

Human prion-like proteins often correspond to nucleic acid binding proteins, displaying both globular domains and long intrinsically disordered regions (IDRs) (Harrison and Shorter, 2017). Their IDRs are of low complexity and resemble in amino acid composition to the disordered yeast prion domains, being usually enriched in Gln and Asn residues and depleted in hydrophobic and charged residues. Accordingly, these sequence stretches are named prion-like domains (PrLDs). Prion-like proteins can aggregate into amyloid fibrils, which can accommodate incoming protein monomers, propagating thus the polymeric fold, both processes being driven by their PrLDs. Human prion-like proteins are attracting attention because they are found in the insoluble inclusions identified in an increasing number of neurodegenerative diseases (Harrison and Shorter, 2017). Some well-characterized examples are FUS, TDP43, TAF15, EWSR1, TIA1, hnRNPA1, and hnRNPA2 proteins. Importantly, mutations in the genes that encode for these polypeptides are connected with degenerative diseases, such as amyotrophic lateral sclerosis, frontotemporal dementia or multisystem proteinopathy. A significant proportion of these mutations map in the PrLD of the prion-like protein, and often they result in their accelerated aggregation, both in vitro and in vivo. These proteins shuttle between the nucleus and the cytoplasm and are involved in the formation of membraneless organelles, like stress granules, through liquid-liquid phase separation (LLPS) (Boeynaems et al., 2018). Their aggregation typically occurs after protein mislocalization to the cytoplasm, where they form the insoluble inclusions observed in patients. It has been hypothesized that LLPS, which creates a high local protein density, is the first step towards a liquid-to-solid state transition that initiates aggregation. Membraneless organelles are highly dynamic in order to sense environmental changes and generate adequate adaptative responses. This property obeys to the fact that prion-like proteins phase separate via transient and weak non-covalent interactions. Nevertheless, genetic mutations can strengthen LLPS interactions, increasing the kinetic barrier for dissociation, leading to the population of an irreversible aberrant state, which resolves into the aggregates observed in the above-described diseases. Thus, mutations in these prion-like proteins have been suggested to result in a gain of toxic function phenotype similar to the one occurring in the brain of patients with neurodegenerative diseases like Alzheimer’s and Parkinson’s diseases (Harrison and Shorter, 2017).
Limb-girdle muscular dystrophy (LGMD) is a rare genetic disease of late childhood to adult-onset, characterized by progressive pelvic or shoulder girdle muscle weakness and wasting (Liewluck and Milone, 2018). It is the fourth most common muscular dystrophy. There are autosomal dominant or recessive inherited forms of the disease, referred to as LGMD1 or LGMD2, respectively. LGMD1G is an autosomal dominant variant caused by mutations in the hnRNPDL prion-like protein (Vieira et al., 2014; Berardo et al., 2019; Sun et al., 2019). hnRNPDL is a ribonucleoprotein displaying two canonical RNA recognition motifs and is involved in mRNA biogenesis, including alternative splicing (AS) and transcriptional regulation (Batlle et al., 2020). LGMD1G-linked mutations involve a single conserved Asp residue in the PrLD of hnRNPDL. In LGMD1G patients, this residue is substituted by either His or Asn. As observed for other prion-like proteins, these mutations accelerate the in vitro aggregation kinetics of hnRNPDL (Batlle et al., 2020). It is important to note that the association between the mutation of a single and well-conserved Asp residue in a PrLD and disease also occurs in the case of hnRNPA1, hnRNPA2, and hnRNPD prion-like proteins (Harrison and Shorter, 2017; Prakash et al., 2017). It has been suggested that these Asp residues act as gatekeepers, increasing the energy barrier for self-assembly employing electrostatic repulsion; therefore, their mutation would facilitate the transition towards aggregated states. Importantly, the impact of these mutations cannot be explained simply by a global reduction of the PrLD absolute net charge, but it is instead the specific positions of these Asp residues that determine the increase in aggregation propensities caused by their mutations (Batlle et al., 2017).
Prion-like protein’s associated diseases (i.e., amyotrophic lateral sclerosis, frontotemporal dementia, and multisystem proteinopathy) are characterized by the presence of cytoplasmic inclusions, as mentioned above (Harrison and Shorter, 2017; Batlle et al., 2020). This kind of deposits has, however, not been detected in LGMD1G patients. Batlle et al. (2020) demonstrated recently that the disease-associated mutations impact hnRNPDL solubility in vivo, making the resulting variants utterly insoluble in the myocytes of a Drosophila disease model, without their coalescence into visible protein inclusions. Therefore, the authors proposed that a loss of function mechanism causes LGMD1G, instead of the toxic gain of function phenotype commonly caused by this kind of mutations (Figure 1A). Consistent with this view, hnRNPDL knockdown in zebrafish results in restricted and uncoordinated movements, typically associated with myopathy (Vieira et al., 2014). In the future, it would be relevant to perform a transcriptome analysis of LGMD1G patients, and compare the obtained results with a recent transcriptome study of hnRNPDL knockdown in HeLa cells (Li et al., 2019), to observe if the transcript profiles and splicing patterns coincide in both cellular contexts, thus supporting a loss of function mechanism for LGMD1G.
Figure 1.
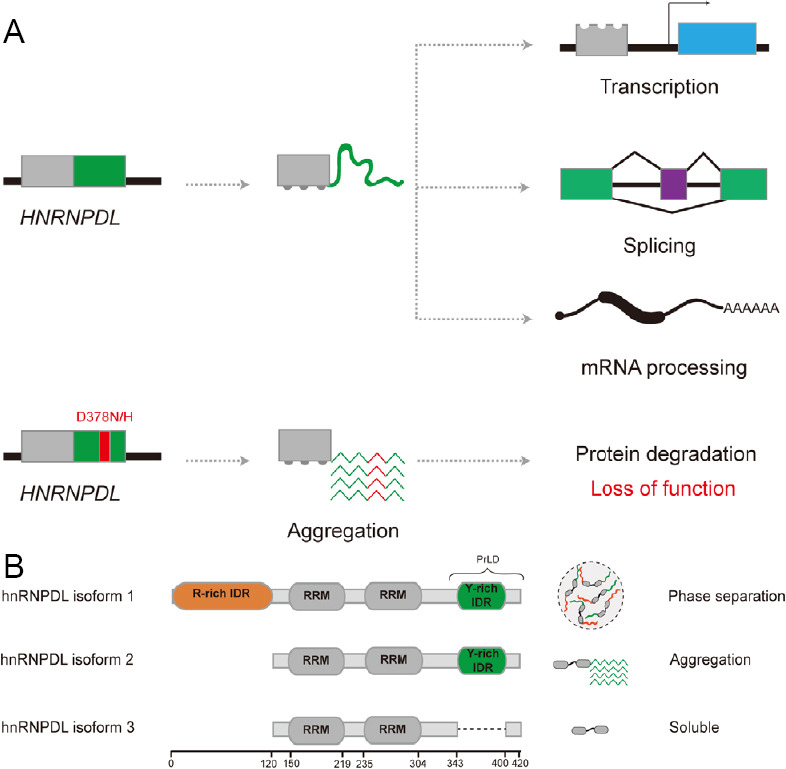
Disease-causing mutations and alternative splicing effects in hnRNPDL.
(A) Schematic representation of hnRNPDL loss of function by LGMD1G-linked mutations. Genome sequencing of LGMD1G patients detected Asn (N) and His (H) substitutions in the Asp (D) residue at position 378 (red). This residue is located in the prion-like domain (PrLD) of hnRNPDL (green), and mutations enhance hnRNPDL aggregation propensity. hnRNPDL functions in transcription, splicing, and mRNA processing are lost because the mutated protein becomes insoluble and is eventually degraded by the protein quality control machinery. (B) Schematic diagram of hnRNPDL isoforms and their behavior. There exist three hnRNPDL isoforms produced by alternative splicing. hnRNPDL consists of two RNA recognition motifs (RRM, dark grey) and two, one or none predicted intrinsically disordered regions (IDRs). hnRNPDL isoform 1 contains an N-terminus Arg-rich IDR (orange) and a C-terminus Tyr-rich IDR (green), which is also predicted to be a PrLD. hnRNPDL isoform 2 only contains the C-terminus Tyr-rich IDR, and isoform 3 does not have any long IDR. The behavior of these hnRNPDL isoforms is connected to their composition: isoform 1 phase separates into liquid droplets, isoform 2 aggregates into amyloid fibrils, and isoform 3 remains soluble in solution.
In their recent work, Batlle et al. (2020) not only show how disease-causing mutations impact protein aggregation, but they also focus on the effect of AS in the properties of prion-like proteins. It is becoming increasingly clear that AS plays a vital role in degenerative disorders. Many prion-like proteins experiment AS events at their PrLDs, generating isoforms with or without these domains, which critically influence their self-assembly properties. It has been reported that the presence of the PrLD in the spliced forms of ribonucleoproteins (hnRNPs) promotes the formation of assemblies that regulate the AS of other genes (Gueroussov et al., 2017; Feng et al., 2019). Therefore, the upstream regulation of hnRNPs’ AS resulting in isoforms with or without PrLDs, regulates at the same time their downstream splicing program. Misregulation of this AS events would lead to an improper intracellular balance of a variety of isoforms with different functionality and aggregation propensities, causing disease.
Three isoforms are produced by AS of the HNRNPDL gene, named hnRNPDL isoform 1, 2, and 3 (DL1, DL2, and DL3). The three isoforms consist of two RNA recognition motifs, but they differ in the number of IDRs present in their sequence (Figure 1B). DL2 is the predominant isoform, and it only contains a C-terminus Tyr-rich IDR, predicted to be a PrLD. DL1 is the longer isoform containing an additional N-terminus Arg-rich IDR, and DL3 does not contain any IDR. Batlle et al. (2020) showed that hnRNPDL isoforms have different LLPS and aggregation behavior, that can be explained according to their IDRs composition. The presence of Arg and Tyr residues at the two distal IDRs in DL1 mediates cation-π interactions promoting LLPS. Mutation of the Arg to Lys or Tyr to Phe decreases DL1 phase separation, indicating that long-distance and specific cation-π interactions govern LLPS in hnRNPDL. Accordingly, DL2, missing the Arg-rich IDR, has lower LLPS propensity, but, in contrast, it is the isoform with higher potential to aggregate into amyloid fibrils. DL3, without IDRs, remains soluble in solution, indicating that the IDRs are the responsible regions for both LLPS and aggregation. LLPS has usually been suggested to be the first step towards amyloid aggregation in prion-like proteins associated with diseases. However, the authors show that this would not be the case for hnRNPDL and LGMD1G. DL1 is the isoform with higher LLPS propensity, but it is instead DL2 the isoform with higher amyloid potential. This observation indicates that the N-terminus Arg-rich IDR in DL1 may act as a protective mechanism against aggregation by promoting LLPS, something not previously described for other prion-like proteins.
The distinct self-assembly properties of hnRNPDL isoforms result in a differential behavior in mammalian cells, impacting their localization, dynamics, and interactions (Batlle et al., 2020). The PrLD in DL1 and DL2 isoforms determines their intranuclear compartmentalization, being both forms excluded from the nucleolus. Moreover, the presence of one or both IDRs in the protein sequence increases protein multivalency, favors protein-protein or protein-nucleic acids interactions, promotes the formation of more rigid and bigger complexes, and decreases protein mobility, being DL1 the less dynamic isoform. The formation of larger or more stable complexes in DL1, due to highly connected interaction networks, affects its nuclear-cytoplasmic shuttling, precluding its transport to the cytoplasm. Interestingly enough, other prion-like proteins, such as FUS, hnRNPD and TAF15 exhibit a similar dependence of their cellular properties on IDRs presence in their isoforms (Gueroussov et al., 2017; Batlle et al., 2020), indicating that AS is an essential and generic mechanism that regulates prion-like proteins’ biological function in cells.
In conclusion, it is now clear that disease-causing mutations in prion-like proteins can promote both loss and gain of function phenotypes, AS controlling the self-assembly properties of these polypeptides, with significant downstream functional consequences. It is still not well understood the exact mechanism by which disease-linked protein aggregates appear and cause neurodegenerative and muscular human disorders since LLPS seems to be able to both promote and prevent amyloid formation. Important open questions remain: Why among a large number of potential mutations in the PrLDs of proteins like hnRNPA1, hnRNPA2, and hnRNPDL, only specific Asp substitutions elicit disease onset? How and why PrLDs inclusion or exclusion is regulated by AS? The answer to these questions might allow developing generic therapeutic approaches for these diseases and provide new insights into human evolution complexity.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Batlle C, Fernandez MR, Iglesias V, Ventura S. Perfecting prediction of mutational impact on the aggregation propensity of the ALS-associated hnRNPA2 prion-like protein. FEBS Lett. 2017;591:1966–1971. doi: 10.1002/1873-3468.12698. [DOI] [PubMed] [Google Scholar]
- 2.Batlle C, Yang P, Coughlin M, Messing J, Pesarrodona M, Szulc E, Salvatella X, Kim HJ, Taylor JP, Ventura S. hnRNPDL phase separation is regulated by alternative splicing and disease-causing nutations accelerate its aggregation. Cell Rep. 2020;30:1117–1128. doi: 10.1016/j.celrep.2019.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berardo A, Lornage X, Johari M, Evangelista T, Cejas C, Barroso F, Dubrovsky A, Bui MT, Brochier G, Saccoliti M, Bohm J, Udd B, Laporte J, Romero NB, Taratuto AL. HNRNPDL-related muscular dystrophy: expanding the clinical, morphological and MRI, phenotypes. J Neurol. 2019;266:2524–2534. doi: 10.1007/s00415-019-09437-3. [DOI] [PubMed] [Google Scholar]
- 4.Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L, Tompa P, Fuxreiter M. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 2018;28:420–435. doi: 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng H, Bao S, Rahman MA, Weyn-Vanhentenryck SM, Khan A, Wong J, Shah A, Flynn ED, Krainer AR, Zhang C. Modeling RNA-binding protein specificity in vivo by precisely registering protein-RNA crosslink sites. Mol Cell. 2019;74:1189–1204. doi: 10.1016/j.molcel.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gueroussov S, Weatheritt RJ, O’Hanlon D, Lin ZY, Narula A, Gingras AC, Blencowe BJ. Regulatory expansion in mammals of multivalent hnRNP assemblies that globally control alternative splicing. Cell. 2017;170:324–339. doi: 10.1016/j.cell.2017.06.037. [DOI] [PubMed] [Google Scholar]
- 7.Harrison AF, Shorter J. RNA-binding proteins with prion-like domains in health and disease. Biochem J. 2017;474:1417–1438. doi: 10.1042/BCJ20160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li RZ, Hou J, Wei Y, Luo X, Ye Y, Zhang Y. hnRNPDL extensively regulates transcription and alternative splicing. Gene. 2019;687:125–134. doi: 10.1016/j.gene.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 9.Liewluck T, Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018;58:167–177. doi: 10.1002/mus.26077. [DOI] [PubMed] [Google Scholar]
- 10.Prakash T, Veerappa A, Ramachandra NB. Complex interaction between HNRNPD mutations and risk polymorphisms is associated with discordant Crohn’s disease in monozygotic twins. Autoimmunity. 2017;50:275–276. doi: 10.1080/08916934.2017.1300883. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Chen H, Lu Y, Duo J, Lei L, OuYang Y, Hao Y, Da Y, Shen XM. Limb girdle muscular dystrophy D3 HNRNPDL related in a Chinese family with distal muscle weakness caused by a mutation in the prion-like domain. J Neurol. 2019;266:498–506. doi: 10.1007/s00415-018-9165-4. [DOI] [PubMed] [Google Scholar]
- 12.Vieira NM, Naslavsky MS, Licinio L, Kok F, Schlesinger D, Vainzof M, Sanchez N, Kitajima JP, Gal L, Cavacana N, Serafini PR, Chuartzman S, Vasquez C, Mimbacas A, Nigro V, Pavanello RC, Schuldiner M, Kunkel LM, Zatz M. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G) Hum Mol Genet. 2014;23:4103–4110. doi: 10.1093/hmg/ddu127. [DOI] [PubMed] [Google Scholar]