

In response to the Song et al. comments, which raised concerns about the methodologies and conclusions of our work, 1 we would like to clarify some points that should not be misinterpreted. Song et al. partially analysed some of the data described in our paper to support their comments. Despite the criticism, our methodology is based on relevant literature, and we provide a detailed method description to enable replication of our study. By contrast, the letter by Song et al. did not include a methodology that would allow the readers to reproduce the data therein mentioned. The authors suggest a different parameter and cut‐off thresholds to re‐analyse some data sets used in our study, ultimately providing different results. It is known that changing parameters and analysis workflows produce diverse results.
The dilemma of selecting bioinformatics strategies or statistical criteria to analyse gene expression remains a matter of no consensus. The approach suggested by Song et al. is based on the selection of different cut‐offs for each cancer type to ensure that different types of cancers present a similar and very high number of differentially expressed genes (DEGs). The authors also suggest that our fixed criterion of DEGs cut‐off, used as a universal threshold for DEGs determination among all tested cancers, might not be suitable due to intrinsic differences between tumours with different tissue origins. To the best of our knowledge, no previous pan‐cancer study has used the criterion proposed by the authors, which includes, for some tumour types, most of the detected genes as differentially expressed. In contrast, a fixed cut‐off to determine DEGs has been widely used to compare gene expression among cancer types in pan‐cancer studies 2 , 3 , 4 , 5 and in tools for large public multi‐omics data analyses. 6 , 7 , 8 , 9
Besides, as previously demonstrated, each type of normal or tumour tissue has a specific and highly variable dimension of the secretome components 10 , 11 that cannot be shown if we assume the same number of DEGs for all tissues. Accordingly, we found that pancreatic cancer presented the highest number of upregulated secretome genes (1267) across all tumour types. This result is expected for the pancreatic tissues, which have more than 70% of the transcripts encoding secreted proteins in the normal tissues. 11 Thus, our data show that these tissue‐specific characteristics, captured by the analyses that we performed considering the same statistical cut‐off for all tumour types, may influence the prevalence and severity of the cachexia in different tumour types.
Song et al. did not mention the data sets and samples tested, as well as the statistical analysis and tools used to re‐analyse the data. Tables and figures do not have captions, which hinders a careful examination and interpretation of their study. Regarding the criticism of the statistical analyses conducted in our study, we would like to clarify that we used Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/) 9 to integrate the TCGA (The Cancer Genome Atlas) and GTEx (Genotype‐Tissue Expression) data. In this case, if the authors had used the same tool, their option for analysing the data would be the four‐way analysis of variance (ANOVA) or LIMMA. We calculated the DEGs between tumour and normal samples by four‐way ANOVA using disease state (tumour or normal), ethnicity, age, and gender as variables. ANOVA measures the strength of the relationship between the log‐transformed expression of a gene and all variables mentioned above, between tumour and normal tissues, as previously described. 4
We used the standard Benjamini and Hochberg false discovery rate (FDR) method to adjust the P value in each factor to obtain the multiple testing adjusted q value, and not fold change = 2 and P < 0.01 as statistical cut‐offs, as reported by Song et al. The DEGs between tumour and normal samples were determined by ANOVA, applying the statistical cut‐offs of log2 fold change >1 and q value <0.01. FDR is recommended in high‐throughput experiments to correct random events that falsely appear significant, exerting robust control over the error rate even when the hypotheses have dependencies. 12 Thus, despite the fixed criterion of DEGs cut‐off used by the authors, the number of DEGs identified in their analyses includes many false positives. Moreover, considering the extremely high number of DEGs (~9000) found by the authors for each tumour type, nearly all cachexia‐inducing factors (CIFs) are altered. Consequently, their data fail to identify CIFs tumour‐specific expression profiles correlated with the prevalence of weight loss and cachexia in different tumour types.
The authors also raised concerns about our survival analysis using cancer gene expression data. Access to SurvExpress database 13 has been lost since October 2019, and the platform is currently out of funds (https://tinyurl.com/y42v5ovf). However, the data sets have recently been made available for analysis. We analysed three additional pancreatic cancer data sets 14 , 15 to further validate the expression of CIFs genes as predictors of cancer survival outcomes. The CIFs gene expression profile for each of these data sets was significantly associated with poor prognosis (Figure 1A). Indeed, the prognostic value (survival) of the CIFs gene expression was further confirmed in pancreatic cancer data sets by the area under receiver operating characteristic (ROC) curves statistics (area under the curve, AUC > 0.6 for all data sets) (Figure 1B). We did not carry out the survival analysis for individual CIFs because our data indicate that cachexia outcome is related to a specific set of CIFs rather than a single factor. These results explain our option in performing the survival analysis using the SurvExpress tool, which was developed to assess the prognostic performance of gene expression signatures.
Figure 1.
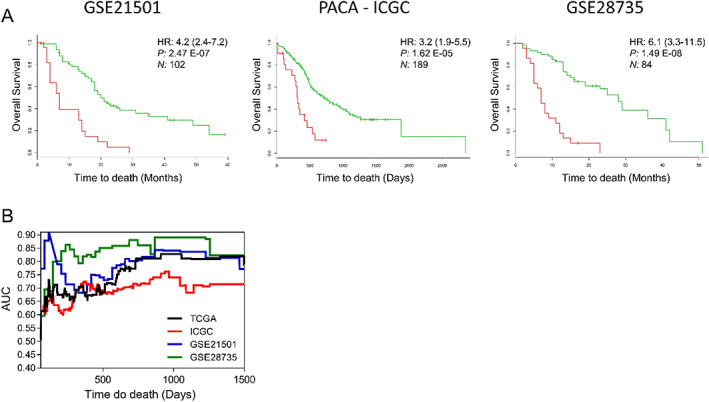
(A) Survival analysis based on the tumour gene expression of 25 cachexia‐inducing factors retrieved from three data sets (GSE21501, 14 GSE28735, 15 and PACA‐ICGC https://dcc.icgc.org/releases/current/Projects/PACA-CA) of pancreatic ductal adenocarcinoma patients in the SurvExpress. 13 This web‐based tool stratified cancer patients into high‐ or low‐risk groups (red and green, respectively). The adjusted hazard ratio (HR) with corresponding 95% confidence intervals, log‐rank P value (P), and the number of patients successfully stratified (N) determined by univariate Cox regression analyses are shown on each survival Kaplan–Meier curve. (B) Analysis of the predictive accuracy of the CIFs gene expression on survival rate tested in four transcriptomic pancreatic cancer data sets (TCGA, 20 GSE21501, 14 GSE28735, 15 and PACA‐ICGC https://dcc.icgc.org/releases/current/Projects/PACA-CA). The predictive accuracy was accessed by the area under the curve (AUC) using time‐dependent receiver operating characteristic (ROC) curves analysis for censored survival data retrieved from SurvExpress tool 13 and computed using R package SurvivalROC. 21
The RNA‐Seq data from TCGA and GTEx databases constitute a rich source of information for cachexia research. Though we lack a validation set for CIFs gene expression profile, the interpretations of our transcriptomics results are clinically and biologically relevant. Previous pan‐cancer studies that used samples exclusively from the TCGA data set have provided a uniquely comprehensive, in‐depth, and interconnected understanding of several aspects of human tumours. 10 , 16 , 17 , 18 , 19 Thus, the lack of further validation does not negatively affect the value of our findings. Moreover, one must consider the broad spectrum of tumours that were compared by robust statistical analysis applied to integrate such big data, including 12 tumour types (4651 and 2737 tumour and normal samples, respectively).
Finally, the molecular analyses of several primary tumours and normal samples have great potential to impact cachexia research positively. The integration of multi‐omics data, including single‐cell data, brings the significant potential to understand the syndrome's complexity. However, it should be emphasized that the repertoire of different analyses of such big data might lead to different interpretations and results, as argued before. These aspects should be considered by researchers when interpreting results from systems biology studies that interrogate cachexia comprehensively.
Acknowledgements
This study was supported by the São Paulo Research Foundation—FAPESP (grants 12/13961‐6, 13/50343‐1, 14/13941‐0, and 18/19695‐2) and by the National Council for Scientific and Technological Development, CNPq (Process 311530/2019‐2 to RFC). The results published here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The authors certify that the manuscript complies with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle. 22
Freire P. P., Fernandez G. J., de Moraes D., Cury S. S., Dal Pai‐Silva M., Dos Reis P. P., Rogatto S. R., and Carvalho R. F. (2020) The authors reply: Comment on “The expression landscape of cachexia‐inducing factors in human cancers” by Freire et al Journal of Cachexia, Sarcopenia and Muscle, 11, 1854–1857, 10.1002/jcsm.12635
References
- 1. Freire PP, Fernandez GJ, Moraes D, Cury SS, Dal Pai‐Silva M, Reis PP, et al. The expression landscape of cachexia‐inducing factors in human cancers. J Cachexia Sarcopenia Muscle 2020;jcsm.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bailey MH, Tokheim C, Porta‐Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018;173:371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yu K, Chen B, Aran D, Charalel J, Yau C, Wolf DM, et al. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat Commun 2019;10:3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malod‐Dognin N, Petschnigg J, Windels SFL, Povh J, Hemingway H, Ketteler R, et al. Towards a data‐integrated cell. Nat Commun 2019;10:805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Izzi V, Lakkala J, Devarajan R, Kääriäinen A, Koivunen J, Heljasvaara R, et al. Pan‐cancer analysis of the expression and regulation of matrisome genes across 32 tumor types. Matrix Biol Plus 2019;1:100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 2020;38:675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data: Figure 1. Cancer Discov 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J, Bajari R, Andric D, Gerthoffert F, Lepsa A, Nahal‐Bose H, et al. The international cancer genome consortium data portal. Nat Biotechnol 2019;37:367–369. [DOI] [PubMed] [Google Scholar]
- 9. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45:W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson JL, Feizi A, Uhlén M, Nielsen J. A systematic investigation of the malignant functions and diagnostic potential of the cancer secretome. Cell Rep 2019;26:2622–2635.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue‐based map of the human proteome. Science 2015;347:1260419–1260419. [DOI] [PubMed] [Google Scholar]
- 12. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 1995;57:289–300. [Google Scholar]
- 13. Aguirre‐Gamboa R, Gomez‐Rueda H, Martínez‐Ledesma E, Martínez‐Torteya A, Chacolla‐Huaringa R, Rodriguez‐Barrientos A, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013;8:e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA, Marron JS, et al. A six‐gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med 2010;7:e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang G, He P, Tan H, Budhu A, Gaedcke J, Ghadimi BM, et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res Off J Am Assoc Cancer Res 2013;19:4983–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang T‐H, et al. The immune landscape of cancer. Immunity 2018;48:812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saghafinia S, Mina M, Riggi N, Hanahan D, Ciriello G. Pan‐cancer landscape of aberrant DNA methylation across human tumors. Cell Rep 2018;25:1066–1080.e8. [DOI] [PubMed] [Google Scholar]
- 18. Demircioğlu D, Cukuroglu E, Kindermans M, Nandi T, Calabrese C, Fonseca NA, et al. A pan‐cancer transcriptome analysis reveals pervasive regulation through alternative promoters. Cell 2019;178:1465–1477.e17. [DOI] [PubMed] [Google Scholar]
- 19. Luo Z, Wang W, Li F, Songyang Z, Feng X, Xin C, et al. Pan‐cancer analysis identifies telomerase‐associated signatures and cancer subtypes. Mol Cancer 2019;18:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heagerty PJ, Saha‐Chaudhuri P, Saha‐Chaudhuri MP. Package ‘survivalROC’. 2013.
- 22. Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle : update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]