

Abstract
Renal ischemia is the most common cause of acute kidney injury. Damage-associated molecular patterns (DAMPs) initiate an inflammatory response and contribute to ischemia–reperfusion (IR) injury in males, yet the contribution of DAMPs to IR injury in females is unknown. The goal of the current study was to test the hypothesis that males have greater increases in the DAMP high-mobility group box 1 (HMGB1), worsening injury compared with females. Thirteen-week-old male and female spontaneously hypertensive rats (SHR) were subjected to sham or 45-min warm bilateral ischemia followed by 24 h of reperfusion before measurement of HMGB1 and renal function. Additional SHR were pre-treated with control (IgG) or HMGB1 neutralizing antibody (300 μg/rat) 1 h prior to renal ischemia. Blood, urine and kidneys were harvested 24 h post-IR for histological and Western blot analyses. Initial studies confirmed that IR resulted in greater increases in renal HMGB1 in male SHR compared with females. Greater renal HMGB1 in male SHR post-IR resulted in greater increases in serum TNF-α and renal IL-1β, neutrophil infiltration and tubular cell death. Neutralization of HMGB1 attenuated IR-induced increases in plasma creatinine, blood urea nitrogen (BUN), inflammation, tubular damage and tubular cell death only in male SHR. In conclusion, our data demonstrate that there is a sex difference in the contribution of HMGB1 to IR-induced injury, where males exhibit greater increases in HMGB1-mediated renal injury in response to IR compared with females.
Introduction
Renal ischemia is the most common cause of acute kidney injury (AKI), which has mortality rates as high as 80% in humans [1–3]. Patients that recover from ischemic AKI are at increased risk for the development of chronic kidney and cardiovascular diseases [4,5]. Despite decades of research, treatment options for AKI remain limited [6]. This is likely related to the fact that the underlying pathological mechanisms mediating ischemia–reperfusion (IR) injury are poorly understood. A better understanding of the cellular mechanisms that mediate renal injury is required to design therapies to treat ischemic AKI in both males and females.
Clinical and pre-clinical studies indicate that males exhibit greater IR-induced renal injury and reduced survival rates [7] compared with females. Although the mechanisms mediating sex differences in IR-injury are still being investigated, there is growing interest in the role of inflammation in mediating renal injury following an ischemic event. In particular, damage-associated molecular pattern molecules (DAMPs) and the activation of downstream toll-like receptors (TLRs) have gained attention due to their ability to activate the immune system and exacerbate injury [8,9]. During IR injury, tubular cell necrosis leads to the release of a wide range of DAMPs; high-mobility group box 1 (HMGB1) is among the best characterized DAMP. HMGB1 is a nuclear factor involved in DNA folding and transcriptional activation in nearly all cell types [10]. HMGB1 can also serve as an extracellular cytokine, and is known to mediate innate immune responses to injury and infection [11,12]. Interestingly, HMGB1 contributes to organ damage in male rodent kidneys following an ischemic insult [13–15] via the activation of TLR4 [13,16] and the recruitment of inflammatory cells and release of pro-inflammatory cytokines and chemokines [17–19]. Moreover, pharmacological inhibition of HMGB1 release [20,21] or HMGB1 neutralization using antibodies [22,23] has confirmed a critical role for HMGB1 in mediating renal IR-injury in males. The contribution of HMGB1 to IR injury in females is unknown.
We previously reported that male spontaneously hypertensive rats (SHR) have greater renal necrosis than females under baseline conditions [24], and necrotic cell death is associated with the release of DAMPs, including HMGB1. Moreover, male SHR also have a more pro-inflammatory renal T-cell profile than females [25]. However, in response to renal ischemia, no sex differences in renal injury were reported 24 h post-IR [26], suggesting that different pathways may contribute to IR injury in males and females. The goal of the current study was to test the hypothesis that males have elevated levels of HMGB1 release compared with females and that this contributes to greater renal injury. Our results demonstrate the greater HMGB1 release after IR in males contributes to IR-induced inflammation in males but not in females, supporting a sex difference in the pathways driving IR injury.
Materials and methods
Animals
Thirteen-week old age-matched male and female SHR (Envingo Laboratories, Indianapolis, IN) were studied. Rats were housed in temperature- and humidity-controlled, light-cycled quarters. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved and monitored by the Augusta University Institutional Animal Care and Use Committee (Approval number 2014–048). All animal work took place in the Sullivan lab at the Medical College of Georgia at Augutsa University.
Warm bilateral renal IR
Rats (n=6) from each sex were randomized to receive either sham or IR surgery as previously described [26]. Briefly, rats were anesthetized with ~2% isoflurane and placed on a heating pad to maintain body temperature at 37°C. Renal arteries were isolated and clamped for 45 min with microserrefines (Fine Science Tools, Foster City, CA). Reperfusion was confirmed visually upon release of the clamps. Sham rats were subjected to the same surgical procedure, except the renal arteries were not clamped. Surgical wounds were closed, and rats were given 1 ml of warm saline intraperitoneally to replace any fluid loss. After 24 h of reperfusion, all rats were anesthetized with ~2% isoflurane. A terminal blood sample was taken, and kidneys were harvested. Additional rats undergoing IR surgery (n=5–6) were randomized to receive a single intraperitoneal injection of control (chicken IgG; 300 μg/rat) or HMGB1 neutralizing = antibody (chicken anti-HMGB1 polyclonal; IBL International Corp., Toronto, ON, Canada; 300 μg/rat i.p.) 1 h before clamping the renal arteries. All the rats were killed after 24 h of reperfusion; a terminal blood sample was taken, and kidney tissue harvested.
Western blot analysis
Thirty micrograms of renal cortex was homogenized, and the whole homogenate was used for Western blot analysis as previously described [27]. Briefly, samples were resolved on 4–20% Tris-glycine-SDS gels (Bio-Rad, Hercules, CA) and proteins were transferred to PVDF membranes (Millipore Sigma, Burlington, MA). Protein expression was determined using two-color immunoblots using primary antibodies to HMGB1 (cat# MA1–20338, 1:1000 dilution; Thermo Fisher Scientific, Lafayette, CO), and TLR4 (cat# 217274, 1:300 dilution; Abcam, Cambridge, MA). Specific protein bands were detected using an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE) with AlexaFluor 680 and IRDye800 (Molecular Probes, Eugene, OR) conjugated secondary antibodies. Protein concentrations were determined by standard BCA reagent (Thermo Scientific) using BSA as the standard. Protein loading was normalized to glyceraldehyde 3-phosphate dehydrogenase (Cell Signaling Technologies, Danvers, Massachusetts).
Creatinine and blood urea nitrogen measurement
Plasma creatinine and blood urea nitrogen (BUN) were measured using commercially available assay kits according to the manufacturer’s instructions (BioAssay Systems, Hayward, CA; Quantichrome Creatinine Assay Kit, Cat# DICT-500, Lot number CA04A06; Quantichrome Urea Assay Kit, Cat # DIUR-100, Lot number CA02A12) [26].
TNF-α measurement
Plasma levels of TNF-α were measured using a commercial ELISA according to the manufacturer’s instructions (Abcam, Cambridge, MA).
Terminal deoxynucleotidyl-transferase-mediated dUTP nick-end labeling staining
Terminal deoxynucleotidyl-transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed to identify cell death in renal tissue using the ApopTag plus Peroxidation In Situ Apoptosis Detection kit (EMD Millipore; S7101) according to the manufacturer’s instructions. Briefly, 5 μM kidney sections were deparaffinized, hydrated, and washed with PBS. Sections were digested with proteinase K for 15 min at 24°C. Slides were then washed with PBS, and endogenous peroxidase activity was quenched with 3% H2O2 in methanol. Slides were washed again and incubated with TdT-labeling reaction mix at 37°C for 1 h and then with anti-digoxigenin peroxidase. The color was developed using peroxidase substrate solution. Slides were washed, counterstained, and mounted with Permount. Sections were blinded, photographed, and labeled cells were counted and quantified the brown nuclei indicating TUNEL-positive cells.
Assessment of tubular injury
Kidneys from male and female SHR were bisected transversely with a razor blade, fixed in buffered 10% formalin overnight and then embedded in paraffin wax. For assessment of renal injury, 5 μM sections were stained with Hematoxylin and Eosin (H&E) following the protocol provided by the manufacturer (Leica Biosystems; Buffalo Grove, IL). Renal tubular injury was assessed using a semi-quantitative scale [28] in which the percentage of tubules showing epithelial cell necrosis, brush-border loss, cast formation, and apoptotic bodies were assessed. Quantification of renal tubular injury was performed in a blinded manner and assigned a score in terms of the percentage of tubular injury: 0 = normal; 1 = <25%; 2 = 25–50%; 3 = 51–75%; 4 = >75%. 10 fields at 20× magnification were examined and the data averaged for each animal. Stained sections were photographed using an Olympus BX40 microscope (Olympus America, Melville, NY) on a bright-field setting fitted with a digital camera (Olympus DP12; Olympus America).
Myeloperoxidase immunostaining
To quantify renal neutrophil infiltration, formalin-fixed kidneys were sectioned and 5 μm sections were incubated in the absence or presence of primary antibodies to myeloperoxidase (Abcam, cat # ab9535; 1:150 dilution) in humidified chambers overnight at 4°C, followed by incubation with HRP-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. Color was developed after incubation with DAB reagent (Vector Lab, Burlingame, CA). Ten fields at 40× magnification were examined in a blinded manner.
Real-time quantitative reverse transcriptase PCR
Total RNA was extracted from the snap-frozen kidney cortex using RNeasy Mini Kits (Qiagen, Germantown, MD) according to the manufacturer’s protocol as described previously [29]. RNA (2 μg) was reverse transcribed to complement DNA using iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad, Hercules, CA). The product was diluted to a volume of 150 μl, and 5 μl aliquots were used for amplification. Real-time PCR was performed on a StepOne real-time instrument (Applied Biosystems, Foster City, CA) using iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA) and gene specific primers from Qiagen: rat HMGB1 (Cat# QT00368410), rat KIM-1 (Cat# QT00185983), rat CXCR1 (Cat# QT00459592), rat CXCR2 (Cat# QT00416675), rat CXCL2 (Cat# QT00184891) and rat IL1β (Cat # QT00181657) and Bio-Rad: rat CXCL1 (qRnoCED0003672) and rat CCL2 (qRnoCED0009272). The amount of DNA was normalized to the rat GAPDH signal amplified in a separate reaction (Cat# QT00199633).
Statistical analysis
All data are presented as means ± SEM. Statistical analyses were performed using GraphPad Prism version 7.0 software (GraphPad Software, La Jolla, CA). All data were compared using two-way ANOVA. P<0.05 was considered significant.
Results
Male SHR have greater increases in renal HMGB1 24 h post-IR than females despite comparable injury
Renal levels of HMGB1 mRNA and protein were measured in male and female SHR 24 h following sham or IR surgery. HMGB1 mRNA and protein levels significantly increased in response to IR in both sexes compared with respective sham control (PIR = 0.0001). Moreover, male SHR exhibited greater increases in IR-induced HMGB1 mRNA (mRNA: Psex = 0.04, Psex*IR = 0.03) and protein expression (Psex = 0.004, Psex*IR = 0.04) in the kidney compared with females 24 h post-IR (Figure 1A–C). IR injury was also assessed in male and female = SHR 24 h post-IR by measuring plasma creatinine, tubular injury marker (KIM-1) mRNA expression and tubular damage (Figure 1D–G). IR resulted in injury in both males and females compared with respective sham control (PIR = 0.0001 for all comparisions). Plasma creatinine, KIM-1 expression levels and tubular damage were comparable in males and females 24 h post-IR (Pcr: Psex = 0.08, Psex*IR = 0.002; KIM-1: Psex = 0.39, Psex*IR = 0.43; tubular damage: Psex = 0.84, Psex*IR = 0.69), although the IR-induced increase in plasma creatinine was greater in males than in females.
Figure 1. Male SHR have greater increases in renal HMGB1 24 h post-IR than females despite comparable injury.
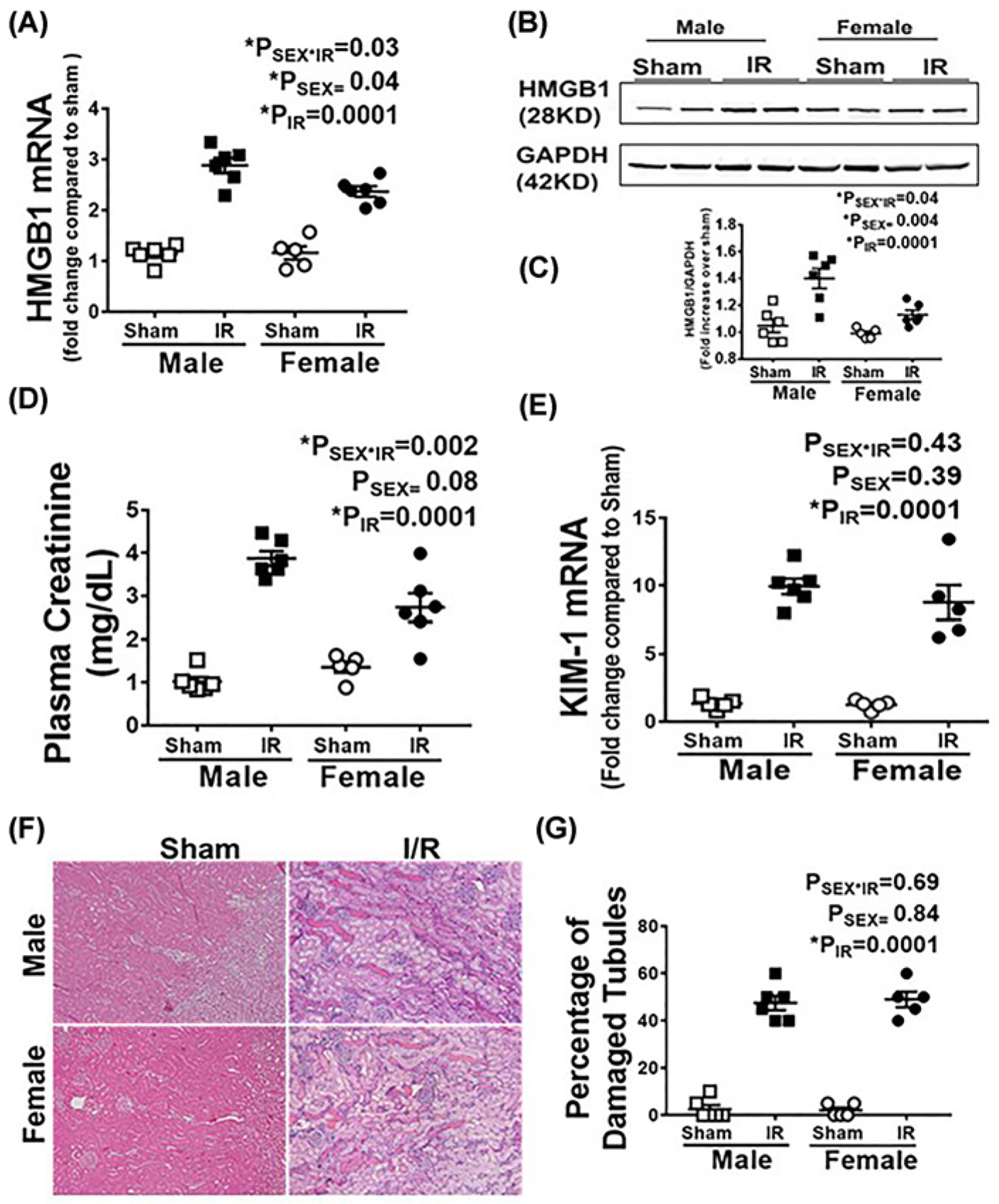
Renal HMGB1 mRNA (A) and protein expression were measured in 13 week old male and female SHR 24 h following sham or 45 min bilateral IR. (B) is a representative Western blot and (C) is the average densitometric analysis. Plasma creatinine (D), renal KIM-1 mRNA levels (E) and tubular damage (F,G) were also measured as indices of renal injury. Representative tubular damage images are shown in (F) (10× magnification, scale bar 200 μM) with the average pathological scoring data in (G). Data are expressed as means ± SEM with individual animal data indicated by the symbols, n=5–6 rats in each group. Open symbols indicate sham animals, filled symbols indicate 45-min ischemia, males are represented by squares and females by circles. Data were compared via two-way ANOVA with P<0.05 considered significant.
HMGB1 neutralizing antibody attenuates IR-induced increases in inflammation
To assess the relative contribution of HMGB1 to IR-induced inflammation and injury, additional male and female SHR were treated with control IgG or HMGB1 neutralizing antibody 1 h prior to IR. HMGB1 induced activation of TLR4 signaling drives immune system activation and promotes renal damage [14]. Therefore, we first measured TLR4 protein expression in kidney homogenates 24 h post-IR by Western blot analysis. Female SHR had less renal TLR4 protein expression than male SHR 24 h post-IR (Psex<0.002), and HMGB1 neutralization significantly attenuated IR-induced increases in TLR4 only in males (PTxt = 0.003; Psex*Txt = 0.11: Figure 2A,B).
Figure 2. TLR4 up-regulated in male but not female SHR 24 h post-IR.
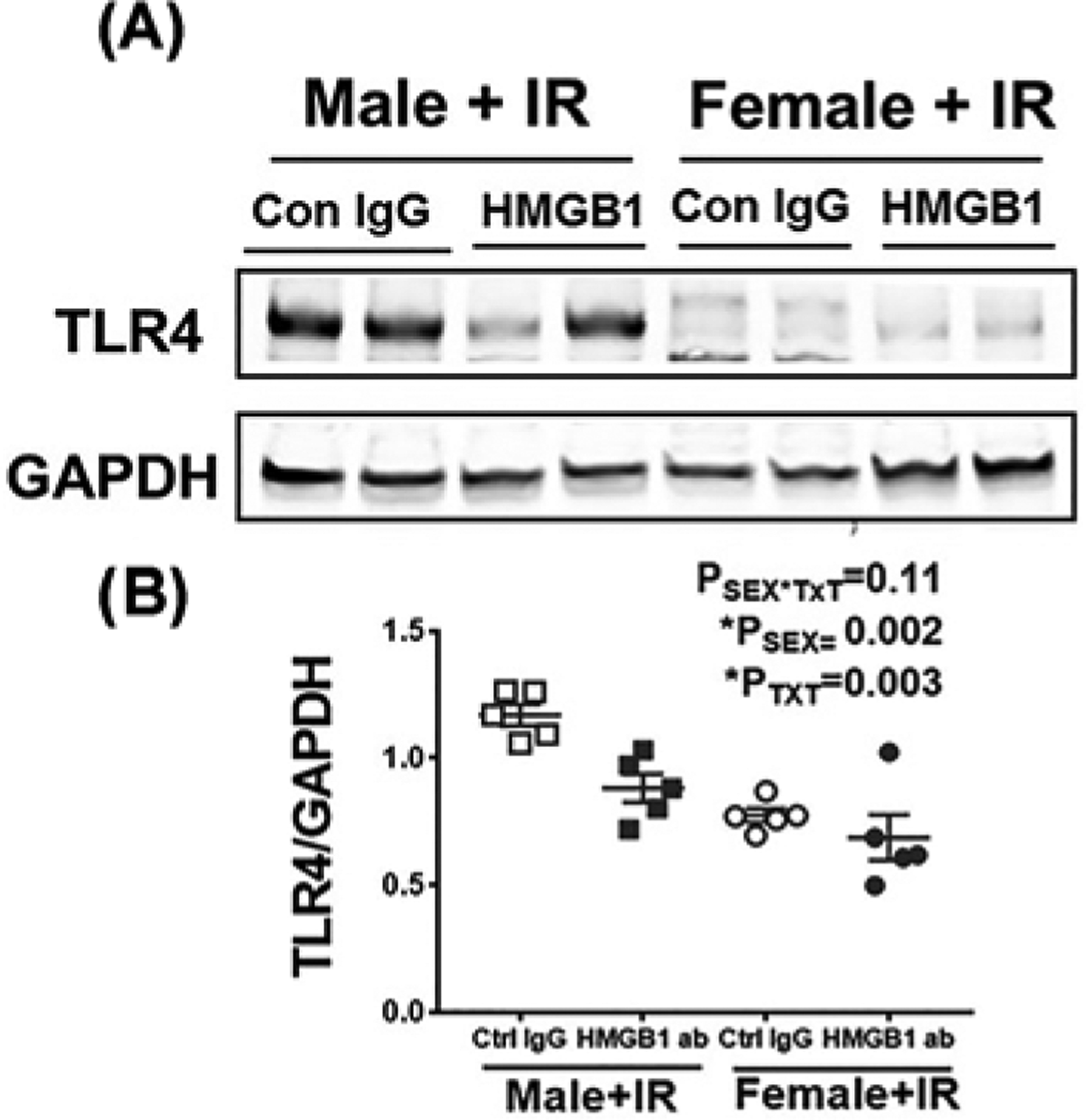
Renal TLR4 expression was assessed by Western blot analysis of kidney homogenates 24 h post-IR in 13 week old male and female SHR treated with control IgG or HMGB1 neutralizing antibody 1 h prior to IR. (A) are representative Western blots and (B) is the average densitometric analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression was used for normalization of protein loading. Data are expressed as means ± SEM, with individual animal data indicated by the symbols, n=5 rats in each group. Open symbols indicate control IgG-treated animals, filled symbols indicate anti-HMGB1-treated animals, males are represented by squares and females by circles. Data were compared via two-way ANOVA with P<0.05 considered significant.
TLR4 activation by HMGB1 induces downstream expression of inflammatory cytokines in the kidney [30]. To investigate the effect of HMGB1 neutralization on IR-induced inflammation, we measured key cytokines downstream of HMGB1 activation (circulating TNF-α and renal IL-1β) and renal neutrophil infiltration in male and female SHR treated with control IgG or HMGB1 neutralizing antibody 24 h post-IR. Consistent with male SHR having greater increases in HMGB1 post-IR than females, levels of TNF-α and IL-1β were greater in control IgG-treated males vs. females (Psex = 0.0001 and Psex = 0.002, respectively; Figure 3A,B). Pretreatment with HMGB1 neutralizing antibody significantly reduced serum TNF-α (PTxT = 0.007; Psex*TxT = 0.001) and renal IL-1β levels (PTxT = 0.024; Psex*TxT = 0.024) in male SHR with no effect in females (Figure 3A,B).
Figure 3. Neutralizing HMGB1 attenuates IR-induced increases in inflammatory cytokine levels 24 h post-IR only in male SHR.
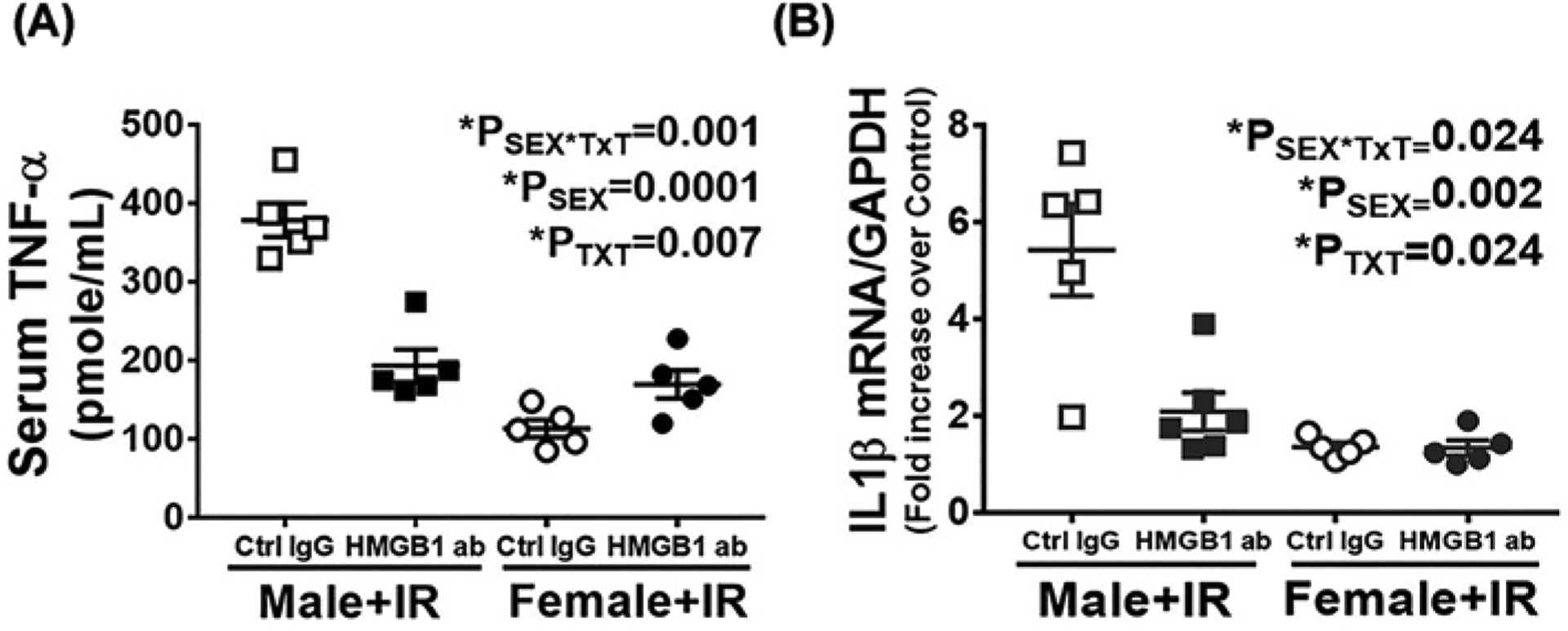
Serum and kidney samples were collected 24 ht-IR in 13 week old male and female SHR treated with control IgG or HMGB1 neutralizing antibody 1 h or to IR. Serum TNF-α was quantified by ELISA (A) and renal IL1β mRNA expression was measured by RT-PCR (B). Data are expressed as means ± SEM; n=5–6 rats in each group with individual animal data indicated by the symbols. Open symbols indicate control IgG-treated animals, filled symbols indicate anti-HMGB1-treated animals, males are represented by squares and females by circles. Data were compared via two-way ANOVA with P<0.05 considered significant.
To further determine if decreased inflammatory cytokine expression with anti-HMGB1 antibody treatment was associated with reductions in inflammatory cell infiltration, we measured neutrophil infiltration in the kidney by myeloperoxidase immunostaining. Control IgG-treated male SHR had significantly greater neutrophil infiltration into the kidney vs. females (Psex = 0.04) (Figure 4A,B). Neutralization of HMGB1 suppressed IR-induced neutrophil infiltration into the kidney of male SHR only (PTxT = 0.08; Psex*Txt = 0.03). To gain additional mechanistic insight underlying differences in neutrophil infiltration, components of the CXCR–CXCL axis were measured. The CXCR–CXCL axis is known to regulate neurophil accumulation in the kidney during ischemic AKI [31,32]. Males exhibited significantly greater expression of CCL2 (Psex = 0.001) and CXCR1 (Psex = 0.0004) than females. Consistent with reduced neurophil accumulation in males treated with anti-HMGB1, neutralization of HMGB1 also attenuated IR-induced increases in mRNA expression of the renal chemokines CCL2 (PTxT = 0.09; Psex*Txt = 0.035), CXCL1 (Psex = 0.07; PTxT = 0.04; Psex*Txt = 0.04) and CXCL2 (Psex = 0.23; PTxT = 0.057; Psex*Txt = 0.005) only in males. Expression of the chemokine receptors CXCR1 (PTxT = 0.014; Psex*Txt = 0.45) and CXCR2 were decreased by HMGB1 neutralization in both sexes (Psex = 0.12; PTxT = 0.02; Psex*Txt = 0.13) (Figure 4C–G).
Figure 4. Neutralizing HMGB1 reduced renal IR-induced neutrophil infiltration 24 h post-IR only in male SHR.
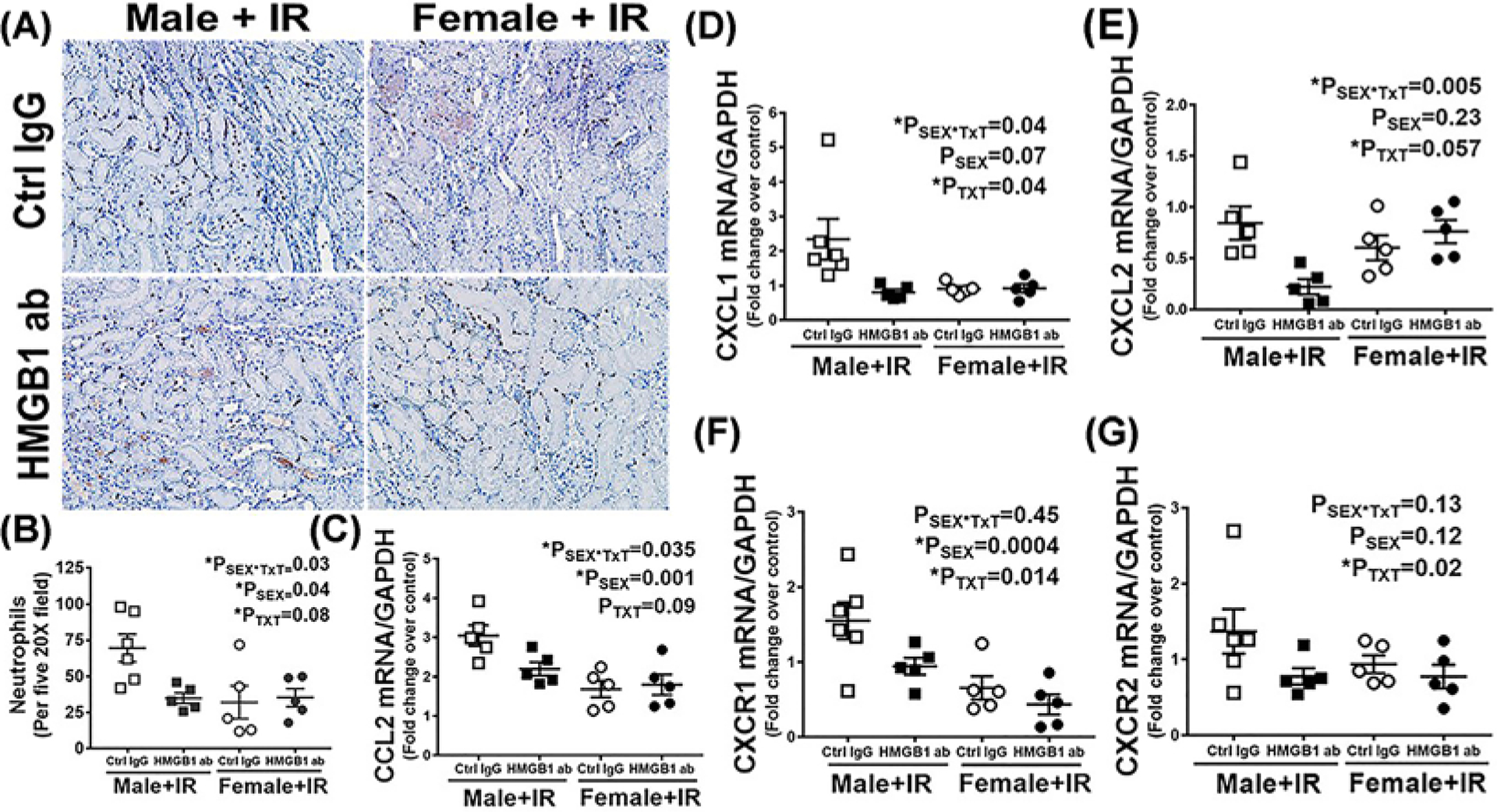
Renal neutrophil infiltration was assessed by in a blinded manner by immunohistochemical analysis of myeloperoxidase staining in kidney samples collected 24 h post-IR in 13 week old male and female SHR treated with control IgG or HMGB1 neutralizing antibody 1 h prior to IR. Brown nuclei staining indicating neutrophil positive cells. Representative images are provided in (A) with the quantification in (B). Renal chemokines and receptor mRNA expression were measured by RT-PCR (C–G). Data are expressed as means ± SEM, n=5–6 rats in each group with individual animal data indicated by the symbols. Open symbols indicate control IgG-treated animals, filled symbols indicate anti-HMGB1-treated animals, males are represented by squares and females by circles. Data were compared via two-way ANOVA with P<0.05 considered significant.
HMGB1 contributes to renal IR injury in male but not female SHR
To assess the relative contribution of HMGB1 to IR-induced renal injury, injury markers were measured 24 h post-IR in male and female SHR pre-treated with control IgG or HMGB1 neutralizing antibody 1 h prior to IR. Plasma creatinine and BUN were comparable in control IgG treated male and female SHR 24 h post-IR (Psex = 0.25 and Psex = 0.084, respectively). HMGB1 neutralizing antibody significantly reduced IR-induced increases in plasma creatinine (PTxT = 0.015, Psex*TxT = 0.01) and BUN (PTxT = 0.0003, Psex*TxT = 0.001), but only in male SHR (Figure 5A,B). We further assessed the effect of HMGB1 neutralization on IR-induced cell death in male and female SHR by measuring renal cell death using TUNEL staining. Administration of HMGB1 neutralizing antibody significantly attenuated cell death only in males (Psex = 0.024; PTxT = 0.004; Psex*TxT = 0.034; Figure 6A,B). Similarly, there was comparable tubular in the kidney of control IgG-treated male and female SHR 24 h post-IR, and HMGB1 neutralization decreased tubular damage (Psex*TxT = 0.016; Psex = 0.009; PTxT = 0.009) and tubular injury marker KIM-1 mRNA expression (Psex*TxT = 0.007; Psex = 0.07; PTxT = 0.38) only in males Figure 7A–C).
Figure 5. Neutralizing HMGB1 attenuates IR injury in male, but not female SHR.
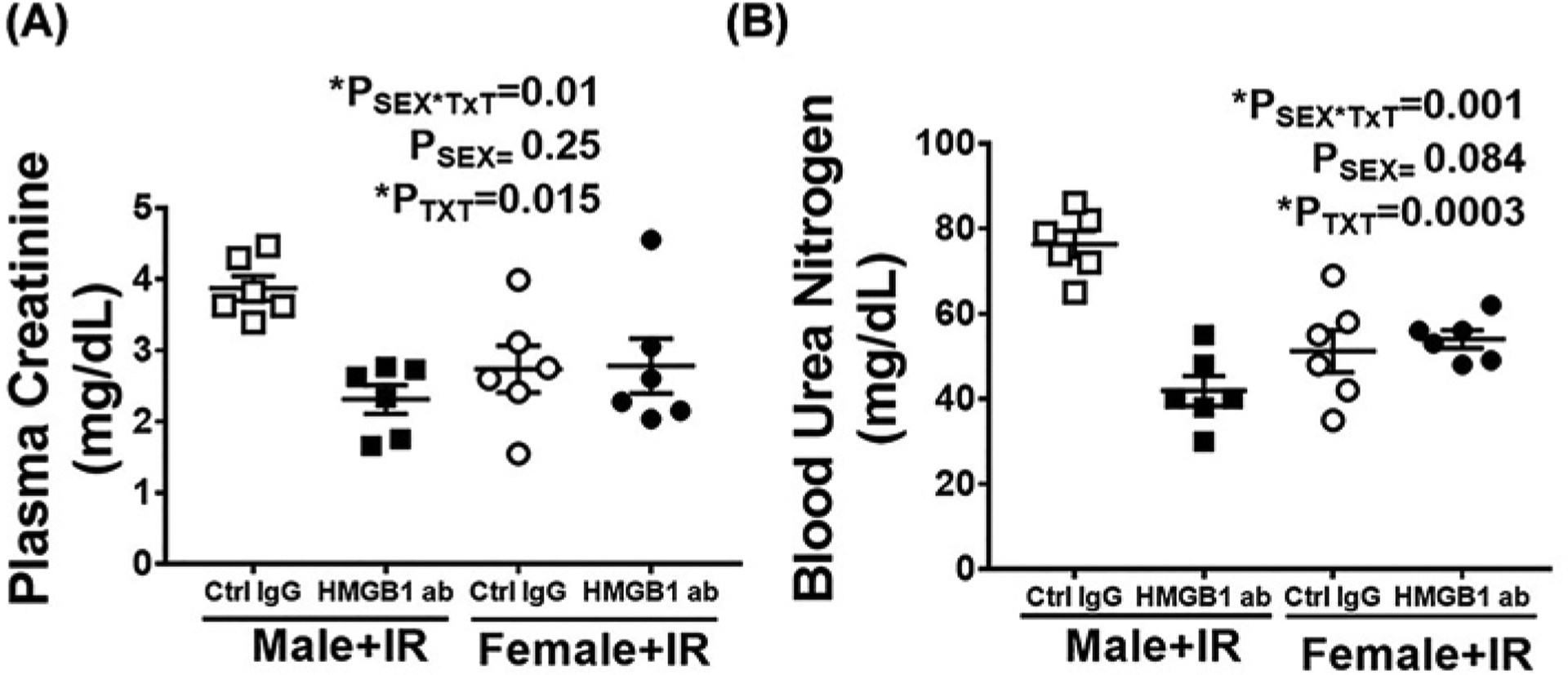
Plasma creatinine (A) and BUN (B) were measured in samples collected 24 h post-IR in 13 week old male and female SHR treated with control IgG or HMGB1 neutralizing antibody 1 h prior to IR. Data are expressed as mean ± SEM, n=6 rats in each group with individual animal data indicated by the symbols. Open symbols indicate control IgG-treated animals, filled symbols indicate anti-HMGB1-treated animals, males are represented by squares and females by circles. Data were compared via two-way ANOVA with P<0.05 considered significant.
Figure 6. Neutralizing HMGB1 attenuates renal cell death 24 h post-IR in male, but not female SHR.
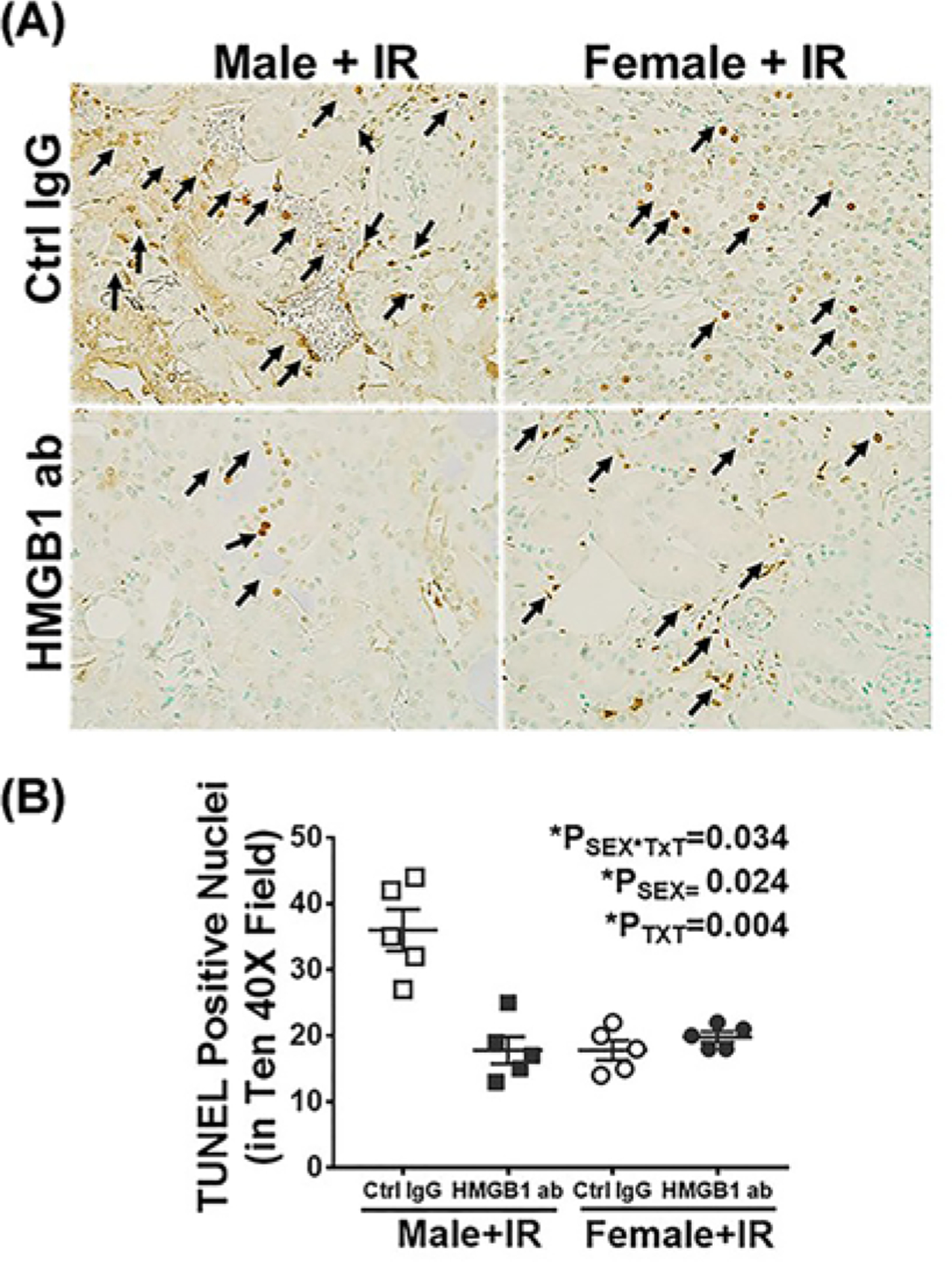
Cell death was measured by TUNEL staining in kidneys collected 24 h post-IR in 13 week old male and female SHR treated with control IgG or HMGB1 neutralizing antibody 1 h prior to IR. Brown nuclei with black arrows indicate TUNEL-positive cells. Representative images are shown in (A) with mean quantification data in (B). Data are expressed as mean ± SEM, n=5 rats in each group with individual animal data indicated by the symbols. Open symbols indicate control IgG-treated animals, filled symbols indicate anti-HMGB1-treated animals, males are represented by squares and females by circles. Data were compared via two-way ANOVA with P<0.05 considered significant.
Figure 7. Neutralization of HMGB1 attenuates tubular injury in male SHR compared with females 24 h post-IR.
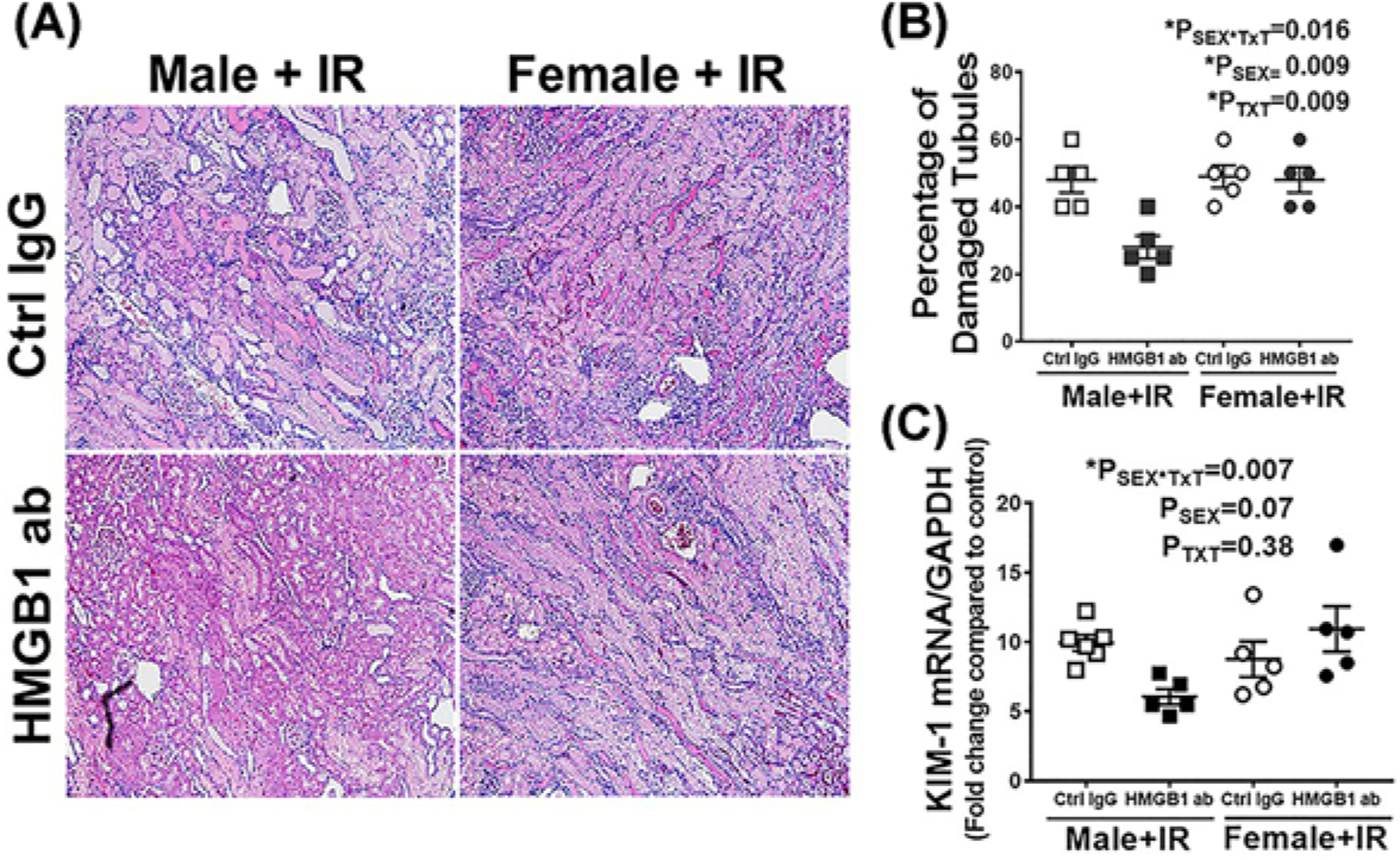
Tubular injury was assessed via histological analysis and measurement of renal KIM-1 mRNA expression via RT-PCR in kidneys collected 24 h post-IR in 13 week old male and female SHR treated with control IgG or HMGB1 neutralizing antibody 1 h prior to IR. Representative images of tubular damage are shown in (A) (10× magnification, scale bar 200 μM) with the mean pathological scoring data in (B). (C) is the renal KIM-1 mRNA expression normalized to GAPDH. Data are expressed as mean ± SEM, n=5–6 rats in each group with individual animal data indicated by the symbols. Open symbols indicate control IgG-treated animals, filled symbols indicate anti-HMGB1-treated animals, males are represented by squares and females by circles. Data were compared via two-way ANOVA with P<0.05 considered significant.
Discussion
HMGB1 contributes to renal IR injury by the induction of a cytokine-driven inflammatory response and exacerbation of tissue injury [11,12]. As such, treatment with HMGB1 inhibitors or neutralizing antibodies reduces inflammation and protects the kidney from ischemic injury in many preclinical animal models, including models of renal IR [14,16]. However, for all these studies, conclusions were drawn based on studies performed exclusively in males. The contribution of HMGB1 to IR-induced renal injury in females was unknown. The role of HMGB1 in IR injury in females is of interest, in part, due to numerous studies supporting sex differences in renal IR injury in normotensive rodent models, where males have greater injury compared with females. However, the mechanisms mediating sex differences in renal IR-injury are unknown. Our studies report for the first time that there is a sex difference in the contribution of HMGB1 to IR-induced renal injury. Males have greater IR-induced HMGB1 release and greater HMGB1-mediated increases in inflammation and injury relative to females. However, male SHR had comparable levels of plasma creatinine and tubular damage to females 24 h post-IR compared with their respective sham controls, further suggesting that the mechanisms mediating IR-injury are distinct between the sexes.
HMGB1 is an evolutionarily conserved protein present in the nucleus of almost all eukaryotic cells. HMGB1 is both secreted from activated macrophages and released from damaged cells to serve as a signal to trigger an inflammatory response during ischemic injury in multiple organs [14,15]. Numerous clinical and basic science studies have implicated HMGB1 in the pathogenesis of renal diseases [33]. In particular, HMGB1 is highly elevated in AKI patients and male rodent models of ischemic AKI [13] and sepsis [34]. Moreover, HMGB1 neutralization before and after renal IR protects the kidney from ischemic [13,14] and sepsis-induced injury [35]. In contrast, administration of recombinant HMGB1 exacerbates renal injury in male mice [14]. These studies clearly establish a pathological role for HMGB1 in ischemic AKI in male rodents, and our studies confirm this finding in male SHR. In addition, we found that renal HMGB1 mRNA and protein levels are also increased in females following IR, although to a lesser extent than in males. Additional studies therefore examined the relative contribution of HMGB1 to IR injury in both sexes.
Although there are numerous reports in the literature of sex differences in renal injury 24 h post-IR [36,37], all of these reports are in normotensive animal models. A previous study by our group in SHR reported that 45 min of bilateral ischemia followed by 24 h of reperfusion resulted in pronounced IR injury in both male and females [26]. Since the SHR is also known to exhibit sex differences in renal inflammation [25] and cell death [24], we used SHR as an animal model to determine the relative contribution of HMGB1 to IR-induced injury in males and females. Although we did not observe significant effects of sex on plasma creatinine, BUN, KIM-1 mRNA levels, or tubular injury 24 h post-IR in male and female SHR, males exhibited greater increases in HMGB1 and greater increases in neutrophil infiltration, chemokine expression, and the inflammatory cytokines TNF-α and IL-1β. Taken together, our data support the hypothesis that the mechanisms mediating IR injury may be distinct between the sexes and injury in males may be more dependent on HMGB1 than in females.
Although the mechanisms mediating the sex difference in HMGB1 release and function were not the primary focus of the present study, we speculate that this may be related to sex differences in cell death. IR injury can lead to cell death [38], and we confirmed in the current study that IR increased cell death (TUNEL-positive) in both males and females, with males having a greater increase in cell death compared with females. Although the TUNEL assay is commonly used to measure apoptotic cell death, it is unable to discriminate among apoptosis, necrosis, and autolytic cell death as it stains for all fragmented DNA [39]. Moreover, cells from males have been shown to be more prone to necrotic cell death compared with females [24,40,41], and we recently published that male SHR have greater renal necrosis than females [24]. In addition, higher level of necrotic cell death were reported in lungs of males with pulmonary arterial hypertension [42]. Therefore, it is plausible that greater necrotic cell death may result in greater HMGB1 release which exacerbated the inflammatory response in the male IR kidney. Future studies will be designed to further test this hypothesis and assess the potential role of sex chromosomes vs. hormones in mediating sex differences in cell death [43–46]. In addition, only the reduced form of HMGB1 is capable of stimulating pro-inflammatory cytokine activity [47], therefore males may also have more reduced HMGB1 further leading to exacerbation of inflammation vs. females.
Recent experimental data suggest that IR rapidly activates an immune response through TLR signaling. TLRs are expressed by a variety of immune and non-immune cells, including kidney tubular epithelial cells, and IR is known to up-regulate TLR4 in tubular epithelial cells [14,16]. Moreover, recombinant HMGB1-induced inflammation in male rodents is blocked by TLR4 deletion, suggesting TLR4 mediates HMGB1 inflammatory responses [48]. Concomitant with increased HMGB1 levels, we found that TLR4 expression increased after IR only in male SHR, and HMGB1-mediated IR injury was apparent only in males. These data suggest that endogenous HMGB1 released from damaged cells after IR may promote kidney injury through TLR4 activation in males. Although not directly assessed in the current study, TLR4 activation by HMGB1 leads to neutrophil recruitment and increased production of pro-inflammatory chemokines and cytokines in the male ischemic kidney [14,16,30]. In addition, neutralization of HMGB1 or TLR4 deletion reduced IR induced inflammatory cytokines and chemokines in male IR mice [14]. Consistent with these previous reports, we found that greater HMGB1 release in males was associated with enhanced neutrophil infiltration and increased circulating TNF-α level and renal IL-1β in males compared with females after 24 h of reperfusion, which were reduced with HMGB1 neutralization. The chemokines CXCL and MCP-1 are up-regulated during ischemic AKI and contribute to renal neutrophil accumulation via the CXCR1 and CXCR2 receptors [32,49]. Further, inhibition of the CXCL1–CXCR2 axis reduced inflammation and protected the kidney from cisplatin-induced AKI [50]. Consistent with reduced neurophil accumulation in male SHR with HMGB1 neutralization in the current study, expression levels of the chemokines CCL2, CXCL1, and CXCL2 were all attenuated by anti-HMGB1 pre-treatment only in males. This observation suggests that HMGB1 mediated activation of inflammatory responses may be directly involved in IR-induced renal injury only in males.
It is important to note that despite the finding that HMGB1 does not significantly contribute to IR-induced injury in female SHR, overall measures of injury were comparable between the sexes. These data beg the question of what is mediating IR-induced injury in the females. We have previously pubished that female SHR have greater numbers of T-regulatory cells compared with male SHR [51], and T-regulatory cells have been shown to protect against IR injury [52]. Greater numbers of Tregs may therefore protect females from IR-mediated inflammatory injury. It should also be noted that injury was assessed at 24 h post-IR, an the impact of HMGB1 may become apparent in females at later time points during recovery. Alternatively, we speculate that females may exhibit greater increases or sensitivity to tubular hypoxia, oxidative stress and lipid peroxidation or mitochondrial dysfunction [53] in the 24 h following IR. More studies are needed to elucidate the mechanisms mediating renal injury in female SHR.
In conclusion, the current study identified a novel pathway that contributes to sex differences in IR injury, where HMGB1 possesses strong pro-inflammatory properties and exacerbates renal damage in male, but not female SHR. This finding opens a completely new avenue in the biology of sex differences in AKI. It suggests a strong requirement need to take the sex of the patient into account, as well as emphasizing the necessity for additional studies aimed to develop sex-based pharmacological approaches. In addition, it is important to further investigate why there are lower levels of HMGB1 release in females compared with males after IR injury. By studying these mechanisms of natural protection, it may become possible to learn to control the level of the immune response to the appropriate benefit of each gender.
Clinical perspectives.
Background as to why the study was undertaken: Renal IR injury is a major cause of AKI, which carries a high mortality rate and increases the risk of later developing hypertension and chronic kidney disease. Recent studies have reported that HMGB1 contributes to renal IR damage in males. However, the contribution to HMGB1 to IR injury in females is unknown, and the mechanisms mediating potential sex differences in renal IR-injury are unknown.
A brief summary of the results: Despite comparable renal injury 24 h post-IR in male and female SHR, male SHR exhibit greater increases in HMGB1 release than females. In addition, neutralization of HMGB1 attenuates IR-induced increases in inflammation and renal injury only in males, suggesting that the mechanisms mediating IR-injury are distinct between the sexes.
The potential significance of the results to human health and disease: Greater understanding of the molecular mechanisms driving IR-induced increases in injury in both males and females will allow for the development of novel therapies to limit AKI severity and mortality.
Funding
This work was supported by the National Institutes of Health [grant numbers 1P01HL134604-01, R01 HL127091 to (J.C.S.), 1P01HL134604-01 (to P.M.O.)].
Abbreviations
- AKI
acute kidney injury
- BUN
blood urea nitrogen
- DAMP
damage-associated molecular pattern
- HMGB1
high-mobility group box 1
- IR
ischemia–reperfusion
- SHR
spontaneously hypertensive rat
- TLR
toll-like receptor
- TUNEL
terminal deoxynucleotidyl-transferase-mediated dUTP nick-end labeling
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Chertow GM, Levy EM, Hammermeister KE, Grover F and Daley J (1998) Independent association between acute renal failure and mortality following cardiac surgery. Am. J. Med 104, 343–348, 10.1016/S0002-9343(98)00058-8 [DOI] [PubMed] [Google Scholar]
- 2.Metnitz PG, Krenn CG, Steltzer H, Lang T, Ploder J et al. (2002) Effect of acute renal failure requiring renal replacement therapy on outcome in critically ill patients. Crit. Care Med 30, 2051–2058, 10.1097/00003246-200209000-00016 [DOI] [PubMed] [Google Scholar]
- 3.Hofmann RM Preventing harm during treatment of acute kidney injury: what do we really know? Adv. Chronic Kidney Dis 19, 142–148, 10.1053/j.ackd.2012.03.006 [DOI] [PubMed] [Google Scholar]
- 4.Coca SG, Singanamala S and Parikh CR Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 81, 442–448, 10.1038/ki.2011.379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B et al. (2003) Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 108, 2154–2169, 10.1161/01.CIR.0000095676.90936.80 [DOI] [PubMed] [Google Scholar]
- 6.Jo SK, Rosner MH and Okusa MD (2007) Pharmacologic treatment of acute kidney injury: why drugs haven’t worked and what is on the horizon. Clin. J. Am. Soc. Nephrol 2, 356–365, 10.2215/CJN.03280906 [DOI] [PubMed] [Google Scholar]
- 7.Muller V, Losonczy G, Heemann U, Vannay A, Fekete A et al. (2002) Sexual dimorphism in renal ischemia-reperfusion injury in rats: possible role of endothelin. Kidney Int. 62, 1364–1371, 10.1111/j.1523-1755.2002.kid590.x [DOI] [PubMed] [Google Scholar]
- 8.Rosin DL and Okusa MD Dangers within: DAMP responses to damage and cell death in kidney disease. J. Am. Soc. Nephrol 22, 416–425, 10.1681/ASN.2010040430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA et al. (2007) The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol. Rev 220, 60–81, 10.1111/j.1600-065X.2007.00579.x [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Liu H, Zeng Q, Imperato GH, Addorisio ME et al. (2019) Inhibition of HMGB1/RAGE-mediated endocytosis by HMGB1 antagonist box A, anti-HMGB1 antibodies, and cholinergic agonists suppresses inflammation. Mol. Med 25, 13, 10.1186/s10020-019-0081-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang H and Tracey KJ (2010) Targeting HMGB1 in inflammation. Biochim. Biophys. Acta 1799, 149–156, 10.1016/j.bbagrm.2009.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson U and Tracey KJ (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol 29, 139–162, 10.1146/annurev-immunol-030409-101323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CB, Liu LS, Zhou J, Wang XP, Han M et al. (2017) Up-Regulation of HMGB1 exacerbates renal ischemia-reperfusion injury by stimulating inflammatory and immune responses through the TLR4 signaling pathway in mice. Cell. Physiol. Biochem 41, 2447–2460, 10.1159/000475914 [DOI] [PubMed] [Google Scholar]
- 14.Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U et al. (2010) HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol 21, 1878–1890, 10.1681/ASN.2009101048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP et al. (2005) The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med 201, 1135–1143, 10.1084/jem.20042614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Xia J, Zhang Y, Xiao F, Wang J et al. (2016) HMGB1-TLR4 signaling participates in renal ischemia reperfusion injury and could be attenuated by dexamethasone-mediated inhibition of the ERK/NF-kappaB pathway. Am. J. Transl. Res 8, 4054–4067 [PMC free article] [PubMed] [Google Scholar]
- 17.Hreggvidsdottir HS, Ostberg T, Wahamaa H, Schierbeck H, Aveberger AC et al. (2009) The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J. Leukoc. Biol 86, 655–662, 10.1189/jlb.0908548 [DOI] [PubMed] [Google Scholar]
- 18.Lotze MT and Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol 5, 331–342, 10.1038/nri1594 [DOI] [PubMed] [Google Scholar]
- 19.Shpacovitch V Mertens PR High mobility group box protein-1 crossing cell borders may incite an inflammatory “tornado” in renal disease. Int. Urol. Nephrol 42, 847–850, 10.1007/s11255-010-9829-1 [DOI] [PubMed] [Google Scholar]
- 20.Rabadi MM, Ghaly T and Goligorksy MS Ratliff BB HMGB1 in renal ischemic injury. Am. J. Physiol. Renal Physiol 303, F873–885, 10.1152/ajprenal.00092.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung KY, Park JJ and Kim YS (2008) The role of high-mobility group box-1 in renal ischemia and reperfusion injury and the effect of ethyl pyruvate. Transplant. Proc 40, 2136–2138, 10.1016/j.transproceed.2008.06.040 [DOI] [PubMed] [Google Scholar]
- 22.Ozrazgat-Baslanti T, Thottakkara P, Huber M, Berg K, Gravenstein N et al. (2016) Acute and chronic kidney disease and cardiovascular mortality after major surgery. Ann. Surg 264, 987–996, 10.1097/SLA.0000000000001582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U et al. (2010) HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol 21, 1878–1890, 10.1681/ASN.2009101048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelbary M, Rafikova O, Gillis EE, Musall JB, Baban B et al. (2019) Necrosis contributes to the development of hypertension in male, but not female, spontaneously hypertensive rats. Hypertension 74, 1524–1531, 10.1161/HYPERTENSIONAHA.119.13477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tipton AJ, Baban B and Sullivan JC (2014) Female spontaneously hypertensive rats have a compensatory increase in renal regulatory T cells in response to elevations in blood pressure. Hypertension 64, 557–564, 10.1161/HYPERTENSIONAHA.114.03512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crislip GR, O’Connor PM, Wei Q and Sullivan JC (2017) Vasa recta pericyte density is negatively associated with vascular congestion in the renal medulla following ischemia reperfusion in rats. Am. J. Physiol. Renal Physiol 313, F1097–F1105, 10.1152/ajprenal.00261.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasser JM, Sullivan JC, Hobbs JL, Yamamoto T, Pollock DM et al. (2007) Endothelin A receptor blockade reduces diabetic renal injury via an anti-inflammatory mechanism. J. Am. Soc. Nephrol 18, 143–154, 10.1681/ASN.2006030208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranganathan PV, Jayakumar C, Mohamed R, Dong Z and Ramesh G (2013) Netrin-1 regulates the inflammatory response of neutrophils and macrophages, and suppresses ischemic acute kidney injury by inhibiting COX-2-mediated PGE2 production. Kidney Int. 83, 1087–1098, 10.1038/ki.2012.423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohamed R, Jayakumar C, Chen F, Fulton D, Stepp D et al. (2016) Low-dose IL-17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J. Am. Soc. Nephrol 27, 745–765, 10.1681/ASN.2014111136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B, Ramesh G, Uematsu S, Akira S and Reeves WB (2008) TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol 19, 923–932, 10.1681/ASN.2007090982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miura M, Fu X, Zhang QW, Remick DG and Fairchild RL (2001) Neutralization of Gro alpha and macrophage inflammatory protein-2 attenuates renal ischemia/reperfusion injury. Am. J. Pathol 159, 2137–2145, 10.1016/S0002-9440(10)63065-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung AC and Lan HY (2011) Chemokines in renal injury. J. Am. Soc. Nephrol 22, 802–809, 10.1681/ASN.2010050510 [DOI] [PubMed] [Google Scholar]
- 33.Chen Q, Guan X, Zuo X, Wang J and Yin W (2016) The role of high mobility group box 1 (HMGB1) in the pathogenesis of kidney diseases. Acta Pharm. Sin. B 6, 183–188, 10.1016/j.apsb.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei S, Gao Y, Dai X, Fu W, Cai S et al. (2019) SIRT1-mediated HMGB1 deacetylation suppresses sepsis-associated acute kidney injury. Am. J. Physiol. Renal Physiol 316, F20–F31, 10.1152/ajprenal.00119.2018 [DOI] [PubMed] [Google Scholar]
- 35.Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD et al. (2017) Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci. Rep 7, 5850, 10.1038/s41598-017-06205-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka R, Tsutsui H, Ohkita M, Takaoka M, Yukimura T et al. (2013) Sex differences in ischemia/reperfusion-induced acute kidney injury are dependent on the renal sympathetic nervous system. Eur. J. Pharmacol 714, 397–404, 10.1016/j.ejphar.2013.07.008 [DOI] [PubMed] [Google Scholar]
- 37.Aufhauser DD Jr, Wang Z, Murken DR, Bhatti TR, Wang Y et al. (2016) Improved renal ischemia tolerance in females influences kidney transplantation outcomes. J. Clin. Invest 126, 1968–1977, 10.1172/JCI84712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S et al. (2014) Regulated cell death in AKI. J. Am. Soc. Nephrol 25, 2689–2701, 10.1681/ASN.2014030262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W et al. (1995) In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21, 1465–1468 [DOI] [PubMed] [Google Scholar]
- 40.Jog NR Caricchio R Differential regulation of cell death programs in males and females by Poly (ADP-Ribose) Polymerase-1 and 17beta estradiol. Cell Death Dis. 4, e758, 10.1038/cddis.2013.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brinson KN, Elmarakby AA, Tipton AJ, Crislip GR, Yamamoto T et al. Female SHR have greater blood pressure sensitivity and renal T cell infiltration following chronic NOS inhibition than males. Am. J. Physiol. Regul. Integr. Comp. Physiol 305, R701–710, 10.1152/ajpregu.00226.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zemskova M, Kurdyukov S, James J, McClain N, Rafikov R et al. (2020) Sex-specific stress response and HMGB1 release in pulmonary endothelial cells. PLoS ONE 15, e0231267, 10.1371/journal.pone.0231267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maselli A, Matarrese P, Straface E, Canu S, Franconi F et al. (2009) Cell sex: a new look at cell fate studies. FASEB J. 23, 978–984, 10.1096/fj.08-114348 [DOI] [PubMed] [Google Scholar]
- 44.Ortona E, Matarrese P and Malorni W Taking into account the gender issue in cell death studies. Cell Death Dis. 5, e1121, 10.1038/cddis.2014.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malorni W, Campesi I, Straface E, Vella S and Franconi F (2007) Redox features of the cell: a gender perspective. Antioxid. Redox Signal 9, 1779–1801, 10.1089/ars.2007.1596 [DOI] [PubMed] [Google Scholar]
- 46.Kher A, Meldrum KK, Wang M, Tsai BM, Pitcher JM et al. (2005) Cellular and molecular mechanisms of sex differences in renal ischemia-reperfusion injury. Cardiovasc. Res 67, 594–603, 10.1016/j.cardiores.2005.05.005 [DOI] [PubMed] [Google Scholar]
- 47.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A et al. (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med 209, 1519–1528, 10.1084/jem.20120189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu M, Wang H, Ding A, Golenbock DT, Latz E et al. (2006) HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26, 174–179, 10.1097/01.shk.0000225404.51320.82 [DOI] [PubMed] [Google Scholar]
- 49.Bihorac A, Baslanti TO, Cuenca AG, Hobson CE, Ang D et al. (2013) Acute kidney injury is associated with early cytokine changes after trauma. J. Trauma Acute Care Surg 74, 1005–1013, 10.1097/TA.0b013e31828586ec [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu P, Li X, Lv W and Xu Z (2020) Inhibition of CXCL1-CXCR2 axis ameliorates cisplatin-induced acute kidney injury by mediating inflammatory response. Biomed. Pharmacother 122, 109693, 10.1016/j.biopha.2019.109693 [DOI] [PubMed] [Google Scholar]
- 51.Tipton AJ, Baban B and Sullivan JC (2012) Female spontaneously hypertensive rats have greater renal anti-inflammatory T lymphocyte infiltration than males. Am. J. Physiol. Regul. Integr. Comp. Physiol 303, R359–367, 10.1152/ajpregu.00246.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kinsey GR, Sharma R, Huang L, Li L, Vergis AL et al. (2009) Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol 20, 1744–1753, 10.1681/ASN.2008111160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malek M and Nematbakhsh M (2015) Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Renal Inj. Prev 4, 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]