: 10.1080/15548627.2019.1644076. doi: 101080/1554862720191644076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Z, Purtell K, Lachance V, Wold MS, Chen S, Yue Z. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 2017;27:491–504. doi: 10.1016/j.tcb.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doi H, Adachi H, Katsuno M, Minamiyama M, Matsumoto S, Kondo N, Miyazaki Y, Iida M, Tohnai G, Qiang Q, Tanaka F, Yanagawa T, Warabi E, Ishii T, Sobue G. p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci. 2013;33:7710–7727. doi: 10.1523/JNEUROSCI.3021-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Durán A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J, Diaz-Meco MT. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004;6:303–309. doi: 10.1016/s1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- 36.Falchetti A, Di Stefano M, Marini F, Del Monte F, Mavilia C, Strigoli D, De Feo ML, Isaia G, Masi L, Amedei A, Cioppi F, Ghinoi V, Bongi SM, Di Fede G, Sferrazza C, Rini GB, Melchiorre D, Matucci-Cerinic M, Brandi ML. Two novel mutations at exon 8 of the Sequestosome 1 (SQSTM1) gene in an Italian series of patients affected by Paget’s disease of bone (PDB) J Bone Miner Res. 2004;19:1013–1017. doi: 10.1359/JBMR.040203. [DOI] [PubMed] [Google Scholar]
- 37.Farrawell NE, Lambert-Smith I, Mitchell K, McKenna J, McAlary L, Ciryam P, Vine KL, Saunders DN, Yerbury JJ. SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J Cell Sci. 2018 doi: 10.1242/jcs.209122. doi: 101242/jcs209122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud-Driss S, Sufit RL, Heller SL, Deng HX, Siddique T. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 39.Foster A, Scott D, Layfield R, Rea SL. An FTLD-associated SQSTM1 variant impacts Nrf2 and NF-kappaB signaling and is associated with reduced phosphorylation of p62. Mol Cell Neurosci. 2019;98:32–45. doi: 10.1016/j.mcn.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, 2, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, Pinto S, Press R, Millecamps S, Molko N, Bernard E, Desnuelle C, Soriani MH, Dorst J, Graf E, Nordström U, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- 42.Fu Y, Wu P, Pan Y, Sun X, Yang H, Difiglia M, Lu B. A toxic mutant huntingtin species is resistant to selective autophagy. Nat Chem Biol. 2017;13:1152–1154. doi: 10.1038/nchembio.2461. [DOI] [PubMed] [Google Scholar]
- 43.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gal J, Ström AL, Kilty R, Zhang F, Zhu H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem. 2007;282:11068–11077. doi: 10.1074/jbc.M608787200. [DOI] [PubMed] [Google Scholar]
- 45.Gal J, Ström AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem. 2009;111:1062–1073. doi: 10.1111/j.1471-4159.2009.06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garner TP, Long J, Layfield R, Searle MS. Impact of p62/SQSTM1 UBA domain mutations linked to Paget’s disease of bone on ubiquitin recognition. Biochemistry. 2011;50:4665–4674. doi: 10.1021/bi200079n. [DOI] [PubMed] [Google Scholar]
- 47.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 48.Giordana MT, Piccinini M, Grifoni S, De Marco G, Vercellino M, Magistrello M, Pellerino A, Buccinnà B, Lupino E, Rinaudo MT. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 2010;20:351–360. doi: 10.1111/j.1750-3639.2009.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goode A, Rea S, Sultana M, Shaw B, Searle MS, Layfield R. ALS-FTLD associated mutations of SQSTM1 impact on Keap1-Nrf2 signaling. Mol Cell Neurosci. 2016;76:52–58. doi: 10.1016/j.mcn.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Götzl JK, Lang CM, Haass C, Capell A. Impaired protein degradation in FTLD and related disorders. Ageing Res Rev. 2016;32:122–139. doi: 10.1016/j.arr.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 51.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo Y, Zhang Y, Wen D, Duan W, An T, Shi P, Wang J, Li Z, Chen X, Li C. The modest impact of transcription factor Nrf2 on the course of disease in an ALS animal model. Lab Invest. 2013;93:825–833. doi: 10.1038/labinvest.2013.73. [DOI] [PubMed] [Google Scholar]
- 53.Hadano S, Mitsui S, Pan L, Otomo A, Kubo M, Sato K, Ono S, Onodera W, Abe K, Chen X, Koike M, Uchiyama Y, Aoki M, Warabi E, Yamamoto M, Ishii T, Yanagawa T, Shang HF, Yoshii F. Functional links between SQSTM1 and ALS2 in the pathogenesis of ALS: cumulative impact on the protection against mutant SOD1-mediated motor dysfunction in mice. Hum Mol Genet. 2016;25:3321–3340. doi: 10.1093/hmg/ddw180. [DOI] [PubMed] [Google Scholar]
- 54.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 55.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heck S, Lezoualc’h F, Engert S, Behl C. Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB. J Biol Chem. 1999;274:9828–9835. doi: 10.1074/jbc.274.14.9828. [DOI] [PubMed] [Google Scholar]
- 57.Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, Miyamoto K, Akamatsu M, Mitsui Y, Kusunoki S. Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology. 2013;80:458–463. doi: 10.1212/WNL.0b013e31827f0fe5. [DOI] [PubMed] [Google Scholar]
- 58.Hocking LJ, Lucas GJ, Daroszewska A, Cundy T, Nicholson GC, Donath J, Walsh JP, Finlayson C, Cavey JR, Ciani B, Sheppard PW, Searle MS, Layfield R, Ralston SH. Novel UBA domain mutations of SQSTM1 in Paget’s disease of bone: genotype phenotype correlation, functional analysis, and structural consequences. J Bone Miner Res. 2004;19:1122–1127. doi: 10.1359/JBMR.0403015. [DOI] [PubMed] [Google Scholar]
- 59.Ichimura Y, Komatsu M. Activation of p62/SQSTM1-keap1-nuclear factor erythroid 2-related factor 2 pathway in cancer. Front Oncol. 2018;8:210. doi: 10.3389/fonc.2018.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 61.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, Hoshii T, Hirao A, Takagi K, Mizushima T, Motohashi H, Lee MS, Yoshimori T, Tanaka K, Yamamoto M, Komatsu M. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 62.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 63.Isogai S, Morimoto D, Arita K, Unzai S, Tenno T, Hasegawa J, Sou YS, Komatsu M, Tanaka K, Shirakawa M, Tochio H. Crystal structure of the ubiquitin-associated (UBA) domain of p62 and its interaction with ubiquitin. J Biol Chem. 2011;286:31864–31874. doi: 10.1074/jbc.M111.259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itakura E, Mizushima N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 66.Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin W, Chang M, Paul EM, Babu G, Lee AJ, Reiley W, Wright A, Zhang M, You J, Sun SC. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118:1858–1866. doi: 10.1172/JCI34257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jin YN, Yu YV, Gundemir S, Jo C, Cui M, Tieu K, Johnson GV. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS One. 2013;8:e57932. doi: 10.1371/journal.pone.0057932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kabashi E, Agar JN, Strong MJ, Durham HD. Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:367–371. doi: 10.3109/17482968.2012.686511. [DOI] [PubMed] [Google Scholar]
- 70.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 71.Kia A, McAvoy K, Krishnamurthy K, Trotti D, Pasinelli P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia. 2018;66:1016–1033. doi: 10.1002/glia.23298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 75.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 76.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol. 2003;62:1241–1253. doi: 10.1093/jnen/62.12.1241. [DOI] [PubMed] [Google Scholar]
- 78.Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001a;12:2085–2090. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- 79.Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol Appl Neurobiol. 2002;28:228–237. doi: 10.1046/j.1365-2990.2002.00394.x. [DOI] [PubMed] [Google Scholar]
- 80.Kuusisto E, Suuronen T, Salminen A. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem Biophys Res Commun. 2001b;280:223–228. doi: 10.1006/bbrc.2000.4107. [DOI] [PubMed] [Google Scholar]
- 81.Kwok CT, Morris A, de Belleroche JS. Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur J Hum Genet. 2014;22:492–496. doi: 10.1038/ejhg.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, Nicolas G, Gabelle A, Didic M, De Septenville A, Millecamps S, Lenglet T, Latouche M, Kabashi E, Campion D, Hannequin D, Hardy J, Brice A. French Clinical and Genetic Research Network on FTD/FTD-ALS (2013) Sqstm1 mutations in french patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 70:1403–1410. doi: 10.1001/jamaneurol.2013.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Le Ber I, De Septenville A, Millecamps S, Camuzat A, Caroppo P, Couratier P, Blanc F, Lacomblez L, Sellal F, Fleury MC, Meininger V, Cazeneuve C, Clot F, Flabeau O, LeGuern E, Brice A. French Clinical and Genetic Research Network on FTLD/FTLD-ALS (2015) TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging. 36:3116e5–3116e8. doi: 10.1016/j.neurobiolaging.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 86.Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in Parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE, Gallo JM, Weller RO, Anderton BH. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain. 1991;114:775–788. doi: 10.1093/brain/114.2.775. [DOI] [PubMed] [Google Scholar]
- 88.Lillo P, Mioshi E, Burrell JR, Kiernan MC, Hodges JR, Hornberger M. Grey and white matter changes across the amyotrophic lateral sclerosis-frontotemporal dementia continuum. PLoS One. 2012;7:e43993. doi: 10.1371/journal.pone.0043993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2003;32:1091–1105. doi: 10.1023/B:NEUR.0000021904.61387.95. [DOI] [PubMed] [Google Scholar]
- 90.Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE, Grafman J, Liang Y, St George-Hyslop PH, Trojanowski JQ, Lee VM. Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol. 2000;48:850–858. [PubMed] [Google Scholar]
- 91.Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C, Liu HF. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21:29. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 93.Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal. Neurology. 2003;60:1094–1097. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- 94.Long J, Garner TP, Pandya MJ, Craven CJ, Chen P, Shaw B, Williamson MP, Layfield R, Searle MS. Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF-kappaB signaling. J Mol Biol. 2010;396:178–194. doi: 10.1016/j.jmb.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 95.Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. 2016;138:54–70. doi: 10.1111/jnc.13588. [DOI] [PubMed] [Google Scholar]
- 96.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, Davidson Y, Pickering-Brown S. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1:68. doi: 10.1186/2051-5960-1-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mann DM, South PW, Snowden JS, Neary D. Dementia of frontal lobe type: neuropathology and immunohistochemistry. J Neurol Neurosurg Psychiatry. 1993;56:605. doi: 10.1136/jnnp.56.6.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24:4429–4442. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- 100.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 101.Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 102.Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- 103.McCombe PA, Ngo ST, Guo CC, Fazlollahi A, Bollmann S, Wang L, Hu X, Barth M, Salvado O, Davis M, Ceslis A, Robinson G, Henderson RD, Steyn FJ. Patient with ALS with a novel TBK1 mutation, widespread brain involvement, behaviour changes and metabolic dysfunction. J Neurol Neurosurg Psychiatry. 2018;90:952–954. doi: 10.1136/jnnp-2018-318823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mimoto T, Miyazaki K, Morimoto N, Kurata T, Satoh K, Ikeda Y, Abe K. Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res. 2012;1446:109–118. doi: 10.1016/j.brainres.2011.12.064. [DOI] [PubMed] [Google Scholar]
- 105.Minakaki G, Menges S, Kittel A, Emmanouilidou E, Schaeffner I, Barkovits K, Bergmann A, Rockenstein E, Adame A, Marxreiter F, Mollenhauer B, Galasko D, Buzás EI, Schlötzer-Schrehardt U, Marcus K, Xiang W, Lie DC, Vekrellis K, Masliah E, Winkler J, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14:98–119. doi: 10.1080/15548627.2017.1395992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mitsui S, Otomo A, Nozaki M, Ono S, Sato K, Shirakawa R, Adachi H, Aoki M, Sobue G, Shang HF, 8, Hadano S. Systemic overexpression of SQSTM1/p62 accelerates disease onset in a SOD1H46R-expressing ALS mouse model. Mol Brain. 2018;11:30–30. doi: 10.1186/s13041-018-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morris HR, Khan MN, Janssen JC, Brown JM, Perez-Tur J, Baker M, Ozansoy M, Hardy J, Hutton M, Wood NW, Lees AJ, Revesz T, Lantos P, Rossor MN. The genetic and pathological classification of familial frontotemporal dementia. Arch Neurol. 2001;58:1813–1816. doi: 10.1001/archneur.58.11.1813. [DOI] [PubMed] [Google Scholar]
- 108.Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, Duff KE. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med. 2016;22:46–53. doi: 10.1038/nm.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286:22426–22440. doi: 10.1074/jbc.M110.149252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Najat D, Garner T, Hagen T, Shaw B, Sheppard PW, Falchetti A, Marini F, Brandi ML, Long JE, Cavey JR, Searle MS, Layfield R. Characterization of a non-UBA domain missense mutation of sequestosome 1 (SQSTM1) in Paget’s disease of bone. J Bone Miner Res. 2009;24:632–642. doi: 10.1359/jbmr.081204. [DOI] [PubMed] [Google Scholar]
- 111.Nakaso K, Yoshimoto Y, Nakano T, Takeshima T, Fukuhara Y, Yasui K, Araga S, Yanagawa T, Ishii T, Nakashima K. Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson’s disease. Brain Res. 2004;1012:42–51. doi: 10.1016/j.brainres.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 112.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 114.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 115.Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, Tanaka K, Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ouali Alami N, Schurr C, Olde Heuvel F, Tang L, Li Q, Tasdogan A, Kimbara A, Nettekoven M, Ottaviani G, Raposo C, Röver S, Rogers-Evans M, Rothenhäusler B, Ullmer C, Fingerle J, Grether U, Knuesel I, Boeckers TM, Ludolph A, Wirth T, et al. NF-kappaB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018 doi: 10.15252/embj.201798697. doi: 1015252/embj201798697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 118.Perera ND, Sheean RK, Lau CL, Shin YS, Beart PM, Horne MK, Turner BJ. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy. 2018;14:534–551. doi: 10.1080/15548627.2017.1385674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Picher-Martel V, Dutta K, Phaneuf D, Sobue G, Julien JP. Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Molecular Brain. 2015;8:71. doi: 10.1186/s13041-015-0162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Piras A, Collin L, Grüninger F, Graff C, Rönnbäck A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. 2016;4:22. doi: 10.1186/s40478-016-0292-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pottier C, Rampersaud E, Baker M, Wu G, Wuu J, McCauley JL, Zuchner S, Schule R, Bermudez C, Hussain S, Cooley A, Wallace M, Zhang J, Taylor JP, Benatar M, Rademakers R. Identification of compound heterozygous variants in OPTN in an ALS-FTD patient from the CReATe consortium: a case report. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:469–471. doi: 10.1080/21678421.2018.1452947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations--bridging Paget disease of bone and ALS/FTLD. Exp Cell Res. 2014;325:27–37. doi: 10.1016/j.yexcr.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 124.Rea SL, Walsh JP, Ward L, Magno AL, Ward BK, Shaw B, Layfield R, Kent GN, Xu J, Ratajczak T. Sequestosome 1 mutations in Paget's disease of bone in Australia: prevalence, genotype/phenotype correlation, and a novel non-UBA domain mutation (P364S) associated with increased NF-kappaB signaling without loss of ubiquitin binding. J Bone Miner Res. 2009;24:1216–1223. doi: 10.1359/jbmr.090214. [DOI] [PubMed] [Google Scholar]
- 125.Rea SL, Walsh JP, Ward L, Yip K, Ward BK, Kent GN, Steer JH, Xu J, Ratajczak T. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res. 2006;21:1136–1145. doi: 10.1359/jbmr.060405. [DOI] [PubMed] [Google Scholar]
- 126.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–590. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 128.Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, Feiwell R, Kramer JH, Miller BL. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208. doi: 10.1212/wnl.58.2.198. [DOI] [PubMed] [Google Scholar]
- 129.Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, Bruni AC, St George-Hyslop PH, Scarpini E, Gallone S, Pinessi L TODEM Study Group. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–1562. doi: 10.1212/WNL.0b013e31826e25df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM, Maniatis T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci U S A. 2017;114:E8294–E8303. doi: 10.1073/pnas.1704294114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66:10–16. doi: 10.1097/nen.0b013e31802c396b. [DOI] [PubMed] [Google Scholar]
- 133.Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry. 2011;82:476–486. doi: 10.1136/jnnp.2010.212225. [DOI] [PubMed] [Google Scholar]
- 134.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Seo H, Sonntag KC, Isacson O. Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann Neurol. 2004;56:319–328. doi: 10.1002/ana.20207. [DOI] [PubMed] [Google Scholar]
- 136.Sha Z, Schnell HM, Ruoff K, Goldberg A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J Cell Biol. 2018;217:1757–1776. doi: 10.1083/jcb.201708168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 138.Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci. 2015;8:77–77. doi: 10.3389/fnmol.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shimizu H, Toyoshima Y, Shiga A, Yokoseki A, Arakawa K, Sekine Y, Shimohata T, Ikeuchi T, Nishizawa M, Kakita A, Onodera O, Takahashi H. Sporadic ALS with compound heterozygous mutations in the SQSTM1 gene. Acta Neuropathol. 2013;126:453–459. doi: 10.1007/s00401-013-1150-5. [DOI] [PubMed] [Google Scholar]
- 140.Strong MJ, Kesavapany S, Pant HC. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy. J Neuropathol Exp Neurol. 2005;64:649–664. doi: 10.1097/01.jnen.0000173889.71434.ea. [DOI] [PubMed] [Google Scholar]
- 141.Swarup V, Phaneuf D, Dupré N, Petri S, Strong M, Kriz J, Julien JP. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB–mediated pathogenic pathways. J Exp Med. 2011;208:2429–2447. doi: 10.1084/jem.20111313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Synofzik M, Maetzler W, Grehl T, Prudlo J, Vom Hagen JM, Haack T, Rebassoo P, Munz M, Schöls L, Biskup S. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol Aging. 2012;33:2949. doi: 10.1016/j.neurobiolaging.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 143.Tang Z, Bereczki E, Zhang H, Wang S, Li C, Ji X, Branca RM, Lehtiö J, Guan Z, Filipcik P, Xu S, Winblad B, Pei JJ. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: implication for Alzheimer disease. J Biol Chem. 2013;288:15556–15570. doi: 10.1074/jbc.M112.435123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tanji K, Odagiri S, Miki Y, Maruyama A, Nikaido Y, Mimura J, Mori F, Warabi E, Yanagawa T, Ueno S, Itoh K, Wakabayashi K. p62 deficiency enhances alpha-synuclein pathology in mice. Brain Pathol. 2015;25:552–564. doi: 10.1111/bpa.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H, Sakimura K, Ito H, Takahashi R. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem. 2012;287:42984–42994. doi: 10.1074/jbc.M112.417600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Teyssou E, Takeda T, Lebon V, Boillée S, Doukouré B, Bataillon G, Sazdovitch V, Cazeneuve C, Meininger V, LeGuern E, Salachas F, Seilhean D, Millecamps S. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125:511–522. doi: 10.1007/s00401-013-1090-0. [DOI] [PubMed] [Google Scholar]
- 147.Ticozzi N, Ratti A, Silani V. Protein aggregation and defective RNA metabolism as mechanisms for motor neuron damage. CNS Neurol Disord Drug Targets. 2010;9:285–296. doi: 10.2174/187152710791292585. [DOI] [PubMed] [Google Scholar]
- 148.van der Zee J, Gijselinck I, Van Mossevelde S, Perrone F, Dillen L, Heeman B, Bäumer V, Engelborghs S, De Bleecker J, Baets J, Gelpi E, Rojas-García R, Clarimón J, Lleó A, Diehl-Schmid J, Alexopoulos P, Perneczky R, Synofzik M, Just J, Schöls L, et al. TBK1 mutation spectrum in an extended European patient cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Hum Mutat. 2017;38:297–309. doi: 10.1002/humu.23161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.van der Zee J, Van Langenhove T, Kovacs GG, Dillen L, Deschamps W, Engelborghs S, Matěj R, Vandenbulcke M, Sieben A, Dermaut B, Smets K, Van Damme P, Merlin C, Laureys A, Van Den Broeck M, Mattheijssens M, Peeters K, Benussi L, Binetti G, Ghidoni R, et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014;128:397–410. doi: 10.1007/s00401-014-1298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, Shaw CE. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 151.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Visconti MR, Langston AL, Alonso N, Goodman K, Selby PL, Fraser WD, Ralston SH. Mutations of SQSTM1 are associated with severity and clinical outcome in paget disease of bone. J Bone Miner Res. 2010;25:2368–2373. doi: 10.1002/jbmr.132. [DOI] [PubMed] [Google Scholar]
- 153.Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CK. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 2012;109:15024. doi: 10.1073/pnas.1206362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wooten MW, Geetha T, Babu JR, Seibenhener ML, Peng J, Cox N, Diaz-Meco MT, Moscat J. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J Biol Chem. 2008;283:6783–6789. doi: 10.1074/jbc.M709496200. [DOI] [PubMed] [Google Scholar]
- 155.Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625–35629. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- 156.Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, Martens S. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife. 2015;4:e08941. doi: 10.7554/eLife.08941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH. Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS mice. Neuron. 2015;87:355–370. doi: 10.1016/j.neuron.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang W, Sopper MM, Leystra-Lantz C, Strong MJ. Microtubule-associated tau protein positive neuronal and glial inclusions in ALS. Neurology. 2003;61:1766. doi: 10.1212/01.wnl.0000099372.75786.f8. [DOI] [PubMed] [Google Scholar]
- 159.Yang Y, Tang L, Zhang N, Pan L, Hadano S, Fan D. Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:378–384. doi: 10.3109/21678421.2015.1009466. [DOI] [PubMed] [Google Scholar]
- 160.Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6:171. doi: 10.4103/2152-7806.169561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, Wang Z, Le W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:412–425. doi: 10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- 163.Zhao W, Beers DR, Bell S, Wang J, Wen S, Baloh RH, Appel SH. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp Neurol. 2015;273:24–35. doi: 10.1016/j.expneurol.2015.07.019. [DOI] [PubMed] [Google Scholar]