

Abstract
Background
The majority of patients with pancreatic cancer develops cachexia. The mechanisms underlying cancer cachexia development and progression remain elusive, although tumour‐derived factors are considered to play a major role. Pancreatic tumour organoids are in vitro three‐dimensional organ‐like structures that retain many pathophysiological characteristics of the in vivo tumour. We aimed to establish a pancreatic tumour organoid biobank from well‐phenotyped cachectic and non‐cachectic patients to enable identification of tumour‐derived factors driving cancer cachexia.
Methods
Organoids were generated from tumour tissue of eight pancreatic cancer patients. A comprehensive pre‐operative patient assessment of cachexia‐related parameters including nutritional status, physical performance, body composition, and inflammation was performed. Tumour‐related and cachexia‐related characteristics of the organoids were analysed using histological stainings, targeted sequencing, and real‐time–quantitative PCR. Cachexia‐related factors present in the circulation of the patients and in the tumour organoid secretome were analysed by enzyme‐linked immunosorbent assay.
Results
The established human pancreatic tumour organoids presented typical features of malignancy corresponding to the primary tumour (i.e. nuclear enlargement, multiple nucleoli, mitosis, apoptosis, and mutated KRAS and/or TP53). These tumour organoids also expressed variable levels of many known cachexia‐related genes including interleukin‐6 (IL‐6), TNF‐α, IL‐8, IL‐1α, IL‐1β, Mcp‐1, GDF15, and LIF. mRNA expression of IL‐1α and IL‐1β was significantly reduced in organoids from cachectic vs. non‐cachectic patients (IL‐1α: −3.8‐fold, P = 0.009, and IL‐1β: −4.7‐fold, P = 0.004). LIF, IL‐8, and GDF15 mRNA expression levels were significantly higher in organoids from cachectic vs. non‐cachectic patients (LIF: 1.6‐fold, P = 0.003; IL‐8: 1.4‐fold, P = 0.01; GDF15: 2.3‐fold, P < 0.001). In line with the GDF15 and IL‐8 mRNA expression levels, tumour organoids from cachectic patients secreted more GDF15 and IL‐8 compared with organoids from non‐cachectic patients (5.4 vs. 1.5 ng/mL, P = 0.01, and 7.4 vs. 1.3 ng/mL, P = 0.07, respectively).
Conclusions
This novel human pancreatic tumour organoid biobank provides a valuable tool to increase our understanding of the mechanisms driving cancer cachexia. Our preliminary characterization of the secretome of these organoids supports their application in functional studies including conditioned medium approaches and in vivo transplantation models.
Keywords: Pancreatic cancer, Cancer cachexia, Organoids, GDF15, LIF
Introduction
Pancreatic cancer is one of the deadliest cancer types that is responsible for approximately 4.5% of all cancer deaths worldwide. 1 The high mortality rate of pancreatic cancer is related to the fact that most patients are diagnosed at advanced disease stages, when therapeutic options are limited. Late detection of pancreatic cancer is due to the absence of specific symptoms, a lack of sensitive and specific tumour markers, and difficulties in imaging early‐stage tumours. 2 As a result, most patients with pancreatic cancer present at the clinic because of jaundice and unexplained weight loss, the latter being indicative of the presence of a phenomenon called cancer cachexia.
Cancer cachexia is a severe wasting syndrome with multifactorial causes, involving tumour‐derived and host tissue‐derived signalling factors and alterations in metabolism that ultimately result in skeletal muscle wasting, its key phenotypic feature. 3 , 4 Cachexia is present in up to 80% of pancreatic cancer patients and is a major contributor to their poor survival rate. 5 , 6 It is associated with reduced physical function, diminished tolerance to anticancer treatment regimens, and a marked reduction in quality of life. 3 In view of the lack of effective treatment options for pancreatic cancer patients, managing cachexia is increasingly considered an attractive strategy to improve survival. However, effective pharmacologic options for the treatment of cachexia are currently lacking. 7
To develop effective and targeted anti‐cachexia therapies, more insight into its underlying pathophysiological mechanisms is required. Our current understanding of the cachexia‐inducing factors expressed and released by tumour cells is predominantly derived from both in vitro and in vivo studies with established murine cancer cell lines like C26 and Lewis lung carcinoma. 8 , 9 , 10 , 11 , 12 Over the last 5 years, human pancreatic cancer cell lines such as MiaPaCa‐2, Capan‐1, and Panc‐1 have been increasingly used to model cancer‐induced cachexia 13 , 14 , 15 because of the high prevalence and severity of cachexia in pancreatic cancer patients. However, it is important to recognize that traditional cell culture models are comparatively artificial because cells are maintained on a stiff two‐dimensional (2D) plastic surface in the absence of physiological gradients of oxygen and nutrients and without the potential for cellular–extracellular matrix interactions. 16 These artificial culture conditions hence require important non‐physiological cellular adaptations that are associated with mutational and chromosomal instability, which increase with prolonged culturing. 17 Because most of the cell lines used in cachexia research have been cultured in 2D for decades, currently available strains are genetically different compared with the originally isolated tumour cells, potentially leading to differential activation of gene expression programmes. This implies that many established pancreatic cell lines will likely have gained or lost cachexia‐inducing properties, resulting in inconsistent findings among studies. On top of this, cachexia‐related clinical data of the patients of whom these cells were derived are completely lacking, complicating the analysis of links between in vivo and in vitro phenotypes. Altogether, this underscores the need for better experimental models that facilitate the identification of novel cachexia‐inducing targets and their ultimate translation into clinical benefit.
Recently, several aspects of cancer biology have been shown to be accurately modelled by a so‐called organoid technology. 18 , 19 , 20 Tumour organoids can be efficiently established by culturing primary epithelial tumour cells in basement membrane extract (BME) and a defined, tissue‐specific growth medium. They self‐organize into three‐dimensional (3D) structures mimicking the architecture of the organ of origin and have been shown to closely recapitulate pathophysiologically aspects of pancreatic, colon, breast, and gastric cancer both in vitro and in vivo. 18 , 21 , 22 , 23 Histologic, genetic, and transcriptomic features of the original tumour have been shown to be maintained in patient‐derived tumour organoids, supporting their applicability as a pre‐clinical model to study disease‐specific mechanisms. 21 , 22 , 23 Pancreatic tumour organoids have already proven to be useful in identifying novel genes associated with pancreatic cancer progression. 18 , 24 Moreover, emerging evidence confirms the potential of tumour organoid‐based high‐throughput drug screens to identify novel targeted drugs and to predict patient treatment responses. 22 , 25 , 26 We therefore hypothesized that modelling human pancreatic cancer with organoids could represent a powerful novel approach to study direct cachexia‐inducing properties of pancreatic cancer cells. The high efficiency with which primary tumour organoid cultures can be established in combination with thorough phenotyping of cachexia‐related parameters of donor patients enables the application of organoids for understanding common cachexia‐inducing mechanisms as well as interindividual differences.
In this paper, we describe our systematic approach to generate a pancreatic tumour organoid biobank and the initial characterization of these organoids by means of histology, targeted mutation analysis, and analysis of expression of known cachexia‐related factors. Our data reveal strong interindividual variation in the production of factors known to be involved in cachexia by tumour organoids, demonstrating its power for modelling cachexia.
Materials and methods
Patients
Patients undergoing pancreaticoduodenectomy at the Maastricht University Medical Centre (MUMC+) for suspected adenocarcinoma of the pancreas have been enrolled in this study. Exclusion criteria included the use of systemic glucocorticoids in the past 4 weeks, neoadjuvant chemotherapy and/or radiotherapy, and the presence of another malignancy. All patients provided written informed consent. This study was approved by the local Medical Ethics Committee (METC 13‐4‐107).
Screening of cachexia‐related parameters
The nutritional status of the patients was thoroughly assessed by a trained physician in the outpatient clinic. The screening included measurements of body weight and height, patient‐reported weight loss in the last 6 months, upper arm circumference, 27 triceps skinfold thickness, 27 handgrip strength, 28 patient‐generated subjective global assessment (PG‐SGA), 29 and the malnutrition universal screening tool (MUST). 30 Systemic inflammation was assessed by measuring plasma C‐reactive protein (CRP) and albumin levels pre‐operatively (routine in‐hospital laboratory test, MUMC+). Faecal elastase levels were determined as a measure of pancreatic exocrine insufficiency. The patients provided written informed consent for retrieving the data from their medical chart.
Computed tomography‐based body composition
Body composition was assessed by using computed tomography (CT) imaging and sliceOmatic 5.0 software (TomoVision, Magog, Canada). 31 Adipose tissue and skeletal muscle mass were quantified on a cross‐sectional CT‐image at the third lumbar (L3) vertebra that was pre‐operatively acquired for diagnostic purposes. Using predefined Hounsfield unit (HU) ranges, the total cross‐sectional area (cm2) of skeletal muscle tissue (−29 to 150 HU), visceral adipose tissue (VAT) (−150 to −50 HU), and subcutaneous tissue (SAT) (−190 to −30 HU) was determined. The radiation attenuation for skeletal muscle was assessed by calculating the average HU value of the total muscle area within the specified range of −29 to 150 HU. The total areas of skeletal muscle, VAT, and SAT were normalized for stature to calculate the L3‐muscle index (L3‐SMI), L3‐VAT index, and L3‐SAT index in cm2/m2. Previously published sex‐specific cut‐off values were used for the CT‐derived body composition parameters. 32
Diagnosis of cancer cachexia
Cachexia was defined according to the international consensus definition as (i) weight loss >5% over the past 6 months in the absence of starvation, and/or (ii) body mass index <20 kg/m2 and >2% ongoing weight loss, and/or (iii) sarcopenia and >2% ongoing weight loss. 3 Patients were diagnosed with cancer cachexia if ≥1 of the criteria were met.
Collection of plasma samples and tumour biopsy
Prior to the start of surgery, blood was collected in EDTA tubes and stored on ice until further processing. The blood was centrifuged at ×1150 g at 4°C for 12 min without brake. Plasma aliquots were stored at −80°C until further analysis.
After removal of the pancreas specimen during surgery, the tissue was immediately transferred to the pathology laboratory (Department of Pathology, MUMC+), where a dedicated gastrointestinal pathologist identified the tumour macroscopically and collected a fresh approximately 0.5–1 cm3 tumour containing tissue slice. The tissue slice was transferred into ice‐cold Advanced Dulbecco's Modified Eagle Medium/Ham's F‐12 (AdvDF+++) (Gibco, Cat. No. 12634‐010) supplemented with 1× GlutaMAX (Gibco, Cat. No. 35050‐061), 10 mM HEPES (Gibco, Cat. No. 15630‐080), and Pen/Strep (50 units/mL penicillin and 50 μg/mL streptomycin) (Gibco, Cat. No 15140–122) and was stored on ice until further processing.
Establishment of human pancreatic tumour organoids
Pancreatic tumour organoids were established according to previously described protocols. 18 , 33 The tumour biopsy arrived at the Department of Surgery (Maastricht University) within 2 h after removal of the pancreas specimen from the patient. Upon arrival, the tumour tissue was minced, washed with 10 mL AdvDF+++, and digested with collagenase II (5 mg/mL, Gibco, Cat. No. 17101‐01) in AdvDF+++ supplemented with 50% (v/v) Wnt3a conditioned medium (CM) 33 and 10 μM Rho kinase inhibitor (Y‐27632) on an orbital shaker at 37°C for 1–2 h. The digested tissue suspension was further digested with TrypLE (Gibco, Cat. No. 12605‐010) supplemented with 10 μM Rho kinase inhibitor at 37 °C. TrypLE digestion was stopped by adding ice‐cold AdvDF+++ followed by 5 min centrifugation at ×350 g at 4°C. Subsequently, the pellet was resuspended in ice‐cold BME (Geltrex LDEV‐Free Reduced Growth Factor Basement Membrane Matrix, Gibco, Cat. No. 1413202) and three approximately 15 μL droplets of Geltrex‐cell suspension were allowed to solidify per well of a 24‐well culture plate (Eppendorf) at 37°C for 30 min. When the droplets were solidified, 500 μL of either organoid Medium ‘a’ or Medium ‘b’ (see Supporting Information, Table S1, 18 ) were added to each well. This resulted in the establishment of two organoid cultures from one individual tumour biopsy. The plate was transferred to a humidified 37°C/5% CO2 incubator, and the medium was changed every 2–3 days.
Culturing of pancreatic tumour organoids
The organoids were passaged every 7–10 days. Organoids were collected in 2 mL AdvDF+++ and mechanically sheared through narrowed glass Pasteur pipettes. Following the addition of 10 mL AdvDF+++ and centrifugation at ×350 g (5 min, 4°C), organoid fragments were resuspended in ice‐cold BME and plated as described earlier, allowing the formation of new organoids. Successfully established organoid cultures were cryopreserved after two to five passages. Characterization of the organoids was performed between passage Numbers 6 and 15.
Histological characterization of tumour and tumour organoids
Tissue and organoids were fixed in 4% paraformaldehyde followed by dehydration, paraffin embedding, sectioning, and standard haematoxylin and eosin staining. Stained organoid slides were digitalized using the Ventana iScan HT (Version 1.1, Roche, Ventana Medical Systems, Inc.) using a ×200 magnification. Sections were blindly analysed by a pancreatic cancer pathologist.
Mutation analysis
Total genomic DNA was isolated from organoids using the QIAamp UCP DNA micro kit (QIAGEN). In short, organoids were collected in AdvDF+++ and centrifuged for 5 min at ×750 g, 4°C. The organoid pellet was further processed according to the manufacturer's instructions. Human genomic DNA was isolated from EDTA blood using the DSP DNA midi kit (QIAGEN). Targeted mutation analysis of 31 cancer‐related genes (Table S2) was performed using the single‐molecule molecular inversion probes technique. 34 Sequence data were analysed using SeqNext software from JSI. Validation of conservation of KRAS and TP53 mutations across organoids and the parent tumour was done by analysing total genomic DNA extracted from formalin‐fixed paraffin‐embedded tumour sections using the QIAamp DNA FFPE Tissue Kit (QIAGEN) according to the manufacturer's instructions. Extracted genomic DNA was amplified by PCR using the following primers: KRAS‐exon2: FW 5′‐GATACACGTCTGCAGTCAACTG‐3′, RV 5′‐GGTCCTGCACCAGTAATATGC‐3′; TP53‐exon5: FW 5′‐GCCCTGACTTTCAACTCTGTCTC‐3′, RV 5′‐CATCGCTATCTGAGCAGCGC‐3′; TP53‐exon6: FW 5′‐GCGCTGCTCAGATAGCGATG‐3′, RV 5′‐CCCAGTTGCAAACCAGACCTC‐3′. Purified amplicons were sequenced by Eurofins Genomics (Germany).
Quantitative real‐time PCR
Organoids cultured for 24 h in basal medium consisting of DMEM/F12 supplemented with 1% (v/v) HEPES and 1% (v/v) Pen/Strep were collected for mRNA expression analysis. Total RNA was extracted from organoid cultures using TRI Reagent (Sigma, St. Louis, MO, USA) according to the manufacturer's protocol. Because the organoid samples contained a high concentration of extracellular material (BME), these lysates were centrifuged at ×12 000 g for 10 min at 4°C in order to remove the insoluble material. RNA yield and quality were measured with a DeNovix DS‐11 spectrophotometer. A total of 750 ng RNA was reversed transcribed to cDNA using the SensiFAST cDNA Synthesis Kit according to the manufacturer's instructions (Bioline GmbH, Germany).
cDNA was diluted (1:20) in nuclease‐free H2O. Each quantitative real‐time–PCR (qRT–PCR) reaction contained 4.7 μL diluted cDNA, 5 μL 2× SensiMix SYBR Hi‐Rox Kit (Bioline, cat. No. QT605–05), and 0.3 μL primers containing 10 μM of both the forward and reverse primers. Specific primer pairs for each gene of interest were ordered from Sigma; sequences are listed in Table S3. To quantify mRNA expression levels, qRT–PCR analysis was performed on a LightCycler480 system (Roche) with a three‐step PCR programme (10 min at 95°C followed by 40 cycles of 15 s at 95°C, 15 s at 60°C, and 30 s at 72°C) followed by melting curve analysis. Melt curves were made using gradual increases in temperature of 0.1°C/s with six acquisitions per second within a temperature range of 60°C to 95°C. Relative gene expression levels were derived from the LinRegPCR (Version 2016.1) method 35 and normalized to the geometric average of two reference genes, cyclophilin A (CYPA) and β‐2‐microglobulin (β2M).
Secretome analysis
Cachexia‐related factors present in the circulation of patients and in the tumour organoid secretome were analysed by enzyme‐linked immunosorbent assay (ELISA). The tumour organoid secretome was obtained by replacing organoid growth medium by basal medium consisting of DMEM/F12 supplemented with 1% (v/v) HEPES and 1% (v/v) Pen/Strep. The medium was conditioned for 24 h by the pancreatic tumour organoids. After 24 h, CM was collected and centrifuged at ×350 g for 10 min at 4°C. The supernatant was centrifuged for another 20 min at ×2000 g at 4°C, and the resulting CM cleared from cellular debris was aliquoted and stored at −80°C.
Concentrations of human IL‐6 (U‐CyTech Biosciences, Cat. No. CT205A) and human GDF15 (Research and Diagnostic Systems, Cat. No. DY957) in plasma and CM were determined by ELISA according to the manufacturer's protocol. Plasma and CM concentrations of human IL‐8 were determined using an in‐house developed ELISA. 36 Absorbance was measured at a wavelength of 450 nm using a Spark™ 10 M multimode microplate reader (Tecan).
Statistical analysis
Patient data and outcome parameters were entered in IBM SPSS 24 for Microsoft Windows®, and statistical analyses were performed using the non‐parametric Mann–Whitney U test to compare differences between the groups. A P value of <0.05 was considered statistically significant.
Results
Phenotyping of cachexia severity in patients with pancreatic cancer
To be able to relate characteristics of pancreatic tumour organoids to the cachexia status of the patient, we designed a work flow that allowed us to systematically and routinely assess the cachexia status of the patient and to collect biological materials following a standardized procedure (Figure 1). The nutritional status of the patient was assessed pre‐operatively by a trained physician in the outpatient clinic. Basic patient characteristics and cachexia‐related parameters collected from the patient's medical records are presented in Table 1. A total of eight patients, three male and five female, were included with a mean age of 68.5 (±10.9) years and a mean body mass index of 23.8 (±3.7) kg/m2. All patients were diagnosed with a tumour in the head of the pancreas, including pancreatic ductal adenocarcinoma (PANCO‐5, PANCO‐9, PANCO‐11, PANCO‐12, PANCO‐17, and PANCO‐22), ampullary carcinoma (PANCO‐6), and cholangiocarcinoma (PANCO‐3); neither received neoadjuvant chemotherapy. According to the international consensus definition, five patients (PANCO‐3, PANCO‐5, PANCO‐6, PANCO‐9, and PANCO‐17) were diagnosed with cancer cachexia. Although this definition is mainly based on the percentage weight loss, we observed considerable heterogeneity among additional cachexia‐related parameters that are often reported in the literature. Additional insight into the nutritional status of the patient was obtained by using nutrition‐related screening tools and anthropometric measurements. The PG‐SGA questionnaire revealed that patients in both the no‐cachexia (PANCO‐11 and PANCO‐22) and cachexia groups (PANCO‐3, PANCO‐5, PANCO‐6, and PANCO‐9) scored ≥9, which is, according to the guidelines, indicative of a critical need of improved symptom management and/or nutrient intervention. The PG‐SGA score of patients PANCO‐6 and PANCO‐22 was accompanied by PG‐SGA global assessment Category B, indicating that they were moderately malnourished. The PG‐SGA score of patient PANCO‐3 was accompanied by Category C, indicating that the patient was severely malnourished. The MUST screening tool, which is used to screen for patients at risk of malnutrition, identified patients PANCO‐3, PANCO‐9, and PANCO‐17 at high risk of malnutrition. In line, using sex‐specific and age‐specific percentiles, patients PANCO‐3 (2568 mm2, <p5), PANCO‐9 (4182 mm2, <p5), and PANCO‐17 (3092 mm2, <p5) were found to have a low upper arm muscle area, which, together with the aforementioned nutritional data, indicates that these patients had a poor nutritional status.
Figure 1.
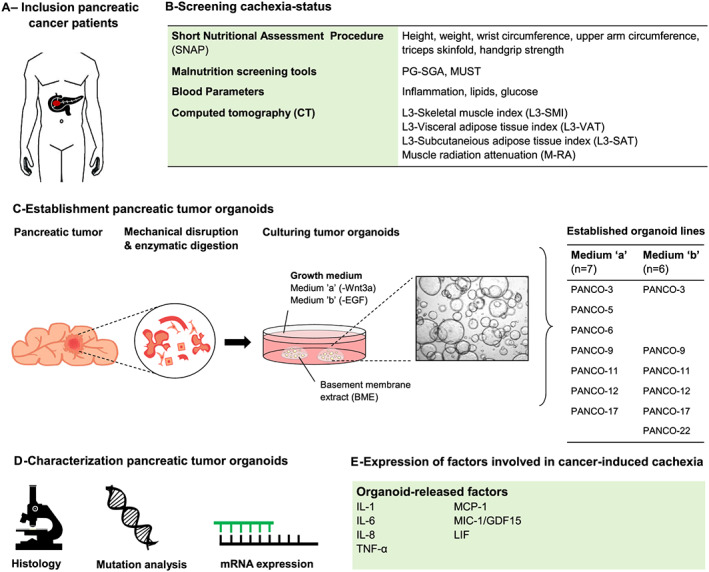
Establishment of pancreatic tumour organoids from cachectic pancreatic cancer patients. Flowchart used to systematically and routinely assess the cachexia status of the patient and to collect biological materials following a standardized procedure. (A) Upon inclusion, (B) the nutritional status of the patients is thoroughly assessed by a trained physician in the outpatient clinic. Additional cachexia‐related parameters are collected from the patient's medical records. (C) Schematic representation of the generation of human pancreatic tumour organoid culture from a tumour section from the surgically resected tumour tissue. Upon establishment, tumour cells are cultured in organoid Medium ‘a’ or Medium ‘b’, resulting in the establishment of two organoid cultures from one individual tumour biopsy. (D, E) Pancreatic tumour organoids are characterized by means of histology, targeted mutation analysis, and analysis of expression of known cachexia‐related factors. EGF, epidermal growth factor; MUST, malnutrition universal screening tool; PG‐SGA, patient‐generated subjective global assessment.
Table 1.
Patient characteristics
| No cachexia | Cachexia | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PANCO‐11 | PANCO‐12 | PANCO‐22 | Average | PANCO‐3 | PANCO‐5 | PANCO‐6 | PANCO‐9 | PANCO‐17 | Average | |
| Gender (male/female) | Male | Female | Female | 1/2 | Female | Female | Female | Male | Male | 2/3 |
| Age (years) | 80 | 72 | 59 | 70.3 (±10.6) | 64 | 53 | 84 | 61 | 75 | 67.4 (±12.2) |
| Nutritional | ||||||||||
| BMI (kg/m2) | 27.1 | 23.7 | 23.6 | 24.8 (±2.0) | 16.7 | 21.9 | 22.3 | 26.5 | 28.4 | 23.2 (±4.5) |
| Weight loss over the last 6 months (%) | −1.9 | 1.2 | 1.9 | 0.4 (±2.0) | 11.3 | 7.0 | 10 | 13.4 | 10.9 | 10.5 (±2.3) |
| PG‐SGA score | 12 | 8 | 11 | 10.3 (±2.1) | 14 | 11 | 14 | 9 | 7 | 11.0 (±3.1) |
| PG‐SGA global | A | A | B | C | A | B | A | B | ||
| MUST score | 0 | 0 | 0 | 0.0 (±0.0) | 5 | 1 | 1 | 2 | 2 | 2.2 (±1.6) |
| Upper arm muscle area (mm2) | 5231 | 4255 | 3169 | 4218 (±1031) | 2568 | 3931 | 3736 | 4182 | 3092 | 3502 (±660) |
| Upper arm muscle area percentile | p15–p90 | p15–p90 | p15–p90 | <p5 | p15–p90 | p15–p90 | <p5 | <p5 | ||
| Upper arm fat area (mm2) | 1651 | 1635 | 3396 | 2227 (±1012) | 617 | 2536 | 2068 | 1622 | 3375 | 2044 (±1028) |
| Upper arm fat area percentile | p15‐p90 | p5–p15 | p15–p90 | <p5 | p15–p90 | p15–p90 | p15–p90 | >p90 | ||
| Handgrip strength (kg) | 35 | 25 | 28 | 29 (±5) | 29 | 31 | 18 | 46 | 40 | 33 (±11) |
| Handgrip strength percentile | >p90 | p15–p90 | p15–p90 | p15–p90 | p15–p90 | p15–p90 | p15–p90 | p15–p90 | ||
| Body composition | ||||||||||
| L3‐SMI (cm2/m2) | 34.2 | 36.1 | 32.7 | 33.2 | 42.4 | 36.0 | 43.0 | 48.3 | ||
| L3‐VAT index (cm2/m2) | 67.5 | 25.8 | 24.4 | 0.9 | 10.6 | 39.3 | 61.9 | 77.7 | ||
| L3‐SAT index (cm2/m2) | 45.7 | 87.1 | 50.6 | 3.0 | 50.6 | 58.6 | 72.7 | 55.9 | ||
| Muscle radiation attenuation (HU) | 33.3 | 39.4 | 41.6 | 46.9 | 51.4 | 32.6 | 34.6 | 30.6 | ||
| Biochemistry | ||||||||||
| C‐reactive protein (mg/L) | 18 | 1 | 13 | 10.7 (±8.7) | 21 | 0 | 33 | 20 | 0 | 15 (±14) |
| Albumin (g/L) | 28.0 | 36.4 | 36.7 | 33.7 (±4.9) | 27.3 | 40.5 | 32.7 | 33.2 | 36.8 | 34.1 (±4.9) |
| mGPS | 2 | 0 | 1 | 2 | 0 | 2 | 2 | 0 | ||
| Total protein (g/L) | 57.1 | 67.5 | 69.9 | 64.8 (±6.8) | 63.6 | 74.4 | 72.2 | 58.1 | 70.4 | 67.7 (±6.7) |
| Neutrophils (%) | 69 | MD | 57 | 63.0 (±8.5) | 77 | 68 | 69 | 78 | 65 | 71.4 (±5.8) |
| Lymphocytes (%) | 13 | MD | 33 | 23.0 (±14.1) | 14 | 21 | 19 | 11 | 20 | 17.0 (±4.3) |
| N/L ratio | 5.3 | MD | 1.7 | 3.5 (±2.5) | 5.5 | 3.2 | 3.6 | 7.1 | 3.2 | 4.5 (±1.7) |
| Haemoglobin (mmol/L) | 7.6 | 7.5 | 8.3 | 7.8 (±0.4) | 8.2 | 7.5 | 6.9 | 8.1 | 9.0 | 7.9 (±0.8) |
| Glucose (mmol/L) | 6.7 | 5.8 | 7.0 | 6.5 (±0.6) | 5.9 | 5.1 | 5.5 | 14.3 | 7.0 | 7.3 (±3.8) |
| Insulin (pmol/L) | 74.6 | MD | MD | MD | <12.0 | 45.5 | 85 | 296 | ||
| HOMA‐IR | 12.4 | MD | MD | MD | 0.5 | 1.9 | 9 | 15.3 | 6.7 (±6.8) | |
| HbA1c (mmol/mol) | 36 | MD | MD | 38 | 40 | 31 | 42 | 38 | 37.8 (±4.1) | |
| HbA1c% (%) | 5.4 | MD | MD | 5.6 | 5.8 | 5.0 | 6.0 | 5.6 | 5.6 (±0.4) | |
| Lipase (U/L) | 16 | MD | 61 | 38.5 (±31.8) | 35 | 23 | 33 | 85 | 37 | 42.6 (±24.3) |
| Amylase (U/L) | 54 | 53 | 93 | 66.7 (±22.8) | 116 | 63 | 69 | 79 | 72 | 79.8 (±21.0) |
| Cholesterol (mmol/L) | 5.4 | MD | MD | 7.4 | 5.3 | 4.8 | 3.3 | 3.6 | 4.9 (±1.6) | |
| HDL (mmol/L) | 0.7 | MD | MD | 1.1 | 1.5 | 1.6 | 0.9 | 0.5 | 1.1 (±0.4) | |
| LDL (mmol/L) | 4.0 | MD | MD | 5.5 | 3 | 2.9 | 1.9 | 2.6 | 3.2 (±1.4) | |
| Triglycerides (mmol/L) | 1.4 | MD | MD | 1.8 | 1.8 | 0.8 | 1.2 | 1.1 | 1.3 (±0.4) | |
| Free fatty acids (mmol/L) | 0.3 | MD | MD | 0.7 | 0.3 | 1.0 | 0.5 | 0.08 | 0.5 (±0.4) | |
| Bilirubin (μmol/L) | 24 | 3.7 | 3.3 | 10.3 (±11.8) | 26.8 | 2.5 | 21.3 | 34.0 | 23.6 | 21.6 (±11.7) |
| Elastase (μg/g) | MD | <15 | 458 | 200 | 480 | 498 | <15 | 27 | ||
| Diagnosis | ||||||||||
| Histopathological diagnosis | PDAC | PDAC (ASCP) | PDAC | Distal cholangiocarcinoma | PDAC | Ampullary carcinoma | PDAC | PDAC | ||
| TNM classification (7th AJCC edition) | T3N1 | T3N1 | T3N1 | T3N0 | T3N1 | T2N1 | T3N1 | T3N1 | ||
| Stage | IIB | IIB | IIB | IIA | IIB | IIB | IIB | IIB | ||
| Adjuvant treatment | MD | No | MD | No | CTx | No | No | CTx | ||
| Survival post‐operative (days) | 312 | 311 | 257 | 293 (±32) | 200 | 377 | >1000 a | 609 | 170 | 339 (±202) |
The data are presented as mean ± standard deviation. Values within the reference range are marked in green. Values outside the reference range are marked in red. ASCP, adenosquamous carcinoma of the pancreas (subtype of pancreatic ductal adenocarcinoma); BMI, body mass index; CTx, chemotherapy, HDL, high‐density lipoprotein; HOMA‐IR, homeostatic model assessment for insulin resistance; HU, Hounsfield unit; L3‐SMI, L3‐skeletal muscle index; L3‐VAT, L3‐visceral adipose tissue; L3‐SAT, L3‐subcutaneous adipose tissue; LDL, low‐density lipoprotein; MD, missing data; mGPS, modified Glasgow prognostic score; MUST, malnutrition universal screening tool; N/L ratio, neutrophil to lymphocyte ratio; PDAC, pancreatic ductal adenocarcinoma; PG‐SGA, patient‐generated subjective global assessment.
Post‐operative survival: 1124 days and still alive at final update (09‐08‐2019).
Whereas anthropometric measurements cannot distinguish between lean muscle mass and fat mass, CT imaging can discriminate between adipose tissue, bone, organs, and muscles including the degree of fatty infiltration, making it the current gold standard for body composition evaluation. 37 Remarkably, CT‐based body composition analysis revealed that patients who did not lose >5% weight over the last 6 months and were not classified as cachectic (PANCO‐11, PANCO‐12, and PANCO‐22) were nevertheless found to have both a low L3‐SMI and low L3‐VAT. Conversely, in the cachectic group, only patients PANCO‐3 and PANCO‐9 had both a low L3‐SMI and a low L3‐VAT.
Furthermore, substantial heterogeneity in cachexia‐related biochemical parameters was observed among both cachectic and non‐cachectic patients. For example, altered levels of inflammatory markers (CRP >10 mg/L, albumin <35 g/L, total protein <60 g/L, and neutrophil to lymphocyte (N/L) ratio >3.5) associated with an inflammatory state in cancer cachexia were observed in patients PANCO‐3, PANCO‐6, PANCO‐9, and PANCO‐11. In addition, patients PANCO‐5, PANCO‐6, PANCO‐9, and PANCO‐11 presented with anaemia (haemoglobin <8.2 mmol/L).
Establishment of 3D tumour organoids from tumour biopsies of pancreatic cancer patients
For the establishment of tumour organoids, a section from the surgically resected tumour tissue was obtained by a pathologist. Through a combination of mechanical disruption and enzymatic digestion, pancreatic tumour cells were isolated and plated in BME droplets and overlaid with optimized pancreatic cancer organoid culture medium (Figure 1). When considering the tumour heterogeneity and the ligand‐independent activation of Ras signalling in pancreatic cancer as a consequence of the KRAS mutation in >90% of pancreatic cancer patients, 38 the organoids were placed in either a Wnt3a‐depleted medium (Medium ‘a’) or epidermal growth factor‐depleted medium (Medium ‘b’). This allows selection of specific tumour clones resulting in two individual organoid cultures originating from the same tumour tissue specimen from one individual patient. Whereas we were able to establish organoid cultures from tumour tissues of all eight individual patients, from 3/8 tumour specimens, we could only establish pancreatic tumour organoids in either ‘Medium a’ (without Wnt3a) or ‘Medium b’ (without epidermal growth factor). The resulting 13 pancreatic tumour organoid cultures were readily expanded and cryopreserved. Within this study, we further characterized these organoids by means of histology, targeted mutation analysis, and by analysing the expression of cachexia‐related factors.
Morphological characterization of pancreatic tumour organoids
The growth and morphology of pancreatic tumour organoids varied considerably among the organoid cultures (Figures 2A and S1, and Video S1‐S10). After passaging, organoids self‐organized into 3D structures within 24 h and continued to grow to 200–400 μm in diameter by Day 3. Interestingly, the diameter of organoids of PANCO‐12 never exceeded 200 μm. Furthermore, PANCO‐12 organoids tended to grow in solid cell clusters whereas in general, all organoid cultures formed cohesive glandular structures with varying morphologies ranging from thin‐walled cystic structures to compact organoids devoid of a lumen. No systematic differences in growth pattern were observed between the organoids cultured in either Medium a or Medium b.
Figure 2.
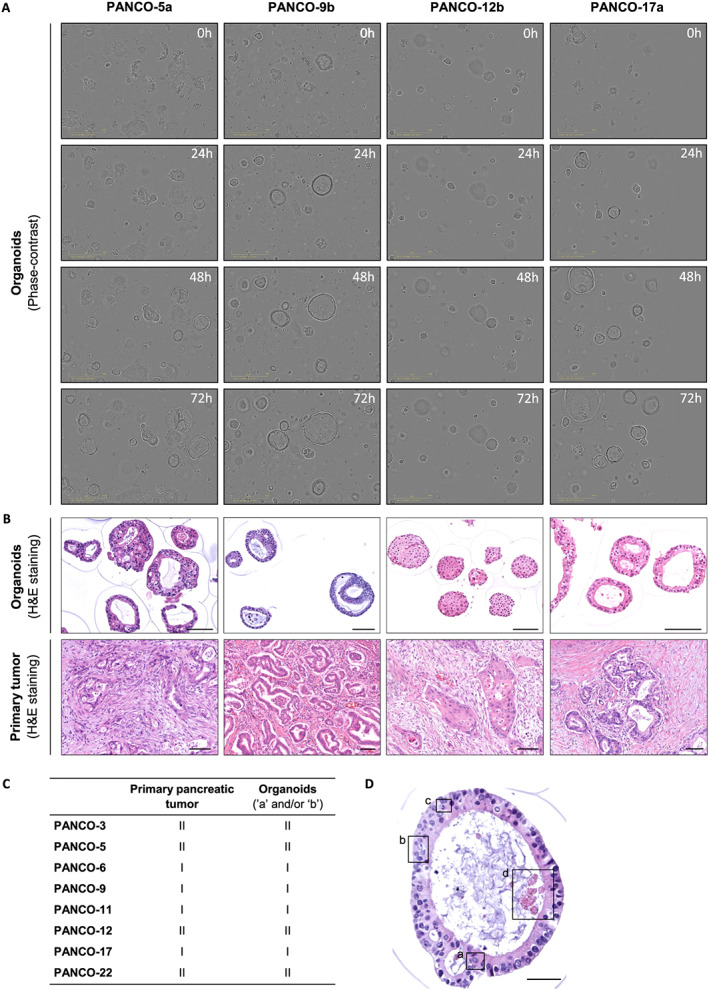
Morphological characterization of pancreatic tumour organoids. (A) Representative phase‐contrast images of pancreatic tumour organoids cultured for 72 h after passaging. (B) Representative haematoxylin and eosin stainings of pancreatic tumour organoids (upper panel) and the primary tumour (lower panel). Scale bar = 100 μm. (C) Differentiation grade of the primary tumours and their corresponding tumour organoids. Tumours are either well‐differentiated (I) or moderately differentiated (II). (D) Representative haematoxylin and eosin staining of a pancreatic tumour organoid representing typical nuclear features of malignancy, including (a) nuclear enlargement, (b) multiple nucleoli, (c) mitoses, and (d) apoptosis. Scale bar = 50 μm.
Next, we examined the morphology of paraffin‐embedded organoids and compared them with the corresponding primary tumour specimen (Figures 2B and S1). From a histologic point of view, the organoids showed clear similarities to their parent tumour. Pancreatic tumour organoids represented an architectural design of mostly well‐formed ducts with occasionally a cribriform growth pattern (e.g. PANCO‐5a) (Figures 2B and S1). To date, pancreatic cancer is still diagnosed based on histology and graded as well differentiated, moderately differentiated, or poorly differentiated. 39 Assessment of the differentiation grades of the primary tumours and their corresponding organoids revealed a similar differentiation grade, with either well‐differentiated or moderately differentiated tumours or organoids (Figure 2C). Additionally, typical nuclear features of malignancy were observed in all organoids, including nuclear enlargement, multiple nucleoli, mitoses, and apoptosis (Figure 2D). Interestingly, we sampled an adenosquamous carcinoma of the pancreas, which is a rare variant of pancreatic ductal adenocarcinoma that represents only 1–4% of the exocrine pancreatic malignancies. 40 , 41 The organoids of this specific patient (PANCO‐12) showed remarkable resemblance to the parent tumour and exhibited a mixture of glandular and squamous differentiation (Figure 2B). Similar to the parental tumour, the organoids formed ducts with a cribriform growth pattern and cytonuclear atypia. Besides these glandular arrangements, fields of closely packed atypical cells with large abundant cytoplasm were observed. Although the squamous component of the primary tumour revealed keratinization, this component was less present in the corresponding organoid cultures (Figures 2B and S1).
Genetic characterization of pancreatic tumour organoids
Pancreatic cancer shows genetic homogeneity on one level with mutations in KRAS, which is detected in >90% of pancreatic cancers. Activating KRAS point mutations at Codon 12, the most common in pancreatic cancer, results in constitutively active RAS protein. 42 Targeted sequencing of KRAS in pancreatic tumour organoids identified oncogenic KRAS mutations in all tumour‐derived organoid cultures (Figure 3 and Table S4). Whereas KRAS point mutations at Codon 12 were present in all organoid cultures, the allele frequency of oncogenic KRAS variants ranged from 21.0% to 100%, indicating differences in zygosity among the organoid cultures. Although mutations in the KRAS oncogene are typically heterozygous, organoid cultures PANCO‐5a and PANCO‐9b were homozygous, which has been suggested to be associated with a more aggressive tumour phenotype. 43 Of note, we have established both a heterozygous (PANCO‐9a) and a homozygous KRAS mutated (PANCO‐9b) organoid culture originating from the same tumour section from patient PANCO‐9.
Figure 3.
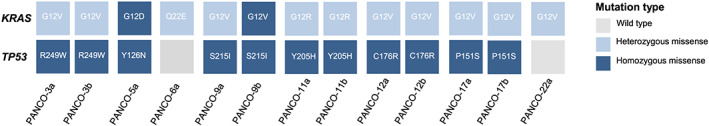
Targeted sequencing analysis of human pancreatic tumour organoids. Targeted sequencing analysis of KRAS and TP53 including the identified substitution mutation is presented for each gene. A colour legend for the type of genetic alterations is shown.
Additional well‐characterized gene mutations that predominate in pancreatic cancer include TP53, SMAD4, and CDKN2A of which TP53 is mutated in >50% of the patients. 44 Homozygous point mutations in the TP53 gene were identified in tumour‐derived organoid cultures from 6/8 patients and were detected in the known hotspots, namely in Exons 5–8 (Figure 3 and Table S4). Analysis of KRAS and TP53 mutation status of the corresponding parent tumours confirmed the presence of the expected variants found in several organoid cultures, although neoplastic cellularity in the tissue sections and/or DNA quality was not always sufficient to pick them up (Tables S5 and S6).
Cachexia‐related factors expressed by pancreatic tumour organoids
It is thought that cancer‐associated cachexia is driven by a combination of tumour‐derived factors that can directly elicit catabolism in target tissues and the interplay between these tumour factors and the immune system. Together, this results in inflammation and the generation of catabolic pro‐inflammatory factors. 45 Tumour‐derived factors previously associated with cancer cachexia mainly include pro‐inflammatory cytokines such as IL‐1, IL‐6, IL‐8, TNF‐α, LIF, MIC‐1/GDF15, and Mcp‐1. 45 To investigate whether pancreatic tumour organoids from cachectic pancreatic cancer patients expressed higher levels of these cachexia‐related factors as compared with tumour organoids from non‐cachectic patients, we first assessed their gene expression levels by quantitative PCR. Organoids were cultured for 24 h in basal medium to exclude potential effects of the two different medium compositions on gene expression. Pronounced differences were observed between the ‘no cachexia’ and ‘cachexia’ groups (Figure 4A and 4B). Unexpectedly, significantly reduced mRNA expression levels of IL‐1α and IL‐1β were detected in pancreatic tumour organoids from cachectic patients compared with non‐cachectic patients (IL‐1α: −3.8‐fold, P = 0.009, and IL‐1β: −4.7‐fold, P = 0.004) (Figure 4B). In contrast, significantly increased mRNA expression levels of IL‐8 (1.4‐fold, P = 0.01), GDF15 (2.3‐fold, P < 0.001), and LIF (1.6‐fold, P = 0.003) were detected in tumour organoids from cachectic pancreatic cancer patients. In parallel, concentrations of secreted IL‐6, IL‐8, and GDF15 were measured in tumour organoid‐derived CM (Table 2) and compared with the systemically circulating levels of these cytokines in the corresponding patients. Whereas IL‐6 was only secreted by organoid culture PANCO‐6a (2.6 pg/mL) and PANCO‐9a (5.4 pg/mL), circulating levels of IL‐6 could be measured in the plasma of all non‐cachectic and cachectic patients (7.0 ± 2.3 pg/mL vs. 15.6 ± 17.8 pg/mL, P = 0.7, respectively). In contrast, all tumour organoids secreted relatively high levels of IL‐8 and GDF15. The concentrations of these factors were significantly higher in organoids from cachectic vs. non‐cachectic patients (IL‐8: 1.3 ± 0.9 vs. 7.4 ± 14.3 ng/mL, P = 0.07, and GDF15: 1.5 ± 1.4 vs. 5.4 ± 3.6 ng/mL, P = 0.01).
Figure 4.

Levels of cachexia‐related factors produced by pancreatic tumour organoids. (A) mRNA expression of cachexia‐related genes was determined in human pancreatic tumour organoids from each individual patient. When two organoid cultures were established from one individual tumour biopsy (organoid Medium ‘a’ or Medium ‘b’), the mRNA expression of both organoid cultures are shown. Data were normalized to CYPA and B2M reference genes. (B) mRNA expression of cachexia‐related genes grouped by the cachexia status of the patients. Results are presented as mean ± SE (Mann–Whitney U test, *P < 0.05).
Table 2.
Cytokine levels in the circulation of patients and in the conditioned media of their corresponding organoid cultures
| No cachexia | Cachexia | P value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PANCO‐11 | PANCO‐12 | PANCO‐22 | Average | PANCO‐3 | PANCO‐5 | PANCO‐6 | PANCO‐9 | PANCO‐17 | Average | ||||
| IL‐6 (pg/mL) | CM | ‘a’ | ND | ND | — | ND | ND | ND | 2.6 | 5.4 | ND | 4.0 ± 2.0 | — |
| ‘b’ | ND | ND | ND | ND | — | — | ND | ND | |||||
| Plasma | ND | 8.6 | 5.4 | 7.0 ± 2.3 | 45.7 | 2.2 | 16.3 | 10.8 | 2.8 | 15.6 ± 17.8 | 0.7 | ||
| IL‐8 (ng/mL) | CM | ‘a’ | 0.7 | 2.0 | — | 1.3 ± 0.9 | 1.3 | 4.1 | 42.7 | 2.5 | 0.8 | 7.4 ± 14.3 | 0.07 |
| ‘b’ | 0.7 | 0.5 | 2.3 | 2.6 | — | — | 3.3 | 0.7 | |||||
| Plasma | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | — | ||
| GDF15 (ng/mL) | CM | ‘a’ | 1.1 | 1.0 | — | 1.5 ± 1.4 | 2.2 | 2.3 | 6.1 | 5.7 | 1.6 | 5.4 ± 3.6 | 0.01 |
| ‘b’ | 1.1 | 0.4 | 4.0 | 10.2 | — | — | 11.2 | 4.2 | |||||
| Plasma | 1.2 | 0.5 | 0.5 | 0.7 ± 0.4 | 0.2 | 0.3 | 0.9 | 1.5 | 0.7 | 0.7 ± 0.5 | 1.0 | ||
CM, conditioned medium; IL, interleukin; ND, not detectable.
Discussion
Cancer cachexia remains a challenging problem with an important impact on quality of life and response to therapy. Despite many efforts to unravel its complex biology, the translation of novel findings into effective therapeutic targets has been hampered by a lack of experimental models that closely resemble human cancer‐induced cachexia. In this study, we have generated a human pancreatic tumour organoid biobank from patients of whom the cachexia status was thoroughly assessed pre‐operatively. We demonstrated that the established organoid cultures retain characteristic malignant features of the original tumour. In addition, we showed that tumour organoids express variable levels of known cachexia‐associated factors including IL‐6, TNF‐α, IL‐8, IL‐1α, IL‐1β, Mcp‐1, GDF15, and LIF. Interestingly, the interindividual variation of the production of these cachexia‐associated factors was not necessarily in line with the cachexia status of the donor patient.
Several pancreatic tumour organoid libraries have been established since the development of the pancreatic tumour organoid model in 2015. 18 To show the validity of using these organoids as a representative model of the in vivo tumour, these studies have extensively characterized their morphology and transcriptional and genetic profile in comparison with the parental tumour. 38 , 46 , 47 In line with these studies, the pancreatic tumour organoids established in the current study recapitulate key morphological features of the original tumour and harbour common pancreatic cancer‐specific driver mutations, including KRAS and TP53. The use of these patient‐derived organoid cultures to study cancer‐induced cachexia has several advantages compared with currently used in vitro models. Organoids provide a better representation of the tissue of origin in comparison with monolayer cultured cells. In contrast to the stiff plastic surface that provides supraphysiological mechanical signals to cells cultured in 2D, the low stiffness and biological nature of the BME used in organoid culture allows physiological cell–matrix protein interactions that mimic the cellular environment in tissues. This environment supports the cells to self‐organize into 3D multicellular structures with apical–basal polarity that is not enforced by the culture surface as it is in traditional 2D monolayer cultures. In combination with the addition of a set of growth factors, organoid cultures are genetically stable in the long term and do not require immortalization for indefinite expansion. Before the methodology of generating organoids was developed, most 3D culture systems were spheroid based. Whereas spheroids better recapitulate in vivo tissue features than 2D cultures and are a simple and easy to use platform, 48 , 49 there are important differences with organoids. For example, spheroids consist of cell aggregates that develop primarily via cell‐to‐cell adhesion, while organoid formation is primarily driven by internal developmental processes that support the long‐term expansion of these structures, retaining the genetic features of the tissue of origin. 18 , 49 , 50 Patient‐derived organoid cultures also offer several benefits over genetically engineered mouse models (GEMMs) 51 , 52 and patient‐derived xenograft models, 53 , 54 which are increasingly used to study the pathogenesis of cancer cachexia. First of all, organoids have been reported to retain a wide spectrum of genetic mutations underlying pancreatic cancer, 18 in contrast to GEMMs, which usually harbour only few of the driver mutations. Second, organoids can be generated and applied in experimental studies in a timeframe of months, in contrast to the development of GEMMs and patient‐derived xenograft models, which takes years. Furthermore, organoid models are much cheaper than mouse studies and have the potential to reduce and replace the use of animal models in translational research. Still, the possibility to transplant patient‐derived organoids into mice offers additional opportunities to study the direct and indirect effects of tumour‐derived factors involved in cancer‐induced cachexia. The potential of this approach was recently demonstrated by Boj and colleagues who showed that orthotopically transplanted neoplastic organoids recapitulate the full spectrum of tumour development and progression. 18 Importantly, transplanting organoids into mice allows detailed comparisons of the effects of human tumour cells on cachexia development and progression with corresponding comprehensive cachexia‐related patient data. Despite these benefits of using organoids, some limitations of the organoid model should be acknowledged, including the labour‐intensive aspects of maintaining organoid cultures in comparison with traditional cell culture and the higher costs of culture reagents. Current organoid models also lack other cell types present in the tumour micro‐environment, although recent developments indicate that tumour associated lymphocytes and fibroblasts can be included in the culture to provide an even better reflection of the original tumour. 55 , 56
In the past two decades, several pro‐inflammatory cytokines have been shown to be increased in the circulation of tumour‐bearing hosts, and they are now widely accepted to be major drivers of cancer cachexia. 45 Nevertheless, it is still largely unknown which tissues and cell types are responsible for the increase in circulating cytokines in cancer cachexia, despite evidence that tumours are an important source. 57 To our surprise, IL‐6, which has emerged as a critical cytokine related to the maintenance of body mass, 58 , 59 , 60 was only secreted in detectable amounts by the organoids derived from cachectic patients PANCO‐6 and PANCO‐9. In contrast, variable levels of IL‐6 were readily measured in the plasma of both cachectic and non‐cachectic patients. This suggests that tumour cells may not be the main source of circulating IL‐6 in patients with pancreatic cancer. Consistent with our data, Öhlund et al. recently showed that pancreatic tumour organoids did not secrete detectable levels of IL‐6 whereas increased IL‐6 levels were measured when tumour organoids and cancer‐associated fibroblasts were co‐cultured in a trans‐well system. 61 Besides cancer‐associated fibroblasts, peripheral blood mononuclear cells (PBMCs) might also be important sources of IL‐6 in cachectic patients. PBMCs from cachectic pancreatic cancer patients stimulated with the secretome of IL‐6 producing pancreatic cancer cell lines have been shown to produce more IL‐6 mRNA compared with PBMCs from non‐cachectic patients or healthy controls. 62 More recently, Moses et al. showed that PBMCs from cachectic patients are primed to produce significantly higher levels of IL‐6 when compared with PBMCs from healthy controls. 63 Thus, tumour‐derived factors may promote IL‐6 secretion through interactions with cells in the micro‐environment or with distant tissues or cells.
In contrast to IL‐6, high levels of IL‐8 were secreted by all pancreatic tumour organoids. This is in line with a recent study from Callaway and co‐workers who showed that IL‐8 was released at high rates from human pancreatic cancer cell lines, including primary human pancreatic cancer cells. 64 In addition, there is emerging evidence that IL‐8 is strongly associated with worse survival of cancer patients and with muscle wasting in patients with different tumour types. 65 , 66 Increased concentrations of IL‐8 have been detected in the serum of cachectic compared with non‐cachectic patients, and treatment of C2C12 myotubes with recombinant IL‐8 was sufficient to induce myotube atrophy, reinforcing its potential as a new mediator of cancer cachexia. 67 In the current study, pancreatic tumour organoids of cachectic patients showed the highest secretion of IL‐8, highlighting the potential of the organoid model to study the direct effects of tumour‐derived factors on target cells. Next to IL‐8, GDF15 has gained considerable attention in the cachexia field ever since Johnen et al. discovered dramatic weight loss in mice bearing tumours engineered to overexpress GDF15. 68 GDF15 is one of the key regulators of lean body mass and body weight, and several studies have shown increased levels of GDF15 in the serum of cachectic vs. non‐cachectic patients, 69 , 70 , 71 suggesting that GDF15 is a potential novel target for treating cancer cachexia. Interestingly, we observed that pancreatic tumour organoids of cachectic patients produced significantly more GDF15 than those of non‐cachectic patients. This suggests that pancreatic tumour cell‐derived GDF15 may directly or indirectly contribute to tissue wasting in cachexia. However, the number of organoid cultures studied here is still relatively small, and the significance of the observed increases and decreases of cachexia‐related factors in organoids from cachectic vs. non‐cachectic patients should be confirmed by expanding the biobank as well as by functional studies with relevant target cells or animal models.
In this study, we used the international consensus definition to define cancer cachexia. 3 However, several slightly different definitions of cancer cachexia have been published, 3 , 72 , 73 underscoring that diagnosing cancer cachexia remains a challenge both in research and in clinical practice. In line with this notion, we encountered difficulties in categorizing patients into the cachectic vs. the non‐cachectic group. Whereas the international consensus definition is predominantly based on body weight loss, other criteria often used to classify cachexia, such as decreased muscle strength and abnormal biochemistry (high CRP, low albumin, and low haemoglobin), 73 were heterogeneously presented by the patients in our study. Interestingly, Vanhoutte et al. compared two definitions of cachexia using different diagnostic guidelines applied on the same patient population and showed that putting the focus on weight loss overrates the assignment of the diagnosis of cachexia resulting in survival rates with less prognostic value. 74 Thorough screening of cachexia‐related parameters and factors, as was performed in the current study, will be essential to get insight into the cachexia status of the patient and will ultimately aid in the translation of in vitro findings to the actual cachectic status of the patient.
In conclusion, our systematic approach of assessing the cachexia status of a patient before establishing and characterizing tumour organoid cultures has generated an organoid biobank which has the potential to be a valuable tool for increasing our understanding of the mechanisms driving human cancer‐induced cachexia. The organoid biobank will be made available for academic research upon reasonable request to support the use of human pre‐clinical models in translational research. Our current research focuses on the functional impact of organoid‐derived factors on various cell types implicated in the pathogenesis of cachexia using in vitro as well as mouse models. These experiments could be the prelude to proteomics‐based and genomics‐based identification of tumour factors that drive key processes underlying cachexia‐related metabolic changes. In addition, our ongoing expansion of the number of organoid cultures established from well‐phenotyped patients will enable more robust analyses of the differences in expression of cachexia‐related factors by organoids from cachectic vs. non‐cachectic patients.
Conflict of interest
None declared.
Funding
R.D.W. Vaes is supported as a PhD candidate by the NUTRIM Graduate Programme.
Supporting information
Table S1: Organoid growth medium
Table S2: Composition of smMIPs panel*
Table S3: qPCR human primers
Table S4: smMIPs data
Table S5: KRAS mutation analysis of parent tumours in relation to KRAS mutation status of organoid cultures
Table S6: TP53 mutation analysis of parent tumours in relation to TP53 mutation status of organoid cultures
Figure S1: Morphological characterization of pancreatic tumour organoids. Representative phase‐contrast images (scale bar = 400 μm) and haematoxylin and eosin stainings (scale bar = 100 μm) of pancreatic tumour organoids cultured for 72 h after passaging.
Video S1 – PANCO‐5a
Video S2 – PANCO‐9a
Video S3 – PANCO‐9b
Video S4 – PANCO‐11a
Video S5 – PANCO‐11b
Video S6 – PANCO‐12a
Video S7 – PANCO‐12b
Video S8 – PANCO‐17a
Video S9 – PANCO‐17b
Video S10 – PANCO‐22b
Acknowledgements
We thank Cathy van Himbeeck and Annemarie van Bijnen from the Department of Surgery (Maastricht University) for their help with the laboratory analyses. We acknowledge the excellent contribution of Wanwisa van Dijk and Arthur van den Wijngaard from the Department of Clinical Genetics (Maastricht University Medical Center, MUMC+) to the mutation analyses. Also, we thank the Surgery, Anesthesiology, and Pathology teams of the MUMC+ for their assistance in taking biopsies and blood samples. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle. 75
Vaes R. D. W., van Dijk D. P. J., Welbers T. T. J., Blok M. J., Aberle M. R., Heij L., Boj S. F., Olde Damink S. W. M., and Rensen S. S. (2020) Generation and initial characterization of novel tumour organoid models to study human pancreatic cancer‐induced cachexia, Journal of Cachexia, Sarcopenia and Muscle, 11, 1509–1524, 10.1002/jcsm.12627
[Correction added on 5 November 2020, after first online publication: The supplementary video files were previously incorrect and have been replaced in this current version.]
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 3. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 4. Mueller TC, Bachmann J, Prokopchuk O, Friess H, Martignoni ME. Molecular pathways leading to loss of skeletal muscle mass in cancer cachexia—can findings from animal models be translated to humans? BMC Cancer 2016;16:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013;10:90–99. [DOI] [PubMed] [Google Scholar]
- 6. Mueller TC, Burmeister MA, Bachmann J, Martignoni ME. Cachexia and pancreatic cancer: are there treatment options? World J Gastroenterol 2014;20:9361–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dev R, Wong A, Hui D, Bruera E. The evolving approach to management of cancer cachexia. Oncology (Williston Park) 2017;31:23–32. [PubMed] [Google Scholar]
- 8. Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 2004;114:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonetto A, Rupert JE, Barreto R, Zimmers TA. The colon‐26 carcinoma tumor‐bearing mouse as a model for the study of cancer cachexia. J Vis Exp 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown JL, Lee DE, Rosa‐Caldwell ME, Brown LA, Perry RA, Haynie WS, et al. Protein imbalance in the development of skeletal muscle wasting in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 2018;9:987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Judge SM, Wu CL, Beharry AW, Roberts BM, Ferreira LF, Kandarian SC, et al. Genome‐wide identification of FoxO‐dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 2014;14:997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour‐derived PTH‐related protein triggers adipose tissue browning and cancer cachexia. Nature 2014;513:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delitto D, Judge SM, Delitto AE, Nosacka RL, Rocha FG, DiVita BB, et al. Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget 2017;8:1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parajuli P, Kumar S, Loumaye A, Singh P, Eragamreddy S, Nguyen TL, et al. Twist1 activation in muscle progenitor cells causes muscle loss akin to cancer cachexia. Dev Cell 2018;45:712–725, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Togashi Y, Kogita A, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett 2015;356:819–827. [DOI] [PubMed] [Google Scholar]
- 16. Baker BM, Chen CS. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci 2012;125:3015–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ben‐David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018;560:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clevers H. Modeling development and disease with organoids. Cell 2016;165:1586–1597. [DOI] [PubMed] [Google Scholar]
- 20. Lancaster MA, Huch M. Disease modelling in human organoids. Dis Model Mech 2019;12:dmm039347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 2018;172:373–386, e10. [DOI] [PubMed] [Google Scholar]
- 22. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015;161:933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yan HHN, Siu HC, Law S, Ho SL, Yue SSK, Tsui WY, et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 2018;23:882–897, e11. [DOI] [PubMed] [Google Scholar]
- 24. Ponz‐Sarvise M, Corbo V, Tiriac H, Engle DD, Frese KK, Oni TE, et al. Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin Cancer Res 2019;25:6742–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scognamiglio G, De Chiara A, Parafioriti A, Armiraglio E, Fazioli F, Gallo M, et al. Patient‐derived organoids as a potential model to predict response to PD‐1/PD‐L1 checkpoint inhibitors. Br J Cancer 2019;121:979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weeber F, Ooft SN, Dijkstra KK, Voest EE. Tumor organoids as a pre‐clinical cancer model for drug discovery. Cell Chem Biol 2017;24:1092–1100. [DOI] [PubMed] [Google Scholar]
- 27. Frisancho AR. New norms of upper limb fat and muscle areas for assessment of nutritional status. Am J Clin Nutr 1981;34:2540–2545. [DOI] [PubMed] [Google Scholar]
- 28. Dodds RM, Syddall HE, Cooper R, Benzeval M, Deary IJ, Dennison EM, et al. Grip strength across the life course: normative data from twelve British studies. PLoS ONE 2014;9:e113637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ottery FD. Definition of standardized nutritional assessment and interventional pathways in oncology. Nutrition 1996;12:S15–S19. [DOI] [PubMed] [Google Scholar]
- 30. Elia M. The ‘MUST’ report. Nutritional screening for adults: a multidisciplinary responsibility. Development and use of the ‘Malnutrition Universal Screening Tool’ (MUST) for adults. British Association for Parenteral and Enteral Nutrition (BAPEN) 2003. [Google Scholar]
- 31. Mourtzakis M, Prado CM, Lieffers JR, Reiman T, McCargar LJ, Baracos VE. A practical and precise approach to quantification of body composition in cancer patients using computed tomography images acquired during routine care. Appl Physiol Nutr Metab 2008;33:997–1006. [DOI] [PubMed] [Google Scholar]
- 32. van Dijk DP, Bakens MJ, Coolsen MM, Rensen SS, van Dam RM, Bours MJ, et al. Low skeletal muscle radiation attenuation and visceral adiposity are associated with overall survival and surgical site infections in patients with pancreatic cancer. J Cachexia Sarcopenia Muscle 2017;8:317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Broutier L, Andersson‐Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, et al. Culture and establishment of self‐renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc 2016;11:1724–1743. [DOI] [PubMed] [Google Scholar]
- 34. Hiatt JB, Pritchard CC, Salipante SJ, O'Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high‐accuracy detection of low‐frequency variation. Genome Res 2013;23:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 2009;37:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dentener MA, Bazil V, Von Asmuth EJ, Ceska M, Buurman WA. Involvement of CD14 in lipopolysaccharide‐induced tumor necrosis factor‐alpha, IL‐6 and IL‐8 release by human monocytes and alveolar macrophages. J Immunol 1993;150:2885–2891. [PubMed] [Google Scholar]
- 37. Dev R. Measuring cachexia‐diagnostic criteria. Ann Palliat Med 2019;8:24–32. [DOI] [PubMed] [Google Scholar]
- 38. Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell 2018;22:454–467, e6. [DOI] [PubMed] [Google Scholar]
- 39. Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fitzgerald TL, Hickner ZJ, Schmitz M, Kort EJ. Changing incidence of pancreatic neoplasms: a 16‐year review of statewide tumor registry. Pancreas 2008;37:134–138. [DOI] [PubMed] [Google Scholar]
- 41. Simone CG, Zuluaga Toro T, Chan E, Feely MM, Trevino JG, George TJ Jr. Characteristics and outcomes of adenosquamous carcinoma of the pancreas. Gastrointest Cancer Res 2013;6:75–79. [PMC free article] [PubMed] [Google Scholar]
- 42. Dunne RF, Hezel AF. Genetics and biology of pancreatic ductal adenocarcinoma. Hematol Oncol Clin North Am 2015;29:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krasinskas AM, Moser AJ, Saka B, Adsay NV, Chiosea SI. KRAS mutant allele‐specific imbalance is associated with worse prognosis in pancreatic cancer and progression to undifferentiated carcinoma of the pancreas. Mod Pathol 2013;26:1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2019;16:207–220. [DOI] [PubMed] [Google Scholar]
- 45. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 46. Romero‐Calvo I, Weber CR, Ray M, Brown M, Kirby K, Nandi RK, et al. Human organoids share structural and genetic features with primary pancreatic adenocarcinoma tumors. Mol Cancer Res 2019;17:70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 2018;8:1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Duval K, Grover H, Han LH, Mou Y, Pegoraro AF, Fredberg J, et al. Modeling physiological events in 2D vs. 3D cell culture. Physiology (Bethesda) 2017;32:266–277. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 49. Nunes AS, Barros AS, Costa EC, Moreira AF, Correia IJ. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol Bioeng 2019;116:206–226. [DOI] [PubMed] [Google Scholar]
- 50. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 2009;459:262–265. [DOI] [PubMed] [Google Scholar]
- 51. Ballaro R, Costelli P, Penna F. Animal models for cancer cachexia. Curr Opin Support Palliat Care 2016;10:281–287. [DOI] [PubMed] [Google Scholar]
- 52. Talbert EE, Cuitino MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, et al. Modeling human cancer‐induced cachexia. Cell Rep 2019;28:1612–1622, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gerber MH, Underwood PW, Judge SM, Delitto D, Delitto AE, Nosacka RL, et al. Local and systemic cytokine profiling for pancreatic ductal adenocarcinoma to study cancer cachexia in an era of precision medicine. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Go KL, Delitto D, Judge SM, Gerber MH, George TJ Jr, Behrns KE, et al. Orthotopic patient‐derived pancreatic cancer xenografts engraft into the pancreatic parenchyma, metastasize, and induce muscle wasting to recapitulate the human disease. Pancreas 2017;46:813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cattaneo CM, Dijkstra KK, Fanchi LF, Kelderman S, Kaing S, van Rooij N, et al. Tumor organoid‐T‐cell coculture systems. Nat Protoc 2020;15:15–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid modeling of the tumor immune microenvironment. Cell 2018;175:1972–1988, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogene 2016;5:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Carson JA, Baltgalvis KA. Interleukin‐6 as a key regulator of muscle mass during cachexia. Exerc Sport Sci Rev 2010;38:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fujimoto‐Ouchi K, Tamura S, Mori K, Tanaka Y, Ishitsuka H. Establishment and characterization of cachexia‐inducing and‐non‐inducing clones of murine colon 26 carcinoma. Int J Cancer 1995;61:522–528. [DOI] [PubMed] [Google Scholar]
- 60. Han J, Lu C, Meng Q, Halim A, Yean TJ, Wu G. Plasma concentration of interleukin‐6 was upregulated in cancer cachexia patients and was positively correlated with plasma free fatty acid in female patients. Nutr Metab (Lond) 2019;16:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ohlund D, Handly‐Santana A, Biffi G, Elyada E, Almeida AS, Ponz‐Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martignoni ME, Kunze P, Hildebrandt W, Kunzli B, Berberat P, Giese T, et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer‐related cachexia. Clin Cancer Res 2005;11:5802–5808. [DOI] [PubMed] [Google Scholar]
- 63. Moses AG, Maingay J, Sangster K, Fearon KC, Ross JA. Pro‐inflammatory cytokine release by peripheral blood mononuclear cells from patients with advanced pancreatic cancer: relationship to acute phase response and survival. Oncol Rep 2009;21:1091–1095. [DOI] [PubMed] [Google Scholar]
- 64. Callaway CS, Delitto AE, Patel R, Nosacka RL, D'Lugos AC, Delitto D, et al. IL‐8 released from human pancreatic cancer and tumor‐associated stromal cells signals through a CXCR2‐ERK1/2 axis to induce muscle atrophy. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hou YC, Wang CJ, Chao YJ, Chen HY, Wang HC, Tung HL, et al. Elevated serum interleukin‐8 level correlates with cancer‐related cachexia and sarcopenia: an indicator for pancreatic cancer outcomes. J Clin Med 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sanmamed MF, Carranza‐Rua O, Alfaro C, Onate C, Martin‐Algarra S, Perez G, et al. Serum interleukin‐8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin Cancer Res 2014;20:5697–5707. [DOI] [PubMed] [Google Scholar]
- 67. Cury SS, de Moraes D, Freire PP, de Oliveira G, Marques DVP, Fernandez GJ, et al. Tumor transcriptome reveals high expression of IL‐8 in non‐small cell lung cancer patients with low pectoralis muscle area and reduced survival. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR, et al. Tumor‐induced anorexia and weight loss are mediated by the TGF‐beta superfamily cytokine MIC‐1. Nat Med 2007;13:1333–1340. [DOI] [PubMed] [Google Scholar]
- 69. Lerner L, Hayes TG, Tao N, Krieger B, Feng B, Wu Z, et al. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J Cachexia Sarcopenia Muscle 2015;6:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, et al. MAP 3K11/GDF15 axis is a critical driver of cancer cachexia. J Cachexia Sarcopenia Muscle 2016;7:467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tsai VW, Brown DA, Breit SN. Targeting the divergent TGFbeta superfamily cytokine MIC‐1/GDF15 for therapy of anorexia/cachexia syndromes. Curr Opin Support Palliat Care 2018;12:404–409. [DOI] [PubMed] [Google Scholar]
- 72. Bozzetti F, Mariani L. Defining and classifying cancer cachexia: a proposal by the SCRINIO Working Group. JPEN J Parenter Enteral Nutr 2009;33:361–367. [DOI] [PubMed] [Google Scholar]
- 73. Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr 2008;27:793–799. [DOI] [PubMed] [Google Scholar]
- 74. Vanhoutte G, van de Wiel M, Wouters K, Sels M, Bartolomeeussen L, De Keersmaecker S, et al. Cachexia in cancer: what is in the definition? BMJ Open Gastroenterol 2016;3:e000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Organoid growth medium
Table S2: Composition of smMIPs panel*
Table S3: qPCR human primers
Table S4: smMIPs data
Table S5: KRAS mutation analysis of parent tumours in relation to KRAS mutation status of organoid cultures
Table S6: TP53 mutation analysis of parent tumours in relation to TP53 mutation status of organoid cultures
Figure S1: Morphological characterization of pancreatic tumour organoids. Representative phase‐contrast images (scale bar = 400 μm) and haematoxylin and eosin stainings (scale bar = 100 μm) of pancreatic tumour organoids cultured for 72 h after passaging.
Video S1 – PANCO‐5a
Video S2 – PANCO‐9a
Video S3 – PANCO‐9b
Video S4 – PANCO‐11a
Video S5 – PANCO‐11b
Video S6 – PANCO‐12a
Video S7 – PANCO‐12b
Video S8 – PANCO‐17a
Video S9 – PANCO‐17b
Video S10 – PANCO‐22b